
HbAdrian (α1:c.251del, p.Leu84Argfs*19)—A Novel Pathogenic Variant in the α1-Globin Gene Associated with Microcytosis from the North of Iran

Abstract
:1. Introduction
2. Materials and Methods
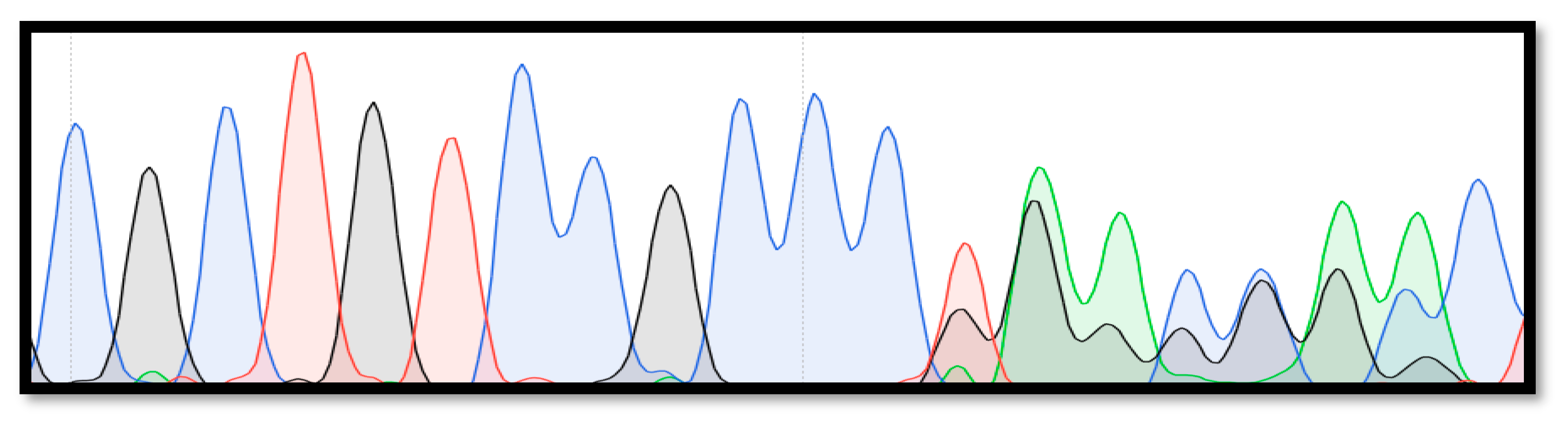
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Higgs, D.R.; Engel, J.D.; Stamatoyannopoulos, G. Thalassaemia. Lancet 2012, 379, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Weatherall, D.J.; Cappellini, M.D. Thalassaemia. Lancet 2018, 391, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Mettananda, S.; Higgs, D.R. Molecular basis and genetic modifiers of thalassemia. Hematol./Oncol. Clin. 2018, 32, 177–191. [Google Scholar] [CrossRef]
- Mettananda, S.; Gibbons, R.J.; Higgs, D.R. α-Globin as a molecular target in the treatment of β-thalassemia. Blood J. Am. Soc. Hematol. 2015, 125, 3694–3701. [Google Scholar] [CrossRef]
- Galanello, R.; Cao, A. Alpha-thalassemia. Genet. Med. 2011, 13, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Jalali, H.; Mahdavi, M.R.; Roshan, P.; Kosaryan, M.; Karami, H.; Mahdavi, M. Alpha thalassemia gene mutations in neonates from Mazandaran, Iran, 2012. Hematology 2014, 19, 192–195. [Google Scholar] [CrossRef]
- Farashi, S.; Harteveld, C.L. Molecular basis of α-thalassemia. Blood Cells Mol. Dis. 2018, 70, 43–53. [Google Scholar] [CrossRef]
- Giardine, B.; van Baal, S.; Kaimakis, P.; Riemer, C.; Miller, W.; Samara, M.; Kollia, P.; Anagnou, N.P.; Chui, D.H.; Wajcman, H.; et al. HbVar database of human hemoglobin variants and thalassemia mutations: 2007 update. Hum. Mutat. 2007, 28, 206. [Google Scholar] [CrossRef]
- Grosso, M.; Sessa, R.; Puzone, S.; Storino, M.R.; Izzo, P. Molecular basis of Thalassemia. Anemia 2012, 2012, 341–358. [Google Scholar]
- Weatherall, D.J.; Clegg, J.B. The Thalassaemia Syndromes; John Wiley & Sons: Hoboken, NJ, USA, 2008. [Google Scholar]
- Hashemieh, M.; Sessa, R.; Puzone, S.; Storino, M.R.; Izzo, P. The Iran thalassemia prevention program: Success or failure? Iran. J. Pediatr. Hematol. Oncol. 2015, 5, 161. [Google Scholar]
- Chong, S.S.; Boehm, C.D.; Higgs, D.R.; Cutting, G.R. Single-tube multiplex-PCR screen for common deletional determinants of α-thalassemia. Blood J. Am. Soc. Hematol. 2000, 95, 360–362. [Google Scholar]
- Tamaddoni, A.; Hadavi, V.; Nejad, N.H.; Khosh-Ain, A.; Siami, R.; Aghai-Meibodi, J.; Almadani, N.; Oberkanins, C.; Law, H.Y.; Najmabadi, H. α-Thalassemia mutation analyses in Mazandaran province, North Iran. Hemoglobin 2009, 33, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, M.R.; Jalali, H.; Kosaryan, M.; Roshan, P.; Mahdavi, M. β-Globin gene cluster haplotypes of Hb D-Los Angeles in Mazandaran province, Iran. Genes Genet. Syst. 2015, 90, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, M.R.; Bayat, N.; Hadavi, V.; Karami, H.; Roshan, P.; Najmabadi, H.; Rohanizadeh, H. Report of haemoglobin J-Toronto and alpha thalassemia in a family from North of Iran. JPMA-J. Pak. Med. Assoc. 2012, 62, 396. [Google Scholar] [PubMed]
- Mahdavi, M.R.; Karimi, M.; Yavarian, M.; Roshan, P.; Kosaryan, M.; Siami, R. Detection of Hb Setif in north Iran and the question of its origin: Iranian or multiethnic? Hemoglobin 2011, 35, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Jalali, H.; Rasouli, S.; Najafi, M.; Karami, H.; Mahdavi, M. A report of Hb Fontainebleau [α21 (B2) Ala> Pro] as a result of founder effect phenomenon. Gene Rep. 2020, 19, 100587. [Google Scholar] [CrossRef]
- Jalali, H.; Mahdavi, M.R.; Karami, H. Hemoglobin Daneshgah-Tehran (HBA1: c. 218A> G p. His72Arg): A Rare α1-Globin Variant from Iran. Iran. J. Pediatr. Hematol. Oncol. 2020, 10, 200–202. [Google Scholar]
- Aghajani, F.; Mahdavi, M.R.; Kosaryan, M.; Mahdavi, M.; Hamidi, M.; Jalali, H. Identification of β-globin haplotypes linked to sickle hemoglobin (Hb S) alleles in Mazandaran province, Iran. Genes Genet. Syst. 2016, 91, 311–313. [Google Scholar] [CrossRef]
- Jalali, H.; Mahdavi, M.R.; Mahdavi, M.; Abbasi, A. Hb Mazandaran (α1) α51 Gly> Cys (CE9), c. 154 GGC> TGC: A Novel Haemoglobin Variant of α1-Globin Gene. Thalass. Rep. 2022, 12, 51–54. [Google Scholar]
- Jalali, H.; Karami, H.; Mahdavi, M.R.; Mahdavi, M. Co-Inheritance of Heterozygous β0-Thalassemia with Single Functional α-Globin Gene: Challenges of Carrier Detection in Pre-Marital Screening Program for Thalassemia. Thalass. Rep. 2022, 12, 101–104. [Google Scholar] [CrossRef]
- Fucharoen, S.; Viprakasit, V. Hb H disease: Clinical course and disease modifiers. ASH Educ. Program Book 2009, 2009, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Papassotiriou, I.; Traeger-Synodinos, J.; Kanavakis, E.; Karagiorga, M.; Stamoulakatou, A.; Kattamis, C. Erythroid marrow activity and hemoglobin H levels in hemoglobin H disease. J. Pediatr. Hematol./Oncol. 1998, 20, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Kalle Kwaifa, I.; Lai, M.I.; Md Noor, S. Non-deletional alpha thalassaemia: A review. Orphanet J. Rare Dis. 2020, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Age (y) | RBC (×106/µL) | Hb (g/dL) | MCV (fl) | MCH (pg) | Hb-A (%) | Hb-A2 (%) | Hb-F (%) | |
|---|---|---|---|---|---|---|---|---|
| Subject | 29 | 5.11 | 11.9 | 74 | 23.3 | 97.4 | 2.6 | <0.5 |
| Mother | 51 | 3.83 | 10 | 74 | 23 | 97.2 | 2.8 | <0.5 |
| Father | 54 | 4.93 | 13 | 90.1 | 26.5 | 97.3 | 2.7 | <0.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jalali, H.; Karami, H.; Mahdavi, M.; Mahdavi, M.R. HbAdrian (α1:c.251del, p.Leu84Argfs*19)—A Novel Pathogenic Variant in the α1-Globin Gene Associated with Microcytosis from the North of Iran. Thalass. Rep. 2023, 13, 152-156. https://doi.org/10.3390/thalassrep13020014
Jalali H, Karami H, Mahdavi M, Mahdavi MR. HbAdrian (α1:c.251del, p.Leu84Argfs*19)—A Novel Pathogenic Variant in the α1-Globin Gene Associated with Microcytosis from the North of Iran. Thalassemia Reports. 2023; 13(2):152-156. https://doi.org/10.3390/thalassrep13020014
Chicago/Turabian StyleJalali, Hossein, Hossein Karami, Mahan Mahdavi, and Mohammad Reza Mahdavi. 2023. "HbAdrian (α1:c.251del, p.Leu84Argfs*19)—A Novel Pathogenic Variant in the α1-Globin Gene Associated with Microcytosis from the North of Iran" Thalassemia Reports 13, no. 2: 152-156. https://doi.org/10.3390/thalassrep13020014