
Intranasal Microemulsion as an Innovative and Promising Alternative to the Oral Route in Improving Stiripentol Brain Targeting
 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Solubility Studies
2.3. Preparation of the Most Promising Nanometric Emulsions
2.4. Pharmaceutic Characterisation of the Selected Formulations
2.4.1. Mean Droplet Size and Polydispersity Index
2.4.2. Osmolarity, pH, and Rheology
2.4.3. Assessment of STP In Vitro Release
2.5. In Vivo Pharmacokinetic Studies
2.5.1. Animals
2.5.2. Single-Dose Pharmacokinetic Studies
2.5.3. Processing of Biological Samples and HPLC Analysis
2.5.4. Pharmacokinetics Analysis and Calculation
2.6. Statistical Analysis
3. Results
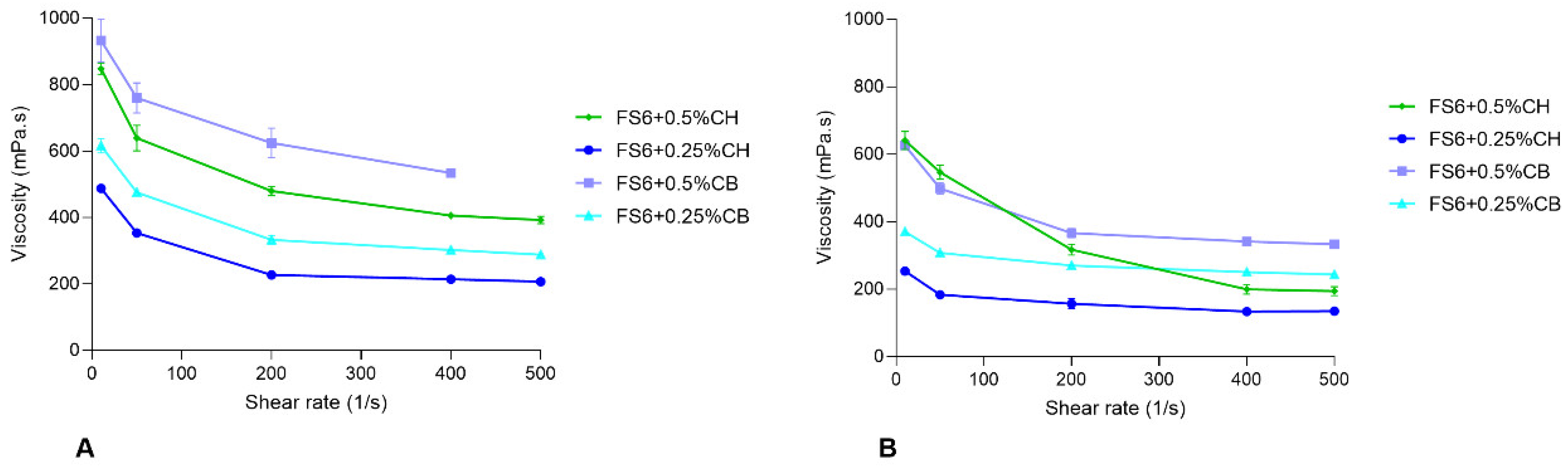
3.1. Characterization of the Selected Nanometric Emulsions
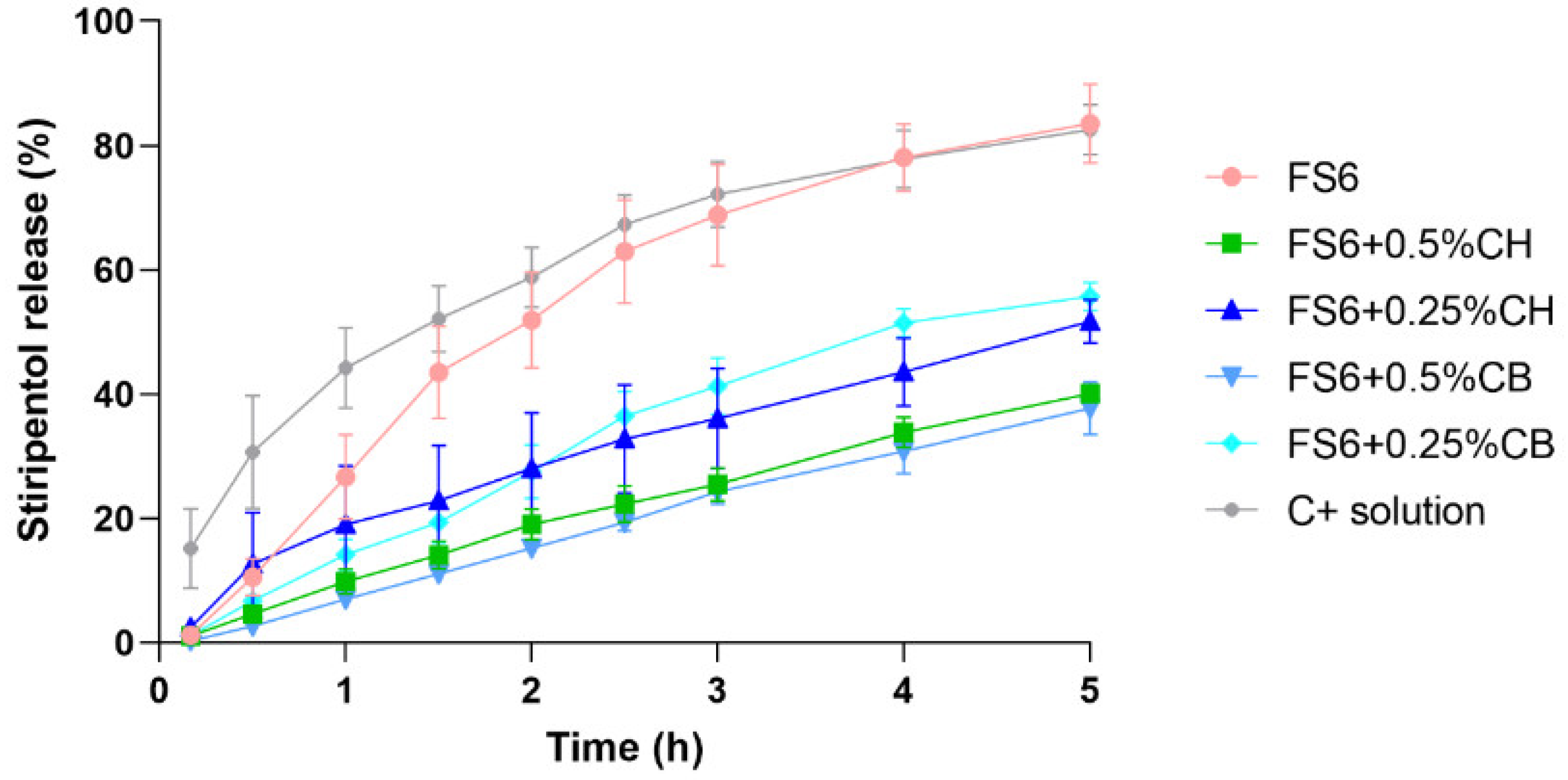
3.2. Assessment of STP In Vitro Release
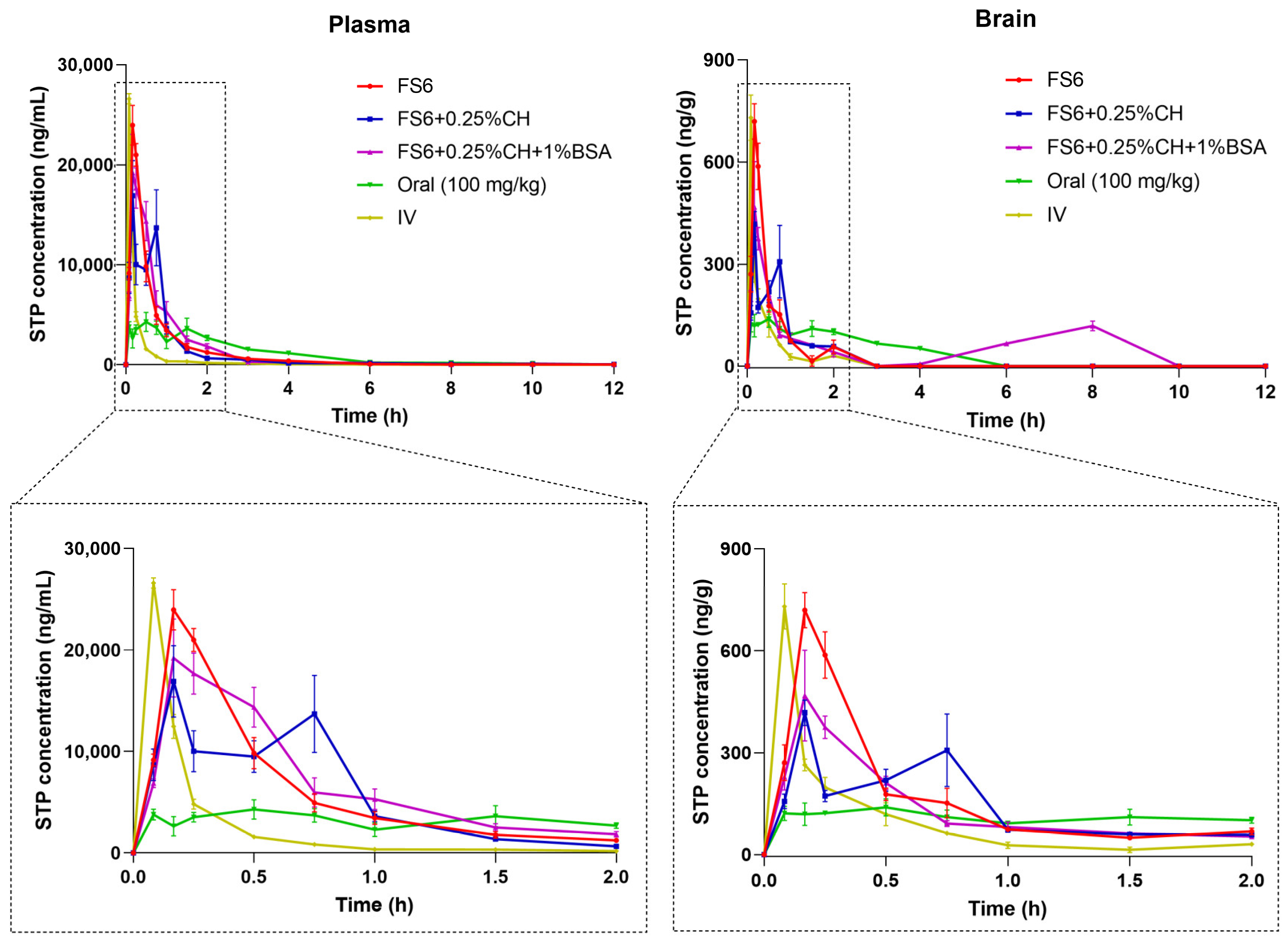
3.3. Stiripentol Pharmacokinetic Results
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Epilepsy. Available online: https://www.who.int/news-room/fact-sheets/detail/epilepsy (accessed on 14 April 2023).
- Wirrell, E.; Tinuper, P.; Perucca, E.; Moshé, S.L. Introduction to the Epilepsy Syndrome Papers. Epilepsia 2022, 63, 1330–1332. [Google Scholar] [CrossRef]
- Buck, M.L.; Goodkin, H.P. Stiripentol: A Novel Antiseizure Medication for the Management of Dravet Syndrome. Ann. Pharmacother. 2019, 53, 1136–1144. [Google Scholar] [CrossRef]
- Isom, L.L.; Knupp, K.G. Dravet Syndrome: Novel Approaches for the Most Common Genetic Epilepsy. Neurotherapeutics 2021, 18, 1524–1534. [Google Scholar] [CrossRef]
- Chiron, C. Stiripentol for the Treatment of Seizures Associated with Dravet Syndrome. Expert Rev. Neurother. 2019, 19, 301–310. [Google Scholar] [CrossRef]
- Wheless, J.W.; Fulton, S.P.; Mudigoudar, B.D. Dravet Syndrome: A Review of Current Management. Pediatr. Neurol. 2020, 107, 28–40. [Google Scholar] [CrossRef]
- Balestrini, S.; Doccini, V.; Boncristiano, A.; Lenge, M.; De Masi, S.; Guerrini, R. Efficacy and Safety of Long-Term Treatment with Stiripentol in Children and Adults with Drug-Resistant Epilepsies: A Retrospective Cohort Study of 196 Patients. Drugs Real World Outcomes 2022, 9, 451–461. [Google Scholar] [CrossRef]
- Strzelczyk, A.; Kortland, L.M.; Knake, S.; Rosenow, F. Stiripentol for the Treatment of Super-Refractory Status Epilepticus. Acta Neurol. Scand. 2015, 132, 435–439. [Google Scholar] [CrossRef]
- Violier, P.; Boyer, O.; Berthaud, R.; Dorval, G. Treatment with Stiripentol in a Patient with Primary Hyperoxaluria Type 1: Lesson for the Clinical Nephrologist. J. Nephrol. 2022, 35, 1049–1051. [Google Scholar] [CrossRef]
- Chen, Q.; Xin, M.; Wang, L.; Li, L.; Shen, Y.; Geng, Y.; Jiang, H.; Wang, Y.; Zhang, L.; Xu, Y.; et al. Inhibition of LDHA to Induce EEF2 Release Enhances Thrombocytopoiesis. Blood 2022, 139, 2958–2971. [Google Scholar] [CrossRef]
- Shin, M.C.; Lee, T.-K.; Lee, J.-C.; Kim, H.I.; Park, C.W.; Cho, J.H.; Kim, D.W.; Ahn, J.H.; Won, M.-H.; Lee, C.-H. Therapeutic Effects of Stiripentol against Ischemia-Reperfusion Injury in Gerbils Focusing on Cognitive Deficit, Neuronal Death, Astrocyte Damage and Blood Brain Barrier Leakage in the Hippocampus. Korean J. Physiol. Pharmacol. 2022, 26, 47–57. [Google Scholar] [CrossRef]
- Lu, R.; Liu, S.; Wang, Q.; Li, X. Nanoemulsions as Novel Oral Carriers of Stiripentol: Insights into the Protective Effect and Absorption Enhancement. Int. J. Nanomed. 2015, 10, 4937–4946. [Google Scholar] [CrossRef]
- Meirinho, S.; Rodrigues, M.; Fortuna, A.; Falcão, A.; Alves, G. Liquid Chromatographic Methods for Determination of the New Antiepileptic Drugs Stiripentol, Retigabine, Rufinamide and Perampanel: A Comprehensive and Critical Review. J. Pharm. Anal. 2021, 11, 405–421. [Google Scholar] [CrossRef]
- European Medicines Agency. Stiripentol (Diacomit). Scientific Discussion. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Scientific_Discussion/human/000664/WC500036521.pdf (accessed on 14 April 2023).
- Dai, Q.; Zhang, P.; Jin, Y.; Tang, M.; Shen, M.; Xu, S.; Huang, S.; Chen, Y. Using Self-Nanoemulsifying System to Improve Oral Bioavailability of a Pediatric Antiepileptic Agent Stiripentol: Formulation and Pharmacokinetics Studies. AAPS PharmSciTech 2020, 21, 192. [Google Scholar] [CrossRef]
- European Medicines Agency. Stiripentol (Diacomit). Summary of Product Characteristics. Available online: https://www.ema.europa.eu/documents/product-information/diacomit-epar-product-information_en.pdf (accessed on 14 April 2023).
- Reimers, A.; Berg, J.A. Reference Ranges for Antiepileptic Drugs Revisited: A Practical Approach to Establish National Guidelines Reference Ranges for Antiepileptic Drugs Revisited: A Practical Approach to Establish National Guidelines. Drug Des. Dev. Ther. 2018, 12, 271–280. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, H.; Zhang, T.; Zhou, X.; Wu, B. Exploring the Potential of Self-Assembled Mixed Micelles in Enhancing the Stability and Oral Bioavailability of an Acid-Labile Drug. Eur. J. Pharm. Sci. 2014, 62, 301–308. [Google Scholar] [CrossRef]
- Afifi, S. Solid Dispersion Approach Improving Dissolution Rate of Stiripentol: A Novel Antiepileptic Drug. Iran. J. Pharm. Res. 2015, 14, 1001–1014. [Google Scholar] [CrossRef]
- Meirinho, S.; Rodrigues, M.; Santos, A.O.; Falcão, A.; Alves, G. Self-Emulsifying Drug Delivery Systems: An Alternative Approach to Improve Brain Bioavailability of Poorly Water-Soluble Drugs through Intranasal Administration. Pharmaceutics 2022, 14, 1487. [Google Scholar] [CrossRef]
- Kapoor, M.; Cloyd, J.C.; Siegel, R.A. A Review of Intranasal Formulations for the Treatment of Seizure Emergencies. J. Control. Release 2016, 237, 147–159. [Google Scholar] [CrossRef]
- Costa, C.; Moreira, J.N.; Amaral, M.H.; Sousa Lobo, J.M.; Silva, A.C. Nose-to-Brain Delivery of Lipid-Based Nanosystems for Epileptic Seizures and Anxiety Crisis. J. Control. Release 2019, 295, 187–200. [Google Scholar] [CrossRef]
- Keller, L.A.; Merkel, O.; Popp, A. Intranasal Drug Delivery: Opportunities and Toxicologic Challenges during Drug Development. Drug Deliv. Transl. Res. 2021, 12, 735–757. [Google Scholar] [CrossRef]
- De Gaetano, F.; d’Avanzo, N.; Mancuso, A.; De Gaetano, A.; Paladini, G.; Caridi, F.; Venuti, V.; Paolino, D.; Ventura, C.A. Chitosan/Cyclodextrin Nanospheres for Potential Nose-to-Brain Targeting of Idebenone. Pharmaceuticals 2022, 15, 1206. [Google Scholar] [CrossRef]
- Oliveira, P.; Fortuna, A.; Alves, G.; Falcão, A. Drug-Metabolizing Enzymes and Efflux Transporters in Nasal Epithelium: Influence on the Bioavailability of Intranasally Administered Drugs. Curr. Drug Metab. 2016, 17, 628–647. [Google Scholar] [CrossRef]
- Froelich, A.; Osmałek, T.; Jadach, B.; Puri, V.; Michniak-Kohn, B. Microemulsion-Based Media in Nose-to-Brain Drug Delivery. Pharmaceutics 2021, 13, 201. [Google Scholar] [CrossRef]
- Bonferoni, M.C.; Rossi, S.; Sandri, G.; Ferrari, F.; Gavini, E.; Rassu, G.; Giunchedi, P. Nanoemulsions for “Nose-to-Brain” Drug Delivery. Pharmaceutics 2019, 11, 84. [Google Scholar] [CrossRef]
- Gupta, S.; Kesarla, R.; Omri, A. Formulation Strategies to Improve the Bioavailability of Poorly Absorbed Drugs with Special Emphasis on Self-Emulsifying Systems. ISRN Pharm. 2013, 2013, 848043. [Google Scholar] [CrossRef]
- Bahadur, S.; Pardhi, D.M.; Rautio, J.; Rosenholm, J.M.; Pathak, K. Intranasal Nanoemulsions for Direct Nose-to-Brain Delivery of Actives for Cns Disorders. Pharmaceutics 2020, 12, 1230. [Google Scholar] [CrossRef]
- Feng, Y.; He, H.; Li, F.; Lu, Y.; Qi, J.; Wu, W. An Update on the Role of Nanovehicles in Nose-to-Brain Drug Delivery. Drug Discov. Today 2018, 23, 1079–1088. [Google Scholar] [CrossRef]
- Popescu, R.; Ghica, M.V.; Dinu-Pîrvu, C.E.; Anuța, V.; Lupuliasa, D.; Popa, L. New Opportunity to Formulate Intranasal Vaccines and Drug Delivery Systems Based on Chitosan. Int. J. Mol. Sci. 2020, 21, 5016. [Google Scholar] [CrossRef]
- Florence, K.; Manisha, L.; Kumar, B.A.; Ankur, K.; Kumar, M.A.; Ambikanandan, M. Intranasal Clobazam Delivery in the Treatment of Status Epilepticus. Pharm. Nanotechnol. 2011, 100, 692–703. [Google Scholar] [CrossRef]
- Shah, V.; Sharma, M.; Pandya, R.; Parikh, R.K.; Bharatiya, B.; Shukla, A.; Tsai, H.C. Quality by Design Approach for an in Situ Gelling Microemulsion of Lorazepam via Intranasal Route. Mater. Sci. Eng. C 2017, 75, 1231–1241. [Google Scholar] [CrossRef]
- Czapp, M.; Bankstahl, J.P.; Zibell, G.; Potschka, H. Brain Penetration and Anticonvulsant Efficacy of Intranasal Phenobarbital in Rats. Epilepsia 2008, 49, 1142–1150. [Google Scholar] [CrossRef]
- Ramreddy, S.; Janapareddi, K. Brain Targeting of Chitosan-Based Diazepam Mucoadhesive Microemulsions via Nasal Route: Formulation Optimization, Characterization, Pharmacokinetic and Pharmacodynamic Evaluation. Drug Dev. Ind. Pharm. 2019, 45, 147–158. [Google Scholar] [CrossRef]
- Pires, P.C.; Santos, L.T.; Rodrigues, M.; Alves, G.; Santos, A.O. Intranasal Fosphenytoin: The Promise of Phosphate Esters in Nose-to-Brain Delivery of Poorly Soluble Drugs. Int. J. Pharm. 2021, 592, 120040. [Google Scholar] [CrossRef]
- Falcone, J.A.; Salameh, T.S.; Yi, X.; Cordy, B.J.; Mortell, W.G.; Kabanov, A.V.; Banks, W.A. Intranasal Administration as a Route for Drug Delivery to the Brain: Evidence for a Unique Pathway for Albumin. J. Pharmacol. Exp. Ther. 2014, 351, 54–60. [Google Scholar] [CrossRef]
- Piazzini, V.; Landucci, E.; D’Ambrosio, M.; Tiozzo Fasiolo, L.; Cinci, L.; Colombo, G.; Pellegrini-Giampietro, D.E.; Bilia, A.R.; Luceri, C.; Bergonzi, M.C. Chitosan Coated Human Serum Albumin Nanoparticles: A Promising Strategy for Nose-to-Brain Drug Delivery. Int. J. Biol. Macromol. 2019, 129, 267–280. [Google Scholar] [CrossRef]
- Iqbal, R.; Ahmed, S.; Jain, G.K.; Vohora, D. Design and Development of Letrozole Nanoemulsion: A Comparative Evaluation of Brain Targeted Nanoemulsion with Free Letrozole against Status Epilepticus and Neurodegeneration in Mice. Int. J. Pharm. 2019, 565, 20–32. [Google Scholar] [CrossRef]
- Kumar, M.; Misra, A.; Mishra, A.K.; Mishra, P.P.; Pathak, K. Mucoadhesive Nanoemulsion-Based Intranasal Drug Delivery System of Olanzapine for Brain Targeting. J. Drug Target. 2008, 16, 806–814. [Google Scholar] [CrossRef]
- Zhao, F.; Tang, H.; Zhang, Q.; Yang, J.; Davey, A.K.; Wang, J. Salting-out Homogeneous Liquid–Liquid Extraction Approach Applied in Sample Pre-Processing for the Quantitative Determination of Entecavir in Human Plasma by LC–MS. J. Chromatogr. B 2012, 881–882, 119–125. [Google Scholar] [CrossRef]
- Patel, M.H.; Sawant, K.K. Self Microemulsifying Drug Delivery System of Lurasidone Hydrochloride for Enhanced Oral Bioavailability by Lymphatic Targeting: In Vitro, Caco-2 Cell Line and in Vivo Evaluation. Eur. J. Pharm. Sci. 2019, 138, 105027. [Google Scholar] [CrossRef]
- Meirinho, S.; Campos, G.; Rodrigues, M.; Fortuna, A.; Falcão, A.; Alves, G. Salting-out Assisted Liquid–Liquid Extraction Method Optimized by Design of Experiments for the Simultaneous High-Performance Liquid Chromatography Analysis of Perampanel and Stiripentol in Mouse Matrices. J. Sep. Sci. 2020, 43, 4289–4304. [Google Scholar] [CrossRef]
- Meirinho, S.; Rodrigues, M.; Ferreira, C.L.; Oliveira, R.C.; Fortuna, A.; Santos, A.O.; Falcão, A.; Alves, G. Intranasal Delivery of Lipid-Based Nanosystems as a Promising Approach for Brain Targeting of the New-Generation Antiepileptic Drug Perampanel. Int. J. Pharm. 2022, 622, 121853. [Google Scholar] [CrossRef]
- Pires, P.C.; Santos, A.O. Nanosystems in Nose-to-Brain Drug Delivery: A Review of Non-Clinical Brain Targeting Studies. J. Control. Release 2018, 270, 89–100. [Google Scholar] [CrossRef]
- Kozlovskaya, L.; Abou-Kaoud, M.; Stepensky, D. Quantitative Analysis of Drug Delivery to the Brain via Nasal Route. J. Control. Release 2014, 189, 133–140. [Google Scholar] [CrossRef]
- Bruschi, M. Mathematical Models of Drug Release. In Strategies to Modify the Drug Release from Pharmaceutical Systems; Bruschi, M.L., Ed.; Woodhead Publishing: Sawston, UK, 2015; pp. 63–86. ISBN 978-0-08-100092-2. [Google Scholar]
- Singhvi, G.; Singh, M. In-Vitro Drug Release Characterisation Models. Int. J. Pharm. Stud. Res. 2011, 2, 77–84. [Google Scholar]
- Deruyver, L.; Rigaut, C.; Lambert, P.; Haut, B.; Goole, J. The Importance of Pre-Formulation Studies and of 3D-Printed Nasal Casts in the Success of a Pharmaceutical Product Intended for Nose-to-Brain Delivery. Adv. Drug Deliv. Rev. 2021, 175, 113826. [Google Scholar] [CrossRef]
- Witika, B.A.; Poka, M.S.; Demana, P.H.; Matafwali, S.K.; Melamane, S.; Khamanga, S.M.M.; Makoni, P.A. Lipid-Based Nanocarriers for Neurological Disorders: A Review of the State-of-the-Art and Therapeutic Success to Date. Pharmaceutics 2022, 14, 836. [Google Scholar] [CrossRef]
- Kumar, A.; Pandey, A.N.; Jain, S.K. Nasal-Nanotechnology: Revolution for Efficient Therapeutics Delivery. Drug Deliv. 2016, 23, 681–693. [Google Scholar] [CrossRef]
- Motaleb, M.A.; Ibrahim, I.T.; Sayyed, M.E.; Awad, G.A.S. 131I-Trazodone: Preparation, Quality Control and in Vivo Biodistribution Study by Intranasal and Intravenous Routes as a Hopeful Brain Imaging Radiopharmaceutical. Rev. Esp. Med. Nucl. Imagen Mol. 2017, 36, 371–376. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formula Code | Oil | Hydrophilic Surfactant | Cosurfactant | Aqueous Phase (%) | STP Solubility ± SD (mg/mL) | |||
|---|---|---|---|---|---|---|---|---|
| Name | % | Name | % | Name | % | |||
| FS1 | Imwitor 988 | 5 | Tween 80 | 8 | PEG 400 | 2 | 85 | 1.93 ± 0.12 |
| FS2 | Imwitor 988 | 5 | Tween 80 | 25 | Propylene glycol | 25 | 45 | 4.51 ± 0.32 |
| FS3 | Imwitor 988 | 15 | Tween 80 | 35 | Ethanol + Propylene glycol | 8.5 + 8.5 | 33 | 75.62 ± 6.63 |
| FS4 | Capryol 90 | 5.6 | Kolliphor EL | 31.5 | Transcutol HP | 32.9 | 30 | 41.74 ± 3.25 |
| FS5 | Capmul MCM | 5 | Acconon CC-6 + Tween 20 | 22.5 + 7.5 | --- | --- | 65 | 31.27 ± 3.33 |
| FS6 | Capmul MCM | 15 | Kolliphor RH 40 | 26.25 | Transcutol HP | 8.75 | 50 | 79.06 ± 4.87 |
| FS7 | Mygliol 812 | 10 | Kolliphor RH 40 | 40 | Transcutol HP | 40 | 10 | 75.05 ± 4.89 |
| Formulation | Mean Size ± SEM (nm) a | PDI ± SEM a | pH | Osmolality (mOsmol/kg) b | Zero Shear Viscosity at 25 °C (mPa·s) | Zero Shear Viscosity at 32 °C (mPa·s) | ||
|---|---|---|---|---|---|---|---|---|
| Mean ± SEM | R2 | Mean ± SEM | R2 | |||||
| FS6 | 13.21 ± 0.09 | 0.066 ± 0.009 | 6.2 | 865.0 ± 16.86 | 124.1 ± 0.47 | -- | 78.9 ± 0.18 | -- |
| FS6 + 0.5%CH | n.d. | n.d. | 5.1 | 1034.3 ± 12.20 | 898.8 ± 17.43 | 0.9727 | 673.9 ± 8.41 | 0.9902 |
| FS6 + 0.25%CH | 14.15 ± 0.12 | 0.103 ± 0.003 | 5.5 | 951.3 ± 22.18 | 535.6 ± 3.86 | 0.9971 | 281.5 ± 7.82 | 0.9471 |
| FS6 + 0.5%CB | n.d. | n.d. | 6.5 | 610.7 ± 36.15 | 966.0 ± 28.50 | 0.9062 | 666.3 ± 7.35 | 0.9892 |
| FS6 + 0.25%CB | 104.86 ± 3.12 | 0.243 ± 0.014 | 6.1 | 503.0 ± 34.70 | 658.3 ± 8.15 | 0.9889 | 388.4 ± 7.08 | 0.9482 |
| FS6 + 0.25%CH + 1%BSA | 15.01 ± 0.11 | 0.222 ± 0.007 | 5.6 | 943.2 ± 12.03 | ND | ND | ND | ND |
| Formulation | Percentual Release (%) | Significance Matrix between Release Rates of the Different Formulations (p-Values) | |||||
|---|---|---|---|---|---|---|---|
| R2 | Drug Release Rate (%∙cm−2∙h) ± SD | FS6 + 0.5%CH | FS6 + 0.25%CH | FS6 + 0.5%CB | FS6 + 0.25%CB | STP Solution | |
| FS6 | 0.9099 | 27.15 ± 3.23 | 0.0005 | 0.0024 | 0.0004 | 0.0248 | 0.4367 (NS) |
| FS6 + 0.5%CH | 0.9930 | 12.56 ± 0.40 | -- | 0.0856 (NS) | 0.6229 (NS) | 0.0003 | <0.0001 |
| FS6 + 0.25%CH | 0.9655 | 14.63 ± 1.05 | -- | -- | 0.0497 | 0.0276 | 0.0002 |
| FS6 + 0.5%CB | 0.9973 | 12.32 ± 0.24 | -- | -- | -- | 0.0002 | <0.0001 |
| FS6 + 0.25%CB | 0.9722 | 18.51 ± 1.18 | -- | -- | -- | -- | 0.0016 |
| STP solution | 0.9505 | 32.71 ± 3.73 | -- | -- | -- | -- | -- |
| Pharmacokinetic Parameters a | Plasma | Brain | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| FS6 | FS6 + 0.25%CH | FS6 + 0.25%CH + 1%BSA | Oral | IV | FS6 | FS6 + 0.25%CH | FS6 + 0.25%CH + 1%BSA | Oral | IV | |
| tmax (min) | 10 | 10 | 10 | 30 | 5 | 10 | 10 | 10 | 30 | 5 |
| Cmax (ng/mL) | 23,956 | 16,905 | 19,218 | 4282 | 26,602 | 719 b | 417 b | 468 b | 39 b | 730 b |
| Cmax/Dose (ng/mL)/(mg/kg)d | 1916.5 | 1352.4 | 1537.4 | 42.8 | 2128.2 | 57.5 b | 33.4 b | 37.4 b | 0.4 b | 58.4 b |
| AUC0-t (ng.h/mL) | 14,541 | 13,485 | 16,193 | 12,267 | 5374 | 313 | 280 c | 540 c | 361 c | 153 c |
| AUC0-t/Dose (ng.h/mL)/(mg/kg)d | 1163.3 | 1078.8 | 1295.4 | 122.7 | 429.9 | 25.0 c | 22.4 c | 43.2 c | 3.6 c | 12.3 c |
| AUCinf (ng.h/mL) | 14,593 | 13,760 | 16,241 | 12,333 | 5385 | 364 c | 346 c | -- | 526 c | 180 c |
| AUCinf/Dose (ng.h/mL)/(mg/kg)d | 1167.5 | 1100.76 | 1299.25 | 123.33 | 430.80 | 29.10 c | 27.65 c | -- | 5.26 c | 14.37 c |
| AUCextrap (%) | 0.359 | 1.99 | 0.29 | 0.53 | 0.20 | 14.02 | 18.97 | -- | 31.4 | 14.60 |
| kel (h−1) | 0.489 | 0.174 | 0.544 | 0.409 | 0.575 | 1.12 | 0.886 | -- | 0.316 | 2.389 |
| t1/2el (h) | 1.42 | 3.97 | 1.27 | 1.69 | 1.21 | 0.62 | 0.78 | -- | 2.19 | 0.29 |
| MRT (h) | 1.03 | 1.44 | 1.17 | 2.48 | 0.51 | 0.83 | 1.17 | -- | 3.43 | 0.38 |
| F (%) | Frel (%) | AUC0-t Brain/Plasma | DTE (%) | DTP (%) | %Bbrain IN/IV | %Bbrain IN/Oral | |
|---|---|---|---|---|---|---|---|
| IN FS6 | 271.0 | 946.6 | 0.0215 | 75.3 | −32.7 | 203.8 | 693.4 |
| IN FS6 + 0.25%CH | 255.5 | 892.6 | 0.0208 | 72.6 | −37.5 | 182.6 | 620.9 |
| IN FS6 + 0.25%CH + 1%BSA | 301.6 | 1053.5 | 0.0334 | 116.9 | 14.5 | 352.2 | 1198.2 |
| Oral | 26.6 | -- | 0.0285 | -- | -- | -- | -- |
| IV | -- | -- | 0.0294 | -- | -- | -- | -- |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meirinho, S.; Rodrigues, M.; Santos, A.O.; Falcão, A.; Alves, G. Intranasal Microemulsion as an Innovative and Promising Alternative to the Oral Route in Improving Stiripentol Brain Targeting. Pharmaceutics 2023, 15, 1641. https://doi.org/10.3390/pharmaceutics15061641
Meirinho S, Rodrigues M, Santos AO, Falcão A, Alves G. Intranasal Microemulsion as an Innovative and Promising Alternative to the Oral Route in Improving Stiripentol Brain Targeting. Pharmaceutics. 2023; 15(6):1641. https://doi.org/10.3390/pharmaceutics15061641
Chicago/Turabian StyleMeirinho, Sara, Márcio Rodrigues, Adriana O. Santos, Amílcar Falcão, and Gilberto Alves. 2023. "Intranasal Microemulsion as an Innovative and Promising Alternative to the Oral Route in Improving Stiripentol Brain Targeting" Pharmaceutics 15, no. 6: 1641. https://doi.org/10.3390/pharmaceutics15061641