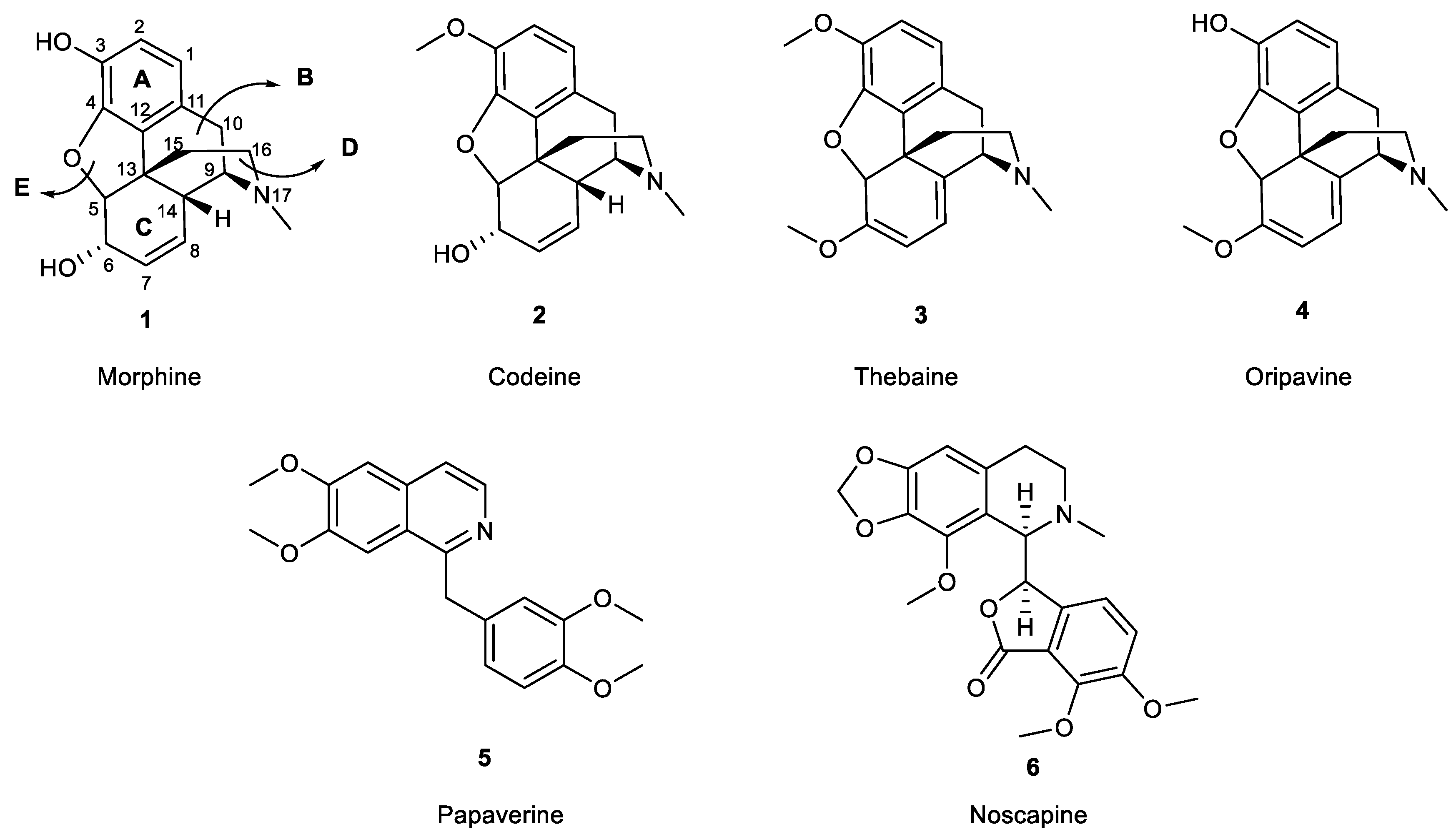
Figure 1.
Structures of the most common opium alkaloids.
Figure 1.
Structures of the most common opium alkaloids.
Figure 2.
Metabolism of morphine and codeine.
Figure 2.
Metabolism of morphine and codeine.
Scheme 1.
Reagents and conditions: (i) HX (Cl or Br), KIO3, or H2O2; (ii) diazomethane, methanol/ether (1:1); (iii) HNO3, glacial acetic acid; (iv) SnCl2, HCl; (v) NaNO2, HCl; (vi) CuCl.
Scheme 1.
Reagents and conditions: (i) HX (Cl or Br), KIO3, or H2O2; (ii) diazomethane, methanol/ether (1:1); (iii) HNO3, glacial acetic acid; (iv) SnCl2, HCl; (v) NaNO2, HCl; (vi) CuCl.
Scheme 2.
Reagents and conditions: (i) HBr, 30%, HCOOH, 30%. H2O2, 80 °C. 53%; (ii) NBA (1.08 eq.), TFAA (1.0 eq), THF, 0–15 °C, 24 h, 81%.
Scheme 2.
Reagents and conditions: (i) HBr, 30%, HCOOH, 30%. H2O2, 80 °C. 53%; (ii) NBA (1.08 eq.), TFAA (1.0 eq), THF, 0–15 °C, 24 h, 81%.
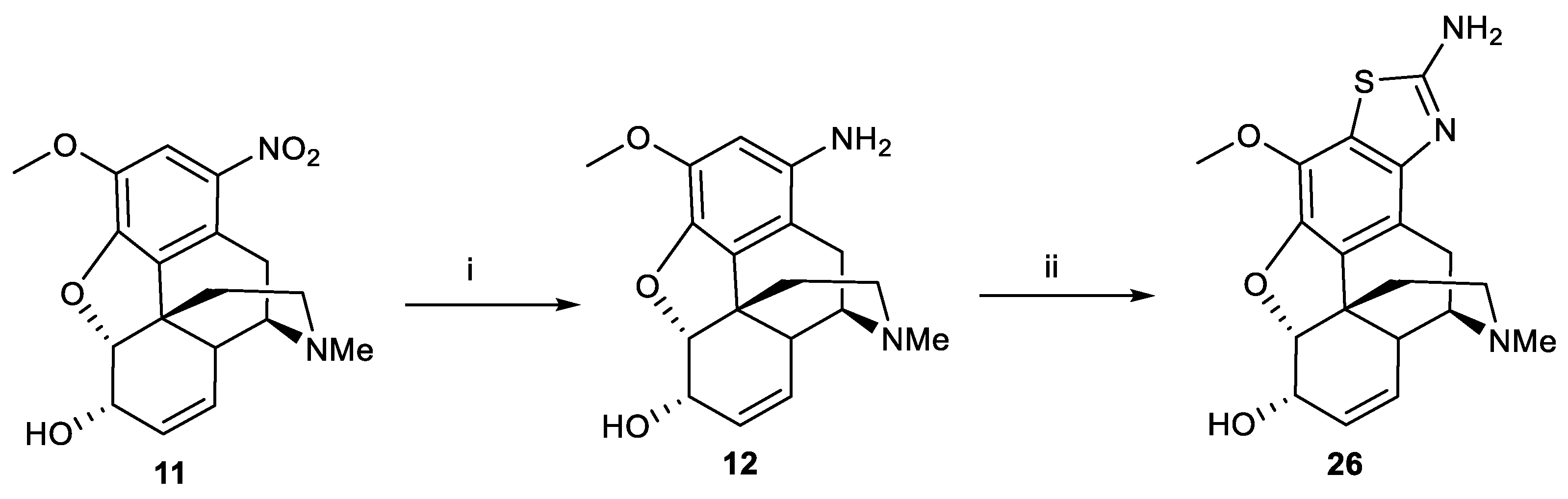
Scheme 3.
Reagents and conditions: (i) Ac2O, reflux; (ii) HNO3, glacial acetic acid, (1) 5 °C, 1 h, (2) room temperature, 1 h; (iii) 10% NaOH, ethanol, reflux, 1 h; (iv) SnCl2, HCl, r.t, 2 h; (v) 48% aqueous HBF4, EtOH, 2 M NaNO2, −15 °C to r.t; (vi) MgO, 170 °C, 1 h, in vacuo.
Scheme 3.
Reagents and conditions: (i) Ac2O, reflux; (ii) HNO3, glacial acetic acid, (1) 5 °C, 1 h, (2) room temperature, 1 h; (iii) 10% NaOH, ethanol, reflux, 1 h; (iv) SnCl2, HCl, r.t, 2 h; (v) 48% aqueous HBF4, EtOH, 2 M NaNO2, −15 °C to r.t; (vi) MgO, 170 °C, 1 h, in vacuo.
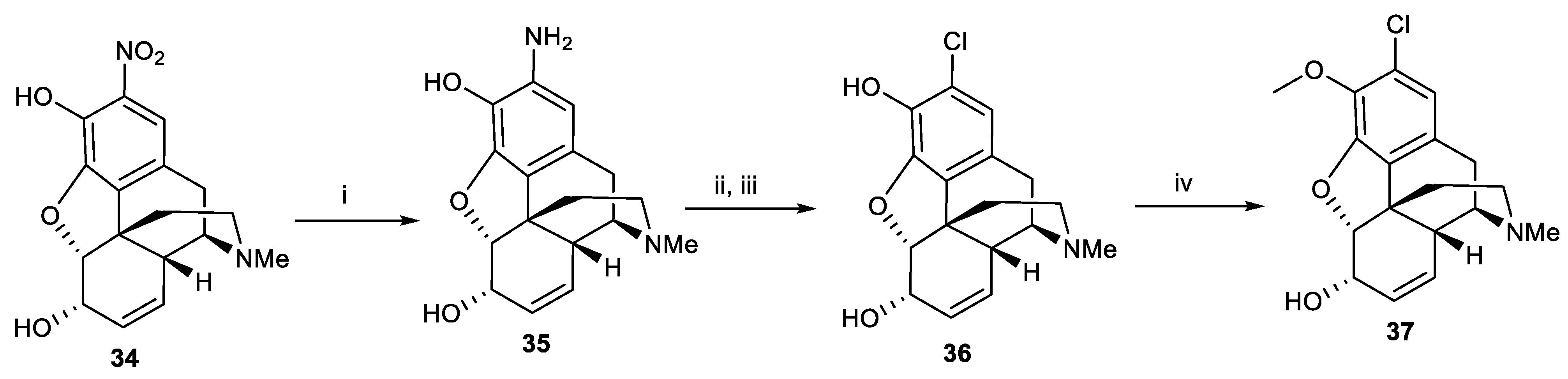
Scheme 4.
Reagents and conditions: (i) NaI, chloramine T, 0.1 M hydrochloric acid; (ii) Ac2O, reflux, 4 h; (iii) BBr3, CH2Cl2, r.t, 1 h.
Scheme 4.
Reagents and conditions: (i) NaI, chloramine T, 0.1 M hydrochloric acid; (ii) Ac2O, reflux, 4 h; (iii) BBr3, CH2Cl2, r.t, 1 h.
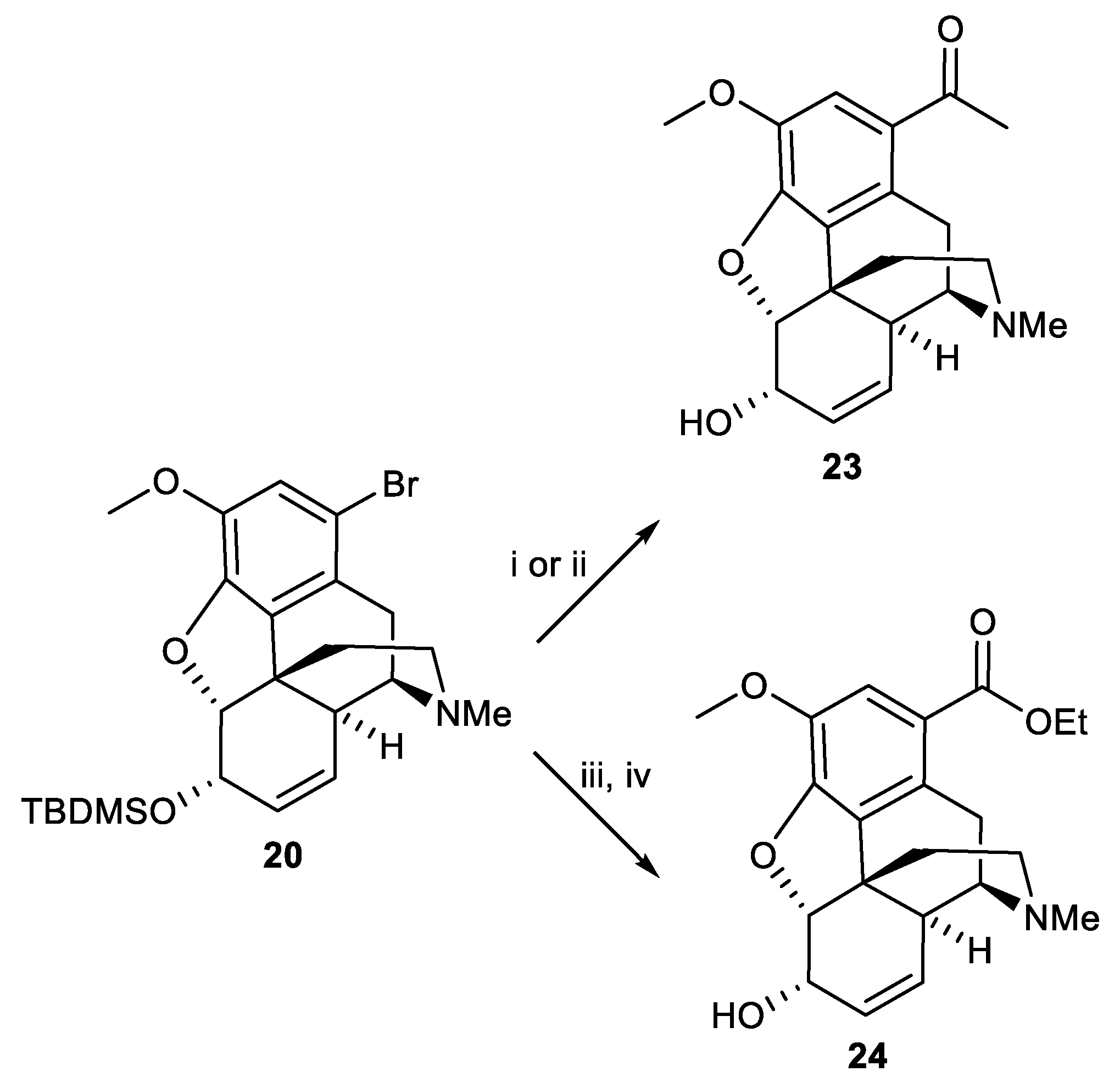
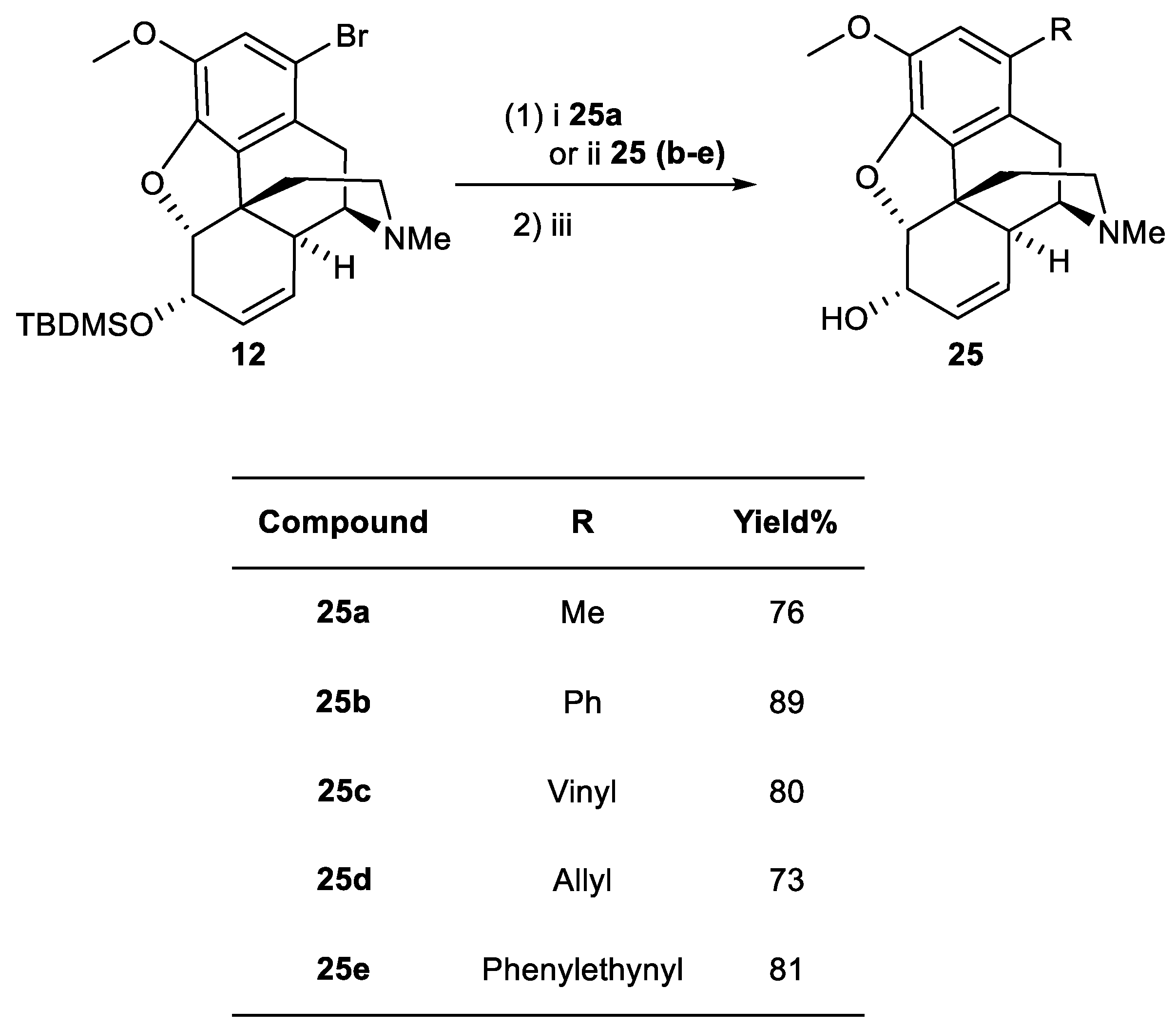
Scheme 5.
Reagents and conditions: (i) TBDMSCl (1.2 eq.), imidazole (1.2 eq.), DMF; (ii) Pd(OAc)2 (2 mol%), methyl acrylate (4 eq.), tri-o-tolylphosphine (8 mol%), NEt3 (1.4 eq.), DMF; (iii) TBAF (2 eq.), THF, r.t.
Scheme 5.
Reagents and conditions: (i) TBDMSCl (1.2 eq.), imidazole (1.2 eq.), DMF; (ii) Pd(OAc)2 (2 mol%), methyl acrylate (4 eq.), tri-o-tolylphosphine (8 mol%), NEt3 (1.4 eq.), DMF; (iii) TBAF (2 eq.), THF, r.t.
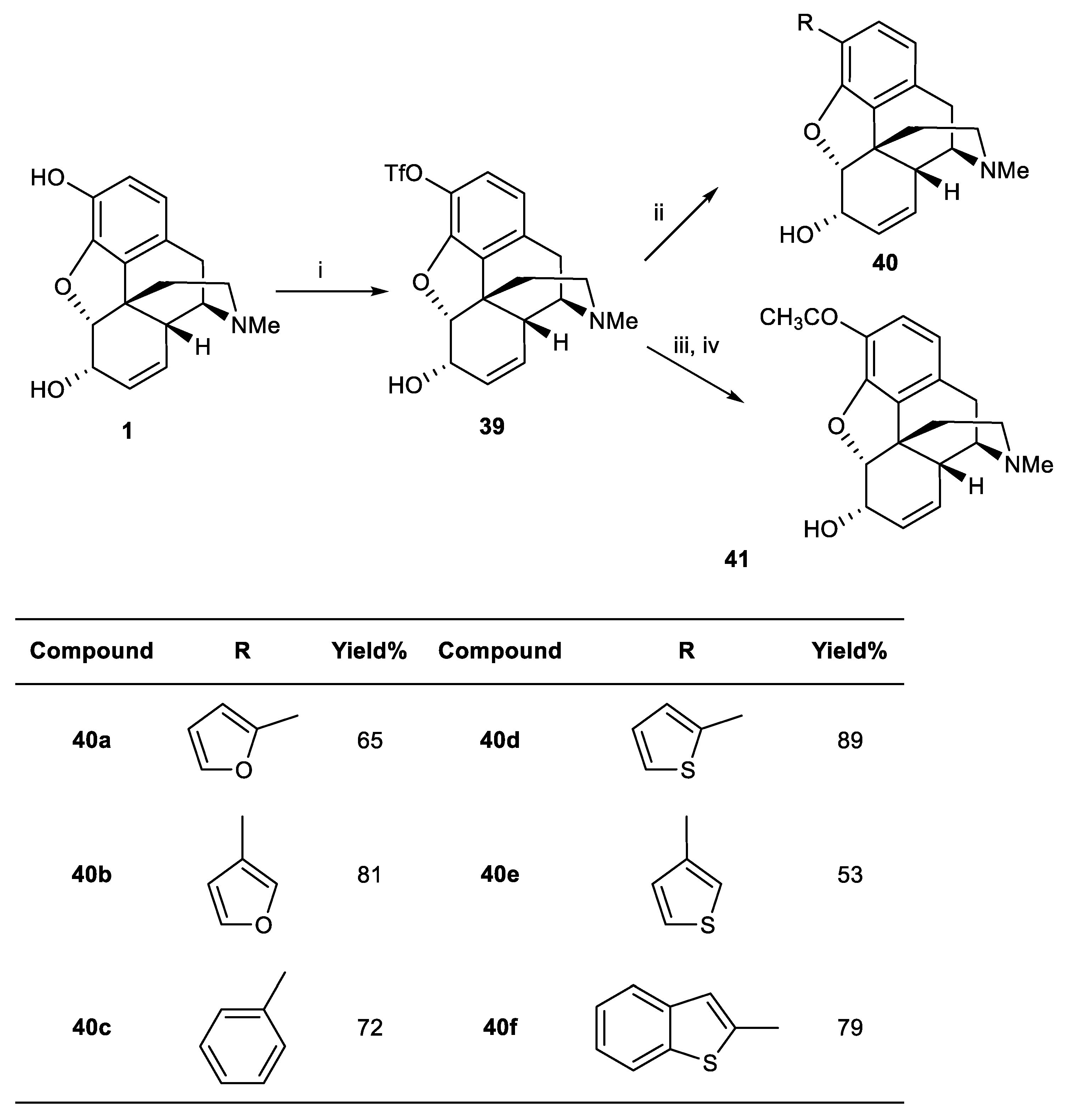
Scheme 6.
Reagents and conditions: (i) Pd(OAc)2 (2 mol%), ethyl vinyl ether (4 eq.), tri-o-tolylphosphine (8 mol%), NEt3 (1.4 eq.), DMF, then HCl (aq), 39%; (ii) Pd(OAc)2 (2 mol%), (α-ethoxyvinyl)tributyltin (4 eq.), PPh3 (8 mol%), NEt3 (1.4 eq.), DMF, then HCl, 85% (aq); (iii) PdCl2 (2 mol%), PPh3 (8 mol%), CO (3 atm), NEt3–EtOH; (iv) TBAF (2 eq.), THF.
Scheme 6.
Reagents and conditions: (i) Pd(OAc)2 (2 mol%), ethyl vinyl ether (4 eq.), tri-o-tolylphosphine (8 mol%), NEt3 (1.4 eq.), DMF, then HCl (aq), 39%; (ii) Pd(OAc)2 (2 mol%), (α-ethoxyvinyl)tributyltin (4 eq.), PPh3 (8 mol%), NEt3 (1.4 eq.), DMF, then HCl, 85% (aq); (iii) PdCl2 (2 mol%), PPh3 (8 mol%), CO (3 atm), NEt3–EtOH; (iv) TBAF (2 eq.), THF.
Scheme 7.
Reagents and conditions: (i) Pd(OAc)2 (2 mol%), tri-o-tolylphosphine (8 mol%), Me4Sn (1.2 eq.), NEt3 (1.4 eq.), DMF; (ii) Pd(OAc)2 (2 mol%), PPh3 (8 mol%), Bu3SnR (1.2 eq.), NEt3; (iii) TBAF (2 eq.), THF, r.t.
Scheme 7.
Reagents and conditions: (i) Pd(OAc)2 (2 mol%), tri-o-tolylphosphine (8 mol%), Me4Sn (1.2 eq.), NEt3 (1.4 eq.), DMF; (ii) Pd(OAc)2 (2 mol%), PPh3 (8 mol%), Bu3SnR (1.2 eq.), NEt3; (iii) TBAF (2 eq.), THF, r.t.
Scheme 8.
Reagents and conditions: (i) FSA, NaOH; (ii) KSCN, Br2, AcOH, 60 W, 130 °C, 15 min.
Scheme 8.
Reagents and conditions: (i) FSA, NaOH; (ii) KSCN, Br2, AcOH, 60 W, 130 °C, 15 min.
Scheme 9.
Reagents and conditions: (i) Bi(NO3)3·5H2O, CH3COOH, r.t, 6 h, 69%; (ii) SnCl2, 35% HCl, r.t, 2 h, 86%.
Scheme 9.
Reagents and conditions: (i) Bi(NO3)3·5H2O, CH3COOH, r.t, 6 h, 69%; (ii) SnCl2, 35% HCl, r.t, 2 h, 86%.
Scheme 10.
Reagents and conditions: (i) NaNO2, HCl, NaBF4, H2O, 5–7 °C, 15 min; (ii) 2-Naphthol, NaOH, H2O, 5–7 °C, 18 h, 97%; (iii) 2-(pyridin-2-yl)acetonitrile, CH3COONa, H2O, 5–7 °C, 18 h, 67%; (iv) malononitrile, CH3COONa, H2O, 3–5 °C, 3 h; (v) N2H4·H2O, MeOH, 65 °C, 3 h, 48% (over two steps); (vi) ethyl (2-cyanoacetyl)carbamate, NaOH, H2O, 3–5 °C, 1 h; (vii) 47% (over two steps).
Scheme 10.
Reagents and conditions: (i) NaNO2, HCl, NaBF4, H2O, 5–7 °C, 15 min; (ii) 2-Naphthol, NaOH, H2O, 5–7 °C, 18 h, 97%; (iii) 2-(pyridin-2-yl)acetonitrile, CH3COONa, H2O, 5–7 °C, 18 h, 67%; (iv) malononitrile, CH3COONa, H2O, 3–5 °C, 3 h; (v) N2H4·H2O, MeOH, 65 °C, 3 h, 48% (over two steps); (vi) ethyl (2-cyanoacetyl)carbamate, NaOH, H2O, 3–5 °C, 1 h; (vii) 47% (over two steps).
Scheme 11.
Reagents and conditions: (i) Sn/HCl, FSA, or electrolytic reduction; (ii) HCl, NaNO2; (iii) CuCl; (iv) ethereal diazomethane, MeOH.
Scheme 11.
Reagents and conditions: (i) Sn/HCl, FSA, or electrolytic reduction; (ii) HCl, NaNO2; (iii) CuCl; (iv) ethereal diazomethane, MeOH.
Scheme 12.
Reagents and conditions: (i) KSCN, Br2, AcOH, 60 W, 130 °C, 15 min.
Scheme 12.
Reagents and conditions: (i) KSCN, Br2, AcOH, 60 W, 130 °C, 15 min.
Scheme 13.
Reagents and conditions: (i) N-phenyltrifluoromethanesulfonimide, CH2Cl2; (ii) RB(OH)2, Pd(PPh3)4, LiCl, 2M Na2CO3, EtOH, DME; (iii) CH2=C(OC2H5)-SnBu3, PPh3, PdCl2(PPh3)2, DMF, 2,6-di-t-butyl-4-methylphenol; (iv) THF, 1 M HCl.
Scheme 13.
Reagents and conditions: (i) N-phenyltrifluoromethanesulfonimide, CH2Cl2; (ii) RB(OH)2, Pd(PPh3)4, LiCl, 2M Na2CO3, EtOH, DME; (iii) CH2=C(OC2H5)-SnBu3, PPh3, PdCl2(PPh3)2, DMF, 2,6-di-t-butyl-4-methylphenol; (iv) THF, 1 M HCl.
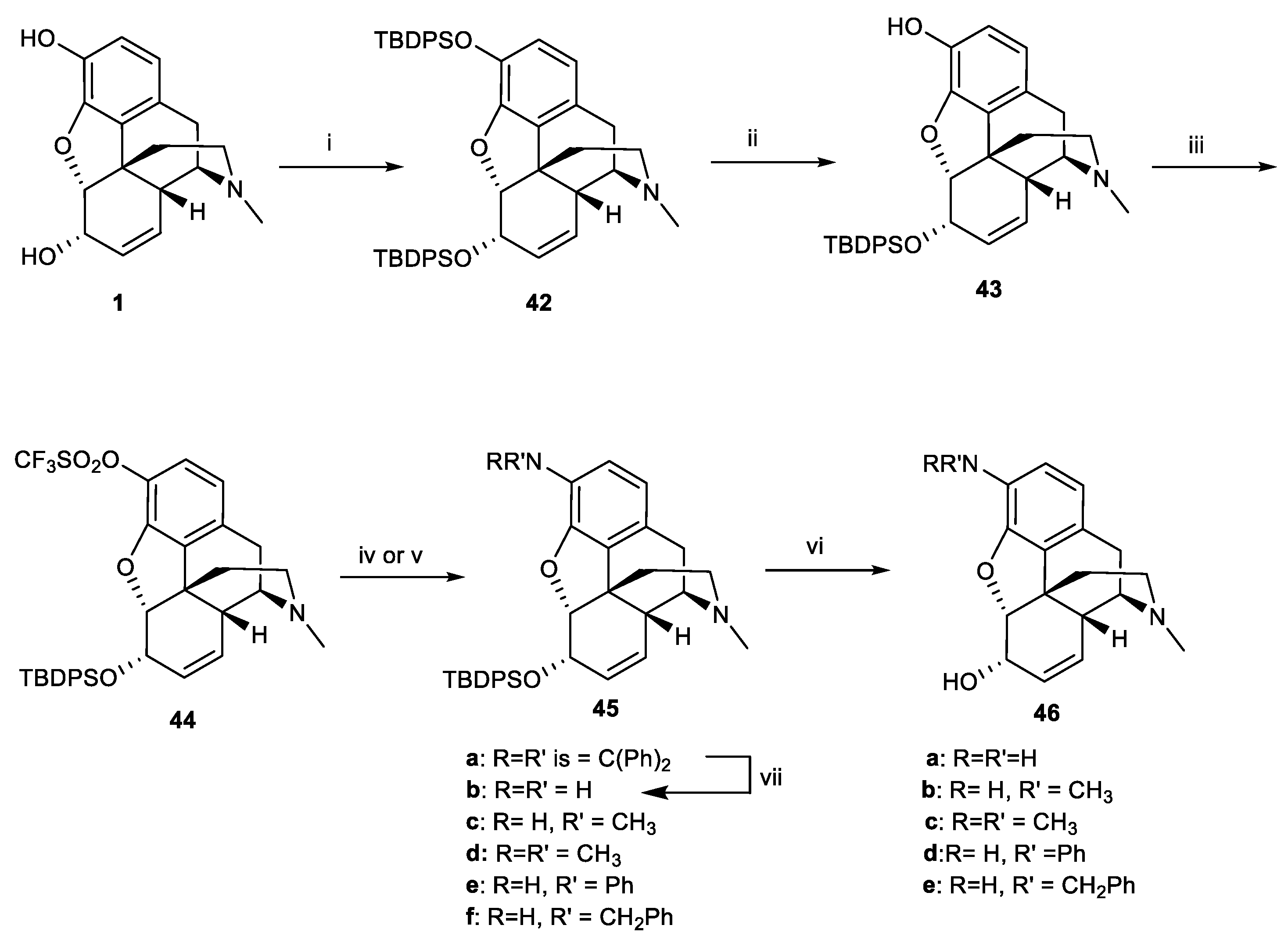
Scheme 14.
Reagents and conditions: (i) TBDPSCl, imidazole, CH2Cl2, 25 °C, 1 h, quant; (ii) 0.25 eq. TBAF, THF/H2O, 25 °C, 1 h, 84%; (iii) Tf2O, pyr, CH2Cl2, 0 °C, 1 h; (iv) RR’NH, t-BuONa, toluene, 80 °C, Pd(OAc)2, BINAP; (v) Pd2(dba)3, DPPF, 26–84%; (vi) 1.5 eq.TBAF, THF/H2O, 25 °C, 6–12 h, 79–96%; (vii) NH2OH.HCl/NaOAc, MeOH, 97%.
Scheme 14.
Reagents and conditions: (i) TBDPSCl, imidazole, CH2Cl2, 25 °C, 1 h, quant; (ii) 0.25 eq. TBAF, THF/H2O, 25 °C, 1 h, 84%; (iii) Tf2O, pyr, CH2Cl2, 0 °C, 1 h; (iv) RR’NH, t-BuONa, toluene, 80 °C, Pd(OAc)2, BINAP; (v) Pd2(dba)3, DPPF, 26–84%; (vi) 1.5 eq.TBAF, THF/H2O, 25 °C, 6–12 h, 79–96%; (vii) NH2OH.HCl/NaOAc, MeOH, 97%.
Scheme 15.
Reagents and conditions: (i) PhNTf2, Et3N; (ii) TBDMSCl, imidazole, THF; (iii) Pd(OAc)2, BINAP, Ph2C=NH, Cs2CO3; (iv) NaOAc, NH2OH.HCl; (v) KSCN, Br2, AcOH, 23.4%; (vi) TBAF, THF, 62.2%.
Scheme 15.
Reagents and conditions: (i) PhNTf2, Et3N; (ii) TBDMSCl, imidazole, THF; (iii) Pd(OAc)2, BINAP, Ph2C=NH, Cs2CO3; (iv) NaOAc, NH2OH.HCl; (v) KSCN, Br2, AcOH, 23.4%; (vi) TBAF, THF, 62.2%.
Scheme 16.
Reagents and conditions: (i) TBDMSCl (3 eq.), imidazole (5.0 eq.), THF; (ii) TBAF (1 eq.), THF, 0 °C; (iii) trifuoromethanesulfonic anhydride (1.2 eq.), 2,6-dimethylpyridine (1 eq.), DCM, 0 °C.
Scheme 16.
Reagents and conditions: (i) TBDMSCl (3 eq.), imidazole (5.0 eq.), THF; (ii) TBAF (1 eq.), THF, 0 °C; (iii) trifuoromethanesulfonic anhydride (1.2 eq.), 2,6-dimethylpyridine (1 eq.), DCM, 0 °C.
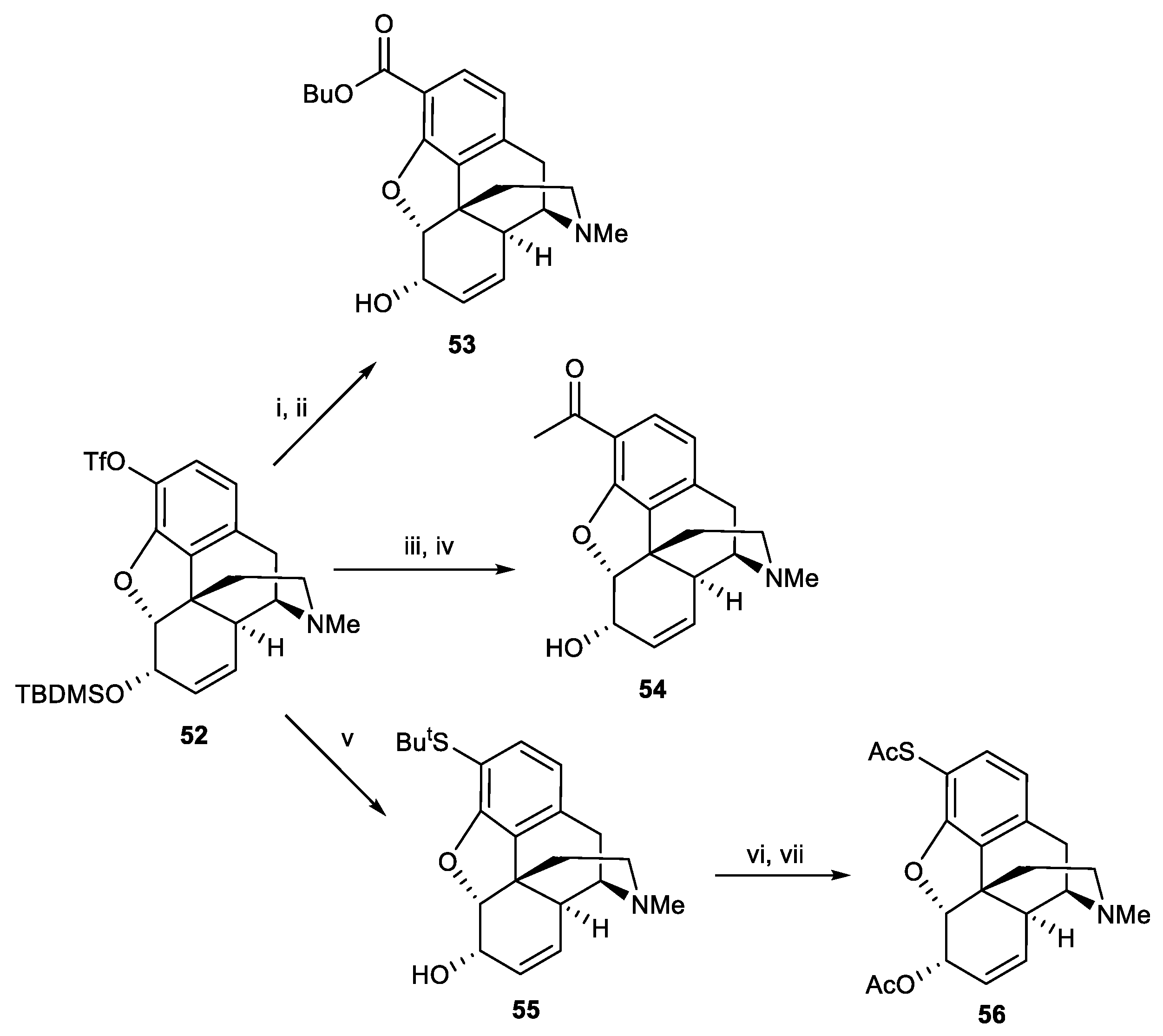
Scheme 17.
Reagents and conditions: (i) CO (3 atm), n-BuOH, Pd(OAc)2 (4 mol%), 1,3-bis(diphenylphosphino)propane (4 mol%), heat (ii) TBAF, THF, 76% (overall two step); (iii) (α-ethoxyvinyl)tributylstannane, Pd(OAc)2 (4 mol%), PPh3 (8 mol%), LiCl, DMF, heat; (iv) HCl(aq), r.t, 80% (overall two steps); (v) Pd(PPh3)4 (0.5 eq.), tributylstannyl tert-butyl sulfide (2 eq.), LiCl (3 eq.), DMF, 83%; (vi) Hg(OAc)2 (1.05 eq.), anisole, TFA, 0 °C, then H2S; (vii) acetic anhydride, NEt3, DMAP, DCM, 47% (overall two step).
Scheme 17.
Reagents and conditions: (i) CO (3 atm), n-BuOH, Pd(OAc)2 (4 mol%), 1,3-bis(diphenylphosphino)propane (4 mol%), heat (ii) TBAF, THF, 76% (overall two step); (iii) (α-ethoxyvinyl)tributylstannane, Pd(OAc)2 (4 mol%), PPh3 (8 mol%), LiCl, DMF, heat; (iv) HCl(aq), r.t, 80% (overall two steps); (v) Pd(PPh3)4 (0.5 eq.), tributylstannyl tert-butyl sulfide (2 eq.), LiCl (3 eq.), DMF, 83%; (vi) Hg(OAc)2 (1.05 eq.), anisole, TFA, 0 °C, then H2S; (vii) acetic anhydride, NEt3, DMAP, DCM, 47% (overall two step).
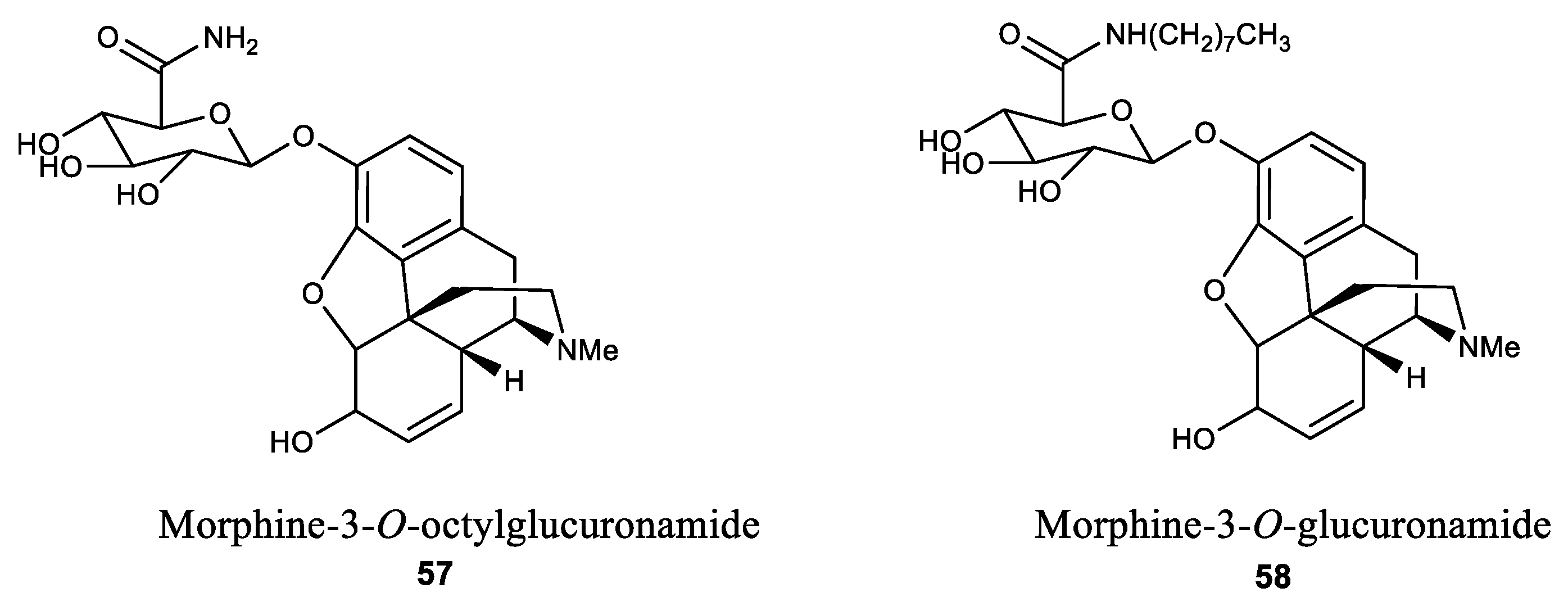
Figure 3.
Structures of morphine-3-O-octylglucuronamide and morphine-3-O-glucuronamide.
Figure 3.
Structures of morphine-3-O-octylglucuronamide and morphine-3-O-glucuronamide.
Scheme 18.
Reagents and conditions. (i) Dimethylchloroformiminium chloride, DMF; (ii) PPh3, diethyl azodicarboxylate (DEAD), or diisopropyl azodicarboxylate (DIAD), benzene or toluene, r.t, 1 h.
Scheme 18.
Reagents and conditions. (i) Dimethylchloroformiminium chloride, DMF; (ii) PPh3, diethyl azodicarboxylate (DEAD), or diisopropyl azodicarboxylate (DIAD), benzene or toluene, r.t, 1 h.
Scheme 19.
Reagents and conditions: (i) AcSH, N,N-dimethylformamide dineopentyl acetal, dry toluene, 80 °C; (ii) AcSH, 2-nitrobenzenesulfenyl chloride, PPh3, dry THF, 0 °C.
Scheme 19.
Reagents and conditions: (i) AcSH, N,N-dimethylformamide dineopentyl acetal, dry toluene, 80 °C; (ii) AcSH, 2-nitrobenzenesulfenyl chloride, PPh3, dry THF, 0 °C.
Scheme 20.
Reagents and conditions: (i) 0.2 N KOH, EtOH, N2 (96%); (ii) 2-nitrobenzenesulfenyl chloride, CH3CN, 0 °C (78%); (iii) L-Cys-OMe.HCl, EtOH, or DMF; (iv) ethyl chloroformate (3 eq), K2CO3 (anh), CH3Cl, 0 °C.
Scheme 20.
Reagents and conditions: (i) 0.2 N KOH, EtOH, N2 (96%); (ii) 2-nitrobenzenesulfenyl chloride, CH3CN, 0 °C (78%); (iii) L-Cys-OMe.HCl, EtOH, or DMF; (iv) ethyl chloroformate (3 eq), K2CO3 (anh), CH3Cl, 0 °C.
Scheme 21.
Reagents and conditions: (i) mesyl chloride, pyridine, 47.5%; (ii) aq.sodium azide, DMF, 24 h, 100 °C, 50.3%.
Scheme 21.
Reagents and conditions: (i) mesyl chloride, pyridine, 47.5%; (ii) aq.sodium azide, DMF, 24 h, 100 °C, 50.3%.
Scheme 22.
Reagents and conditions: (i) Ac2O, NaHCO3, H2O; (ii) PPh3, i-PrO2CN=NCO2i-Pr, phthalimide; (iii) NH2OH.HCl, EtOH, 55 °C; (iv) TIPSCl, imidazole, DMF; (v) NH2NH2, EtOH, 55 °C; (vi) RCOCl, NEt3, CH2Cl2; (vii) TBAF, THF; (viii) LiOH.
Scheme 22.
Reagents and conditions: (i) Ac2O, NaHCO3, H2O; (ii) PPh3, i-PrO2CN=NCO2i-Pr, phthalimide; (iii) NH2OH.HCl, EtOH, 55 °C; (iv) TIPSCl, imidazole, DMF; (v) NH2NH2, EtOH, 55 °C; (vi) RCOCl, NEt3, CH2Cl2; (vii) TBAF, THF; (viii) LiOH.
Scheme 23.
Reagents and conditions: (i) acetic anhydride, NaHCO3, H2O, r.t, 1 h; (ii) phthalimide, DIAD, Ph3P, benzene, r.t, 2h; (iii) hydrazine hydrate, EtOH; (iv) 1.1 eq. thionyl chloride, RCOOH, Et3N, CH2Cl2, r.t, 2 h; (v) Na2CO3, H2O, MeOH, r.t, 12 h.
Scheme 23.
Reagents and conditions: (i) acetic anhydride, NaHCO3, H2O, r.t, 1 h; (ii) phthalimide, DIAD, Ph3P, benzene, r.t, 2h; (iii) hydrazine hydrate, EtOH; (iv) 1.1 eq. thionyl chloride, RCOOH, Et3N, CH2Cl2, r.t, 2 h; (v) Na2CO3, H2O, MeOH, r.t, 12 h.
Figure 4.
Structure of 3-iodobenzoyl naltrexamine.
Figure 4.
Structure of 3-iodobenzoyl naltrexamine.
Scheme 24.
Reagents and conditions: (i) phthalimide, Ph3P, dry THF, DIAD in dry toluene, 16 h, r.t, 93%; (ii) hydrazine hydrate, cis-2-penten-1-ol, MeOH, 16 h, r.t, 100%; (iii) 3-iodobenzoic acid, 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU), N,N-diisopropylethylamine (DIEA), dimethyl formamide (DMF), 15 min, r.t, 76%; (iv) BBr3, DCM, 30 min, 0 °C to r.t, 55%.
Scheme 24.
Reagents and conditions: (i) phthalimide, Ph3P, dry THF, DIAD in dry toluene, 16 h, r.t, 93%; (ii) hydrazine hydrate, cis-2-penten-1-ol, MeOH, 16 h, r.t, 100%; (iii) 3-iodobenzoic acid, 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU), N,N-diisopropylethylamine (DIEA), dimethyl formamide (DMF), 15 min, r.t, 76%; (iv) BBr3, DCM, 30 min, 0 °C to r.t, 55%.
Figure 5.
Structure of compound 81.
Figure 5.
Structure of compound 81.
Scheme 25.
Reagents and conditions: (i) acetic anhydride, aq. NaHCO3, r.t, 2 h; (ii) Ph3P, phthalimide, DIAD, benzene, r.t, 1 h; (iii) ethanol/hydrazine monohydrate, heating, 3 h; (iv) hydrazine monohydrate/RANEY nickel, ethanol, r.t., 2 h; (v) acyl chloride, Et3N, DCM; (vi) K2CO3, methanol, heating, 4 h.
Scheme 25.
Reagents and conditions: (i) acetic anhydride, aq. NaHCO3, r.t, 2 h; (ii) Ph3P, phthalimide, DIAD, benzene, r.t, 1 h; (iii) ethanol/hydrazine monohydrate, heating, 3 h; (iv) hydrazine monohydrate/RANEY nickel, ethanol, r.t., 2 h; (v) acyl chloride, Et3N, DCM; (vi) K2CO3, methanol, heating, 4 h.
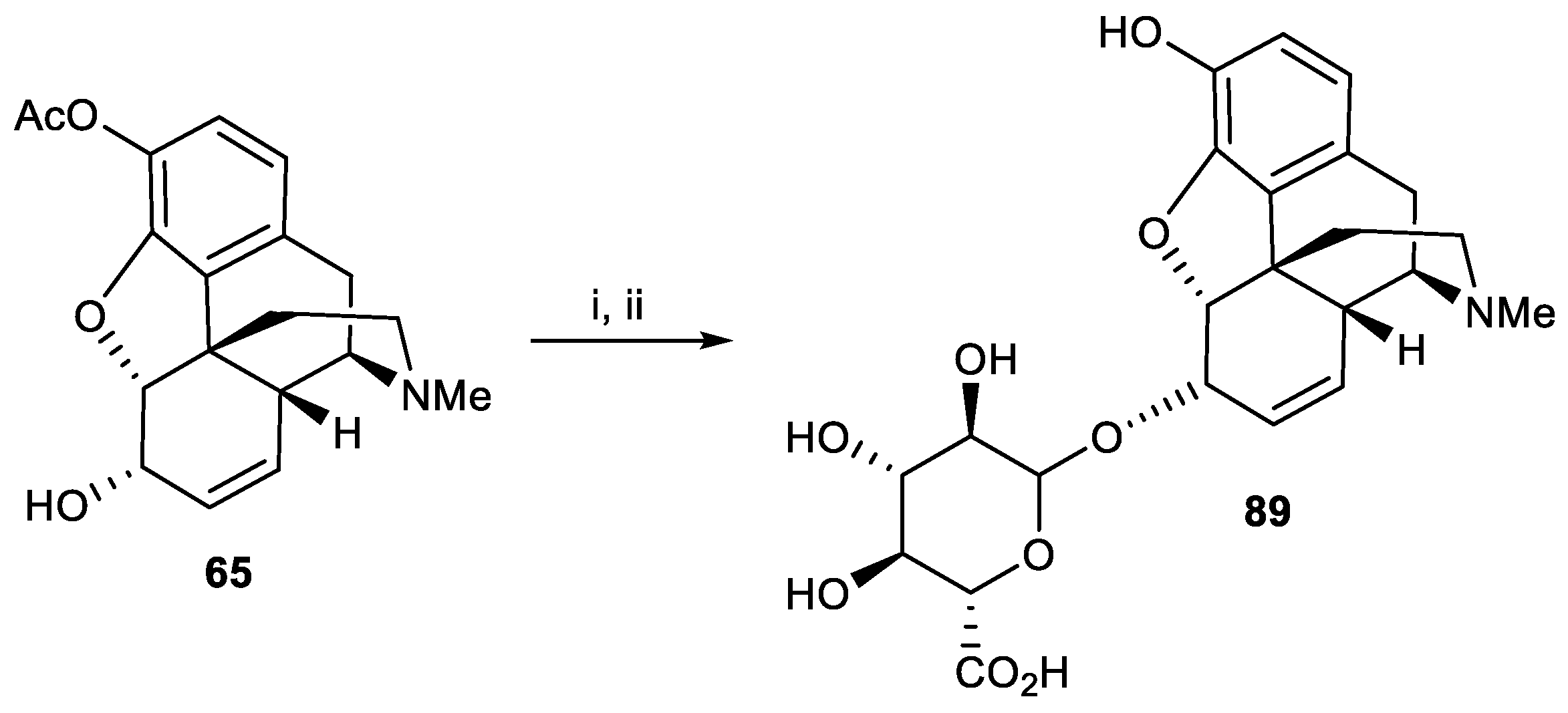
Figure 6.
Structure of potent C6-glycosylated derivatives.
Figure 6.
Structure of potent C6-glycosylated derivatives.
Scheme 26.
Reagents and conditions; (i) methyl 2,3,4-tri-O-acetyl-D-glucopyranosyluronate bromide, ZnBr2, molecular sieves, CHCl3; (ii) NaOH 5%.
Scheme 26.
Reagents and conditions; (i) methyl 2,3,4-tri-O-acetyl-D-glucopyranosyluronate bromide, ZnBr2, molecular sieves, CHCl3; (ii) NaOH 5%.
Figure 7.
Structures of nalorphine and nalodeine.
Figure 7.
Structures of nalorphine and nalodeine.
Figure 8.
N-phenethyl derivative of morphine.
Figure 8.
N-phenethyl derivative of morphine.
Scheme 27.
Reagents and conditions: (i) H2O2, acetonitrile, r.t, 4 h; (ii) FeSO4.7H2O, MeOH, −5–0 °C, 8h, 57%; (iii) propargyl bromide, K2CO3, acetone, r.t, 4 h, 86%; (iv) R-N3, CuSO4, sodium ascorbate, MeOH/DCM (1:1), r.t, 10–60 min.
Scheme 27.
Reagents and conditions: (i) H2O2, acetonitrile, r.t, 4 h; (ii) FeSO4.7H2O, MeOH, −5–0 °C, 8h, 57%; (iii) propargyl bromide, K2CO3, acetone, r.t, 4 h, 86%; (iv) R-N3, CuSO4, sodium ascorbate, MeOH/DCM (1:1), r.t, 10–60 min.
Scheme 28.
Reagents and conditions: (i) pyridine-SO3, pyridine, 55 °C; (ii) 5% MeOH, aq. NaOH, r.t, 1 h; (iii) Pd-C, 5%, H2, 50psig; (iv) CH3I/CH2Cl2, r.t, 12–18 h.
Scheme 28.
Reagents and conditions: (i) pyridine-SO3, pyridine, 55 °C; (ii) 5% MeOH, aq. NaOH, r.t, 1 h; (iii) Pd-C, 5%, H2, 50psig; (iv) CH3I/CH2Cl2, r.t, 12–18 h.
Scheme 29.
Reagents and conditions: (i) acetic anhydride, NaHCO3, H2O, r.t, 1 h; (ii) MeI, acetone, 40 °C, 4 h; (iii) pyridine-SO3, pyridine, 60 °C, 3.5 h; (iv) 20% aq. K2CO3 solution, r.t, 1 h; (v) 2-iodopropane, NaOEt, EtOH, reflux, 4 h; (vi) N,N′-dicyclohexylcarbodiimide, conc. sulfuric acid, DMF, 0 °C, 15 min.
Scheme 29.
Reagents and conditions: (i) acetic anhydride, NaHCO3, H2O, r.t, 1 h; (ii) MeI, acetone, 40 °C, 4 h; (iii) pyridine-SO3, pyridine, 60 °C, 3.5 h; (iv) 20% aq. K2CO3 solution, r.t, 1 h; (v) 2-iodopropane, NaOEt, EtOH, reflux, 4 h; (vi) N,N′-dicyclohexylcarbodiimide, conc. sulfuric acid, DMF, 0 °C, 15 min.
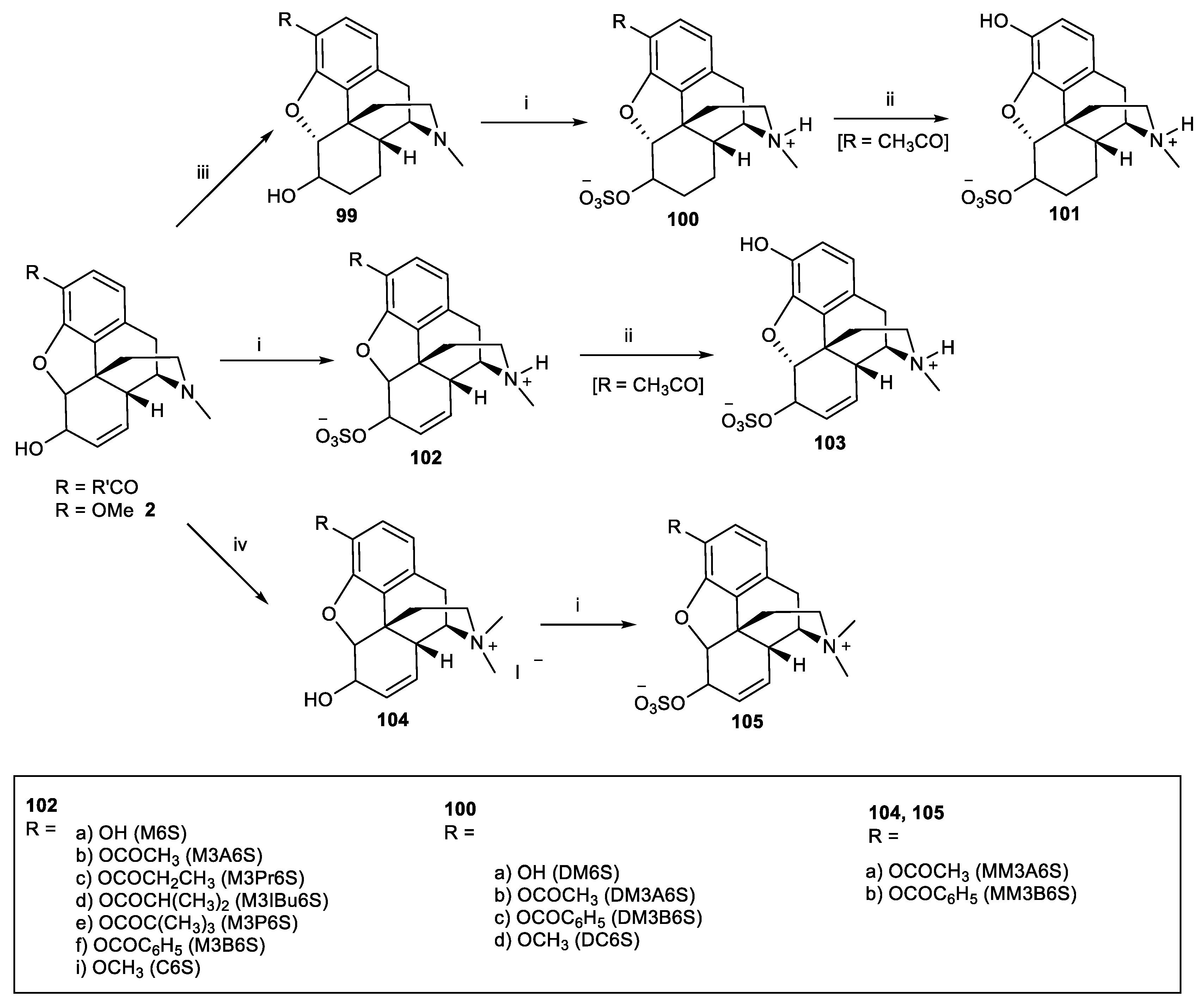
Scheme 30.
Reagents and conditions: (i) hydroxylamine hydrochloride, EtOH, 60 °C, 45 min; (ii) pyridine-SO3, pyridine, 60 °C, 3.5 h; (iii) 10% aq. NaOH, MeOH, r.t, 1 h; (iv) MeI, acetone, 40 °C, 4 h; (v) 20% aq. K2CO3 solution, r.t, 1 h.
Scheme 30.
Reagents and conditions: (i) hydroxylamine hydrochloride, EtOH, 60 °C, 45 min; (ii) pyridine-SO3, pyridine, 60 °C, 3.5 h; (iii) 10% aq. NaOH, MeOH, r.t, 1 h; (iv) MeI, acetone, 40 °C, 4 h; (v) 20% aq. K2CO3 solution, r.t, 1 h.
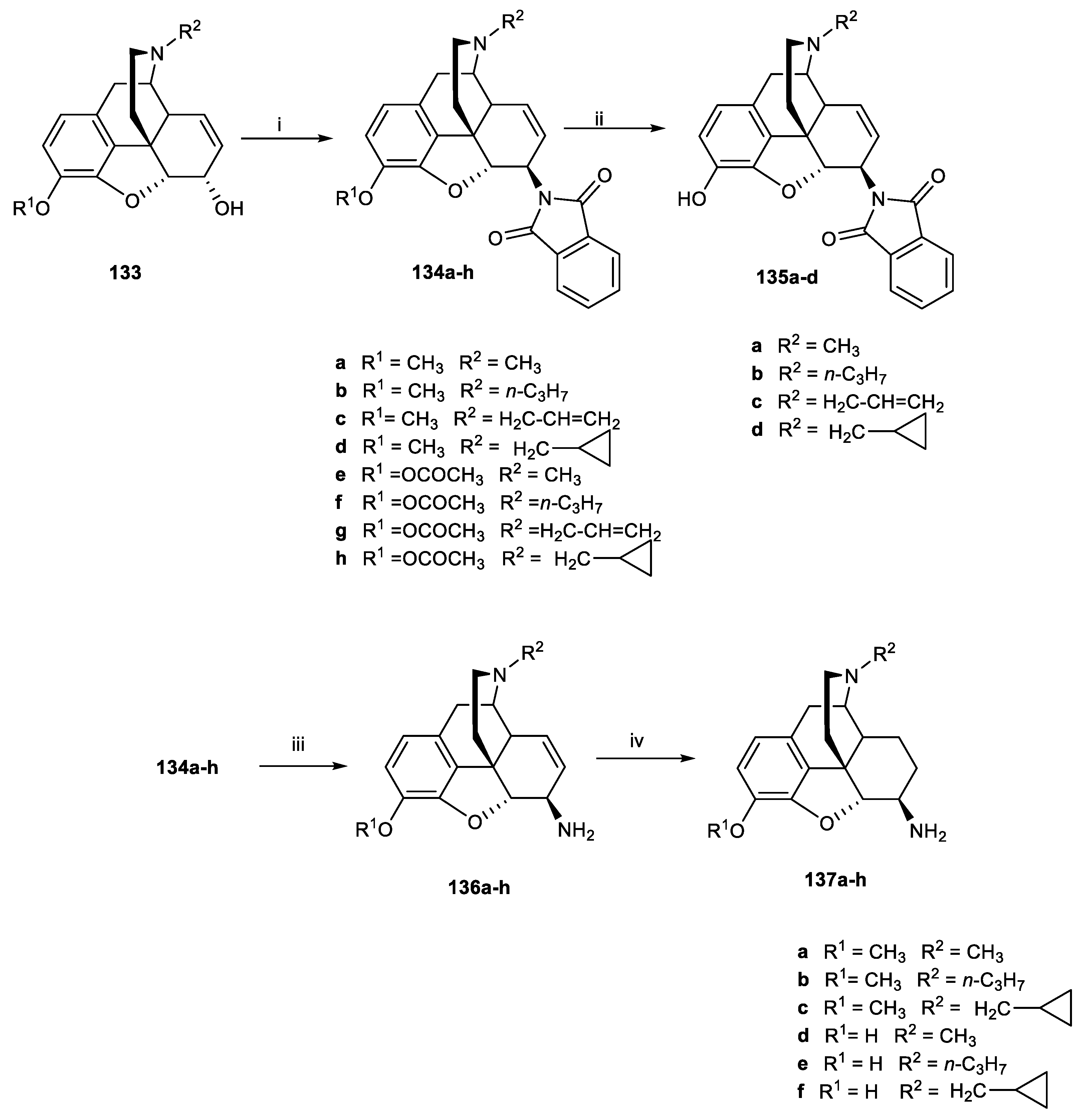
Scheme 31.
Reagents and conditions: (i) Ac2O, reflux, 4 h; (ii) (1) 1-chloroethyl chloroformate, NaHCO3, 1,2-dichloroethane, reflux, 8 h; (2) methanol, 50 °C, 1 h; (3) NaOH, ethanol, reflux, 1 h; (iii) RX (n-propylbromide or allyl bromide), NaHCO3, DMF, 95 °C, 18 h.
Scheme 31.
Reagents and conditions: (i) Ac2O, reflux, 4 h; (ii) (1) 1-chloroethyl chloroformate, NaHCO3, 1,2-dichloroethane, reflux, 8 h; (2) methanol, 50 °C, 1 h; (3) NaOH, ethanol, reflux, 1 h; (iii) RX (n-propylbromide or allyl bromide), NaHCO3, DMF, 95 °C, 18 h.
Scheme 32.
Reagents and conditions: (i) NaI, chloramine T, 0.1 M hydrochloric acid; (ii): Ac2O, reflux, 4 h; (iii): (1) α-chloroethyl chloroformate, NaHCO3, 1,2-dichloroethane; (2) methanol, 50 °C, 1 h; (3) 5% NaOH, ethanol, reflux, 1 h; (iv): RX (n-propylbromide or allyl bromide), NaHCO3, DMF, 95 °C, 18 h.
Scheme 32.
Reagents and conditions: (i) NaI, chloramine T, 0.1 M hydrochloric acid; (ii): Ac2O, reflux, 4 h; (iii): (1) α-chloroethyl chloroformate, NaHCO3, 1,2-dichloroethane; (2) methanol, 50 °C, 1 h; (3) 5% NaOH, ethanol, reflux, 1 h; (iv): RX (n-propylbromide or allyl bromide), NaHCO3, DMF, 95 °C, 18 h.
Figure 9.
N-AIkyl-3-O-alkyl derivatives of normorphine.
Figure 9.
N-AIkyl-3-O-alkyl derivatives of normorphine.
Scheme 33.
Reagents and conditions: (i) PPh3, phthalimide, diethyl azodicarboxylate, benzene; (ii) hydroxylamine hydrochloride, EtOH, 50 °C, 10 min; (iii) 98% hydrazine hydrate, EtOH; (iv) Pd/C 10%, EtOH.
Scheme 33.
Reagents and conditions: (i) PPh3, phthalimide, diethyl azodicarboxylate, benzene; (ii) hydroxylamine hydrochloride, EtOH, 50 °C, 10 min; (iii) 98% hydrazine hydrate, EtOH; (iv) Pd/C 10%, EtOH.
Scheme 34.
Reagents and conditions: (i) PPh3, succinimide, diethyl azodicarboxylate, benzene; (ii) Pd/C 10%, EtOH.
Scheme 34.
Reagents and conditions: (i) PPh3, succinimide, diethyl azodicarboxylate, benzene; (ii) Pd/C 10%, EtOH.
Scheme 35.
Reagents and conditions: (i) PPh3, benzoic acid, anhydrous benzene, diethyl azodicarboxylate, 1 h; (ii) 10% aqueous KOH, EtOH, reflux, 10 min.
Scheme 35.
Reagents and conditions: (i) PPh3, benzoic acid, anhydrous benzene, diethyl azodicarboxylate, 1 h; (ii) 10% aqueous KOH, EtOH, reflux, 10 min.
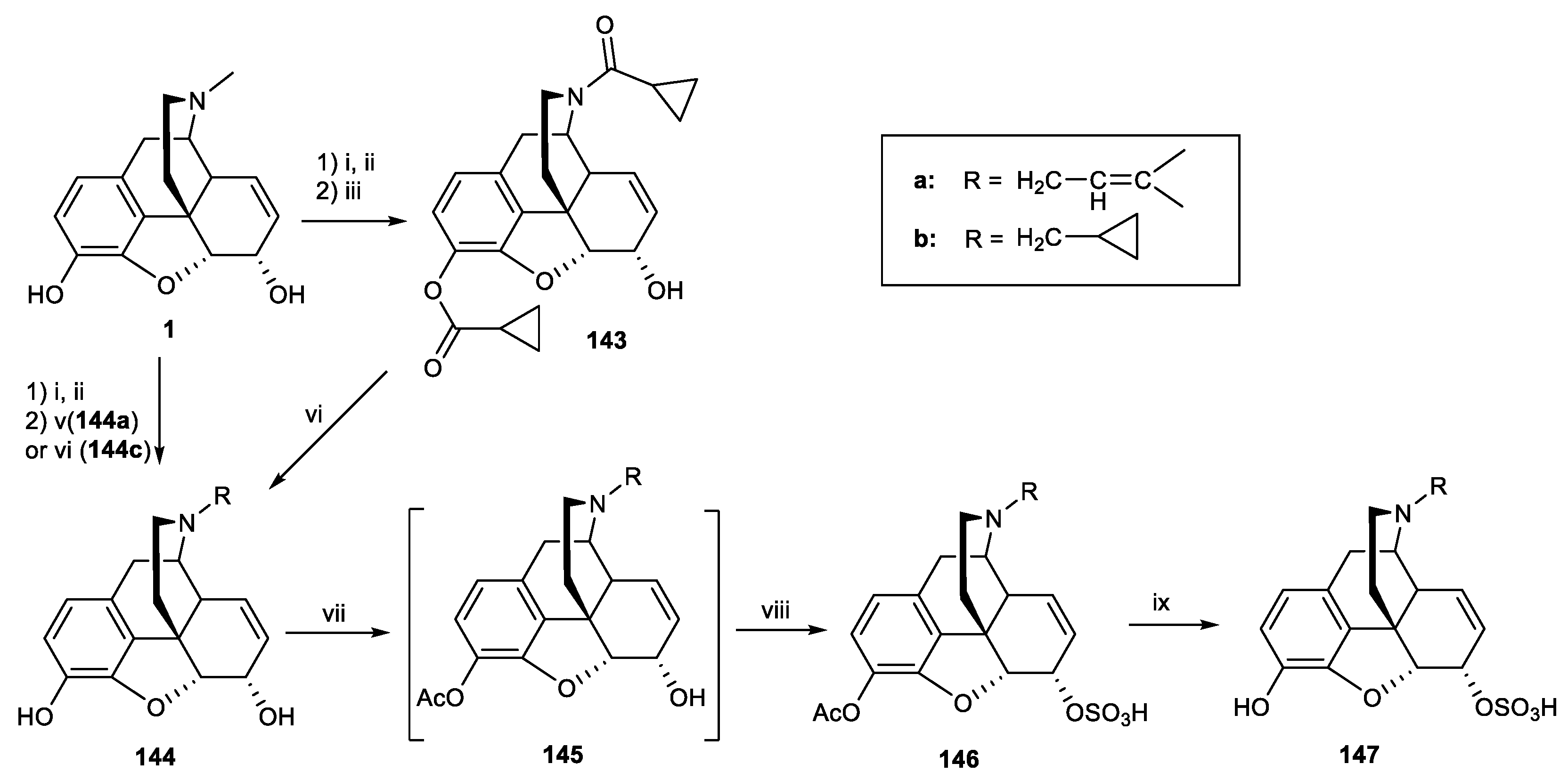
Scheme 36.
Reagents and conditions: (i) acetic anhydride; (ii) BrCN, HCl; (iii) cyclopropanecarbonyl chloride, CHCl3, triethylamine, reflux, 8 h; (iv) LiAlH4, THF, r, t; (v) dimethylallyl bromide, K2CO3, DMF, 90–95 °C, N2, 3 h; (vi) allyl bromide, Na2CO3, dry DMF, 16 h, 90 °C; (vii) Ac2O, aq.NaHCO3, 30 min; (viii) ClSO3H, dry pyridine; (ix) 5% NaOH-MeOH.
Scheme 36.
Reagents and conditions: (i) acetic anhydride; (ii) BrCN, HCl; (iii) cyclopropanecarbonyl chloride, CHCl3, triethylamine, reflux, 8 h; (iv) LiAlH4, THF, r, t; (v) dimethylallyl bromide, K2CO3, DMF, 90–95 °C, N2, 3 h; (vi) allyl bromide, Na2CO3, dry DMF, 16 h, 90 °C; (vii) Ac2O, aq.NaHCO3, 30 min; (viii) ClSO3H, dry pyridine; (ix) 5% NaOH-MeOH.
Scheme 37.
Reagents and conditions: (i) chloroethyl chloroformate, NaHCO3, 1,2-dichloroethane, 30 min, r.t, then 16 h, 85 °C; (ii) allyl bromide, Na2CO3, dry DMF, 16 h, 90 °C, 68%; (iii) phthalimide, Ph3P, dry THF, DIAD in dry toluene, 16 h, r.t, 68%; (iv) hydrazine hydrate, cis-2-penten-1-ol, MeOH, 16 h, r.t, 100%; (v) 3-iodobenzoic acid, HATU, N,N-diisopropylethylamine (DIEA), dimethyl formamide (DMF), 15 min, r.t, 70%; (vi) BBr3, DCM, 30 min, 0 °C to r.t, 50–60%; (vii) DIAD, acetonitrile, 20 h, 65 °C, then pyridine-HCl, 72 h, r.t, 54%; (viii) alkyl halide, Na2CO3, or Cs2CO3, DMF, 16 h, 90 °C.
Scheme 37.
Reagents and conditions: (i) chloroethyl chloroformate, NaHCO3, 1,2-dichloroethane, 30 min, r.t, then 16 h, 85 °C; (ii) allyl bromide, Na2CO3, dry DMF, 16 h, 90 °C, 68%; (iii) phthalimide, Ph3P, dry THF, DIAD in dry toluene, 16 h, r.t, 68%; (iv) hydrazine hydrate, cis-2-penten-1-ol, MeOH, 16 h, r.t, 100%; (v) 3-iodobenzoic acid, HATU, N,N-diisopropylethylamine (DIEA), dimethyl formamide (DMF), 15 min, r.t, 70%; (vi) BBr3, DCM, 30 min, 0 °C to r.t, 50–60%; (vii) DIAD, acetonitrile, 20 h, 65 °C, then pyridine-HCl, 72 h, r.t, 54%; (viii) alkyl halide, Na2CO3, or Cs2CO3, DMF, 16 h, 90 °C.
Table 1.
Potencies of morphine and analogues at μ, δ, and κ-Opioid receptor recognition sites labeled by [3H]DAMGO, [3H]DPDPE, and [3H]U69593, respectively, in the guinea pig brain minus cerebellum (μ and δ) and the guinea pig cerebellum (κ).
Table 1.
Potencies of morphine and analogues at μ, δ, and κ-Opioid receptor recognition sites labeled by [3H]DAMGO, [3H]DPDPE, and [3H]U69593, respectively, in the guinea pig brain minus cerebellum (μ and δ) and the guinea pig cerebellum (κ).
| IC50 (nM) |
|---|
| Compound | [3H]DAMGO (µ) | [3H]DPDPE (δ) | [3H]U69593 (κ) |
|---|
| Morphine | 8.5 | 430 | 130 |
| 40a | 2600 | 31% at 10 μM | 9500 |
| 40b | 200 | 7400 | 910 |
| 40c | 43% at 10 μM | 16% at 10 μM | 76,000 |
| 40d | 49% at 10 μM | 17% at 10 μM | 25,000 |
| 40e | 3300 | 10% at 10 μM | 15,000 |
| 40f | 41% at 10 μM | 14% at 10 μM | 126,000 |
| 41 | 640 | 7900 | 5100 |
Table 2.
Opioid receptor binding data for compounds 46a–e.
Table 2.
Opioid receptor binding data for compounds 46a–e.
| | Ki (nM ± SE) | Receptor Selectivity a |
|---|
| Compound | [3H]DAMGO (µ) | [3H]naltrindole (δ) | [3H]U69593 (κ) | µ:δ | µ:κ | κ:δ |
|---|
| Morphine | 0.88 ± 0.14 | 140 ±18 | 24 ± 2.3 | 156 | 27 | 6 |
| 46a | 53 ± 3.0 | 2400 ± 190 | 740 ± 75 | 45 | 14 | 3 |
| 46b | 63 ± 15 | 5700 ± 1100 | 2800 ± 420 | 90 | 44 | 2 |
| 46c | 240 ± 16 | 1600 ± 110 | 290 ± 8.1 | 7 | 1 | 6 |
| 46d | 59 ± 3.7 | 1500 ± 100 | 240 ± 23 | 25 | 4 | 61 |
| 46e | 33 ± 5.1 | 5500 ± 190 | 3400 ± 540 | 67 | 103 | 2 |
Table 3.
Ki value inhibition of δ, κ, and µ opioid binding to Chinese hamster ovary membranes by compound 50 a.
Table 3.
Ki value inhibition of δ, κ, and µ opioid binding to Chinese hamster ovary membranes by compound 50 a.
| | Ki ± SEM (nM) | Selectivity |
|---|
| Compound | [3H]DAMGO (µ) | [3H]naltrindole (δ) | [3H]U69593 (κ) | µ:κ | δ:κ |
|---|
| Morphine | 0.88 ± 0.14 | 140 ±18 | 24 ± 2 | 0.04 | 5.8 |
| Levorphanol | 0.21 ± 0.02 | 4.2 ± 2.3 | 2.3 ± 0.3 | 0.09 | 2 |
| 50 | 30 ± 3 | 32 ± 3 | 220 ± 10 | 0.13 | 0.14 |
Table 4.
The relative analgesic activity of azidomorphine and azidocodeine.
Table 4.
The relative analgesic activity of azidomorphine and azidocodeine.
| Compound | Hot Plate Test
ED50 (mg/kg) | Tail Flick Test
ED50 (mg/kg) |
|---|
| Morphine | 4.7 | 1.8 |
| (2.9–7.8) a | (1.08–3.15) |
| Codeine | 14 | 22 |
| (6.7–29.4) | (15.3–28.8) |
| 67a | 0.016 | 0.012 |
| (0.007–0.038) | (0.009–0.016) |
| 67b | 0.36 | 0.61 |
| (0.16–0.79) | (0.4.1–0.79) |
Table 5.
Ki inhibition values of μ, δ, and κ opioid binding to CHO membranes, and EC50 values of stimulation of [35S]GTPγS binding by amides 72a–h and 73 mediated by μ, δ, and κ opioid receptors.
Table 5.
Ki inhibition values of μ, δ, and κ opioid binding to CHO membranes, and EC50 values of stimulation of [35S]GTPγS binding by amides 72a–h and 73 mediated by μ, δ, and κ opioid receptors.
| | Ki ± SEM (nM) | EC50 |
|---|
| Compound | [3H]DAMGO (µ) | [3H]DPDPE (δ) | [3H]U69593 (κ) | µ | δ | κ |
|---|
| Morphine | 1.1 ± 0.1 | 140 ± 2 | 46.9 ± 14 | | | |
| M6G | 12.85 ± 0.95 | 160.96 ± 0.73 | 4058.75 ± 230 | 72.3 ± 26.7 | 190.35 ± 22.9 | >10K |
| 72a | 0.35 ± 0.08 | 9.5 ± 2.6 | 0.96 ± 0.2 | 1.7 ± 0.2 | 18.8 ± 2.2 | 4.8 ± 0.1 |
| 72b | 0.59 ± 0.19 | 8.89 ± 2.1 | 2.84 ± 0.96 | 4.8 ± 2.1 | 25.2 ± 3.0 | 5.0 ± 1.0 |
| 72c | 0.21 ± 0.07 | 8.96 ± 1.95 | 2.65 ± 1.25 | 2.8 ± 0.2 | 37.8 ± 1.9 | 9.8 ± 0.02 |
| 72d | 0.40 ± 0.03 | 24.95 ± 2.33 | 4.13 ± 1.21 | 5.5 ± 0.9 | 62.7 ± 24.0 | 5.3 ± 1.3 |
| 72e | 0.23 ± 0.05 | 3.39 ± 0.03 | 1.53 ± 0.2 | 2.4 ± 0.3 | 6.20 ± 0.42 | 11.22 ± 3.51 |
| 72f | 0.20 ± 0.04 | 18.0 ± 5.63 | 2.63 ± 1.1 | 1.9 ± 0.2 | 24.5 ± 3.0 | 1.2 ± 0.1 |
| 72g | 0.20 ± 0.04 | 0.94 ± 0.02 | 0.75 ± 0.2 | 0.1 ± 0.0 | 1.3 ± 0.3 | 0.03 ± 0.02 |
| 72h | 0.33 ± 0.02 | 14.42 ± 1.26 | 0.58 ± 0.17 | 6.0 ± 2.1 | 32.4 ± 2.1 | 4.5 ± 0.2 |
| 73 | 19.92 ± 4.29 | 50.78 ± 8.86 | >10K | 65.1 ± 21.8 | 74.0 ± 12.7 | 5096 |
Table 6.
Receptor binding and in vivo analgesia data of selected 6β-acylaminomorphinans (75a–d) a.
Table 6.
Receptor binding and in vivo analgesia data of selected 6β-acylaminomorphinans (75a–d) a.
| | Affinity (Ki (nM) ± SEM) | Analgesia
(ED50, mg/kg, s.c.) |
|---|
| Compound | MOR-1 | KOR-1 | DOR-1 |
|---|
| Morphine | 4.60 ± 1.81 | | | 4.96 ± 0.96 |
| 75a | 0.10 ± 0.02 | 2.90 ± 0.66 | 10.26 ± 6.76 | 3.13 ± 1.09 |
| 75b | 0.15 ± 0.03 | 1.97 ± 0.01 | 9.38 ± 1.53 | >10 |
| 75c | 0.19 ± 0.09 | 10.52 ± 0.90 | 14.52 ± 6.82 | >10 |
| 75d | 0.74 ± 0.12 | 5.43 ± 1.38 | 15.26 ± 1.74 | >10 |
| 75e | 0.12 ± 0.006 | 0.81 ± 0.09 | 5.15 ± 0.75 | >10 |
Table 7.
Binding and analgesia of 79 and 80.
Table 7.
Binding and analgesia of 79 and 80.
| | Ki (nM) ± SEM a | Tail Flick Analgesia (CI) b, ED50 [mg/kg] |
|---|
| Compound | MOR-1 | KOR-1 | DOR-1 | 6TM/E11 |
|---|
| Morphine | 4.6 ± 1.81 | | | >1000 | 4.96 ± 0.96 |
| IBNtxA | 0.11 ± 0.02 | 0.03 ± 0.001 | 0.24 ± 0.05 | 0.16 ± 0.04 | 0.39 (0.15, 0.58) |
| 79 | 1.2 ± 0.48 | 10 ± 3.5 | 49 ± 6.4 | 61 ± 7.7 | >10 |
| 80 | 0.037 ± 0.0051 | 0.21 ± 0.046 | 0.88 ± 0.08 | 27 ± 18 | 6.31 (4.6, 8.5) |
Table 8.
In vitro efficacy of the compounds 82a–d in the [35S]GTPγS assay a.
Table 8.
In vitro efficacy of the compounds 82a–d in the [35S]GTPγS assay a.
| MOR | KOR |
|---|
| Compound | EC50 (nM) | %Emax | EC50 (nM) | %Emax |
|---|
| 82a | 4.62 ± 0.28 | 114.36 ± 7.06 | nd b | nd b |
| 82b | 2.83 ± 0.41 | 113.63 ± 4.54 | nd b | nd b |
| 82c | 8.16 ± 2.64 | 114.85 ± 1.26 | nd b | nd b |
| 82d | 7.98 ± 2.35 | 117.16 ± 1.98 | 58.83 ± 7.49 | 87.88 ± 4.9 |
| DAMGO | 19 ± 7.0 | nd b | nd b | nd b |
| U50,488H | nd b | nd b | 17 ± 6.1 | nd b |
Table 9.
N-substituted normorphine derivatives.
Table 9.
N-substituted normorphine derivatives.
| Compound | | R1 | R2 | R3 |
|---|
![Pharmaceutics 15 01779 i001]() | a | CH2CH2CH3 | H | H |
| b | CH2CONH2 | H | H |
| c | CH2COOEt | H | H |
| d | CH2CH2OC6H5 | H | H |
| e | CH2CH=CH2CH2Br | H | H |
| f | CH2CHBr=CH2 | H | H |
| g | CH2CH2CH3 | CH3 | H |
| h | CH2(CH3)2 | H | H |
| i | CH2(CH3)2 | CH3 | H |
| j | CH2CH2CN | H | H |
| k | CH2CHOHCH3 | H | H |
| l | CH2CH2COOEt | H | H |
| m | CH2C(CH3)=CH2 | H | H |
| n | CH2CH(CH3)2 | CH3 | H |
| o | CH2(CH2)3CH3 | H | H |
| p | CH2C6H5 | H | H |
| q | CH2COC6H5 | H | H |
| r | CH2CH2C6H5 | H | H |
Table 10.
N-substituted dihydronormorphine derivatives.
Table 10.
N-substituted dihydronormorphine derivatives.
| Compound | | R1 | R2 | R3 |
|---|
![Pharmaceutics 15 01779 i002]() | a | CH2CH=CH2 | CH3 | H |
| b | CH2CH=CH2 | Ac | Ac |
| c | CH2CH2CH3 | CH3 | H |
| d | CH2CH2CH3 | Ac | Ac |
| e | CH2C(CH3)=CH2 | H | H |
| f | CH2CH(CH3)2 | H | H |
Table 11.
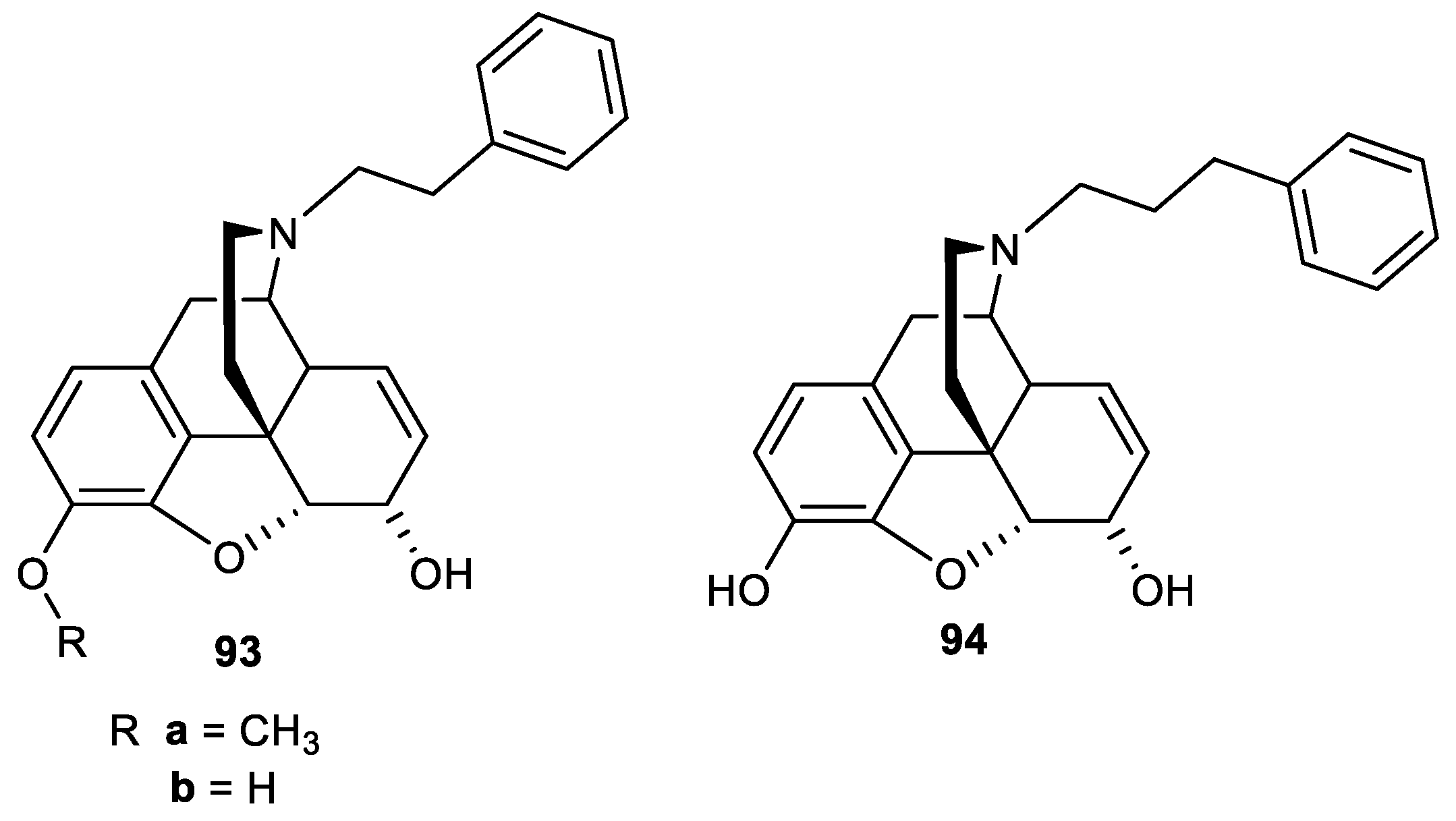
Opioid receptor binding affinity and selectivity of compounds 93a and 94 at MOP, DOP, and KOP receptors.
Table 11.
Opioid receptor binding affinity and selectivity of compounds 93a and 94 at MOP, DOP, and KOP receptors.
| | Ki ± SEM (nM) | Selectivity |
|---|
| Compound | MOP | DOP | KOP | δ:μ | κ:μ |
|---|
| Morphine | 6.556 ± 0.74 | 2176 ± 19 | 1136 ± 9 | 33 | 17 |
| 93a | 0.936 ± 0.14 | 37.06 ± 5.5 | 1076 ± 18 | 40 | 115 |
| 94 | 79.56 ± 1.1 | 8696 ± 171 | 5656 ± 24 | 11 | 7 |
Table 12.
Values of Ki and IC50 of various ligands in radioligand binding assay and ED50 by in vivo studies of synthetic compounds.
Table 12.
Values of Ki and IC50 of various ligands in radioligand binding assay and ED50 by in vivo studies of synthetic compounds.
| | Affinity | Analgesia |
|---|
| Compound | IC50 a (%95 CI b)
(nM) | Ki c (nM) | ED50 d (95% CI)
(mg/kg) |
|---|
| Codeine | 327.4 | 144.2 | 9.55 |
| (152.5 to 702.9) | (67.1 to 309.6) | (8.50 to 10.74) |
| 98d | 363.1 | 159.9 | 8.68 |
| (150.6 to 875.0) | (66.3 to 385.4) | (6.47 to 11.66) |
| 98c | 442.7 | 195.0 | 17.05 |
| (255.3 to 767.7) | (112.4 to 338.1) | (13.72 to 22.49) |
| 98a | 486.6 | 214.3 | 17.57 |
| (381.3 to 620.9) | (167.9 to 273.5) | (14.56 to 19.97) |
| 98f | 559.3 | 246.3 | 26.14 |
| (299.0 to 1046.0) | (131.7 to 460.7) | (19.82 to 34.46) |
| 98r | 536.7 | 236.4 | 30.27 |
| (349.3 to 824.6) | (153.8 to 363.2) | (19.41 to 47.21) |
| 98h | 566.3 | 249.4 | 31.17 |
| (274.7 to 1167.0) | (121.0 to 514.0) | (22.98 to 42.28) |
Table 13.
Structures of synthesized compounds.
Table 13.
Structures of synthesized compounds.
| Compound | | R1 | R2 | R3 | C7–C8 Bond |
|---|
![Pharmaceutics 15 01779 i003]() | 108 | H | SO3− | CH3 | Double |
| 109 | H | SO3− | CH3 | Single |
| 110 | CH3 | SO3− | CH3 | Double |
| 111 | CH3 | SO3− | CH3 | Single |
| 112 | iPr | SO3− | H | Double |
| 113 | Et | SO3− | H | Double |
| 114 | SO3− | SO3− | H | Double |
| 115 | CH3 | SO3− (6β) | H | Double |
| 119 | SO3− | H | H | Double |
| 120 | SO3− | H | H | Single |
| 123 | SO3− | H | CH3 | Double |
| 124 | SO3− | H | CH3 | Single |
Table 14.
Structures of 148a–l derivatives.
Table 14.
Structures of 148a–l derivatives.
| Compound | | X | R1 | R2 | C7–C8 Bond |
|---|
![Pharmaceutics 15 01779 i004]() | a | Br | H | CH2CH=CH2 | Double |
| b | Cl | H | CH2CH=CH2 | Double |
| c | Br | H | n-Pr | Double |
| d | Cl | H | n-Pr | Double |
| e | Br | CH3 | CH2CH=CH2 | Double |
| f | Cl | CH3 | CH2CH=CH2 | Double |
| g | Br | CH3 | n-Pr | Double |
| h | Cl | CH3 | n-Pr | Double |
| i | Br | H | CH2CH=CH2 | Single |
| g | Cl | H | CH2CH=CH2 | Single |
| k | Br | CH3 | CH2CH=CH2 | Single |
| l | Cl | CH3 | CH2CH=CH2 | Single |
Table 15.
Binding and Analgesia of compounds 152, 155a–b.
Table 15.
Binding and Analgesia of compounds 152, 155a–b.
| | Ki a (nM) ± SEM | Tail Flick Analgesia (CI) b, ED50 [mg/kg] |
|---|
| Compound | MOR-1 | KOR-1 | DOR-1 | 6TM/E11 |
|---|
| Morphine | 4.6 ± 1.81 | | | >1000 | 4.96 ± 0.96 |
| 152 | 0.034 ± 0.006 | 0.022 ± 0.0043 | 0.39 ± 0.0041 | 0.78 ± 0.1 | 0.53 (0.45, 0.59) |
| 155a | 0.088 ± 0.028 | 0.14 ± 0.022 | 0.13 ± 0.032 | 14 ± 3.2 | 1.3 (1.03, 2.2) |
| 155b | 0.021 ± 0.0034 | 0.0064 ± 0.002 | 0.08 ± 0.019 | 0.47 ± 0.009 | 0.33 ± 0.09 |
| IBNtxA | 0.11 ± 0.02 | 0.03 ± 0.001 | 0.24 ± 0.05 | 0.16 ± 0.04 | 0.39 (0.15, 0.58) |

















































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



