
Novel Bioequivalent Tablet of Solifenacin Succinate Prepared Using Direct Compression Technique for Improved Chemical Stability

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of SOL-Loaded Tablets Using DC Technique
2.3. Preparation of SOL-Loaded Tablets Using Wet Granulation Method
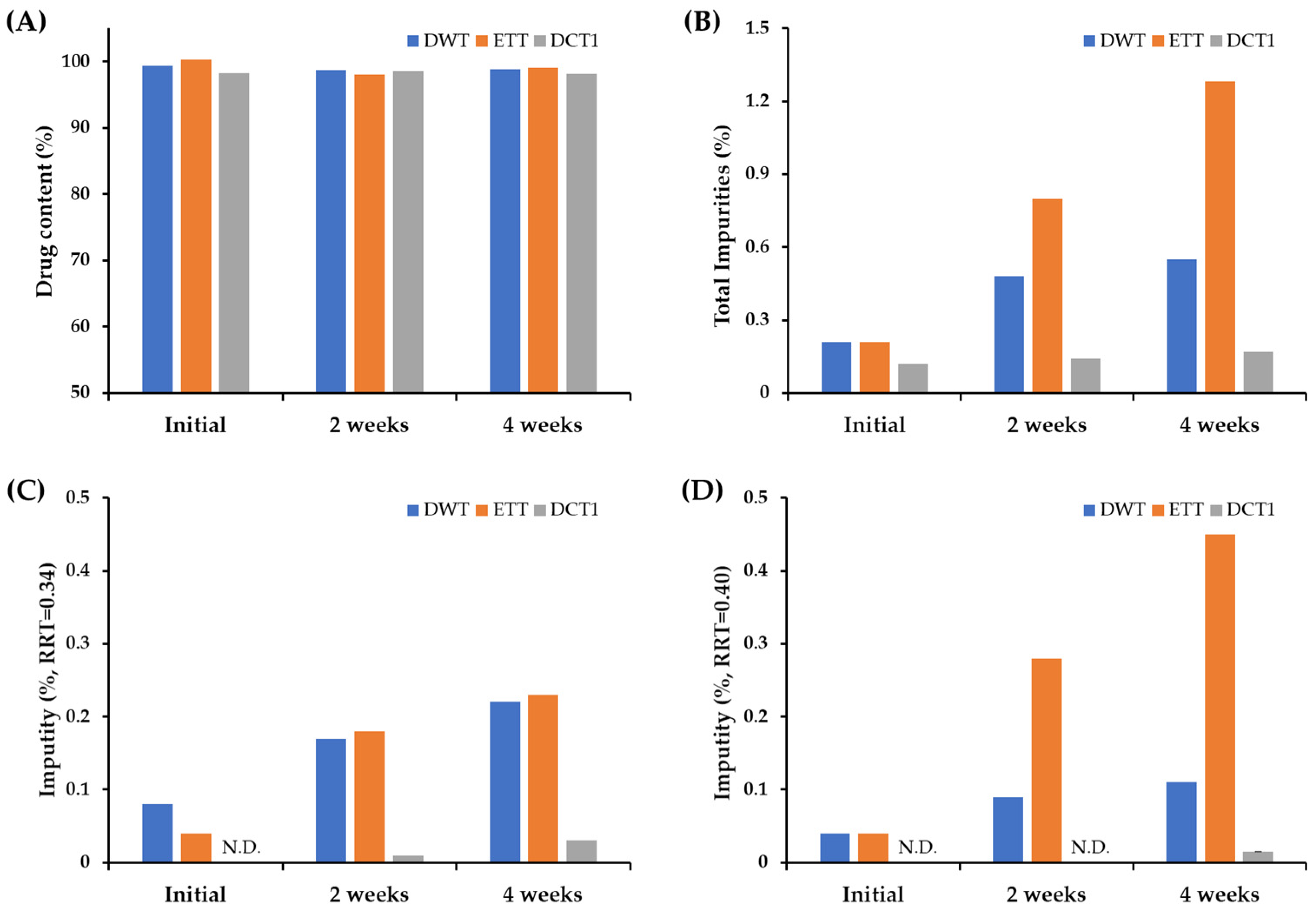
2.4. Drug Content and Degradation Product Analysis
2.5. Disintegration Time of SOL-Loaded Tablets
2.6. Mechanical Properties of SOL-Loaded Tablets
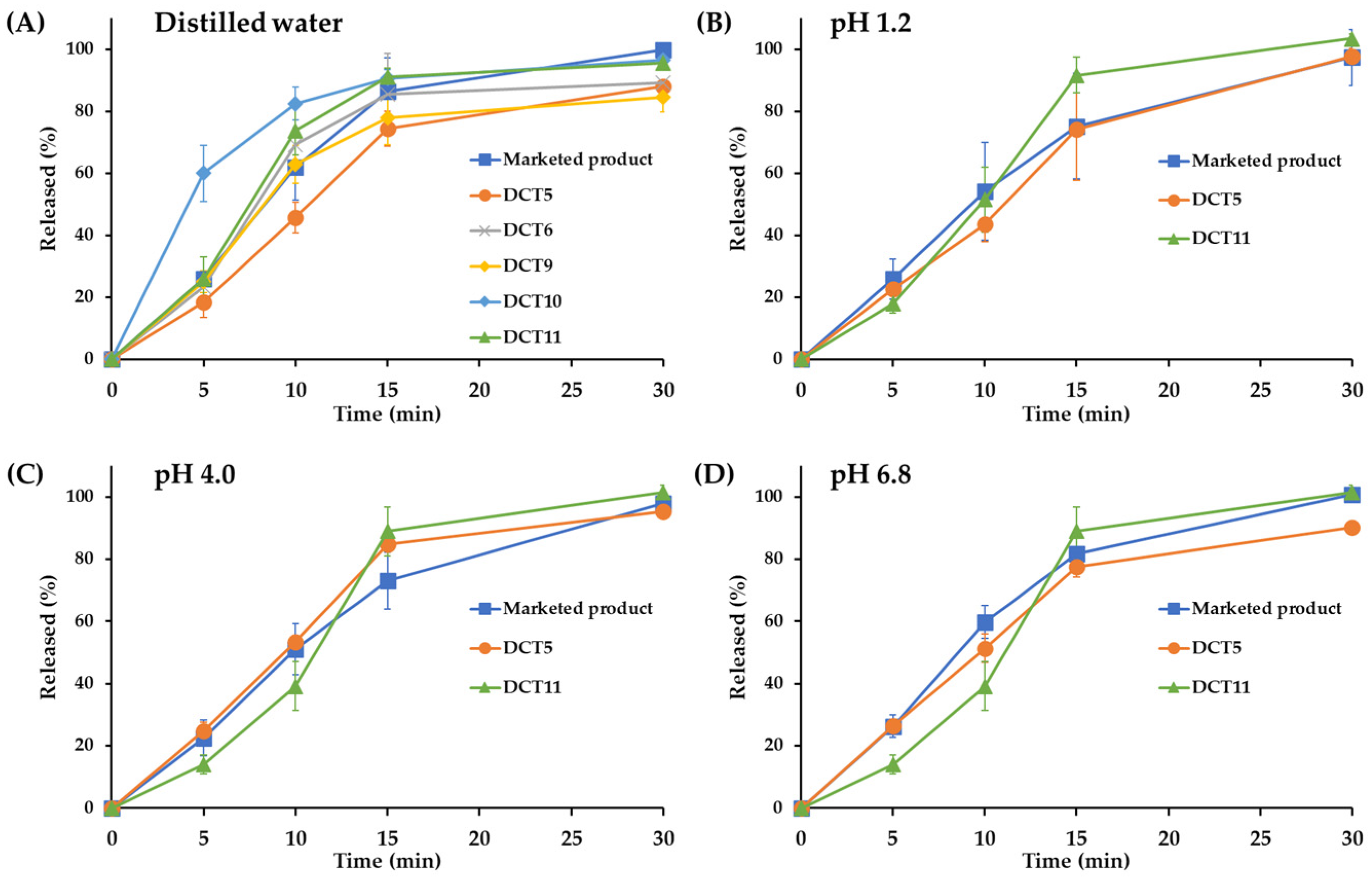
2.7. In Vitro Dissolution Profile of SOL-Loaded Tablets
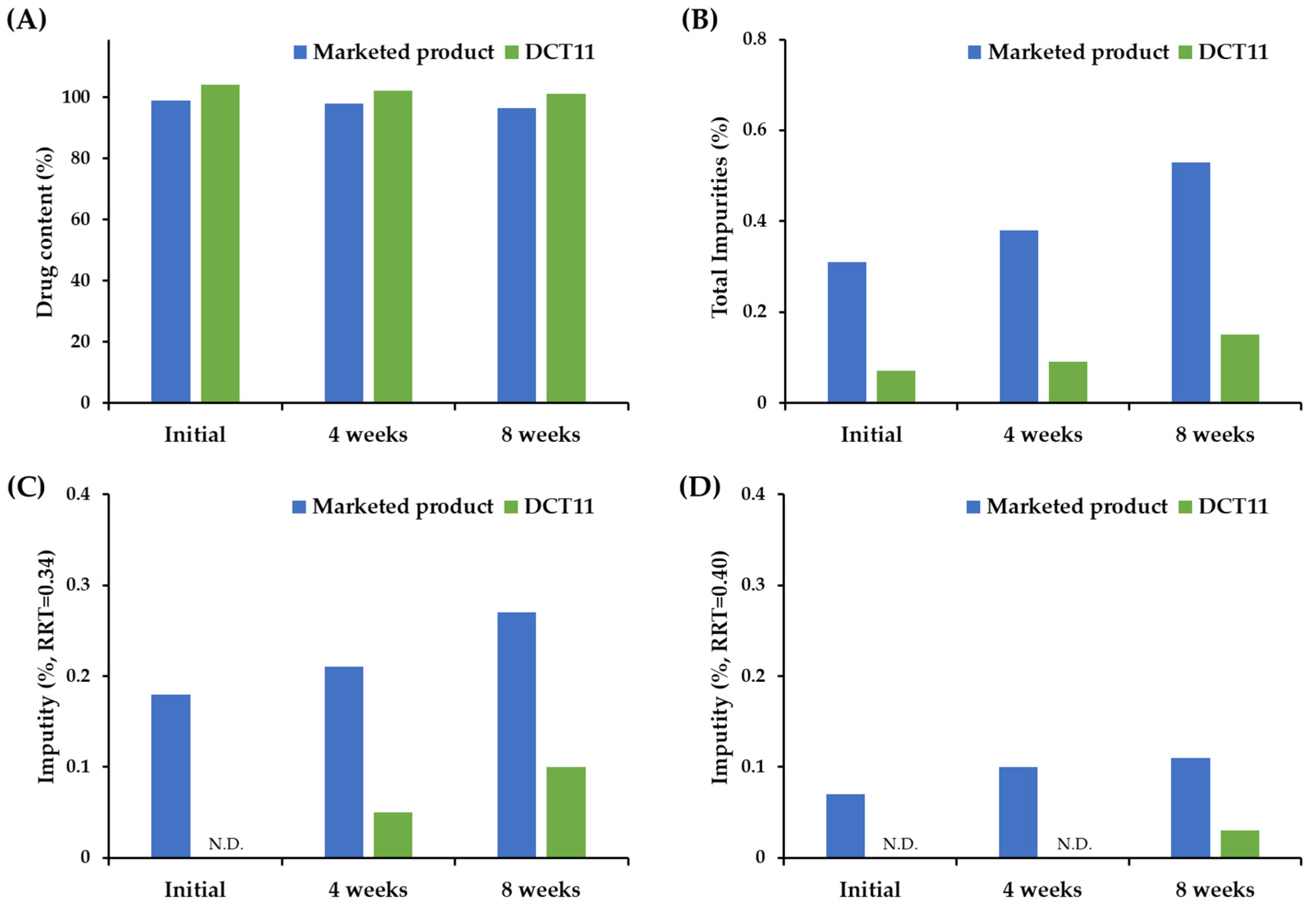
2.8. Physicochemical Stability of SOL Tablets under Accelerated Condition
2.9. Bioequivalence Study in Healthy Volunteers
2.9.1. Volunteers and Protocols
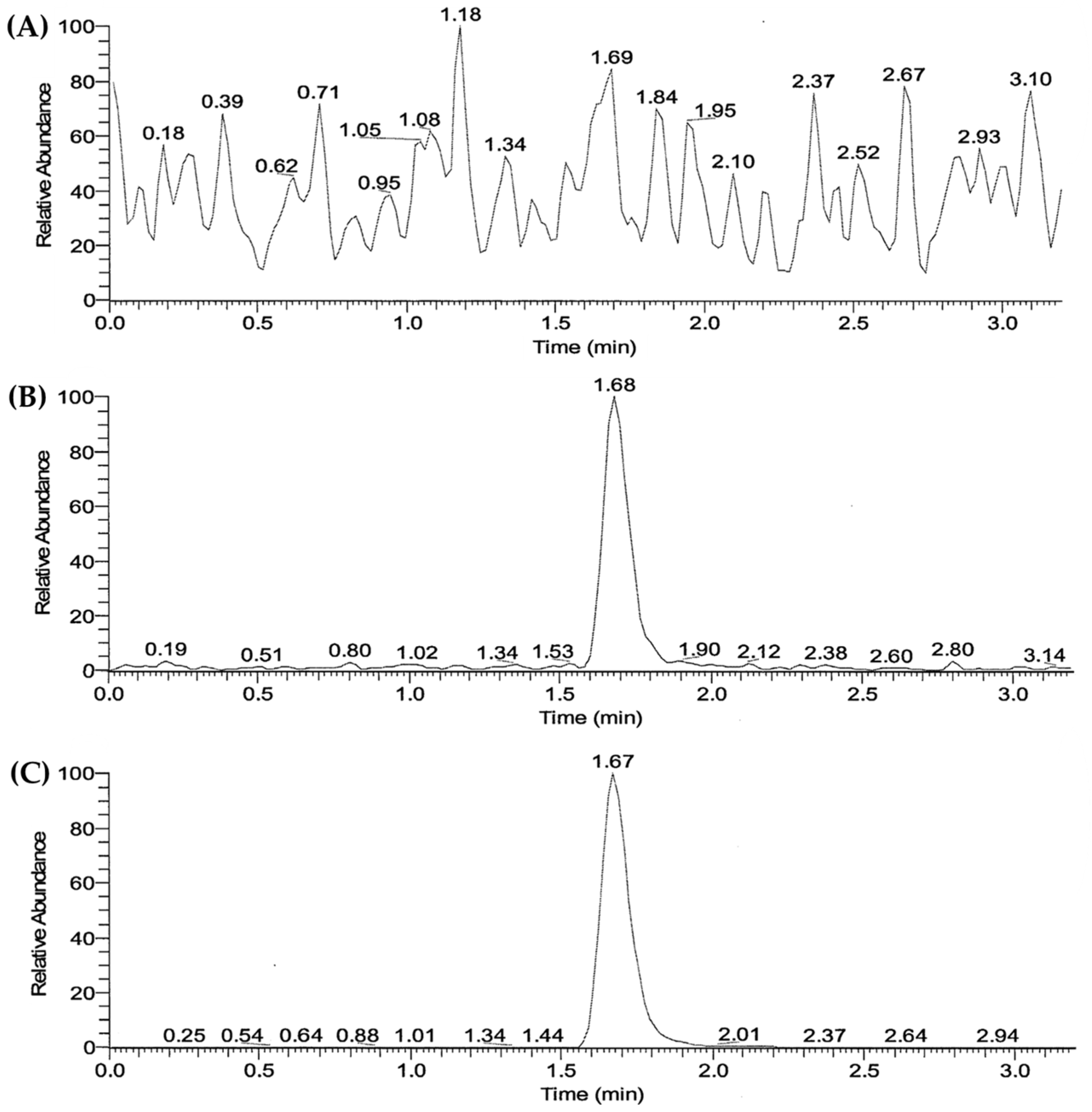
2.9.2. LC/MS-MS Analysis of SOL in Plasma
2.9.3. Pharmacokinetic and Statistical Analysis
3. Results and Discussion
3.1. Effect of Fabrication Process on Chemical Stability of SOL-Loaded Tablets
3.2. In Vitro Dissolution and Optimization of SOL-Loaded DCTs
3.3. Validation of LC/MS-MS Analysis Method of SOL in Human Plasma
3.4. Stability of the Optimized DCT under Accelerated Storage Condition
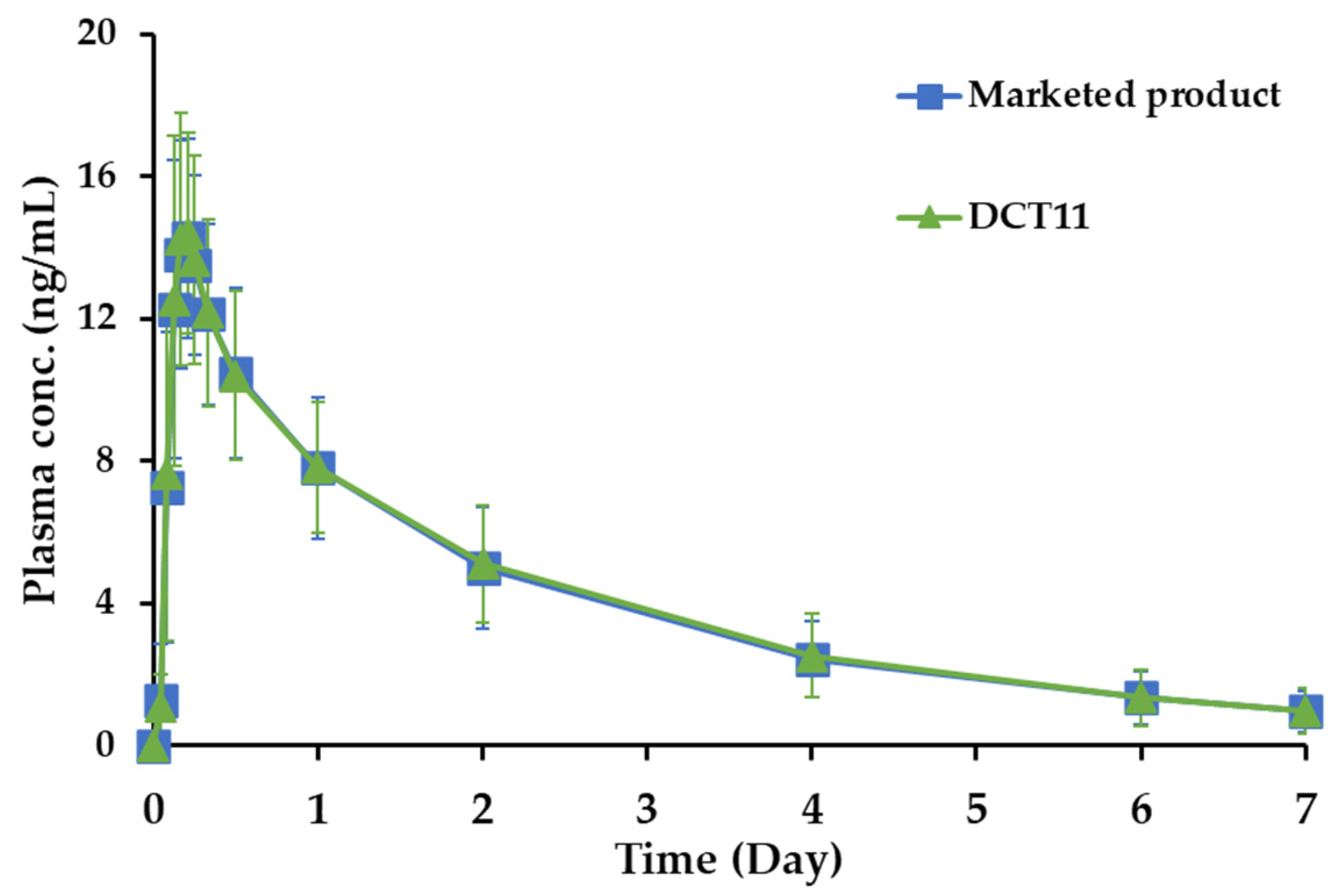
3.5. Pharmacokinetics and Bioequivalence Study in Healthy Volunteers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Franco, I.; Hoebeke, P.; Baka-Ostrowska, M.; Bolong, D.; Davies, L.N.; Dahler, E.; Snijder, R.; Stroosma, O.; Verheggen, F.; Newgreen, D.; et al. Long-Term Efficacy and Safety of Solifenacin in Pediatric Patients Aged 6 Months to 18 Years with Neurogenic Detrusor Overactivity: Results from Two Phase 3 Prospective Open-Label Studies. J. Pediatr. Urol. 2020, 16, 180.e1–180.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melling, C.V.; Goyal, A. Current Pharmacological Management of Idiopathic Overactive Bladder in Children in the UK: A National Survey of Practice. J. Pediatr. Urol. 2020, 16, 37.e1–37.e8. [Google Scholar] [CrossRef] [PubMed]
- Mostafaei, H.; Salehi-Pourmehr, H.; Jilch, S.; Carlin, G.L.; Mori, K.; Quhal, F.; Pradere, B.; Grossmann, N.C.; Laukhtina, E.; Schuettfort, V.M.; et al. Choosing the Most Efficacious and Safe Oral Treatment for Idiopathic Overactive Bladder: A Systematic Review and Network Meta-Analysis. Eur. Urol. Focus 2022, 8, 1072–1089. [Google Scholar] [CrossRef]
- Staskin, D.R.; Te, A.E. Short- and Long-Term Efficacy of Solifenacin Treatment in Patients with Symptoms of Mixed Urinary Incontinence. Br. J. Urol. 2006, 97, 1256–1261. [Google Scholar] [CrossRef] [PubMed]
- Niphade, N.C.; Jagtap, K.M.; Mali, A.C.; Solanki, P.V.; Jachak, M.N.; Mathad, V.T. Efficient and Single Pot Process for the Preparation of Enantiomerically Pure Solifenacin Succinate, an Antimuscarinic Agent. Monatsh. Chem. 2011, 142, 1181–1186. [Google Scholar] [CrossRef]
- Kuipers, M.E.; Krauwinkel, W.J.J.; Mulder, H.; Visser, N. Solifenacin Demonstrates High Absolute Bioavailability in Healthy Men. Drugs R D 2004, 5, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Kumagai, H.; Haneda, M.; Vertzoni, M.; Ouwerkerk, N.; Murayama, D.; Katakawa, Y.; Motonaga, K.; Reppas, C.; Tajiri, T. The Mechanism of Solifenacin Release from a PH-Responsive Ion-Complex Oral Suspension in the Fasted Upper Gastrointestinal Lumen. Eur. J. Pharm. Sci. 2020, 142, 105107. [Google Scholar] [CrossRef]
- Sudha, R.K.V.N.; Kishore, V.S.; Babu, C.H.V.; Jitendranath, E. Design, Development and Evaluation of Solifenacin Succinate Tablets. Rese. J. Pharmaceut. Dosag. Form. Technol. 2015, 7, 111. [Google Scholar] [CrossRef]
- Trasi, N.S.; Bhujbal, S.; Zhou, Q.T.; Taylor, L.S. Amorphous Solid Dispersion Formation via Solvent Granulation—A Case Study with Ritonavir and Lopinavir. Int. J. Pharm. X 2019, 1, 100035. [Google Scholar] [CrossRef]
- Wang, B.; Sun, X.; Xiang, J.; Guo, X.; Cheng, Z.; Liu, W.; Tan, S. A Critical Review on Granulation of Pharmaceuticals and Excipients: Principle, Analysis and Typical Applications. Powder Technol. 2022, 401, 117329. [Google Scholar] [CrossRef]
- Liu, B.; Wang, J.; Zeng, J.; Zhao, L.; Wang, Y.; Feng, Y.; Du, R. A Review of High Shear Wet Granulation for Better Process Understanding, Control and Product Development. Powder Technol. 2021, 381, 204–223. [Google Scholar] [CrossRef]
- Terada, H.; Hattori, Y.; Sasaki, T.; Otsuka, M. Quantitation of Trace Amorphous Solifenacin Succinate in Pharmaceutical Formulations by Transmission Raman Spectroscopy. Int. J. Pharm. 2019, 565, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.; Baxendale, I.R. An Overview of the Synthetic Routes to the Best Selling Drugs Containing 6-Membered Heterocycles. Beilstein J. Org. Chem. 2013, 9, 2265–2319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, S.-W.; Park, J.-H.; Kim, J.-E.; Park, Y.-J. Design of Experiment (DoE)-Based Formulation Design of Bepotastine Sustained-Release Tablet and in vitro-in vivo Pharmacokinetic Correlation. J. Pharm. Investig. 2023, 53, 407–416. [Google Scholar] [CrossRef]
- Abu Fara, D.; Al-Hmoud, L.; Rashid, I.; Chowdhry, B.Z.; Badwan, A. Understanding the Performance of a Novel Direct Compression Excipient Comprising Roller Compacted Chitin. Mar. Drugs 2020, 18, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gohel, M.C.; Jogani, P.D. A Review of Co-Processed Directly Compressible Excipients. J. Pharm. Sci. 2005, 8, 76–93. [Google Scholar]
- Prajapati, S.T.; Patel, P.K.; Patel, M.; Chauhan, V.B.; Patel, C.N. Development and Validation of the Liquid Chromatography-Tandem Mass Spectrometry Method for Quantitative Estimation of Candesartan from Human Plasma. Pharm. Methods 2011, 2, 130–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, C.; Wu, F.; Hong, Y.; Shen, L.; Lin, X.; Zhao, L.; Feng, Y. Updates on Applications of Low-Viscosity Grade Hydroxypropyl Methylcellulose in Coprocessing for Improvement of Physical Properties of Pharmaceutical Powders. Carbohydr. Polym. 2023, 311, 120731. [Google Scholar] [CrossRef]
- Desai, D.; Patel, G.; Shukla, N.; Rajput, S. Development and Validation of Stability-Indicating HPLC Method for Solifenacin Succinate: Isolation and Identification of Major Base Degradation Product. Acta Chromatogr. 2012, 24, 399–418. [Google Scholar] [CrossRef] [Green Version]
- Schmid, K.; Löbenberg, R. Influence of the Changed USP Specifications on Disintegration Test Performance. Dissolution Technol. 2010, 17, 6–10. [Google Scholar] [CrossRef]
- USP 2019. The United States Pharmacopeial Convention <701> DISINTEGRATION Part. 1 May 2020. Available online: https://www.usp.org/sites/default/files/usp/document/harmonization/gen-chapter/april-2019-m99460.pdf (accessed on 20 May 2023).
- Sousa, A.S.; Serra, J.; Estevens, C.; Costa, R.; Ribeiro, A.J. A Quality by Design Approach in Oral Extended Release Drug Delivery Systems: Where We Are and Where We Are Going? J. Pharm. Investig. 2023, 53, 269–306. [Google Scholar] [CrossRef]
- Lee, T.J.; Kim, D.; Kim, J.C.; Ro, S.W.; Na, D.H. Formulation Development and Pharmacokinetic Evaluation of Enteric-Coated Dexrabeprazole Tablets. J. Pharm. Investig. 2023, 53, 323–331. [Google Scholar] [CrossRef]
- Rao, T.; Tirumala, R.; Rao, P. Quantification of Tamsulosin in Human Plasma Using LC-MS/MS. J. Bioanal. Biomed. 2011, 3, 55–58. [Google Scholar] [CrossRef] [Green Version]
- Singh, D.K.; Kurmi, M.; Handa, T.; Singh, S. LC–MS/TOF, LC–MSn and H/D Exchange Studies on Solifenacin Succinate Targeted to Characterize Its Forced Degradation Products. Chromatographia 2016, 79, 159–168. [Google Scholar] [CrossRef]
- Kang, D.W.; Cho, S.; Choi, G.-W.; Cho, H.-Y. Strategies for Developing Alzheimer’s Disease Treatments: Application of Population Pharmacokinetic and Pharmacodynamic Models. J. Pharm. Investig. 2022, 52, 519–538. [Google Scholar] [CrossRef]
- Reddy, B.V.R.; Reddy, B.S.; Raman, N.V.V.S.S.; Reddy, K.S.; Rambabu, C. Development and Validation of a Specific Stability Indicating High Performance Liquid Chromatographic Methods for Related Compounds and Assay of Solifenacin Succinate. J. Chem. 2012, 2013, e412353. [Google Scholar] [CrossRef] [Green Version]
- Sheskey, P.J.; Rowe, S.R.C. Handbook of Pharmaceutical Excipients, 5th ed.; Rowe, R.C., Sheskey, P.J., Owen, S.C., Eds.; American Pharmacists Association: Washington, DC, USA, 2006. [Google Scholar]
- Park, H.; Kim, J.-S.; Hong, S.; Ha, E.-S.; Nie, H.; Zhou, Q.T.; Kim, M.-S. Tableting Process-Induced Solid-State Polymorphic Transition. J. Pharm. Investig. 2022, 52, 175–194. [Google Scholar] [CrossRef]
- Lamešić, D.; Planinšek, O.; Lavrič, Z.; Ilić, I. Spherical Agglomerates of Lactose with Enhanced Mechanical Properties. Int. J. Pharm. 2017, 516, 247–257. [Google Scholar] [CrossRef]
- Kurashima, H.; Uchida, S.; Kashiwagura, Y.; Tanaka, S.; Namiki, N. Evaluation of Weight Variation in Mini-Tablets Manufactured by a Multiple-Tip Tool. Chem. Pharm. Bull. 2020, 68, 981–988. [Google Scholar] [CrossRef]
- Jagtap, P.S.; Tagad, R.R.; Shendge, R.S. A Brief Review on Kollidon. J. Drug Deliv. Ther. 2019, 9, 493–500. [Google Scholar] [CrossRef] [Green Version]
- Seem, T.C.; Rowson, N.A.; Ingram, A.; Huang, Z.; Yu, S.; de Matas, M.; Gabbott, I.; Reynolds, G.K. Twin Screw Granulation—A Literature Review. Powder Technol. 2015, 276, 89–102. [Google Scholar] [CrossRef]
- Scholz, A.; Kostewicz, E.; Abrahamsson, B.; Dressman, J.B. Can the USP Paddle Method Be Used to Represent In-Vivo Hydrodynamics? J. Pharm. Pharmacol. 2003, 55, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.P.; Tsong, Y.; Sathe, P.; Liu, J.-P. In Vitro Dissolution Profile Comparison—Statistics and Analysis of the Similarity Factor, F2. Pharm. Res. 1998, 15, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Center for Drug Evaluation and Research. Bioanalytical Method Validation Guidance for Industry. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioanalytical-method-validation-guidance-industry (accessed on 6 April 2023).
- Smulders, R.A.; Krauwinkel, W.J.; Swart, P.J.; Huang, M. Pharmacokinetics and Safety of Solifenacin Succinate in Healthy Young Men. J. Clin. Pharmacol. 2004, 44, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Center for Drug Evaluation and Research. Bioavailability and Bioequivalence Studies Submitted in NDAs or INDs—General Considerations. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioavailability-and-bioequivalence-studies-submitted-ndas-or-inds-general-considerations (accessed on 6 April 2023).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DWT | ETT | DCT1 | |
|---|---|---|---|
| Compositions (mg) | |||
| SOL | 10.0 | 10.0 | 10.0 |
| Lactose hydrate (200 mesh) | 106.5 | 106.5 | 106.5 |
| Corn starch | 30.0 | 30.0 | 30.0 |
| Hypromellose 2910 | 2.5 | 2.5 | 2.5 |
| Distilled water | 20 | - | - |
| Ethanol | - | 25 | - |
| Magnesium stearate | 1.0 | 1.0 | 1.0 |
| Total (mg) | 150 | 150 | 150 |
| Tablets features | |||
| Drug content (%) 1 | 99.4 | 100.3 | 98.2 |
| Disintegration time (min) 2 | 7.8 ± 1.6 | 6.5 ± 2.1 | 3.5 ± 1.4 |
| Hardness (N) 3 | 87.3 ± 2.2 | 78.5 ± 6.7 | 101.0 ± 8.5 |
| Friability (%) | 0.02 | 0.15 | 0.04 |
| DCT 2 | DCT 3 | DCT 4 | DCT 5 | DCT 6 | DCT 7 | DCT 8 | DCT 9 | DCT 10 | DCT 11 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Compositions (mg) | ||||||||||
| SOL | 10.0 | 10.0 | 10.0 | 10.0 | 10.0 | 10.0 | 10.0 | 10.0 | 10.0 | 10.0 |
| Flowlac100 | 89.5 | 89.5 | - | - | - | - | - | - | - | - |
| Supertab 30GR | - | - | 89.5 | 89.5 | 89.8 | 109.8 | 99.8 | 88.3 | 87.3 | 89.8 |
| Vivapur-12 | 45.0 | - | - | - | - | - | - | - | - | - |
| MCC pH102 | - | 40.5 | - | - | - | - | - | - | - | - |
| Prosolv SMCC50 | - | - | 40.5 | - | - | - | - | - | - | - |
| Prosolv SMCC 90 | - | - | - | 40.5 | 40.0 | 20.0 | 30.0 | 40.0 | 38.0 | 38.5 |
| Sodium croscarmellose | - | 4.5 | - | - | - | - | - | - | - | - |
| Sodium starch glycolate | - | - | 3.0 | - | - | - | - | - | - | - |
| Kollidone CL | - | - | - | 3.0 | 3.0 | 3.0 | 3.0 | 4.5 | 7.5 | 3.0 |
| Aerosil 200 | - | - | - | - | - | - | - | - | - | 1.5 |
| Kollidone VA64 | 6.0 | 6.0 | 6.0 | 6.0 | 4.5 | 4.5 | 4.5 | 4.5 | 4.5 | 4.5 |
| Sodium stearyl fumarate | 2.0 | 2.0 | 2.0 | 2.0 | 2.7 | 2.7 | 2.7 | 2.7 | 2.7 | 2.7 |
| Opadry 03B640016 | 4.0 | 4.0 | 4.0 | 4.0 | 4.0 | 4.0 | 4.0 | 4.0 | 4.0 | 4.0 |
| Total (mg) | - | 154.0 | 154.0 | 154.0 | 154.0 | 154.0 | 154.0 | 154.0 | 154.0 | 154.0 |
| Tablets features | ||||||||||
| Drug content (%) 1 | 92.7 ± 1.3 | 86.7 ± 1.1 | 94.2 ± 1.0 | 97.9 ± 1.4 | 98.4 ± 1.4 | 97.4 ± 1.8 | 97.5 ± 1.0 | 99.6 ± 2.0 | 98.4 ± 0.7 | 100.1 ± 0.7 |
| Disintegration (min) 2 | 12.1 ± 1.4 | 8.6 ± 0.8 | 8.4 ± 0.3 | 7.3 ± 1.1 | 5.7 ± 0.4 | 2.3 ± 0.3 | 1.7 ± 0.3 | 5.3 ± 0.6 | 2.6 ± 0.2 | 6.7 ± 0.8 |
| Hardness (N) 3 | 103.0 ± 5.9 | 99.0 ± 4.9 | 109.8 ± 4.9 | 115.7 ± 5.9 | 98.1 ± 3.9 | 109.8 ± 3.9 | 108.9 ± 2.9 | 104.9 ± 6.9 | 102.0 ± 5.9 | 107.9 ± 9.8 |
| Friability (%) | 0.06 | 0.05 | 0.05 | 0.04 | 0.15 | 0.05 | 0.16 | 0.26 | 0.17 | 0.11 |
| LLOQ (0.2 ng/mL) | Low (0.6 ng/mL) | Medium (4.0 ng/mL) | High (40 ng/mL) | |||
|---|---|---|---|---|---|---|
| Intra | 1 | Accuracy (%) | 112.3 | 105.9 | 105.6 | 102.2 |
| Precision (C.V., %) | 4.8 | 2.2 | 3.5 | 3 | ||
| 2 | Accuracy (%) | 101.7 | 98.5 | 100.1 | 101.3 | |
| Precision (C.V., %) | 8 | 5.4 | 4.6 | 1.8 | ||
| 3 | Accuracy (%) | 99.4 | 98.4 | 96.9 | 96.8 | |
| Precision (C.V., %) | 4.5 | 4.7 | 1.4 | 1.8 | ||
| 4 | Accuracy (%) | 96.7 | 100.8 | 100 | 98 | |
| Precision (C.V., %) | 2.9 | 3.6 | 1.3 | 2.8 | ||
| 5 | Accuracy (%) | 111.2 | 107.1 | 100.7 | 99.8 | |
| Precision (C.V., %) | 2.2 | 5.9 | 1.5 | 2.8 | ||
| Inter | n = 25 | Accuracy (%) | 104.3 | 102.1 | 100.6 | 99.6 |
| Precision (C.V., %) | 7.6 | 5.6 | 3.8 | 3.1 |
| Parameters | Marketed Product | DCT11 |
|---|---|---|
| Cmax (ng/mL) | 14.9 ± 2.9 | 15.3 ± 3.1 |
| Tmax (h) | 4.6 ± 0.9 | 4.3 ± 1.0 |
| AUC0–196 h (ng·h/mL) | 685.6 ± 214.2 | 696.5 ± 220.0 |
| AUC0–∞ (ng·h/mL) | 761.0 ± 267.1 | 772.7 ± 284.0 |
| T1/2 (h) | 49.5 ± 9.4 | 48.1 ± 11.2 |
| Parameters | Marketed Product | DCT11 | T/R Ratio | 90% CI (log0.8–log1.25) |
|---|---|---|---|---|
| Cmax (ng/mL) | 1.166 ± 0.081 | 1.177 ± 0.088 | 1.01 | log 0.98–log 1.07 |
| AUC0–196 h (ng·h/mL) | 2.815 ± 0.139 | 2.822 ± 0.139 | 1.00 | log 0.98–log 1.05 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.H.; Ho, M.J.; Jeong, C.K.; Kang, M.J. Novel Bioequivalent Tablet of Solifenacin Succinate Prepared Using Direct Compression Technique for Improved Chemical Stability. Pharmaceutics 2023, 15, 1723. https://doi.org/10.3390/pharmaceutics15061723
Kim DH, Ho MJ, Jeong CK, Kang MJ. Novel Bioequivalent Tablet of Solifenacin Succinate Prepared Using Direct Compression Technique for Improved Chemical Stability. Pharmaceutics. 2023; 15(6):1723. https://doi.org/10.3390/pharmaceutics15061723
Chicago/Turabian StyleKim, Do Hwan, Myoung Jin Ho, Chan Kyu Jeong, and Myung Joo Kang. 2023. "Novel Bioequivalent Tablet of Solifenacin Succinate Prepared Using Direct Compression Technique for Improved Chemical Stability" Pharmaceutics 15, no. 6: 1723. https://doi.org/10.3390/pharmaceutics15061723