
Increased Bone Marrow Uptake and Accumulation of Very-Late Antigen-4 Targeted Lipid Nanoparticles
 , , and
, , and
Abstract
:1. Introduction
2. Material and Methods
2.1. siRNA LNPs Preparation
2.2. Particle Size Determination by Dynamic Light Scattering (DLS)
2.3. Zeta Potential Determination
2.4. Particle Size Determination by Cryo-Electron Microscopy (cryoEM)
2.5. siRNA Concentration and Encapsulation Efficiency
2.6. Click Chemistry Conjugation and Functionalization of LNPs
2.7. Cell Culture
2.8. LNP Treatment
2.9. LNP Uptake and Internal ETO Visualization
2.10. RNA Extraction, cDNA Synthesis and qPCR
2.11. Protein Extraction and Western Blot for RUNX1/ETO
2.12. Animal Experiments
2.13. In Vivo Biodistribution of LDV-LNPs and LNPs
2.14. In Vivo Circulation Time and Uptake of LDV-LNPs and LNPs by Bone Marrow Cell Populations
2.15. Flow Cytometry
2.16. Statistical Analysis
3. Results
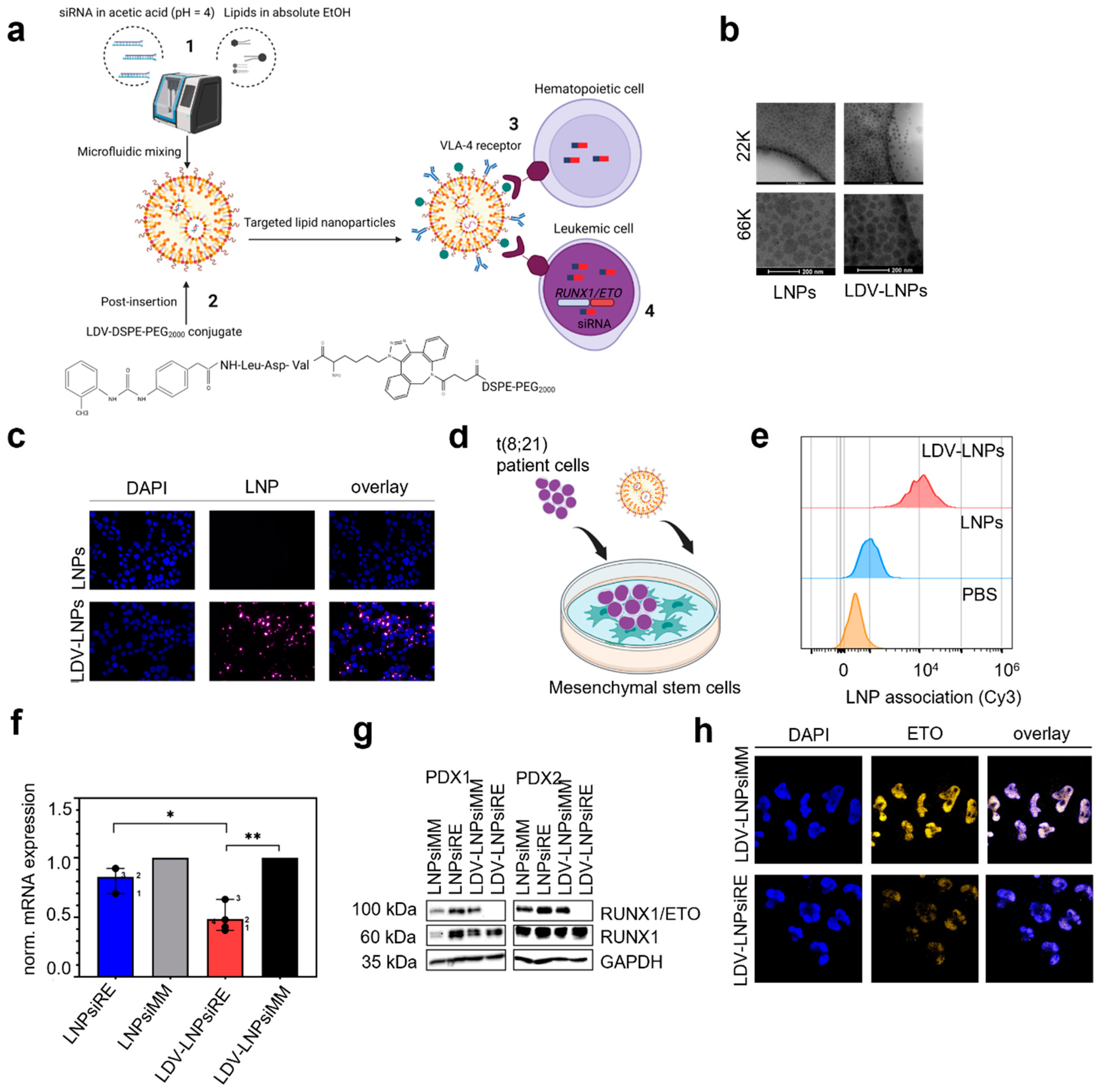
3.1. Characterization of LNPs and LDV-LNPs
3.2. In Vitro Cell Specificity and Delivery Efficacy of LDV-LNPs
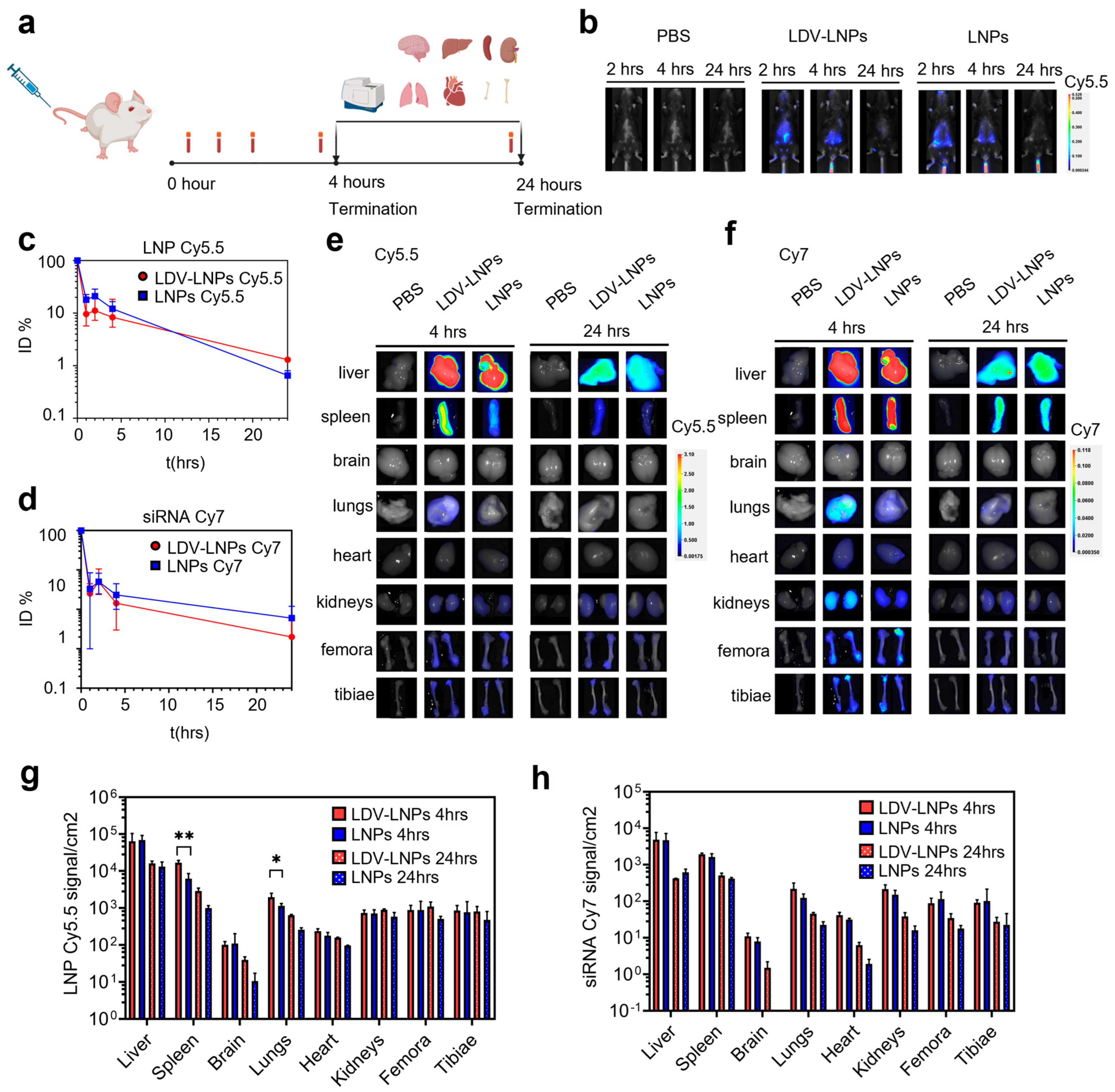
3.3. Pharmacokinetics and In Vivo Biodistribution of LDV-LNPs and LNPs
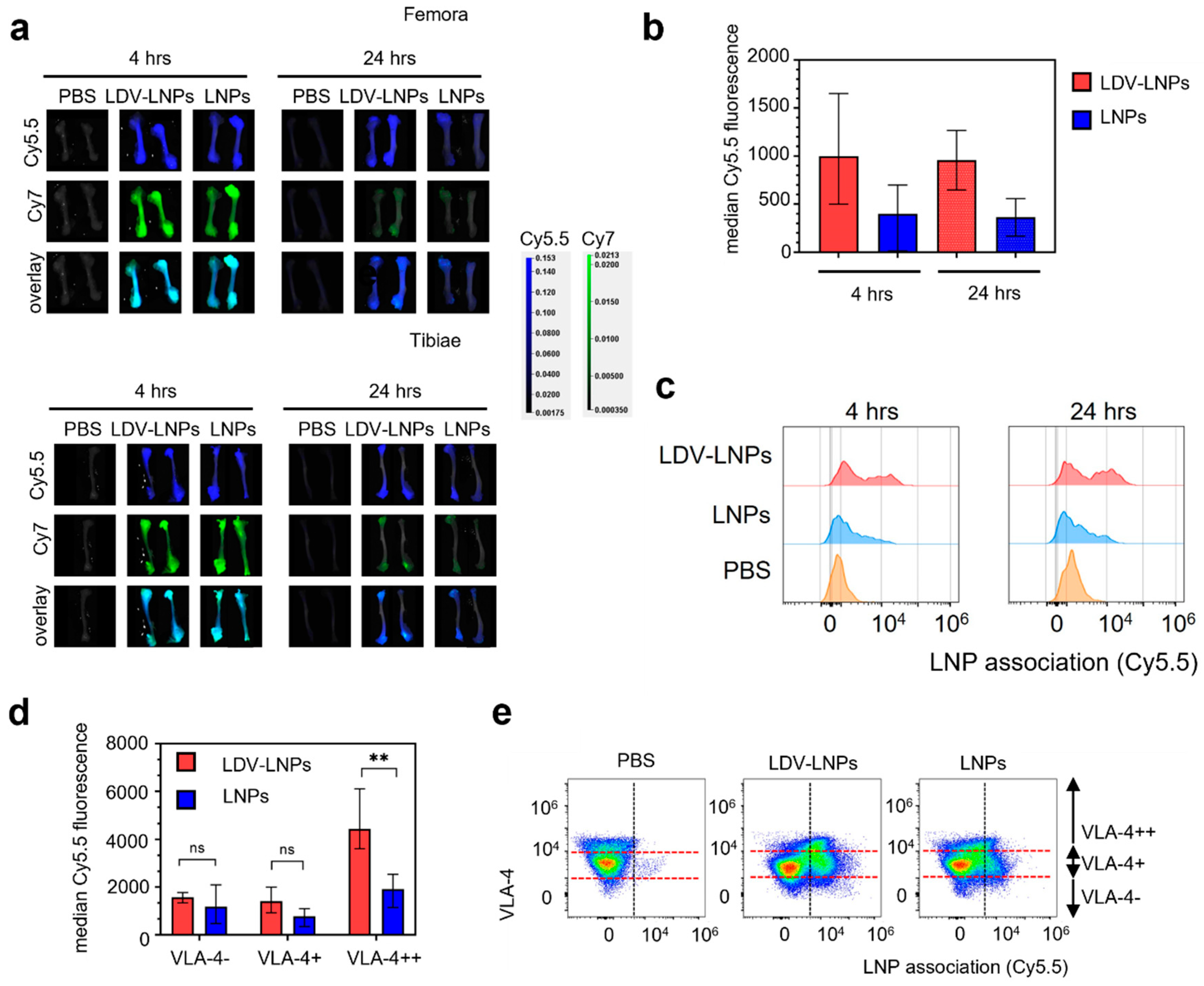
3.4. LDV-Decoration Improves LNP Uptake via VLA-4 in Hematopoietic Bone Marrow Cells
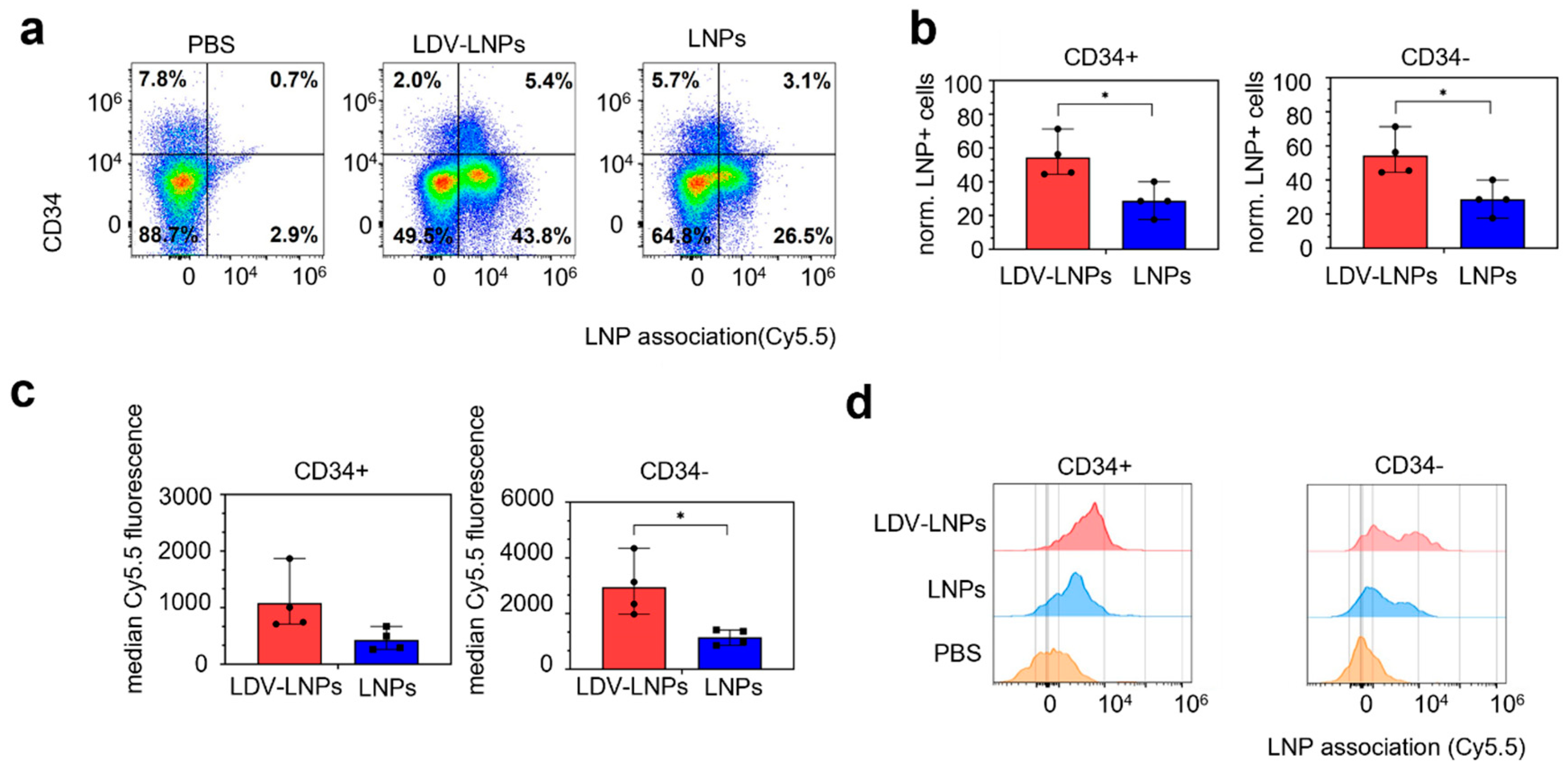
3.5. LDV-LNPs Target VLA-4-Positive Immature Myeloid Cells in the Bone Marrow
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rothdiener, M.; Müller, D.; Castro, P.G.; Scholz, A.; Schwemmlein, M.; Fey, G.; Heidenreich, O.; Kontermann, R.E. Targeted delivery of SiRNA to CD33-positive tumor cells with liposomal carrier systems. J. Control. Release 2010, 144, 251–258. [Google Scholar] [CrossRef]
- Gavrilov, K.; Seo, Y.-E.; Tietjen, G.T.; Cui, J.; Cheng, C.J.; Saltzman, W.M. Enhancing potency of siRNA targeting fusion genes by optimization outside of target sequence. Proc. Natl. Acad. Sci. USA 2015, 112, E6597–E6605. [Google Scholar] [CrossRef] [PubMed]
- Jyotsana, N.; Sharma, A.; Chaturvedi, A.; Budida, R.; Scherr, M.; Kuchenbauer, F.; Lindner, R.; Noyan, F.; Sühs, K.-W.; Stangel, M.; et al. Lipid nanoparticle-mediated siRNA delivery for safe targeting of human CML in vivo. Ann. Hematol. 2019, 98, 1905–1918. [Google Scholar] [CrossRef]
- Mohanty, S.; Jyotsana, N.; Sharma, A.; Kloos, A.; Gabdoulline, R.; Othman, B.; Lai, C.K.; Schottmann, R.; Mandhania, M.; Schmoellerl, J.; et al. Targeted Inhibition of the NUP98-NSD1 Fusion Oncogene in Acute Myeloid Leukemia. Cancers 2020, 12, 2766. [Google Scholar] [CrossRef] [PubMed]
- Issa, H.; Swart, L.E.; Rasouli, M.; Ashtiani, M.; Nakjang, S.; Jyotsana, N.; Schuschel, K.; Heuser, M.; Blair, H.; Heidenreich, O. Nanoparticle-mediated targeting of the fusion gene RUNX1/ETO in t(8;21)-positive acute myeloid leukaemia. Leukemia 2023, 37, 820–834. [Google Scholar] [CrossRef]
- Jackson, A.L.; Burchard, J.; Leake, D.; Reynolds, A.; Schelter, J.; Guo, J.; Johnson, J.M.; Lim, L.; Karpilow, J.; Nichols, K.; et al. Position-specific chemical modification of siRNAs reduces “off-target” transcript silencing. RNA 2006, 12, 1197–1205. [Google Scholar] [CrossRef]
- Soutschek, J.; Akinc, A.; Bramlage, B.; Charisse, K.; Constien, R.; Donoghue, M.; Elbashir, S.; Geick, A.; Hadwiger, P.; Harborth, J.; et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 2004, 432, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Marques, J.T.; Williams, B.R.G. Activation of the mammalian immune system by siRNAs. Nat. Biotechnol. 2005, 23, 1399–1405. [Google Scholar] [CrossRef]
- Kim, D.H.; Rossi, J.J. Strategies for silencing human disease using RNA interference. Nat. Rev. Genet. 2007, 8, 173–184. [Google Scholar] [CrossRef]
- Aigner, A. Nonviral in vivo delivery of therapeutic small interfering RNAs. Curr. Opin. Mol. Ther. 2007, 9, 345–352. [Google Scholar]
- Lee, S.J.; Son, S.; Yhee, J.Y.; Choi, K.; Kwon, I.C.; Kim, S.H.; Kim, K. Structural modification of siRNA for efficient gene silencing. Biotechnol. Adv. 2013, 31, 491–503. [Google Scholar] [CrossRef]
- Liu, P.; Chen, G.; Zhang, J. A Review of Liposomes as a Drug Delivery System: Current Status of Approved Products, Regulatory Environments, and Future Perspectives. Molecules 2022, 27, 1372. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, Z.; Shah, A.; Siddiq, M.; Kraatz, H.-B. Polymeric micelles as drug delivery vehicles. RSC Adv. 2014, 4, 17028–17038. [Google Scholar] [CrossRef]
- Paunovska, K.; Loughrey, D.; Dahlman, J.E. Drug delivery systems for RNA therapeutics. Nat. Rev. Genet. 2022, 23, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Liu, X.-Y.; Lu, A.; Wang, X.-Y.; Jiang, L.-X.; Wang, J.-C. Non-viral vectors for RNA delivery. J. Control. Release 2022, 342, 241–279. [Google Scholar] [CrossRef]
- Sato, Y.; Kinami, Y.; Hashiba, K.; Harashima, H. Different kinetics for the hepatic uptake of lipid nanoparticles between the apolipoprotein E/low density lipoprotein receptor and the N-acetyl-d-galactosamine/asialoglycoprotein receptor pathway. J. Control. Release 2020, 322, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Querbes, W.; De, S.; Qin, J.; Frank-Kamenetsky, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Cantley, W.L.; Dorkin, J.R.; et al. Targeted Delivery of RNAi Therapeutics With Endogenous and Exogenous Ligand-Based Mechanisms. Mol. Ther. 2010, 18, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Kuipers, F.; Havekes, L.M.; Havinga, R.; Dontje, B.; Poelstra, K.; Scherphof, G.L.; Kamps, J.A. The role of apolipoprotein E in the elimination of liposomes from blood by hepatocytes in the mouse. Biochem. Biophys. Res. Commun. 2005, 328, 57–62. [Google Scholar] [CrossRef]
- Zhang, X.; Goel, V.; Attarwala, H.; Sweetser, M.T.; Clausen, V.A.; Robbie, G.J. Patisiran Pharmacokinetics, Pharmacodynamics, and Exposure-Response Analyses in the Phase 3 APOLLO Trial in Patients With Hereditary Transthyretin-Mediated (hATTR) Amyloidosis. J. Clin. Pharmacol. 2020, 60, 37–49. [Google Scholar] [CrossRef]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Sweetenham, J.; Masek, L. Endothelial Cell Culture; Bicknell, R., Ed.; Cambridge University Press: Cambridge, UK, 1996; pp. 23–36. [Google Scholar]
- Lorenzer, C.; Dirin, M.; Winkler, A.-M.; Baumann, V.; Winkler, J. Going beyond the liver: Progress and challenges of targeted delivery of siRNA therapeutics. J. Control. Release 2015, 203, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Abraham, W.D.; Zheng, Y.; López, S.C.B.; Luo, S.S.; Irvine, D.J. Active targeting of chemotherapy to disseminated tumors using nanoparticle-carrying T cells. Sci. Transl. Med. 2015, 7, 291ra94. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Xiao, Z.; Kamaly, N.; Farokhzad, O.C. Self-Assembled Targeted Nanoparticles: Evolution of Technologies and Bench to Bedside Translation. Accounts Chem. Res. 2011, 44, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, N.; Wu, J.; Xu, X.; Kamaly, N.; Farokhzad, O.C. Cancer nanotechnology: The impact of passive and active targeting in the era of modern cancer biology. Adv. Drug Deliv. Rev. 2014, 66, 2–25. [Google Scholar] [CrossRef]
- Peer, D.; Margalit, R. Tumor-Targeted Hyaluronan Nanoliposomes Increase the Antitumor Activity of Liposomal Doxorubicin in Syngeneic and Human Xenograft Mouse Tumor Models. Neoplasia 2004, 6, 343–353. [Google Scholar] [CrossRef]
- Dammes, N.; Goldsmith, M.; Ramishetti, S.; Dearling, J.L.J.; Veiga, N.; Packard, A.B.; Peer, D. Conformation-sensitive targeting of lipid nanoparticles for RNA therapeutics. Nat. Nanotechnol. 2021, 16, 1030–1038. [Google Scholar] [CrossRef]
- Li, H.; Huang, S.Y.; Shi, F.H.; Gu, Z.C.; Zhang, S.G.; Wei, J.F. α(4)β(7) integrin inhibitors: A patent review. Expert Opin. Ther. Pat. 2018, 28, 903–917. [Google Scholar] [CrossRef]
- Petrovic, A.; Alpdogan, O.; Willis, L.M.; Eng, J.M.; Greenberg, A.S.; Kappel, B.J.; Liu, C.; Murphy, G.J.; Heller, G.; Van Den Brink, M.R. LPAM (α4β7 integrin) is an important homing integrin on alloreactive T cells in the development of intestinal graft-versus-host disease. Blood 2004, 103, 1542–1547. [Google Scholar] [CrossRef] [PubMed]
- de la Puente, P.; Azab, A.K. Nanoparticle delivery systems, general approaches, and their implementation in multiple myeloma. Eur. J. Haematol. 2017, 98, 529–541. [Google Scholar] [CrossRef]
- Kiziltepe, T.; Ashley, J.D.; Stefanick, J.F.; Qi, Y.M.; Alves, N.J.; Handlogten, M.W.; Suckow, M.A.; Navari, R.M.; Bilgicer, B. Rationally engineered nanoparticles target multiple myeloma cells, overcome cell-adhesion-mediated drug resistance, and show enhanced efficacy in vivo. Blood Cancer J. 2012, 2, e64. [Google Scholar] [CrossRef]
- Hemler, M.E.; Elices, M.J.; Parker, C.; Takada, Y. Structure of the Integrin VLA-4 and its Cell-Cell and Cell-Matrix Adhesion Functions. Immunol. Rev. 1990, 114, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Härzschel, A.; Zucchetto, A.; Gattei, V.; Hartmann, T.N. VLA-4 Expression and Activation in B Cell Malignancies: Functional and Clinical Aspects. Int. J. Mol. Sci. 2020, 21, 2206. [Google Scholar] [CrossRef]
- Deshantri, A.K.; Fens, M.H.; Ruiter, R.W.; Metselaar, J.M.; Storm, G.; van Bloois, L.; Varela-Moreira, A.; Mandhane, S.N.; Mutis, T.; Martens, A.C.; et al. Liposomal dexamethasone inhibits tumor growth in an advanced human-mouse hybrid model of multiple myeloma. J. Control. Release 2019, 296, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Hemler, M.E. VLA proteins in the integrin family: Structures, functions, and their role on leukocytes. Annu. Rev. Immunol. 1990, 8, 365–400. [Google Scholar] [CrossRef]
- Komoriya, A.; Green, L.; Mervic, M.; Yamada, S.; Yamada, K.; Humphries, M. The minimal essential sequence for a major cell type-specific adhesion site (CS1) within the alternatively spliced type III connecting segment domain of fibronectin is leucine-aspartic acid-valine. J. Biol. Chem. 1991, 266, 15075–15079. [Google Scholar] [CrossRef]
- Baiula, M.; Spampinato, S.; Gentilucci, L.; Tolomelli, A. Novel Ligands Targeting α4β1 Integrin: Therapeutic Applications and Perspectives. Front Chem 2019, 7, 489. [Google Scholar] [CrossRef]
- Lin, K.C.; Ateeq, H.S.; Hsiung, S.H.; Chong, L.T.; Zimmerman, C.N.; Castro, A.; Lee, W.C.; Hammond, C.E.; Kalkunte, S.; Chen, L.L.; et al. Selective, tight-binding inhibitors of integrin alpha4beta1 that inhibit allergic airway responses. J. Med. Chem. 1999, 42, 920–934. [Google Scholar] [CrossRef] [PubMed]
- Gérard, E.; Meulle, A.; Feron, O.; Marchand-Brynaert, J. LDV peptidomimetics equipped with biotinylated spacer-arms: Synthesis and biological evaluation on CCRF-CEM cell line. Bioorg. Med. Chem. Lett. 2012, 22, 586–590. [Google Scholar] [CrossRef]
- Swart, L.E.; Koekman, C.; Seinen, C.; Issa, H.; Rasouli, M.; Schiffelers, R.; Heidenreich, O. A robust post-insertion method for the preparation of targeted siRNA LNPs. Int. J. Pharm. 2022, 620, 121741. [Google Scholar] [CrossRef]
- Heidenreich, O.; Krauter, J.; Riehle, H.; Hadwiger, P.; John, M.; Heil, G.; Vornlocher, H.P.; Nordheim, A. AML1/MTG8 oncogene suppression by small interfering RNAs supports myeloid differentiation of t(8;21)-positive leukemic cells. Blood 2003, 101, 3157–3163. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Tam, Y.Y.C.; Lin, P.J.; Sung, M.M.; Tam, Y.K.; Cullis, P.R. Influence of particle size on the in vivo potency of lipid nanoparticle formulations of siRNA. J. Control. Release 2016, 235, 236–244. [Google Scholar] [CrossRef]
- Pal, D.; Blair, H.J.; Elder, A.; Dormon, K.; Rennie, K.J.; Coleman, D.J.L.; Weiland, J.; Rankin, K.S.; Filby, A.; Heidenreich, O.; et al. Long-term in vitro maintenance of clonal abundance and leukaemia-initiating potential in acute lymphoblastic leukaemia. Leukemia 2016, 30, 1691–1700. [Google Scholar] [CrossRef] [PubMed]
- Lobb, R.R.; Hemler, M.E. The pathophysiologic role of alpha 4 integrins in vivo. J. Clin. Investig. 1994, 94, 1722–1728. [Google Scholar] [CrossRef] [PubMed]
- van de Water, F.M.; Boerman, O.C.; Wouterse, A.C.; Peters, J.G.; Russel, F.G.; Masereeuw, R. Intravenously administered short interfering RNA accumulates in the kidney and selectively suppresses gene function in renal proximal tubules. Drug Metab. Dispos. 2006, 34, 1393–1397. [Google Scholar] [CrossRef]
- Bumcrot, D.; Manoharan, M.; Koteliansky, V.; Sah, D.W.Y. RNAi therapeutics: A potential new class of pharmaceutical drugs. Nat. Chem. Biol. 2006, 2, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Santel, A.; Aleku, M.; Keil, O.; Endruschat, J.; Esche, V.; Fisch, G.; Dames, S.; Löffler, K.; Fechtner, M.; Arnold, W.; et al. A novel siRNA-lipoplex technology for RNA interference in the mouse vascular endothelium. Gene Ther. 2006, 13, 1222–1234. [Google Scholar] [CrossRef]
- Sou, K.; Klipper, R.; Goins, B.; Tsuchida, E.; Phillips, W.T. Circulation Kinetics and Organ Distribution of Hb-Vesicles Developed as a Red Blood Cell Substitute. Experiment 2005, 312, 702–709. [Google Scholar] [CrossRef]
- Miteva, M.; Kirkbride, K.C.; Kilchrist, K.V.; Werfel, T.A.; Li, H.; Nelson, C.E.; Gupta, M.K.; Giorgio, T.D.; Duvall, C.L. Tuning PEGylation of mixed micelles to overcome intracellular and systemic siRNA delivery barriers. Biomaterials 2015, 38, 97–107. [Google Scholar] [CrossRef]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef]
- Zalba, S.; Hagen, T.L.M.T.; Burgui, C.; Garrido, M.J. Stealth nanoparticles in oncology: Facing the PEG dilemma. J. Control. Release 2022, 351, 22–36. [Google Scholar] [CrossRef]
- Curcio, M.; Brindisi, M.; Cirillo, G.; Frattaruolo, L.; Leggio, A.; Rago, V.; Nicoletta, F.P.; Cappello, A.R.; Iemma, F. Smart Lipid–Polysaccharide Nanoparticles for Targeted Delivery of Doxorubicin to Breast Cancer Cells. Int. J. Mol. Sci. 2022, 23, 2386. [Google Scholar] [CrossRef] [PubMed]
- De Leo, V.; Milano, F.; Agostiano, A.; Catucci, L. Recent Advancements in Polymer/Liposome Assembly for Drug Delivery: From Surface Modifications to Hybrid Vesicles. Polymers 2021, 13, 1027. [Google Scholar] [CrossRef] [PubMed]
- LoPresti, S.T.; Arral, M.L.; Chaudhary, N.; Whitehead, K.A. The replacement of helper lipids with charged alternatives in lipid nanoparticles facilitates targeted mRNA delivery to the spleen and lungs. J. Control. Release 2022, 345, 819–831. [Google Scholar] [CrossRef] [PubMed]
- Weber, B.; Seidl, C.; Schwiertz, D.; Scherer, M.; Bleher, S.; Süss, R.; Barz, M. Polysarcosine-Based Lipids: From Lipopolypeptoid Micelles to Stealth-Like Lipids in Langmuir Blodgett Monolayers. Polymers 2016, 8, 427. [Google Scholar] [CrossRef]
- Hu, Y.; Hou, Y.; Wang, H.; Lu, H. Polysarcosine as an Alternative to PEG for Therapeutic Protein Conjugation. Bioconjugate Chem. 2018, 29, 2232–2238. [Google Scholar] [CrossRef]
- Wang, X.; Liu, S.; Sun, Y.; Yu, X.; Lee, S.M.; Cheng, Q.; Wei, T.; Gong, J.; Robinson, J.; Di Zhang, D.; et al. Preparation of selective organ-targeting (SORT) lipid nanoparticles (LNPs) using multiple technical methods for tissue-specific mRNA delivery. Nat. Protoc. 2023, 18, 265–291. [Google Scholar] [CrossRef]
- Dilliard, S.A.; Cheng, Q.; Siegwart, D.J. On the mechanism of tissue-specific mRNA delivery by selective organ targeting nanoparticles. Proc. Natl. Acad. Sci. USA 2021, 118, e2109256118. [Google Scholar] [CrossRef]
- Dahlman, J.E.; Kauffman, K.J.; Xing, Y.; Shaw, T.E.; Mir, F.F.; Dlott, C.C.; Langer, R.; Anderson, D.G.; Wang, E.T. Barcoded nanoparticles for high throughput in vivo discovery of targeted therapeutics. Proc. Natl. Acad. Sci. USA 2017, 114, 2060–2065. [Google Scholar] [CrossRef]
- Moghimi, S.M.; Patel, H.M. Opsonophagocytosis of liposomes by peritoneal macrophages and bone marrow reticuloendothelial cells. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 1992, 1135, 269–274. [Google Scholar] [CrossRef]
- Nagayasu, A.; Uchiyama, K.; Nishida, T.; Yamagiwa, Y.; Kawai, Y.; Kiwada, H. Is control of distribution of liposomes between tumors and bone marrow possible? Biochim. Biophys. Acta 1996, 1278, 29–34. [Google Scholar] [CrossRef]
- Ouyang, B.; Poon, W.; Zhang, Y.-N.; Lin, Z.P.; Kingston, B.R.; Tavares, A.J.; Zhang, Y.; Chen, J.; Valic, M.S.; Syed, A.M.; et al. The dose threshold for nanoparticle tumour delivery. Nat. Mater. 2020, 19, 1362–1371. [Google Scholar] [CrossRef] [PubMed]
- Biscans, A.; Ly, S.; McHugh, N.; Cooper, D.A.; Khvorova, A. Engineered ionizable lipid siRNA conjugates enhance endosomal escape but induce toxicity in vivo. J. Control. Release 2022, 349, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Ashley, J.D.; Stefanick, J.F.; Schroeder, V.A.; Suckow, M.A.; Alves, N.J.; Suzuki, R.; Kikuchi, S.; Hideshima, T.; Anderson, K.C.; Kiziltepe, T.; et al. Liposomal carfilzomib nanoparticles effectively target multiple myeloma cells and demonstrate enhanced efficacy in vivo. J. Control. Release 2014, 196, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Varela-Moreira, A.; van Straten, D.; van Leur, H.F.; Ruiter, R.W.; Deshantri, A.K.; Hennink, W.E.; Fens, M.H.; Groen, R.W.; Schiffelers, R.M. Polymeric micelles loaded with carfilzomib increase tolerability in a humanized bone marrow-like scaffold mouse model. Int. J. Pharm. X 2020, 2, 100049. [Google Scholar] [CrossRef]
- Mitroulis, I.; Kalafati, L.; Bornhäuser, M.; Hajishengallis, G.; Chavakis, T. Regulation of the Bone Marrow Niche by Inflammation. Front. Immunol. 2020, 11, 1540. [Google Scholar] [CrossRef]
- Leimkuhler, N.B.; Schneider, R.K. Inflammatory bone marrow microenvironment. Hematol. Am. Soc. Hematol. Educ. Program 2019, 2019, 294–302. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Components | Particle Size (nm) | PDI | Zeta Potential (mV) | Encapsulation Efficiency (%) |
|---|---|---|---|---|---|
| LNP | Dlin-MC3-DMA/DSPC/Cholesterol/DMG-PEG2000 = 50/10/38.5/1.5 | 66 (57, 83) | 0.14 (0.12, 0.16) | −2.5 (−4.2, −1.0) | 94 (93, 95) |
| LDV-LNP | Dlin-MC3-DMA/DSPC/Cholesterol/DMG-PEG2000/LDV-azide-DBCO-DSPE-PEG2000 = 50/10/38.5/1.5/0.1 | 94 (87, 105) | 0.26 (0.20, 0.35) | −4.1 (−6.3, −2.3) | 93 (91, 94) |
| LNP-Cy7 | Dlin-MC3-DMA/DSPC/Cholesterol/DMG-PEG2000/DBCO-DSPE-PEG2000 = 50/10/38.5/1.5/0.1 | 69 (68, 70) | 0.08 (0.07, 0.09) | −0.3 (−0.2, −0.3) | 97 |
| LDV-LNP-Cy7 | Dlin-MC3-DMA/DSPC/Cholesterol/DMG-PEG2000/LDV-azide-DBCO-DSPE-PEG2000 = 50/10/38.5/1.5/0.1 | 74 (73, 75) | 0.08 (0.07, 0.09) | −3.1 (−2.7, −3.7) | 97 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swart, L.E.; Fens, M.H.A.M.; van Oort, A.; Waranecki, P.; Mata Casimiro, L.D.; Tuk, D.; Hendriksen, M.; van den Brink, L.; Schweighart, E.; Seinen, C.; et al. Increased Bone Marrow Uptake and Accumulation of Very-Late Antigen-4 Targeted Lipid Nanoparticles. Pharmaceutics 2023, 15, 1603. https://doi.org/10.3390/pharmaceutics15061603
Swart LE, Fens MHAM, van Oort A, Waranecki P, Mata Casimiro LD, Tuk D, Hendriksen M, van den Brink L, Schweighart E, Seinen C, et al. Increased Bone Marrow Uptake and Accumulation of Very-Late Antigen-4 Targeted Lipid Nanoparticles. Pharmaceutics. 2023; 15(6):1603. https://doi.org/10.3390/pharmaceutics15061603
Chicago/Turabian StyleSwart, Laura E., Marcel H. A. M. Fens, Anita van Oort, Piotr Waranecki, L. Daniel Mata Casimiro, David Tuk, Martijn Hendriksen, Luca van den Brink, Elizabeth Schweighart, Cor Seinen, and et al. 2023. "Increased Bone Marrow Uptake and Accumulation of Very-Late Antigen-4 Targeted Lipid Nanoparticles" Pharmaceutics 15, no. 6: 1603. https://doi.org/10.3390/pharmaceutics15061603