
The C-Terminus of Panusin, a Lobster β-Defensin, Is Crucial for Optimal Antimicrobial Activity and Serum Stability
 , ,
, ,  , , and
, , and
Abstract
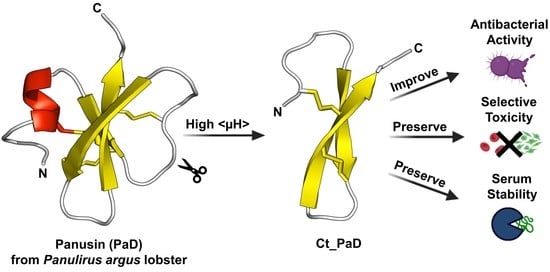
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Peptide Synthesis
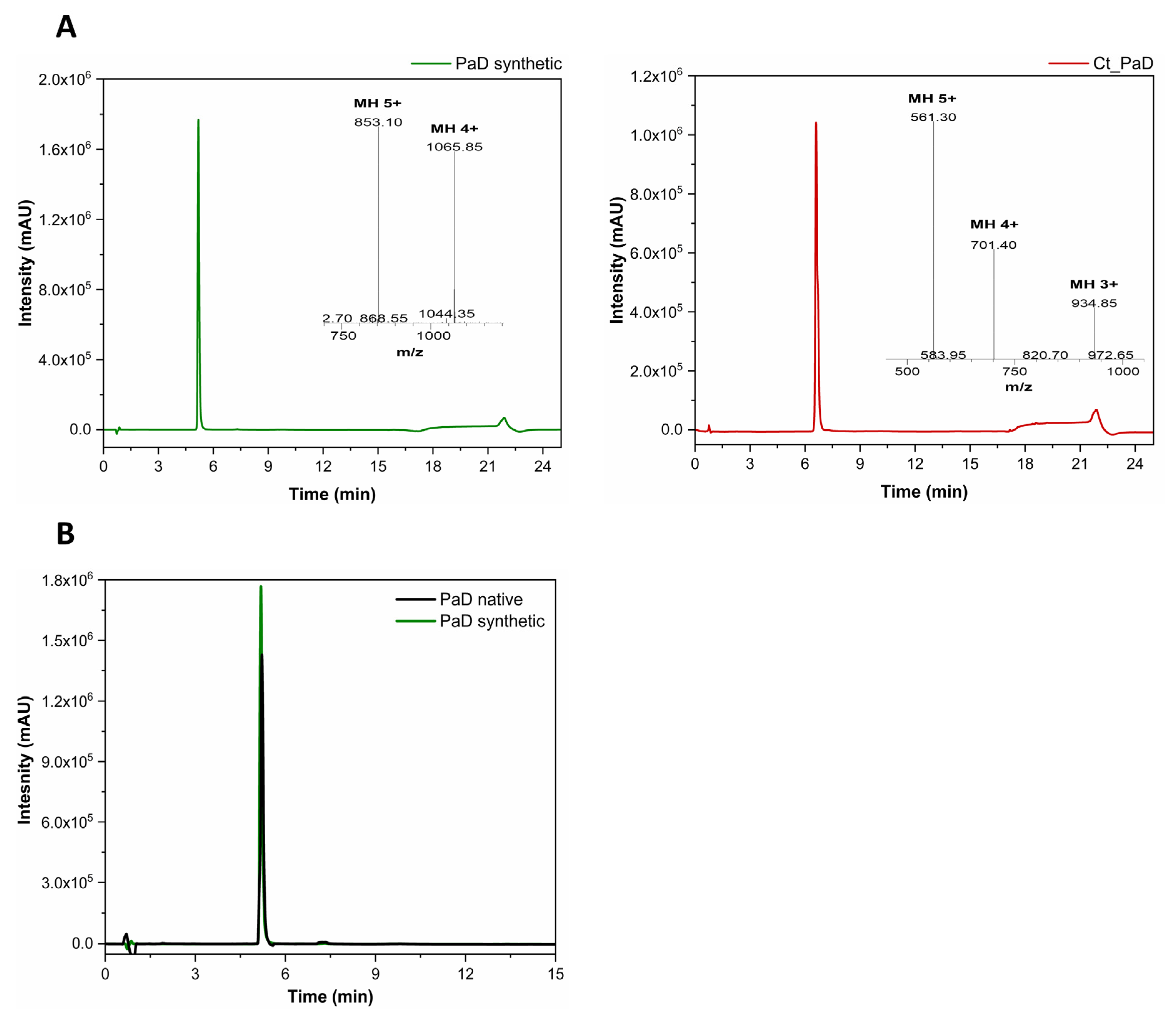
2.3. RP-HPLC and LC-MS
2.4. Anaerobic Oxidative Folding
2.5. Minimum Inhibitory Concentration (MIC) and Minimum Bactericidal Concentration (MBC)
2.6. Hemolytic Activity
2.7. Cytotoxicity in Mammalian Cells
2.8. Serum Stability
2.9. NMR Spectroscopy
2.10. Circular Dichroism Spectroscopy (CD)
3. Results and Discussion
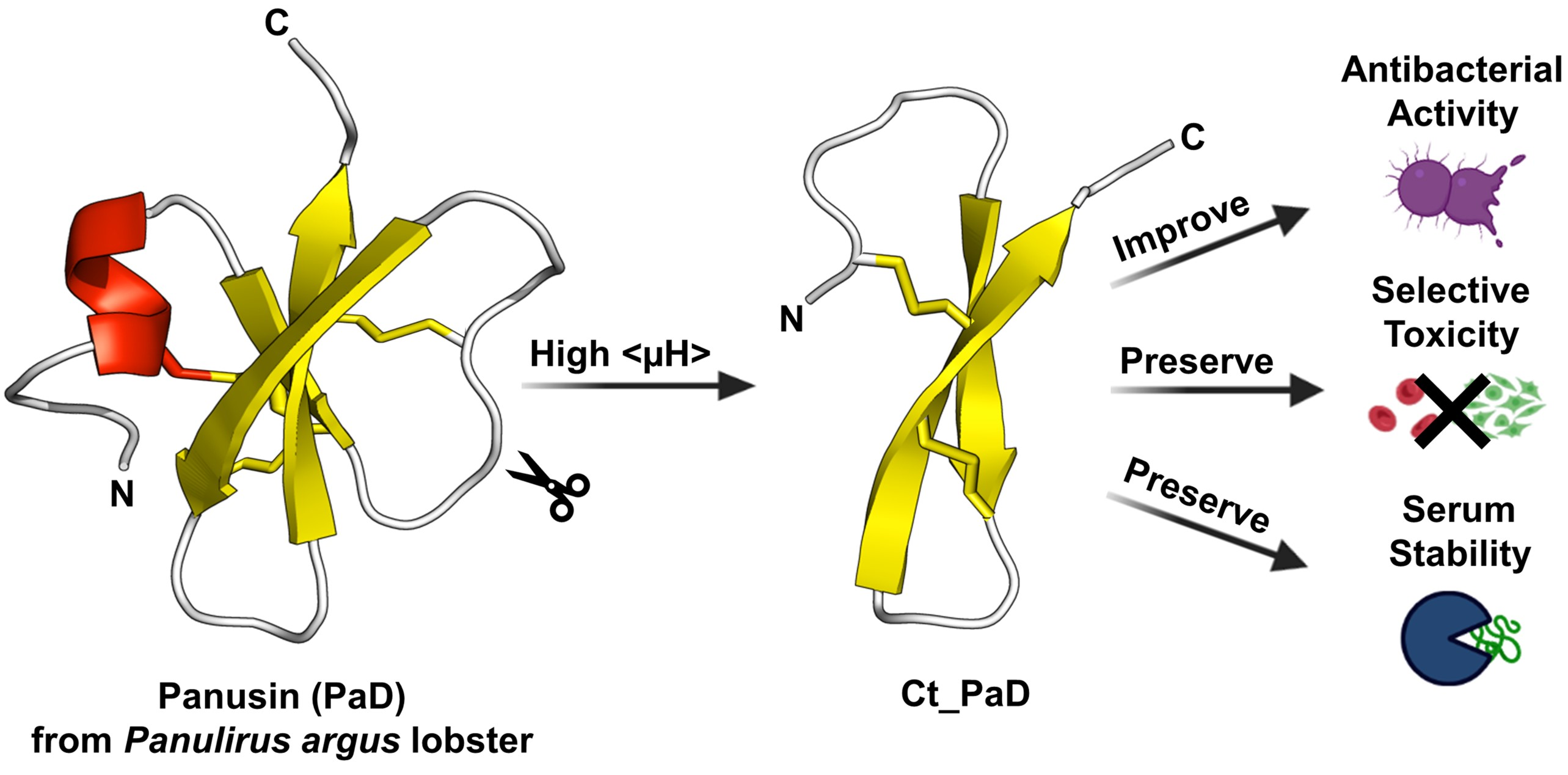
3.1. Peptide Design and Synthesis
3.2. Synthesis and Oxidative Folding of PaD and Ct_PaD
3.3. Antimicrobial Activity
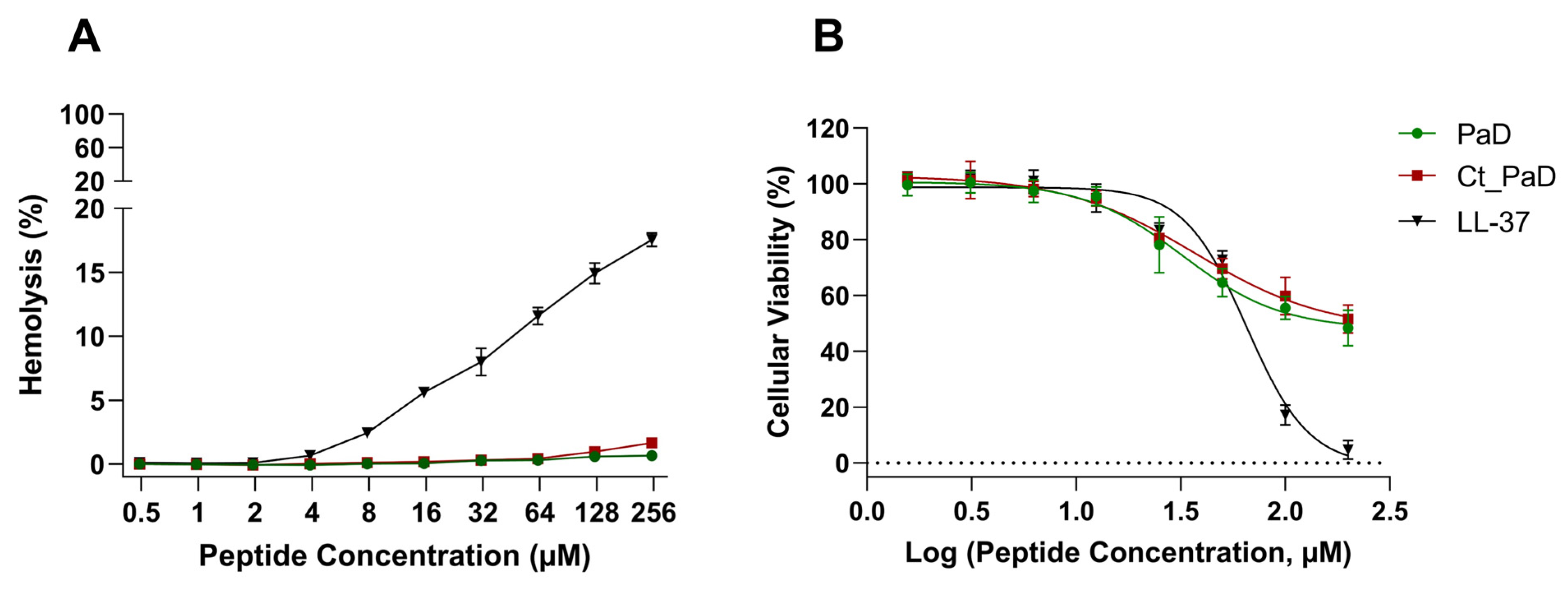
3.4. Hemolytic Activity and Cell Viability
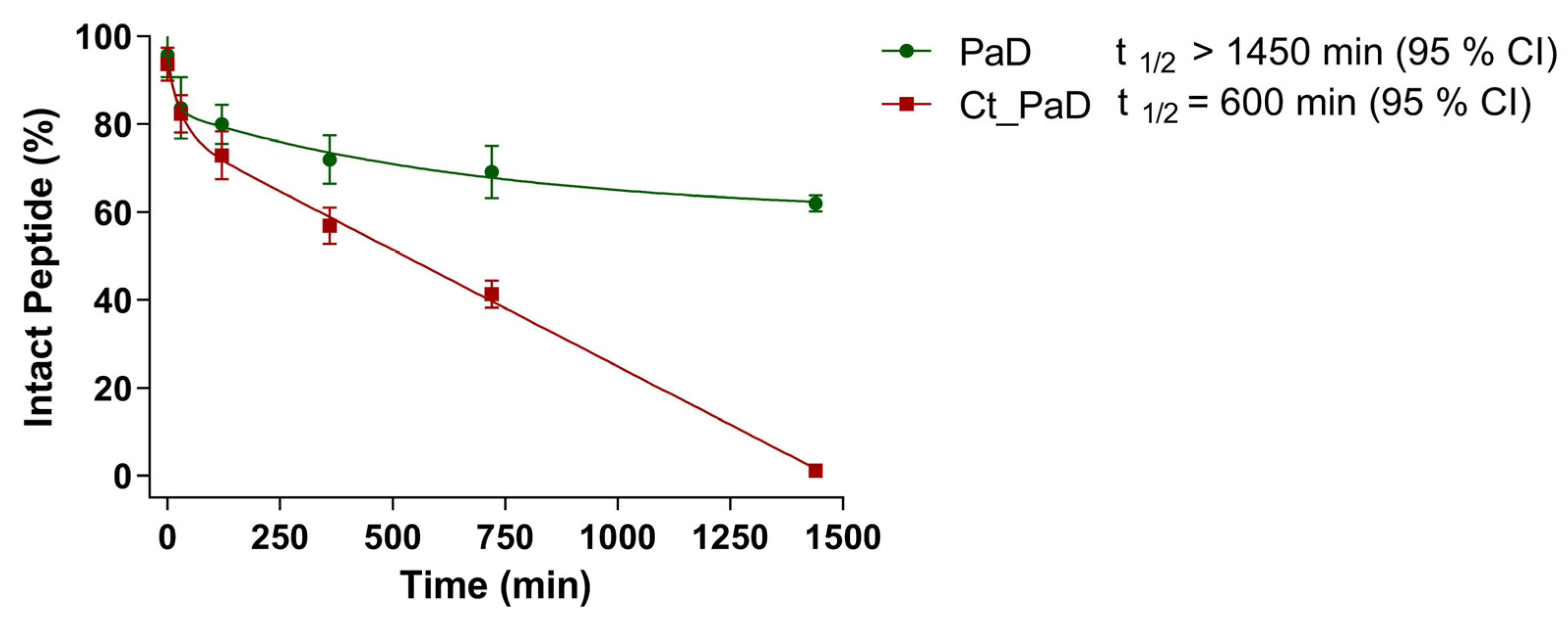
3.5. Serum Stability
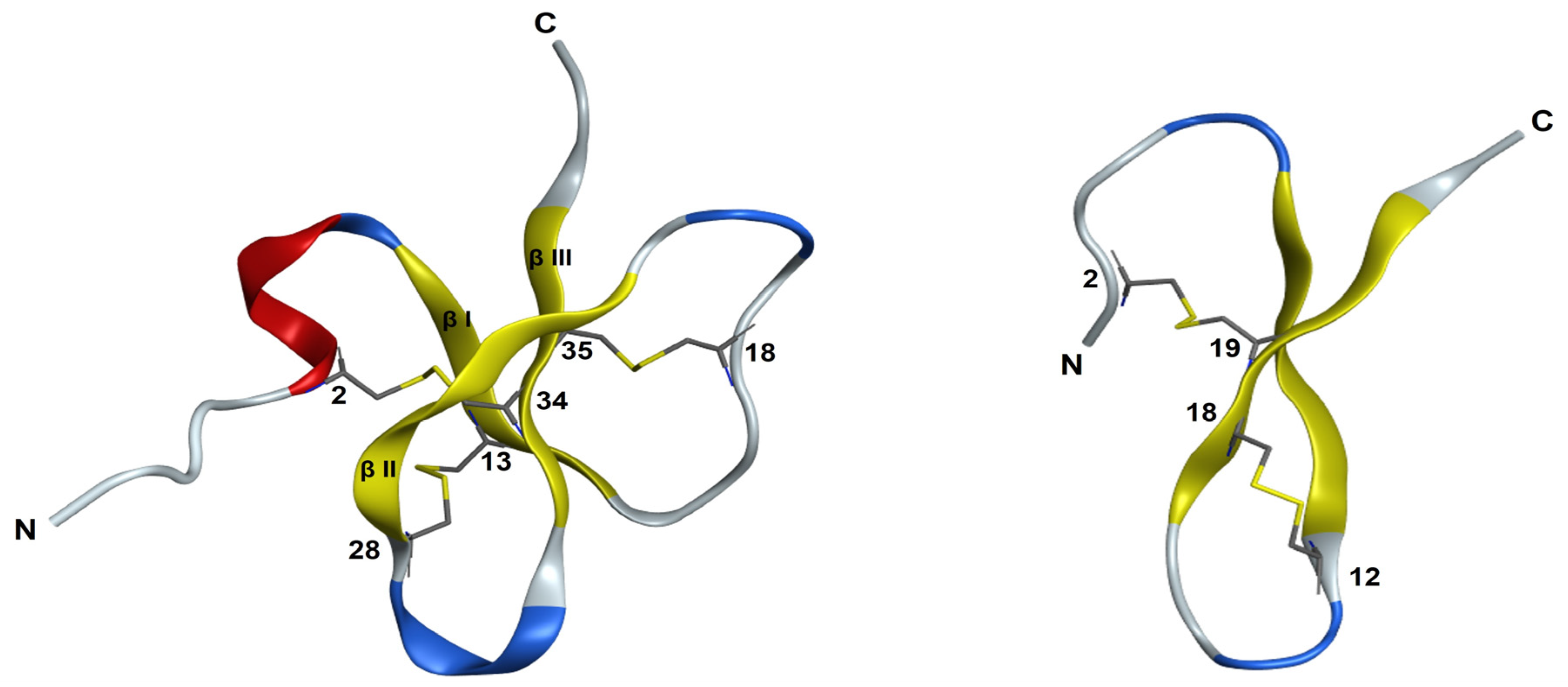
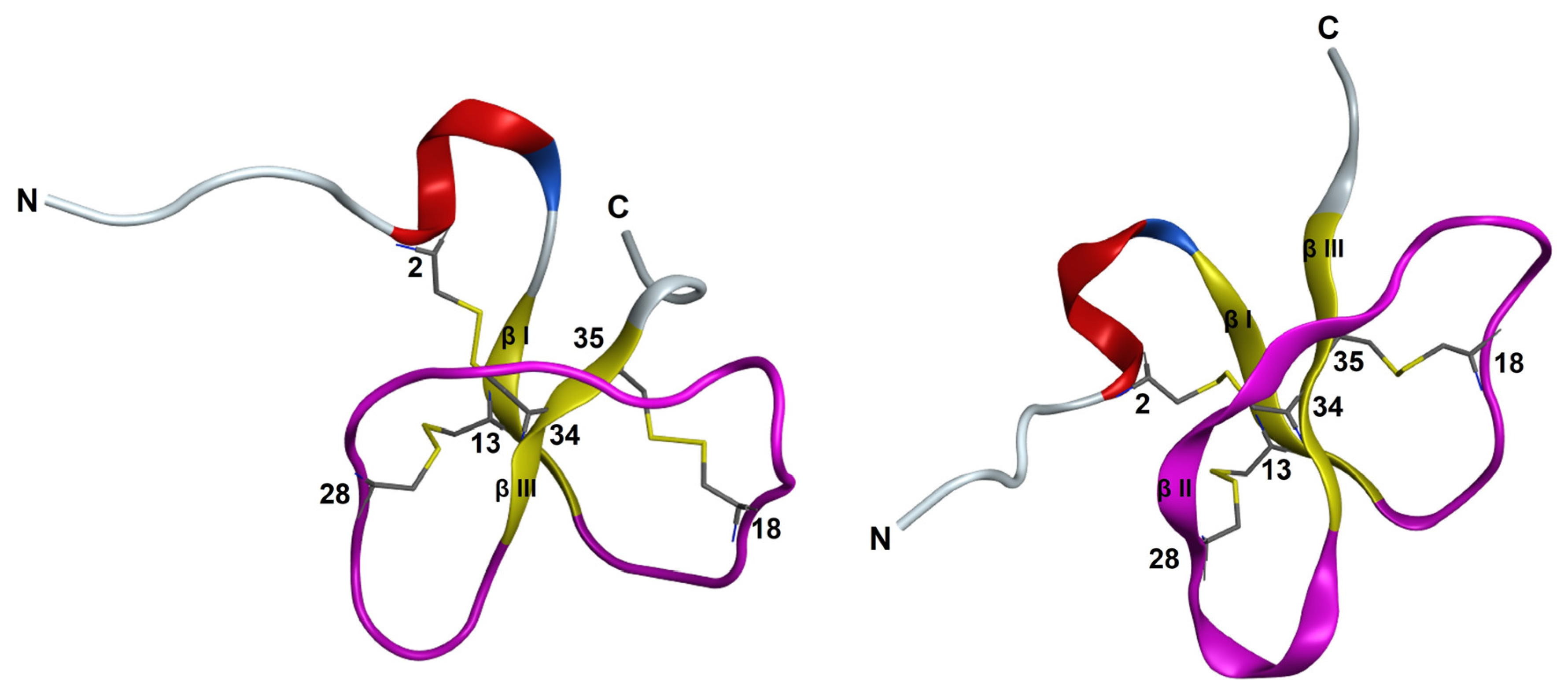
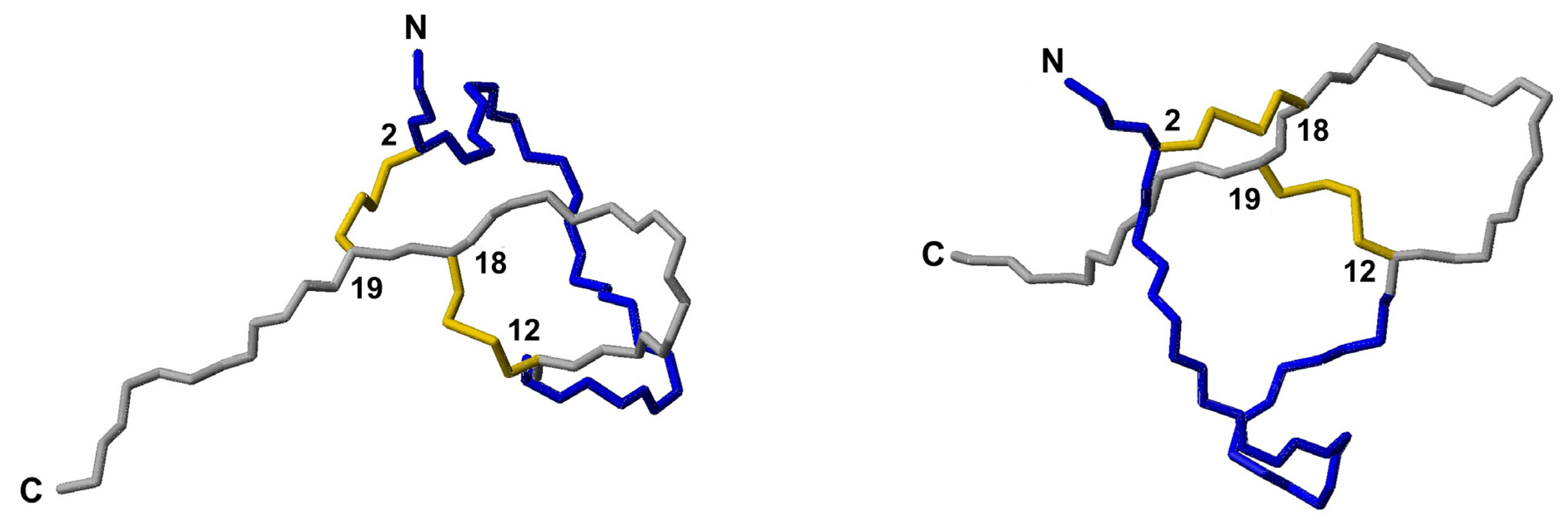
3.6. NMR Structural Study
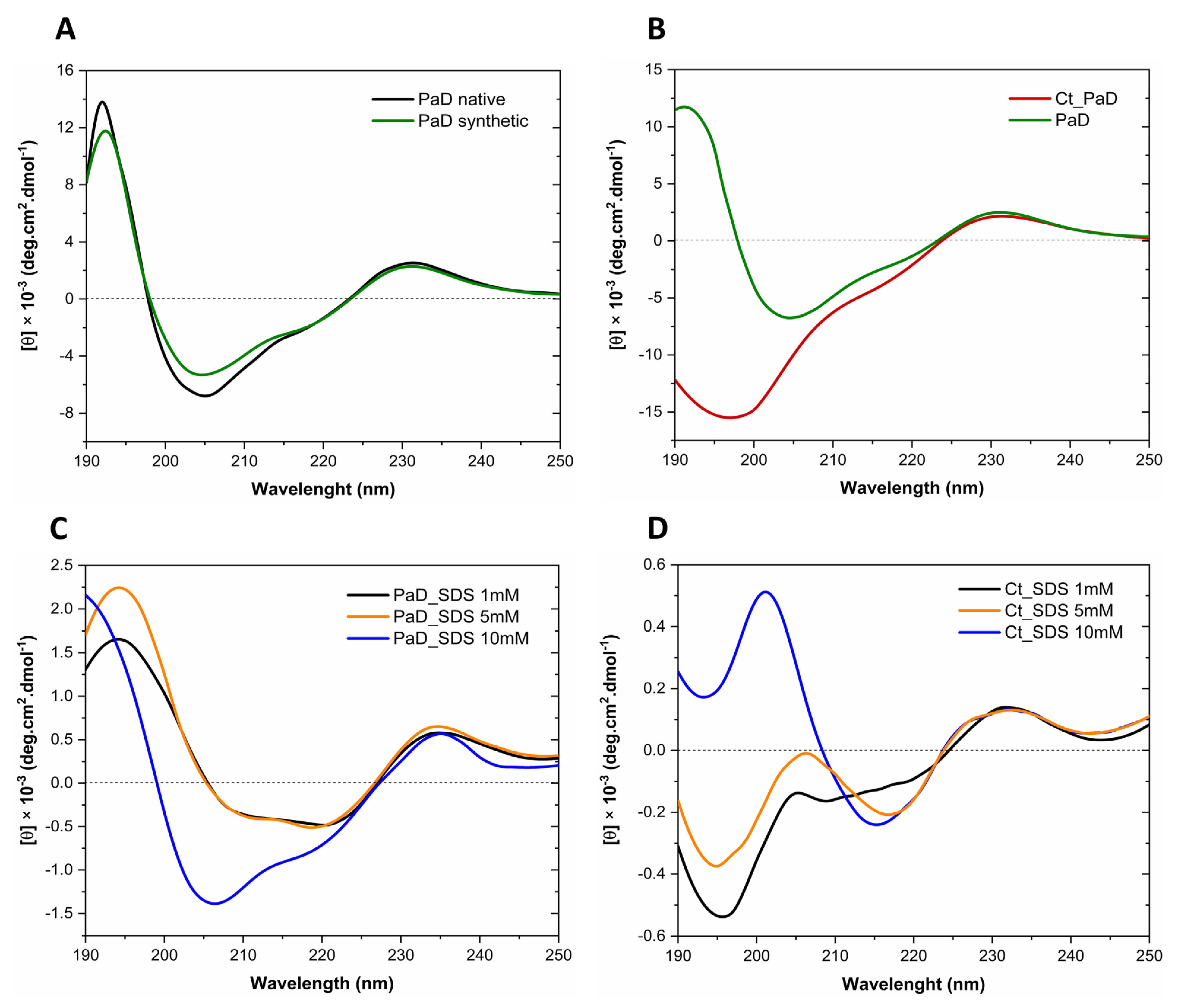
3.7. Analysis of Secondary Structure by Circular Dichroism
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pacios, O.; Blasco, L.; Bleriot, I.; Fernandez-Garcia, L.; González Bardanca, M.; Ambroa, A.; López, M.; Bou, G.; Tomás, M. Strategies to Combat Multidrug-Resistant and Persistent Infectious Diseases. Antibiotics 2020, 9, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahlapuu, M.; Björn, C.; Ekblom, J. Antimicrobial peptides as therapeutic agents: Opportunities and challenges. Crit. Rev. Biotechnol. 2020, 40, 978–992. [Google Scholar] [CrossRef] [PubMed]
- Amerikova, M.; Pencheva El-Tibi, I.; Maslarska, V.; Bozhanov, S.; Tachkov, K. Antimicrobial activity, mechanism of action, and methods for stabilisation of defensins as new therapeutic agents. Biotechnol. Biotechnol. Equip. 2019, 33, 671–682. [Google Scholar] [CrossRef]
- Khurshid, Z.; Zafar, M.S.; Naseem, M.; Khan, R.S.; Najeeb, S. Human oral defensins antimicrobial peptides: A future promising antimicrobial drug. Curr. Pharm. Des. 2018, 24, 1130–1137. [Google Scholar] [CrossRef] [PubMed]
- Montero-Alejo, V.; Corzo, G.; Porro-Suardíaz, J.; Pardo-Ruiz, Z.; Perera, E.; Rodríguez-Viera, L.; Sánchez-Díaz, G.; Hernández-Rodríguez, E.W.; Álvarez, C.; Peigneur, S. Panusin represents a new family of β-defensin-like peptides in invertebrates. Dev. Comp. Immunol. 2017, 67, 310–321. [Google Scholar] [CrossRef]
- Sudheendra, U.S.; Dhople, V.; Datta, A.; Kar, R.K.; Shelburne, C.E.; Bhunia, A.; Ramamoorthy, A. Membrane disruptive antimicrobial activities of human β-defensin-3 analogs. Eur. J. Med. Chem. 2015, 91, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Nehls, C.; Böhling, A.; Podschun, R.; Schubert, S.; Grötzinger, J.; Schromm, A.; Fedders, H.; Leippe, M.; Harder, J.; Kaconis, Y.; et al. Influence of disulfide bonds in human beta defensin-3 on its strain specific activity against Gram-negative bacteria. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183273. [Google Scholar] [CrossRef]
- Sakagami-Yasui, Y.; Shirafuji, Y.; Yamasaki, O.; Morizane, S.; Hamada, T.; Umemura, H.; Iwatsuki, K. Two arginine residues in the COOH-terminal of human β-defensin-3 constitute an essential motif for antimicrobial activity and IL-6 production. Exp. Dermatol. 2017, 26, 1026–1032. [Google Scholar] [CrossRef]
- Liu, H.; Lei, M.; Du, X.; Cui, P.; Zhang, S. Identification of a novel antimicrobial peptide from amphioxus Branchiostoma japonicum by in silico and functional analyses. Sci. Rep. 2015, 5, 18355. [Google Scholar] [CrossRef] [Green Version]
- Chaves-Arquero, B.; Pérez-Cañadillas, J.M.; Jiménez, M.A. Effect of Phosphorylation on the Structural Behaviour of Peptides Derived from the Intrinsically Disordered C-Terminal Domain of Histone H1.0. Eur. J. Chem. 2020, 26, 5970–5981. [Google Scholar] [CrossRef]
- Lee, W.; Tonelli, M.; Markley, J.L. NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 2015, 31, 1325–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wüthrich, K.; Billeter, M.; Braun, W. Polypeptide secondary structure determination by nuclear magnetic resonance observation of short proton-proton distances. J. Mol. Biol. 1984, 180, 715–740. [Google Scholar] [CrossRef] [PubMed]
- Güntert, P. Automated NMR structure calculation with CYANA. Methods Mol. Biol. 2004, 278, 353–378. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Bax, A. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 2013, 56, 227–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Bax, A. Protein structural information derived from NMR chemical shift with the neural network program TALOS-N. Methods. Mol. Biol. 2015, 1260, 17–32. [Google Scholar] [CrossRef] [Green Version]
- Koradi, R.; Billeter, M.; Wüthrich, K. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 1996, 14, 51–55. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Skalska, J.; Andrade, V.M.; Cena, G.L.; Harvey, P.J.; Gaspar, D.; Mello, É.O.; Henriques, S.T.; Valle, J.; Gomes, V.M.; Conceição, K.; et al. Synthesis, Structure, and Activity of the Antifungal Plant Defensin PvD1. J. Med. Chem. 2020, 63, 9391–9402. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar] [CrossRef]
- Boman, H.G. Antibacterial peptides: Basic facts and emerging concepts. J. Intern. Med. 2003, 254, 197–215. [Google Scholar] [CrossRef]
- Dennison, S.R.; Mura, M.; Harris, F.; Morton, L.H.G.; Zvelindovsky, A.; Phoenix, D.A. The role of C-terminal amidation in the membrane interactions of the anionic antimicrobial peptide, maximin H5. Biochim. Biophys. Acta Biomembr. 2015, 1848, 1111–1118. [Google Scholar] [CrossRef] [Green Version]
- Hoover, D.M.; Wu, Z.; Tucker, K.; Lu, W.; Lubkowski, J. Antimicrobial characterization of human beta-defensin 3 derivatives. Antimicrob. Agents Chemother. 2003, 47, 2804–2809. [Google Scholar] [CrossRef] [Green Version]
- Wendler, J.; Schroeder, B.O.; Ehmann, D.; Koeninger, L.; Mailänder-Sánchez, D.; Lemberg, C.; Wanner, S.; Schaller, M.; Stange, E.F.; Malek, N.P.; et al. Proteolytic Degradation of reduced Human Beta Defensin 1 generates a Novel Antibiotic Octapeptide. Sci. Rep. 2019, 9, 3640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greco, I.; Molchanova, N.; Holmedal, E.; Jenssen, H.; Hummel, B.D.; Watts, J.L.; Håkansson, J.; Hansen, P.R.; Svenson, J. Correlation between hemolytic activity, cytotoxicity and systemic in vivo toxicity of synthetic antimicrobial peptides. Sci. Rep. 2020, 10, 13206. [Google Scholar] [CrossRef] [PubMed]
- Zannella, C.; Chianese, A.; Palomba, L.; Marcocci, M.E.; Bellavita, R.; Merlino, F.; Grieco, P.; Folliero, V.; De Filippis, A.; Mangoni, M.; et al. Broad-Spectrum Antiviral Activity of the Amphibian Antimicrobial Peptide Temporin L and Its Analogs. Int. J. Mol. Sci. 2022, 23, 2060. [Google Scholar] [CrossRef] [PubMed]
- Böttger, R.; Hoffmann, R.; Knappe, D. Differential stability of therapeutic peptides with different proteolytic cleavage sites in blood, plasma and serum. PLoS ONE 2017, 12, e0178943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavaco, M.; Andreu, D.; Castanho, M. The Challenge of Peptide Proteolytic Stability Studies: Scarce Data, Difficult Readability, and the Need for Harmonization. Angew. Chem. Int. Ed. Engl. 2021, 60, 1686–1688. [Google Scholar] [CrossRef] [PubMed]
- Falanga, A.; Nigro, E.; De Biasi, M.G.; Daniele, A.; Morelli, G.; Galdiero, S.; Scudiero, O. Cyclic Peptides as Novel Therapeutic Microbicides: Engineering of Human Defensin Mimetics. Molecules 2017, 22, 1217. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Peinado, C.; Dias, S.A.; Mendonça, D.A.; Castanho, M.A.R.B.; Veiga, A.S.; Andreu, D. Structural determinants conferring unusual long life in human serum to rattlesnake-derived antimicrobial peptide Ctn[15–34]. J. Pept. Sci. 2019, 25, e3195. [Google Scholar] [CrossRef]
- Strömstedt, A.A.; Pasupuleti, M.; Schmidtchen, A.; Malmsten, M. Evaluation of strategies for improving proteolytic resistance of antimicrobial peptides by using variants of EFK17, an internal segment of LL-37. Antimicrob. Agents Chemother. 2009, 53, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Fázio, M.A.; Oliveira, V.X., Jr.; Bulet, P.; Miranda, M.T.; Daffre, S.; Miranda, A. Structure-activity relationship studies of gomesin: Importance of the disulfide bridges for conformation, bioactivities, and serum stability. Biopolymers 2006, 84, 205–218. [Google Scholar] [CrossRef]
- Andersson, H.S.; Figueredo, S.M.; Haugaard-Kedström, L.M.; Bengtsson, E.; Daly, N.L.; Qu, X.; Craik, D.J.; Ouellette, A.J.; Rosengren, K.J. The α-defensin salt-bridge induces backbone stability to facilitate folding and confer proteolytic resistance. Amino Acids 2012, 43, 1471–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Musaimi, O.; Lombardi, L.; Williams, D.R.; Albericio, F. Strategies for Improving Peptide Stability and Delivery. Pharmaceuticals 2022, 15, 1283. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.; Gao, X.; Pan, D.; Hua, Y.; He, J.; Liu, Z.; Dang, Y. Advances in the stability challenges of bioactive peptides and improvement strategies. Curr. Res. Food. Sci. 2022, 5, 2162–2170. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.F.; Yang, H.; Zhao, Y.Z.; Xue, M. Metabolism of Peptide Drugs and Strategies to Improve their Metabolic Stability. Curr. Drug Metab. 2018, 19, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.; Labudde, D.; Oschkinat, H.; Schmieder, P. A software tool for the prediction of Xaa-Pro peptide bond conformations in proteins based on 13C chemical shift statistics. J. Biomol. NMR 2002, 24, 149–154. [Google Scholar] [CrossRef]
- Shen, Y.; Bax, A. Prediction of Xaa-Pro peptide bond conformation from sequence and chemical shifts. J. Biomol. NMR 2010, 46, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Sharma, D.; Rajarathnam, K. 13C NMR chemical shifts can predict disulfide bond formation. J. Biomol. NMR 2000, 18, 165–171. [Google Scholar] [CrossRef]
- Perinelli, D.R.; Cespi, M.; Lorusso, N.; Palmieri, G.F.; Bonacucina, G.; Blasi, P. Surfactant Self-Assembling and Critical Micelle Concentration: One Approach Fits All? Langmuir 2020, 36, 5745–5753. [Google Scholar] [CrossRef]
- Greenfield, N.J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2006, 1, 2876–2890. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Sequence | Cys Pairing | Theoretical Mass (Da) 1 | Experimental Mass (Da) 1 | Purity (HPLC, %) |
|---|---|---|---|---|---|
| PaD | SYVGDC1GSNGGSC2VSSYC3PYGNRLNYFC4PLGRTC5C6RRSY-amide | C1–C5 C2–C4 C3–C6 | 4259.76 | 4260.5 | 98.7 |
| Ct_PaD | YC3PYGNRLNYFC4PLGRTC5C6RRSY-amide | C3–C6 C4–C5 | 2801.25 | 2801.6 | 99.8 |
| ID | Net Charge a | pI b | Aliphatic Index c | Hydrophobic Moment d | Boman Index (kcal/mol) e |
|---|---|---|---|---|---|
| PaD | 4+ | 8.64 | 34.87 | 1.86 | 1.99 |
| Ct_PaD | 5+ | 9.20 | 33.91 | 2.56 | 2.59 |
| Peptide | ||||
|---|---|---|---|---|
| PaD | Ct_PaD | LL-37 | ||
| MIC/MBC (µM) | Escherichia coli | 12.5/12.5 | 3.1/3.1 | 1.6/3.1 |
| Pseudomonas aeruginosa | 25/25 | 6.3/6.3 | 1.6/1.6 | |
| Acinetobacter baumannii | 25/25 | 6.3/12.5 | 6.3/6.3 | |
| Staphylococcus aureus | 25/25 | 3.1/6.3 | 25/25 | |
| Enterococcus faecium | 25/25 | 6.3/6.3 | 1.6/3.1 | |
| Micrococcus luteus | 12.5/12.5 | 3.1/3.1 | 1.6/3.1 | |
| Toxicity | Hemolysis % (250 µM peptide) | 0.6 ± 0.02 | 1.7 ± 0.08 | 17.5 ± 0.3 |
| MRC-5 cytotoxicity (IC50, µM) | 145 ± 1.7 | 175 ± 1.5 | 65 ± 0.7 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bello-Madruga, R.; Valle, J.; Jiménez, M.Á.; Torrent, M.; Montero-Alejo, V.; Andreu, D. The C-Terminus of Panusin, a Lobster β-Defensin, Is Crucial for Optimal Antimicrobial Activity and Serum Stability. Pharmaceutics 2023, 15, 1777. https://doi.org/10.3390/pharmaceutics15061777
Bello-Madruga R, Valle J, Jiménez MÁ, Torrent M, Montero-Alejo V, Andreu D. The C-Terminus of Panusin, a Lobster β-Defensin, Is Crucial for Optimal Antimicrobial Activity and Serum Stability. Pharmaceutics. 2023; 15(6):1777. https://doi.org/10.3390/pharmaceutics15061777
Chicago/Turabian StyleBello-Madruga, Roberto, Javier Valle, M. Ángeles Jiménez, Marc Torrent, Vivian Montero-Alejo, and David Andreu. 2023. "The C-Terminus of Panusin, a Lobster β-Defensin, Is Crucial for Optimal Antimicrobial Activity and Serum Stability" Pharmaceutics 15, no. 6: 1777. https://doi.org/10.3390/pharmaceutics15061777