
The Management of Parkinson’s Disease: An Overview of the Current Advancements in Drug Delivery Systems



Abstract
:1. Introduction
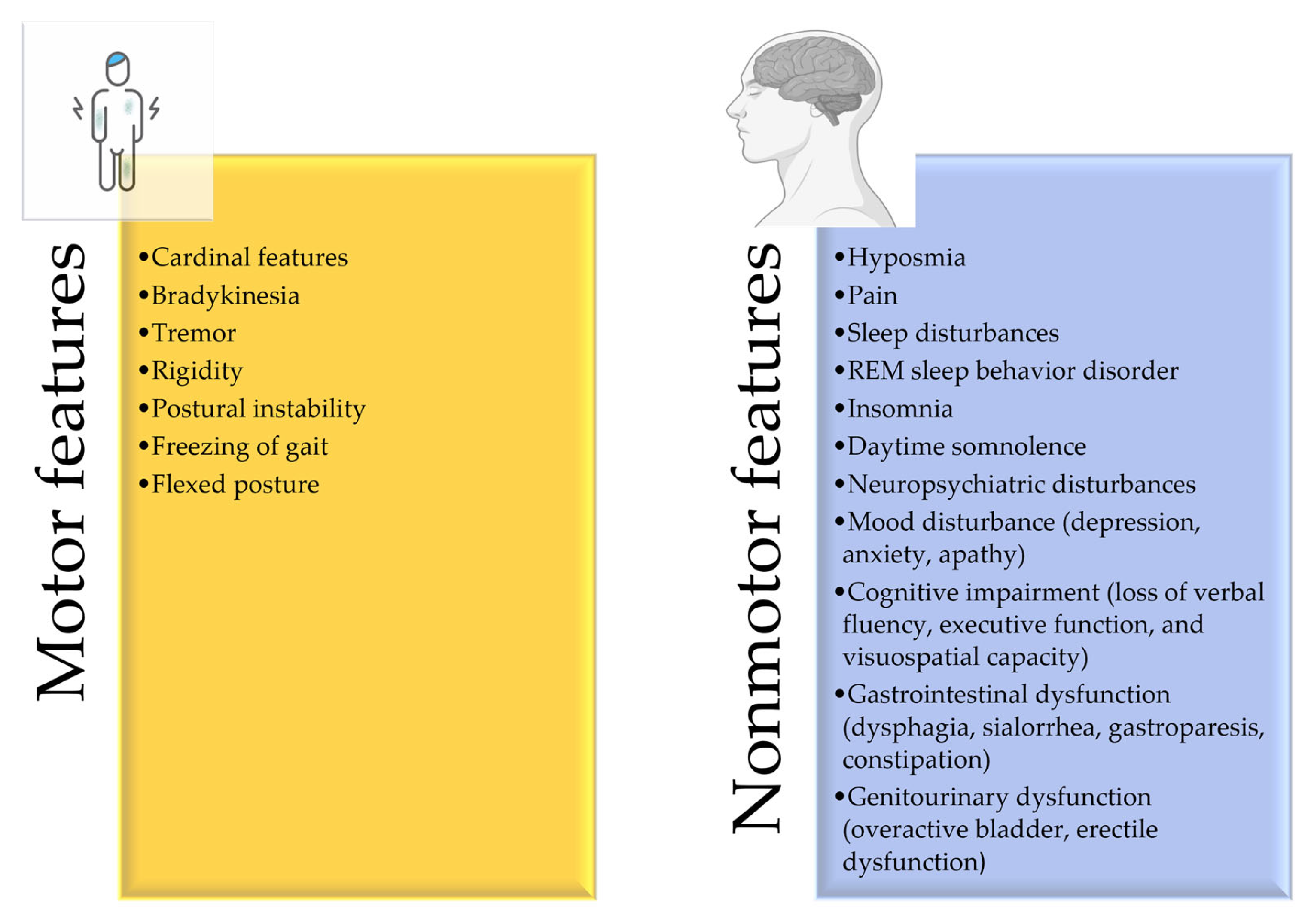
2. Disease Overview and Risk Factors
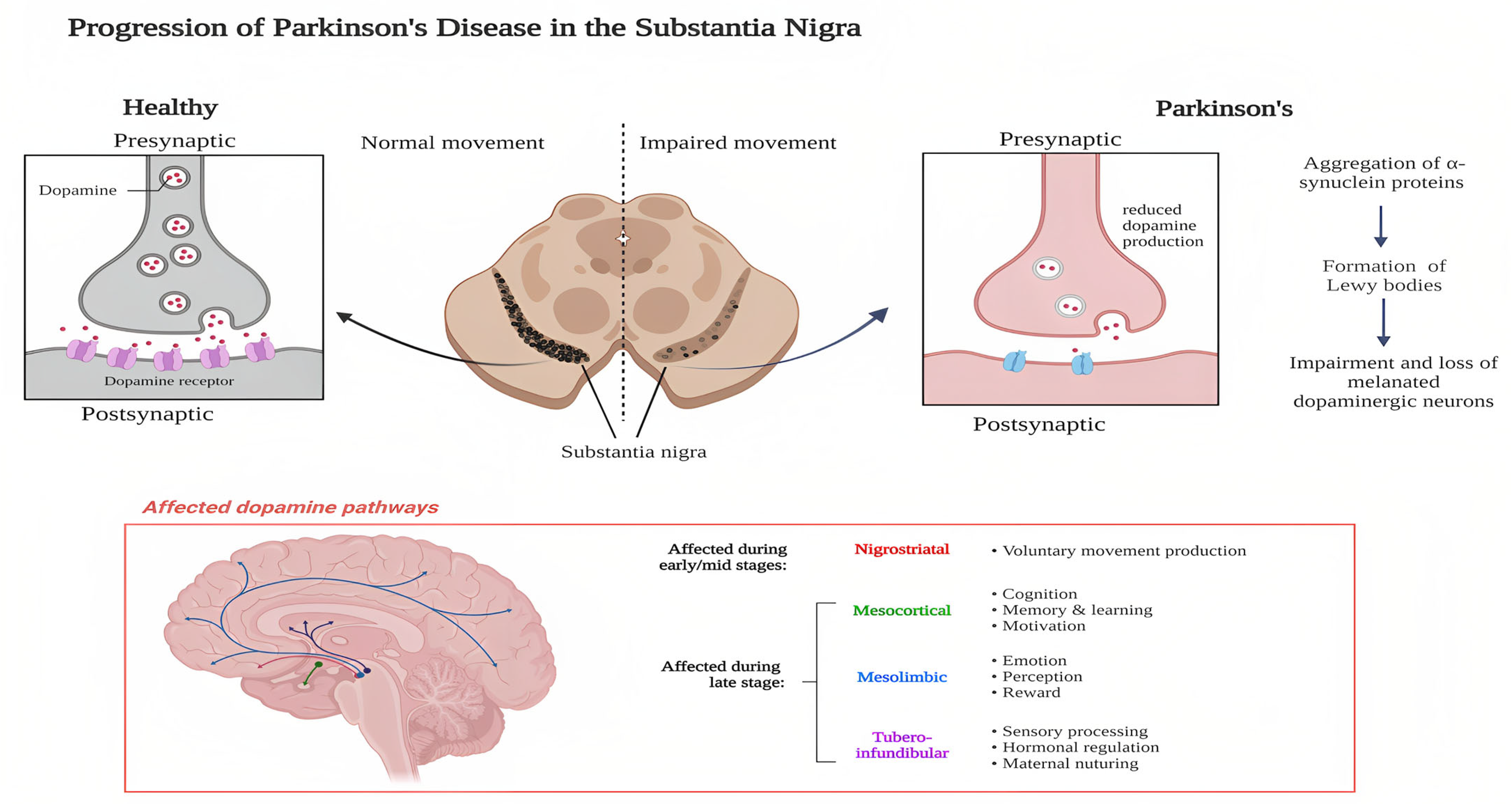
2.1. Pathophysiology
2.2. Etiology
2.3. Age
2.4. Pesticides and Herbicides
2.5. Genetic Predisposition
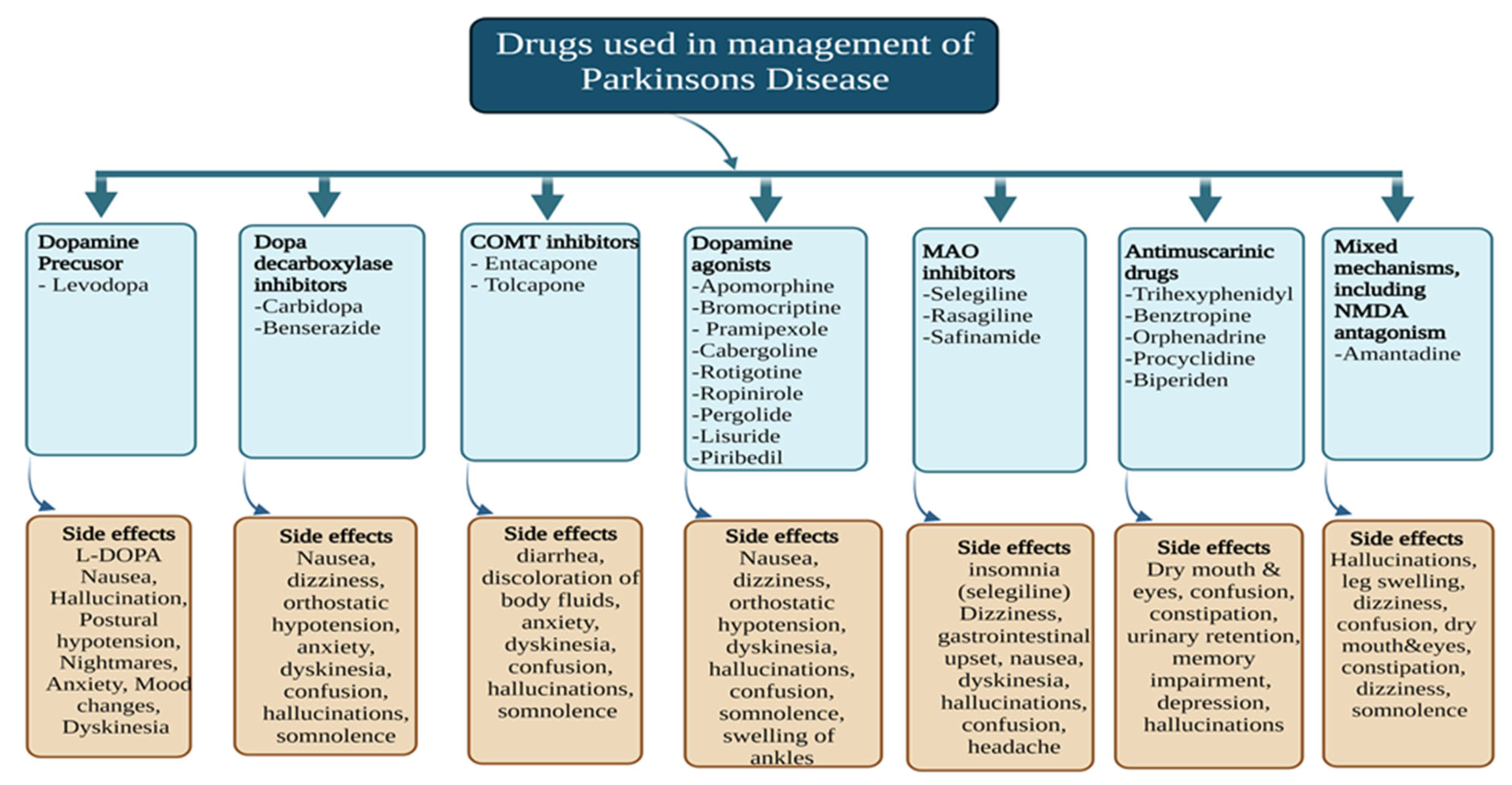
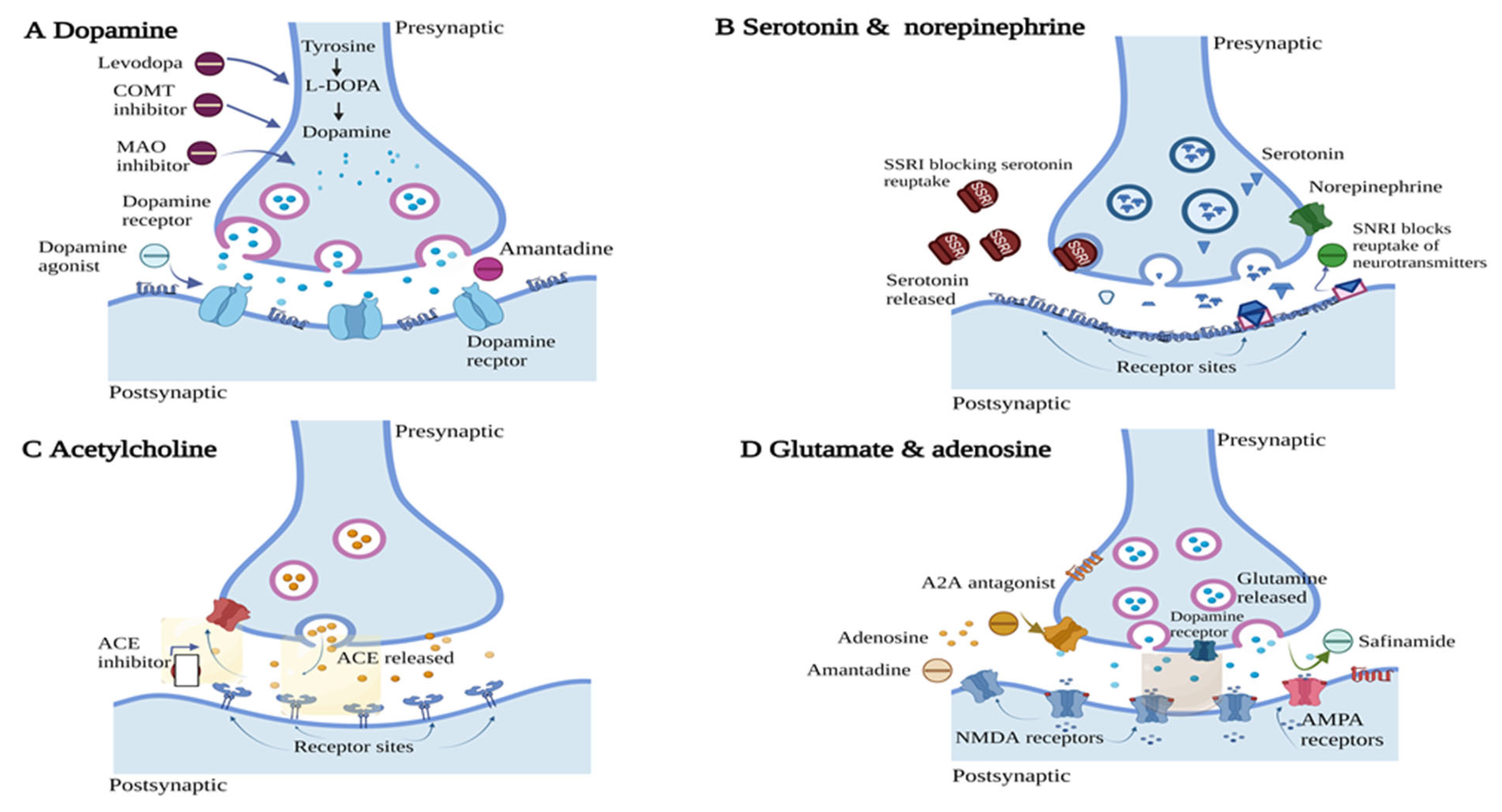
3. Clinically Approved Therapies for PD
3.1. DA Precursor

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Drug | Polymer | Characteristic Feature | Route of Administration | Reference |
|---|---|---|---|---|---|
| Nanoparticle | L-DOPA, curcumin | Block polymer consisting of polyethylene (Average molecular weight (M.w) 5 × 103 Da) and polycaprolactone (Average M.w 10.5 × 103 Da) | Enhancement in the potential of nanoparticles to cross the BBB | NA | [68] |
| Nanoparticle | L-DOPA | D,L-PLGA50:50 (M.w 7000–17,000 Da) | The intranasal formulation was prepared; extended release of up to 9 h was observed. | Intranasal | [69] |
| Crystalsomes | L-DOPA | L-PLGA (M.w 12,000 Da) | L-DOPA crystalsomes showed release for a longer period compared to traditional L-DOPA nanoparticles | Intravenous | [70] |
| Nanogel/ Hydrogel | L-DOPA | ĸ-Carrageenan | pH-dependent drug release was obtained for >11 days | NA | [71] |
| Carboxylated carbon nanotubes | L-DOPA | Single-walled carbon nanotube, chitosan (low molecular chitosan with 75–85% degree of deacetylation) | A surface coating helps in reducing the dosing frequency without increasing the dose of the drug | Oral | [72] |
| Microparticles | L-DOPA methyl ester, benserazide | D,L-PLGA 50:50 (M.w 47,000 Da) | Microspheres showed sustained release without burst release. Reduction in dyskinesia was observed due to continuous stimulation | Subcutaneous | [73] |
| Microspheres | L-DOPA-α-lipoic acid | D,L-PLGA 50:50 (intrinsic viscosity- 0.41 dL/g) | Reduced fluctuation in the level of L-DOPA in plasma | Subcutaneous | [74] |
3.2. Disease-Modifying Treatment
3.2.1. Small Molecules
3.2.2. Repurposing of Other Drugs
3.2.3. Immunotherapies
3.2.4. Gene Transfer/Viral Vector-Mediated
4. The Special Emphasis of Nanoformulations on Controlled Drug Delivery Systems for PD Management
4.1. Polymeric Nanoparticles
| Formulation Type | Drug/API | Polymer | Advantages | Route of Administration | References |
|---|---|---|---|---|---|
| Nanoparticle | Rotigotine | Maleimide-PEG -D,L-PLGA (M.w 3400–20,000 Da) and methoxy-PEG-D,L-PLGA (M.w 2000–20,000 Da) | A higher concentration of rotigotine was observed in the striatum with release up to 48 h | Intranasal | [113] |
| Nanoparticle imprinted film | Selegiline | D,L-PLGA, Ethylene-vinyl acetate | In-vivo drug release for more than 72 h was observed from the film | Transdermal | [117] |
| Nanoparticle | L-DOPA methyl ester/benserazide | D,L-PLGA50:50 (M.w 47,000 Da), PLA (M.w 83,000 Da) | The prepared formulation reduced the L-DOPA-induced dyskinesia in the dyskinetic rats | Subcutaneous | [118] |
| Nanoparticle | Pramipexole 2HCL | Chitosan (>90% deacetylation) | In-vivo and ex-vivo diffusion showed complete drug release in 24 h. Reduced motor deficit in nanoparticle-treated group compared to nasal solution and oral tablet | Intranasal | [119] |
| Nanospheres | VEGF | D,L-PLGA | VEGF encapsulated in the nanosphere could cross the BBB and showed a strong neuroprotective effect | Intravenous | [120] |
| Nanoparticle (Device) | DA | Cellulose acetate phthalate (M.w 49,000 Da) | DA was released for 30 days with a higher concentration in CSF than in plasma, which reduced the side effects of DA | Intracranial | [121] |
| Polysorbate 80 coated nanoparticle | Ropinirole HCL | Chitosan (M.w 150,000 Da) | In-vitro drug release up to 10 h was observed. In-vivo studies revealed the higher concentration of ropinirole in the brain over other highly perfused organs | Intravenous | [122] |
4.2. Solid Lipid Nanoparticles (SLNs)
4.3. Microsphere and Microcapsules
4.4. Liposomes
5. Conclusions and Future Perspective
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- The World Health Organization (WHO). Parkinson’s Disease. Available online: https://www.who.int/news-room/fact-sheets/detail/parkinson-disease (accessed on 31 January 2023).
- Lee, T.K.; Yankee, E.L. A review on Parkinson’s disease treatment. Neurol-Neuroimmunology 2021, 8, 222. [Google Scholar] [CrossRef]
- Kouli, A.; Kelli; Torsney, K.M.; Kuan, W.-L. Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: London, UK, 2021; Chapter 1; p. 3. [Google Scholar]
- Samii, A.; Nutt, J.G.; Ransom, B.R. Parkinson’s disease. Lancet 2004, 363, 1783–1793. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Okun, M.S. Diagnosis and treatment of Parkinson disease: A review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef]
- Tanner, P.W.S.K.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar]
- Cerri, S.; Mus, L.; Blandini, F. Parkinson’s disease in women and men: What’s the difference? J Parkinsons Dis. 2019, 9, 501–515. [Google Scholar] [CrossRef]
- Ben-Joseph, A.; Marshall, C.R.; Lees, A.J.; Noyce, A.J. Ethnic variation in the manifestation of Parkinson’s disease: A narrative review. J. Parkinson’s Dis. 2020, 10, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Ross, G.W.; Abbott, R.D.; Petrovitch, H.; Morens, D.M.; Grandinetti, A.; Tung, K.H.; Tanner, C.M.; Masaki, K.H.; Blanchette, P.L.; Curb, J.D.; et al. Association of coffee and caffeine intake with the risk of Parkinson disease. JAMA 2000, 283, 2674–2679. [Google Scholar] [CrossRef]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- Del Tredici, K.; Rub, U.; De Vos, R.A.; Bohl, J.R.; Braak, H. Where does Parkinson disease pathology begin in the brain? J. Neuropathol. Exp. Neurol. 2002, 61, 413–426. [Google Scholar] [CrossRef]
- Rizek, P.; Kumar, N.; Jog, M.S. An update on the diagnosis and treatment of Parkinson disease. CMAJ 2016, 188, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Steece-Collier, K.; Maries, E.; Kordower, J.H. Etiology of Parkinson’s disease: Genetics and environment revisited. Proc. Natl. Acad. Sci. USA 2002, 99, 13972–13974. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef]
- Payami, H.; Larsen, K.; Bernard, S.; Nutt, J. Increased risk of Parkinson’s disease in parents and siblings of patients. Ann. Neurol. 1994, 36, 659–661. [Google Scholar] [CrossRef] [PubMed]
- Calne, S.; Schoenberg, B.; Martin, W.; Uitti, R.J.; Spencer, P.; Calne, D.B. Familial Parkinson’s disease: Possible role of environmental factors. Can. J. Neurol. Sci. 1987, 14, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- DiPiro, J.T.; Schwinghammer, T.L.; DiPiro, C.V. Pharmacotherapy Handbook; McGraw-Hill Companies: New York, NY, USA, 2009; Section 9; p. 578. [Google Scholar]
- Ellis, J.M.; Fell, M.J. Current approaches to the treatment of Parkinson’s disease. Bioorg. Med. Chem. Lett. 2017, 27, 4247–4255. [Google Scholar] [CrossRef]
- Giugni, J.C.; Okun, M.S. Treatment of advanced Parkinson’s disease. Curr. Opin. Neurol. 2014, 27, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Ovallath, S.; Sulthana, B. Levodopa: History and therapeutic applications. Ann. Indian Acad. Neurol. 2017, 20, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, E.; Marti, M.J.; Valldeoriola, F.; Molinuevo, J.L. History of levodopa and dopamine agonists in Parkinson’s disease treatment. Neurology 1998, 50, S2–S10, discussion S44–S118. [Google Scholar] [CrossRef]
- Ogungbenro, K.; Pertinez, H.; Aarons, L. Empirical and semi-mechanistic modelling of double-peaked pharmacokinetic profile phenomenon due to gastric emptying. AAPS J. 2015, 17, 227–236. [Google Scholar] [CrossRef]
- Hickey, P.; Stacy, M. Available and emerging treatments for Parkinson’s disease: A review. Drug Des. Dev. Ther. 2011, 5, 241–254. [Google Scholar] [CrossRef]
- Vijayakumar, D.; Jankovic, J. Drug-induced dyskinesia, Part 1: Treatment of levodopa-induced dyskinesia. Drugs 2016, 76, 759–777. [Google Scholar] [CrossRef]
- Schrag, A.; Quinn, N. Dyskinesias and motor fluctuations in Parkinson’s disease. A community-based study. Brain 2000, 123, 2297–2305. [Google Scholar] [CrossRef] [PubMed]
- SINEMET® CR (Carbidopa-Levodopa). US Food and Drug Administration (FDA). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/019856s025lbl.pdf (accessed on 10 March 2023).
- LeWitt, P.A.; Fahn, S. Levodopa therapy for Parkinson disease: A look backward and forward. Neurology 2016, 86, S3–S12. [Google Scholar] [CrossRef]
- Pahwa, R.; Lyons, K.E.; Hauser, R.A.; Fahn, S.; Jankovic, J.; Pourcher, E.; Hsu, A.; O’Connell, M.; Kell, S.; Gupta, S.; et al. Randomized trial of IPX066, carbidopa/levodopa extended release, in early Parkinson’s disease. Park. Relat. Disord. 2014, 20, 142–148. [Google Scholar] [CrossRef]
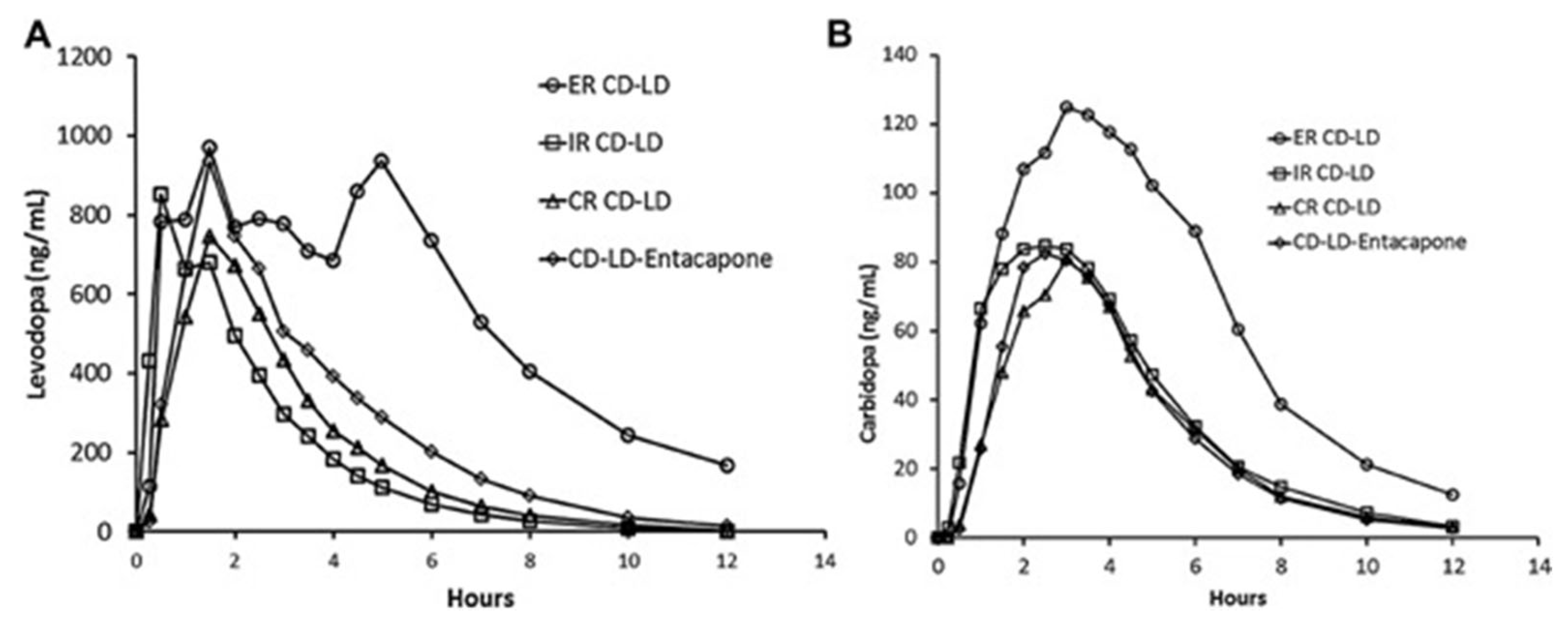
- Hsu, A.; Yao, H.M.; Gupta, S.; Modi, N.B. Comparison of the pharmacokinetics of an oral extended-release capsule formulation of carbidopa-levodopa (IPX066) with immediate-release carbidopa-levodopa (Sinemet(R), sustained-release carbidopa-levodopa (Sinemet((R)) CR), and carbidopa-levodopa-entacapone (Stalevo(R)). J. Clin. Pharmacol. 2015, 55, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W.; Kieburtz, K.; Odin, P.; Espay, A.J.; Standaert, D.G.; Fernandez, H.H.; Vanagunas, A.; Othman, A.A.; Widnell, K.L.; Robieson, W.Z.; et al. LCIG Horizon Study Group. Continuous intrajejunal infusion of levodopa-carbidopa intestinal gel for patients with advanced Parkinson’s disease: A randomised, controlled, double-blind, double-dummy study. Lancet Neurol. 2014, 13, 141–149. [Google Scholar] [CrossRef]
- Yeh, K.C.; August, T.F.; Bush, D.F.; Lasseter, K.C.; Musson, D.G.; Schwartz, S.; Smith, M.E.; Titus, D.C. Pharmacokinetics and bioavailability of Sinemet CR: A summary of human studies. Neurology 1989, 39, 25–38. [Google Scholar]
- Nausieda, P.A.; Pfeiffer, R.F.; Tagliati, M.; Kastenholz, K.V.; DeRoche, C.; Slevin, J.T. A multicenter, open-label, sequential study comparing preferences for carbidopa-levodopa orally disintegrating tablets and conventional tablets in subjects with Parkinson’s disease. Clin. Ther. 2005, 27, 58–63. [Google Scholar] [CrossRef]
- Johnston, T.H.; Fox, S.H.; Brotchie, J.M. Advances in the delivery of treatments for Parkinson’s disease. Expert. Opin. Drug Deliv. 2005, 2, 1059–1073. [Google Scholar] [CrossRef]
- Urso, D.; Chaudhuri, K.R.; Qamar, M.A.; Jenner, P. Improving the delivery of levodopa in Parkinson’s disease: A review of approved and emerging therapies. CNS Drugs 2020, 34, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, H.H.; Standaert, D.G.; Hauser, R.A.; Lang, A.E.; Fung, V.S.; Klostermann, F.; Lew, M.F.; Odin, P.; Steiger, M.; Yakupov, E.Z.; et al. Levodopa-carbidopa intestinal gel in advanced Parkinson’s disease: Final 12-month, open-label results. Mov. Disord. 2015, 30, 500–509. [Google Scholar] [CrossRef]
- Graham, J.S.; Henderson, J.M.; Morris, J.G.; Yiannikas, C. A comparison of standard madopar and controlled release Madopar in Parkinson’s disease. Aust N. Z. J. Med. 1991, 21, 11–15. [Google Scholar] [CrossRef]
- Pahwa, R.; Lyons, K.E.; Hauser, R.A. Ropinirole therapy for Parkinson’s disease. Expert. Rev. Neurother. 2004, 4, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Hitti, M. Parkinson’s Drug Taken Off Market. Available online: https://www.webmd.com/parkinsons-disease/news/20070329/parkinsons-drug-taken-off-market (accessed on 9 January 2023).
- Chwieduk, C.M.; Curran, M.P. Pramipexole extended release: In Parkinson’s disease. CNS Drugs 2010, 24, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Dostinex Tablets. Available online: https://www.medicines.org.uk/emc/product/1691/smpc#gref (accessed on 9 January 2023).
- Somerset Pharma/Cocensys Deal for Eldepryl. The Pharmaletter. Available online: https://www.thepharmaletter.com/article/somerset-pharma-cocensys-deal-for-eldepryl (accessed on 9 January 2023).
- Zelapar (Selegiline). Available online: https://parkinsonsnewstoday.com/zelapar-selegiline/) (accessed on 9 January 2023).
- Navon, N. The Accordion pill(R): Unique oral delivery to enhance pharmacokinetics and therapeutic benefit of challenging drugs. Ther. Deliv. 2019, 10, 433–442. [Google Scholar] [CrossRef]
- Verhagen Metman, L.; Stover, N.; Chen, C.; Cowles, V.E.; Sweeney, M. Gastroretentive carbidopa/levodopa, DM-1992, for the treatment of advanced Parkinson’s disease. Mov. Disord. 2015, 30, 1222–1228. [Google Scholar] [CrossRef]
- Lewitt, P.A.; Ellenbogen, A.; Chen, D.; Lal, R.; McGuire, K.; Zomorodi, K.; Luo, W.; Huff, F.J. Actively transported levodopa prodrug XP21279: A study in patients with Parkinson disease who experience motor fluctuations. Clin. Neuropharmacol. 2012, 35, 103–110. [Google Scholar] [CrossRef]
- Stocchi, F.; Vacca, L.; Grassini, P.; Pawsey, S.; Whale, H.; Marconi, S.; Torti, M. L-Dopa pharmacokinetic profile with effervescent melevodopa/carbidopa versus standard-release levodopa/carbidopa tablets in Parkinson’s disease: A Randomised Study. Parkinson’s Dis. 2015, 2015, 369465. [Google Scholar] [CrossRef]
- Nyholm, D.; Jost, W.H. Levodopa-entacapone-carbidopa intestinal gel infusion in advanced Parkinson’s disease: Real-world experience and practical guidance. Ther. Adv. Neurol. Disord. 2022, 15, 1–15. [Google Scholar] [CrossRef]
- LeWitt, P.A.; Hauser, R.A.; Grosset, D.G.; Stocchi, F.; Saint-Hilaire, M.H.; Ellenbogen, A.; Leinonen, M.; Hampson, N.B.; DeFeo-Fraulini, T.; Freed, M.I.; et al. A randomized trial of inhaled levodopa (CVT-301) for motor fluctuations in Parkinson’s disease. Mov. Disord. 2016, 31, 1356–1365. [Google Scholar] [CrossRef] [PubMed]
- A Study of LY03003 in Patients with Early-Stage Parkinson’s Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT04044547?term=NCT04044547&draw=2&rank=1 (accessed on 10 March 2023).
- A study to Evaluate the Safety and Efficacy of IPX203 in Parkinson’s Disease Patients with Motor Fluctuations. Available online: https://clinicaltrials.gov/ct2/show/NCT03670953?term=IPX203&draw=2&rank=3 (accessed on 10 March 2023).
- Rosebraugh, M.; Voight, E.A.; Moussa, E.M.; Jameel, F.; Lou, X.; Zhang, G.G.Z.; Mayer, P.T.; Stolarik, D.; Carr, R.A.; Enright, B.P.; et al. Foslevodopa/foscarbidopa: A new subcutaneous treatment for Parkinson’s disease. Ann. Neurol. 2021, 90, 52–61. [Google Scholar] [CrossRef]
- Freitas, M.E.; Ruiz-Lopez, M.; Fox, S.H. Novel levodopa formulations for Parkinson’s disease. CNS Drugs 2016, 30, 1079–1095. [Google Scholar] [CrossRef] [PubMed]
- Study Comparing Intravenous and Subcutaneous Infudopa with Intestinal Duodopa in Patients with Parkinson’s Disease (IPO-001). Available online: https://clinicaltrials.gov/ct2/show/NCT03419806?term=Infudopa+SubC&draw=2&rank=1 (accessed on 10 March 2023).
- Relative Bioavailability Study of Ropinirole Implants in Parkinson’s Patients on l-Dopa Switched from Oral Ropinirole. Available online: https://clinicaltrials.gov/ct2/show/NCT03250117?term=ProNeura%E2%84%A2&draw=2&rank=1 (accessed on 10 March 2023).
- Efficacy, Safety and Tolerability of Rotigotine Nasal Spray for the Acute Treatment of Parkinson Symptoms. Available online: https://clinicaltrials.gov/ct2/show/results/NCT00296192?term=SPM+952&draw=2&rank=1s (accessed on 10 March 2023).
- Aradi, S.D.; Hauser, R.A. Medical management and prevention of motor complications in Parkinson’s disease. Neurotherapeutics 2020, 17, 1339–1365. [Google Scholar] [CrossRef] [PubMed]
- Grace, A.A.; Bunney, B.S. The control of firing pattern in nigral dopamine neurons: Single spike firing. J. Neurosci. 1984, 4, 2866–2876. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.M.; Engber, T.M.; Kask, A.M.; Chase, T.N. Motor fluctuations in levodopa treated parkinsonian rats: Relation to lesion extent and treatment duration. Brain Res. 1994, 662, 69–74. [Google Scholar] [CrossRef]
- Larson, D.; Simuni, T. New dopaminergic therapies for PD motor complications. Neuropharmacology 2022, 204, 108869. [Google Scholar] [CrossRef]
- Juncos, J.L.; Engber, T.M.; Raisman, R.; Susel, Z.; Thibaut, F.; Ploska, A.; Agid, Y.; Chase, T.N. Continuous and intermittent levodopa differentially affect basal ganglia function. Ann. Neurol. 1989, 25, 473–478. [Google Scholar] [CrossRef]
- Jenner, P. Factors influencing the onset and persistence of dyskinesia in MPTP-treated primates. Ann. Neurol. 2000, 47, S90–S99, discussion S99–104. [Google Scholar]
- Gomez-Mancilla, B.; Bedard, P.J. Effect of chronic treatment with (+)-PHNO, a D2 agonist in MPTP-treated monkeys. Exp. Neurol. 1992, 117, 185–188. [Google Scholar] [CrossRef]
- Falardeau, P.; Bouchard, S.; Bedard, P.J.; Boucher, R.; Di Paolo, T. Behavioral and biochemical effect of chronic treatment with D-1 and/or D-2 dopamine agonists in MPTP monkeys. Eur. J. Pharmacol. 1988, 150, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Pearce, R.K.; Banerji, T.; Jenner, P.; Marsden, C.D. De novo administration of ropinirole and bromocriptine induces less dyskinesia than L-dopa in the MPTP-treated marmoset. Mov. Disord. 1998, 13, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Blanchet, P.J.; Calon, F.; Martel, J.C.; Bedard, P.J.; Di Paolo, T.; Walters, R.R.; Piercey, M.F. Continuous administration decreases and pulsatile administration increases behavioral sensitivity to a novel dopamine D2 agonist (U-91356A) in MPTP-exposed monkeys. J. Pharmacol. Exp. Ther. 1995, 272, 854–859. [Google Scholar] [PubMed]
- Olanow, C.W.; Watts, R.L.; Koller, W.C. An algorithm (decision tree) for the management of Parkinson’s disease (2001): Treatment guidelines. Neurology 2001, 56, S1–S88. [Google Scholar] [CrossRef] [PubMed]
- Mogharbel, B.F.; Cardoso, M.A.; Irioda, A.C.; Stricker, P.E.F.; Slompo, R.C.; Appel, J.M.; de Oliveira, N.B.; Perussolo, M.C.; Sacaki, C.S.; da Rosa, N.N.; et al. Biodegradable nanoparticles loaded with levodopa and curcumin for treatment of Parkinson’s disease. Molecules 2022, 27, 2811. [Google Scholar] [CrossRef]
- Arisoy, S.; Sayiner, O.; Comoglu, T.; Onal, D.; Atalay, O.; Pehlivanoglu, B. In vitro and in vivo evaluation of levodopa-loaded nanoparticles for nose to brain delivery. Pharm. Dev. Technol. 2020, 25, 735–747. [Google Scholar] [CrossRef]
- Li, X.; Liu, Q.; Zhu, D.; Che, Y.; Feng, X. Preparation of levodopa-loaded crystalsomes through thermally induced crystallization reverses functional deficits in parkinsonian mice. Biomater. Sci. 2019, 7, 1623–1631. [Google Scholar] [CrossRef]
- Bardajee, G.R.; Khamooshi, N.; Nasri, S.; Vancaeyzeele, C. Multi-stimuli responsive nanogel/hydrogel nanocomposites based on κ-carrageenan for prolonged release of levodopa as model drug. Int. J. Biol. Macromol. 2020, 153, 180–189. [Google Scholar] [CrossRef]
- Tan, J.M.; Saifullah, B.; Kura, A.U.; Fakurazi, S.; Hussein, M.Z. Incorporation of levodopa into biopolymer coatings based on carboxylated carbon nanotubes for pH-dependent sustained release drug delivery. Nanomaterials 2018, 8, 389. [Google Scholar] [CrossRef]
- Ren, T.; Yang, X.; Wu, N.; Cai, Y.; Liu, Z.; Yuan, W. Sustained-release formulation of levodopa methyl ester/benserazide for prolonged suppressing dyskinesia expression in 6-OHDA-leisoned rats. Neurosci. Lett. 2011, 502, 117–122. [Google Scholar] [CrossRef]
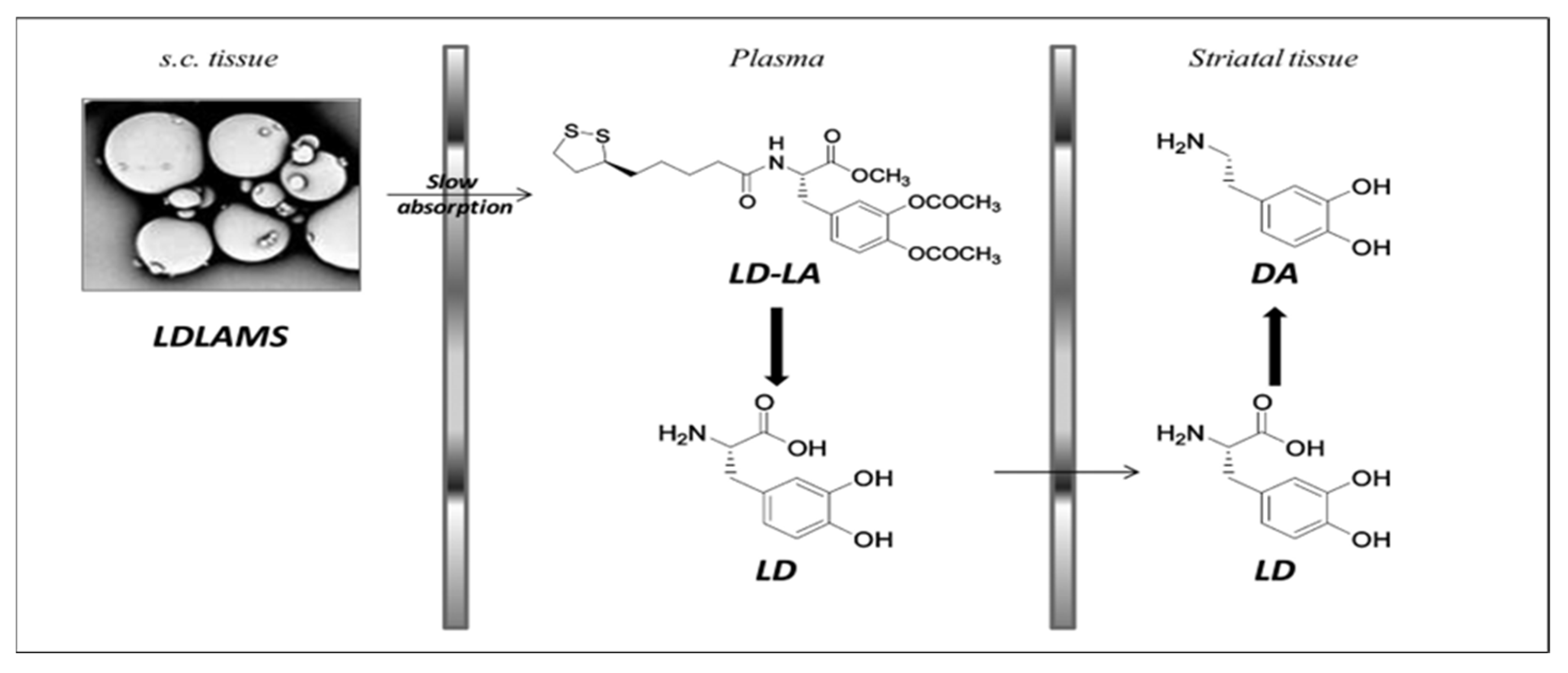
- D’Aurizio, E.; Sozio, P.; Cerasa, L.S.; Vacca, M.; Brunetti, L.; Orlando, G.; Chiavaroli, A.; Kok, R.J.; Hennink, W.E.; Di Stefano, A. Biodegradable microspheres loaded with an anti-parkinson prodrug: An in vivo pharmacokinetic study. Mol. Pharm. 2011, 8, 2408–2415. [Google Scholar] [CrossRef]
- Lang, A.E.; Espay, A.J. Disease modification in Parkinson’s disease: Current approaches, challenges, and future considerations. Mov. Disord. 2018, 33, 660–677. [Google Scholar] [CrossRef]
- Stoker, T.B.; Torsney, K.M.; Barker, R.A. Emerging treatment approaches for Parkinson’s disease. Front. Neurosci. 2018, 12, 693. [Google Scholar] [CrossRef]
- Wrasidlo, W.; Tsigelny, I.F.; Price, D.L.; Dutta, G.; Rockenstein, E.; Schwarz, T.C.; Ledolter, K.; Bonhaus, D.; Paulino, A.; Eleuteri, S.; et al. A de novo compound targeting alpha-synuclein improves deficits in models of Parkinson’s disease. Brain 2016, 139, 3217–3236. [Google Scholar] [CrossRef]
- Phase 1 Study of NPT200-11 in Healthy Subjects. Available online: https://clinicaltrials.gov/ct2/show/NCT02606682?term=NCT02606682&draw=2&rank=1 (accessed on 10 March 2023).
- Teil, M.; Arotcarena, M.L.; Faggiani, E.; Laferriere, F.; Bezard, E.; Dehay, B. Targeting alpha-synuclein for PD therapeutics: A pursuit on all fronts. Biomolecules 2020, 10, 391. [Google Scholar] [CrossRef]
- Lonskaya, I.; Hebron, M.L.; Desforges, N.M.; Schachter, J.B.; Moussa, C.E. Nilotinib-induced autophagic changes increase endogenous parkin level and ubiquitination, leading to amyloid clearance. J. Mol. Med. 2014, 92, 373–386. [Google Scholar] [CrossRef]
- Brahmachari, S.; Ge, P.; Lee, S.H.; Kim, D.; Karuppagounder, S.S.; Kumar, M.; Mao, X.; Shin, J.H.; Lee, Y.; Pletnikova, O.; et al. Activation of tyrosine kinase c-Abl contributes to alpha-synuclein-induced neurodegeneration. J. Clin. Investig. 2016, 126, 2970–2988. [Google Scholar] [CrossRef]
- Lindholm, D.; Pham, D.D.; Cascone, A.; Eriksson, O.; Wennerberg, K.; Saarma, M. c-Abl inhibitors enable insights into the pathophysiology and neuroprotection in Parkinson’s disease. Front. Aging Neurosci. 2016, 8, 254. [Google Scholar] [CrossRef]
- Pagan, F.; Hebron, M.; Valadez, E.H.; Torres-Yaghi, Y.; Huang, X.; Mills, R.R.; Wilmarth, B.M.; Howard, H.; Dunn, C.; Carlson, A.; et al. Nilotinib effects in Parkinson’s disease and dementia with lewy bodies. J. Parkinson’s Dis. 2016, 6, 503–517. [Google Scholar] [CrossRef]
- Smith, L.; Schapira, A.H.V. GBA variants and Parkinson disease: Mechanisms and treatments. Cells 2022, 11, 1261. [Google Scholar] [CrossRef]
- Sanchez-Martinez, A.; Beavan, M.; Gegg, M.E.; Chau, K.Y.; Whitworth, A.J.; Schapira, A.H. Parkinson disease-linked GBA mutation effects reversed by molecular chaperones in human cell and fly models. Sci. Rep. 2016, 6, 31380. [Google Scholar] [CrossRef] [PubMed]
- Bertilsson, G.; Patrone, C.; Zachrisson, O.; Andersson, A.; Dannaeus, K.; Heidrich, J.; Kortesmaa, J.; Mercer, A.; Nielsen, E.; Ronnholm, H.; et al. Peptide hormone exendin-4 stimulates subventricular zone neurogenesis in the adult rodent brain and induces recovery in an animal model of Parkinson’s disease. J. Neurosci. Res. 2008, 86, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Moon, M.; Park, S. Exendin-4 protects dopaminergic neurons by inhibition of microglial activation and matrix metalloproteinase-3 expression in an animal model of Parkinson’s disease. J. Endocrinol. 2009, 202, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Athauda, D.; Maclagan, K.; Skene, S.S.; Bajwa-Joseph, M.; Letchford, D.; Chowdhury, K.; Hibbert, S.; Budnik, N.; Zampedri, L.; Dickson, J.; et al. Exenatide once weekly versus placebo in Parkinson’s disease: A randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1664–1675. [Google Scholar] [CrossRef]
- George, S.; Brundin, P. Immunotherapy in Parkinson’s disease: Micromanaging alpha-synuclein aggregation. J. Parkinson’s Dis. 2015, 5, 413–424. [Google Scholar] [CrossRef]
- Brundin, P.; Dave, K.D.; Kordower, J.H. Therapeutic approaches to target alpha-synuclein pathology. Exp. Neurol. 2017, 298, 225–235. [Google Scholar] [CrossRef]
- Study Assessing Tolerability and Safety of AFFITOPE® PD03A in Patients with Early Parkinson’s Disease (AFF011). Available online: https://clinicaltrials.gov/ct2/show/NCT02267434 (accessed on 10 March 2023).
- Schenk, D.B.; Koller, M.; Ness, D.K.; Griffith, S.G.; Grundman, M.; Zago, W.; Soto, J.; Atiee, G.; Ostrowitzki, S.; Kinney, G.G. First-in-human assessment of PRX002, an anti-alpha-synuclein monoclonal antibody, in healthy volunteers. Mov. Disord. 2017, 32, 211–218. [Google Scholar] [CrossRef]
- Jankovic, J.; Goodman, I.; Safirstein, B.; Marmon, T.K.; Schenk, D.B.; Koller, M.; Zago, W.; Ness, D.K.; Griffith, S.G.; Grundman, M.; et al. Safety and tolerability of multiple ascending doses of PRX002/RG7935, an anti-α-synuclein monoclonal antibody, in patients with Parkinson disease: A randomized clinical trial. JAMA Neurol. 2018, 75, 1206–1214. [Google Scholar] [CrossRef]
- Fajardo-Serrano, A.; Rico, A.J.; Roda, E.; Honrubia, A.; Arrieta, S.; Ariznabarreta, G.; Chocarro, J.; Lorenzo-Ramos, E.; Pejenaute, A.; Vazquez, A.; et al. Adeno-associated viral vectors as versatile tools for Parkinson’s research, both for disease modeling purposes and for therapeutic uses. Int. J. Mol. Sci. 2021, 22, 6389. [Google Scholar] [CrossRef]
- Kells, A.P.; Forsayeth, J.; Bankiewicz, K.S. Glial-derived neurotrophic factor gene transfer for Parkinson’s disease: Anterograde distribution of AAV2 vectors in the primate brain. Neurobiol. Dis. 2012, 48, 228–235. [Google Scholar] [CrossRef]
- Bartus, R.T.; Kordower, J.H.; Johnson, E.M., Jr.; Brown, L.; Kruegel, B.R.; Chu, Y.; Baumann, T.L.; Lang, A.E.; Olanow, C.W.; Herzog, C.D. Post-mortem assessment of the short and long-term effects of the trophic factor neurturin in patients with alpha-synucleinopathies. Neurobiol. Dis. 2015, 78, 162–171. [Google Scholar] [CrossRef]
- Kordower, J.H.; Olanow, C.W.; Dodiya, H.B.; Chu, Y.; Beach, T.G.; Adler, C.H.; Halliday, G.M.; Bartus, R.T. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain 2013, 136, 2419–2431. [Google Scholar] [CrossRef]
- Christine, C.W.; Richardson, R.M.; Van Laar, A.D.; Thompson, M.E.; Fine, E.M.; Khwaja, O.S.; Li, C.; Liang, G.S.; Meier, A.; Roberts, E.W.; et al. Safety of AADC gene therapy for moderately advanced Parkinson disease: Three-year outcomes from the PD-1101 trial. Neurology 2022, 98, e40–e50. [Google Scholar] [CrossRef]
- Marks, W.J., Jr.; Ostrem, J.L.; Verhagen, L.; Starr, P.A.; Larson, P.S.; Bakay, R.A.; Taylor, R.; Cahn-Weiner, D.A.; Stoessl, A.J.; Olanow, C.W.; et al. Safety and tolerability of intraputaminal delivery of CERE-120 (adeno-associated virus serotype 2-neurturin) to patients with idiopathic Parkinson’s disease: An open-label, phase I trial. Lancet Neurol. 2008, 7, 400–408. [Google Scholar] [CrossRef]
- Hawthorne, G.H.; Bernuci, M.P.; Bortolanza, M.; Tumas, V.; Issy, A.C.; Del-Bel, E. Nanomedicine to overcome current Parkinson’s treatment liabilities: A systematic review. Neurotox. Res. 2016, 30, 715–729. [Google Scholar] [CrossRef]
- Itin, C.; Komargodski, R.; Barasch, D.; Domb, A.J.; Hoffman, A. prolonged delivery of apomorphine through the buccal mucosa, towards a noninvasive sustained administration method in Parkinson’s disease: In vivo investigations in pigs. J. Pharm. Sci. 2021, 110, 1824–1833. [Google Scholar] [CrossRef]
- Kulkarni, A.D.; Vanjari, Y.H.; Sancheti, K.H.; Belgamwar, V.S.; Surana, S.J.; Pardeshi, C.V. Nanotechnology-mediated nose to brain drug delivery for Parkinson’s disease: A mini review. J. Drug Target. 2015, 23, 775–788. [Google Scholar] [CrossRef]
- Khatri, D.K.; Preeti, K.; Tonape, S.; Bhattacharjee, S.; Patel, M.; Shah, S.; Singh, P.K.; Srivastav, S.; Gugulothu, D.; Vora, L.; et al. Nanotechnological advances for nose to brain delivery of therapeutics to improve the Parkinson therapy. Curr. Neuropharmacol. 2022, 21, 493–516. [Google Scholar] [CrossRef]
- Allen, T.M.; Cullis, P.R. Drug delivery systems: Entering the mainstream. Science 2004, 303, 1818–1822. [Google Scholar] [CrossRef]
- Farokhzad, O.C.; Langer, R. Impact of nanotechnology on drug delivery. ACS Nano 2009, 3, 16–20. [Google Scholar] [CrossRef]
- Wohlfart, S.; Gelperina, S.; Kreuter, J. Transport of drugs across the blood-brain barrier by nanoparticles. J. Control. Release 2012, 161, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Haddad, F.; Sawalha, M.; Khawaja, Y.; Najjar, A.; Karaman, R. Dopamine and levodopa prodrugs for the treatment of Parkinson’s disease. Molecules 2017, 23, 40. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.; Kim, H.K.; Lee, H. Dopamine-loaded poly(D,L-lactic-co-glycolic acid) microspheres: New strategy for encapsulating small hydrophilic drugs with high efficiency. Biotechnol. Prog. 2014, 30, 215–223. [Google Scholar] [CrossRef]
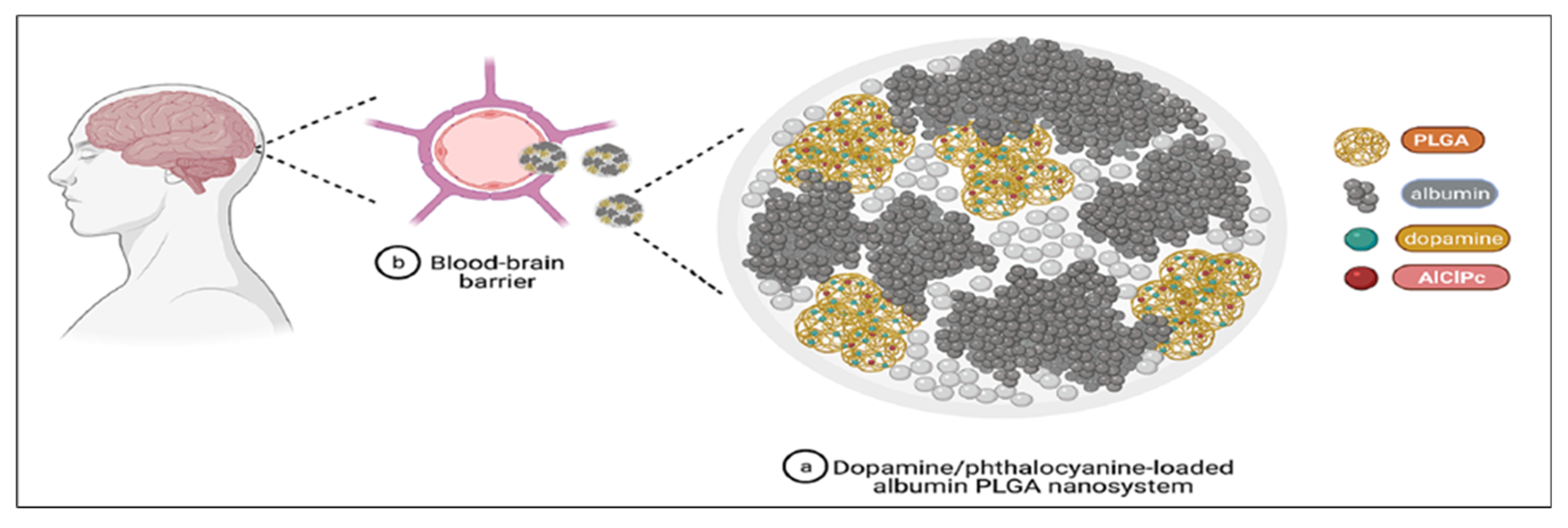
- Pahuja, R.; Seth, K.; Shukla, A.; Shukla, R.K.; Bhatnagar, P.; Chauhan, L.K.; Saxena, P.N.; Arun, J.; Chaudhari, B.P.; Patel, D.K.; et al. Trans-blood brain barrier delivery of dopamine-loaded nanoparticles reverses functional deficits in parkinsonian rats. ACS Nano 2015, 9, 4850–4871. [Google Scholar] [CrossRef] [PubMed]
- Monge-Fuentes, V.; Biolchi Mayer, A.; Lima, M.R.; Geraldes, L.R.; Zanotto, L.N.; Moreira, K.G.; Martins, O.P.; Piva, H.L.; Felipe, M.S.S.; Amaral, A.C.; et al. Dopamine-loaded nanoparticle systems circumvent the blood-brain barrier restoring motor function in mouse model for Parkinson’s disease. Sci. Rep. 2021, 11, 15185. [Google Scholar] [CrossRef]
- Lin, T.; Zhao, P.; Jiang, Y.; Tang, Y.; Jin, H.; Pan, Z.; He, H.; Yang, V.C.; Huang, Y. Blood-brain-barrier-penetrating albumin nanoparticles for biomimetic drug delivery via albumin-binding protein pathways for antiglioma therapy. ACS Nano 2016, 10, 9999–10012. [Google Scholar] [CrossRef]
- Jayme, C.C.; Calori, I.R.; Cunha, E.M.F.; Tedesco, A.C. Evaluation of aluminum phthalocyanine chloride and DNA interactions for the design of an advanced drug delivery system in photodynamic therapy. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2018, 201, 242–248. [Google Scholar] [CrossRef]
- Bi, C.; Wang, A.; Chu, Y.; Liu, S.; Mu, H.; Liu, W.; Wu, Z.; Sun, K.; Li, Y. Intranasal delivery of rotigotine to the brain with lactoferrin-modified PEG-PLGA nanoparticles for Parkinson’s disease treatment. Int. J. Nanomed. 2016, 11, 6547–6559. [Google Scholar] [CrossRef]
- Sridhar, V.; Gaud, R.; Bajaj, A.; Wairkar, S. Pharmacokinetics and pharmacodynamics of intranasally administered selegiline nanoparticles with improved brain delivery in Parkinson’s disease. Nanomedicine 2018, 14, 2609–2618. [Google Scholar] [CrossRef]
- Sharma, S.; Lohan, S.; Murthy, R.S. Formulation and characterization of intranasal mucoadhesive nanoparticulates and thermo-reversible gel of levodopa for brain delivery. Drug Dev. Ind. Pharm. 2014, 40, 869–878. [Google Scholar] [CrossRef]
- Bali, N.R.; Salve, P.S. Impact of rasagiline nanoparticles on brain targeting efficiency via gellan gum based transdermal patch: A nanotheranostic perspective for parkinsonism. Int. J. Biol. Macromol. 2020, 164, 1006–1024. [Google Scholar] [CrossRef]
- Bali, N.R.; Salve, P.S. Selegiline nanoparticle embedded transdermal film: An alternative approach for brain targeting in Parkinson’s disease. J. Drug Deliv. Sci. Technol. 2019, 54, 101299. [Google Scholar] [CrossRef]
- Yang, X.; Zheng, R.; Cai, Y.; Liao, M.; Yuan, W.; Liu, Z. Controlled-release levodopa methyl ester/benserazide-loaded nanoparticles ameliorate levodopa-induced dyskinesia in rats. Int. J. Nanomed. 2012, 7, 2077–2086. [Google Scholar] [CrossRef]
- Raj, R.; Wairkar, S.; Sridhar, V.; Gaud, R. Pramipexole dihydrochloride loaded chitosan nanoparticles for nose to brain delivery: Development, characterization and in vivo anti-parkinson activity. Int. J. Biol. Macromol. 2018, 109, 27–35. [Google Scholar] [CrossRef]
- Meng, X.Y.; Huang, A.Q.; Khan, A.; Zhang, L.; Sun, X.Q.; Song, H.; Han, J.; Sun, Q.R.; Wang, Y.D.; Li, X.L. Vascular endothelial growth factor-loaded poly-lactic-co-glycolic acid nanoparticles with controlled release protect the dopaminergic neurons in Parkinson’s rats. Chem. Biol. Drug Des. 2020, 95, 631–639. [Google Scholar] [CrossRef]
- Pillay, S.; Pillay, V.; Choonara, Y.E.; Naidoo, D.; Khan, R.A.; du Toit, L.C.; Ndesendo, V.M.; Modi, G.; Danckwerts, M.P.; Iyuke, S.E. Design, biometric simulation and optimization of a nano-enabled scaffold device for enhanced delivery of dopamine to the brain. Int. J. Pharm. 2009, 382, 277–290. [Google Scholar] [CrossRef]
- Ray, S.; Sinha, P.; Laha, B.; Maiti, S.; Bhattacharyya, U.K.; Nayak, A.K. Polysorbate 80 coated crosslinked chitosan nanoparticles of ropinirole hydrochloride for brain targeting. J. Drug Deliv. Sci. Technol. 2018, 48, 21–29. [Google Scholar] [CrossRef]
- Duan, Y.; Dhar, A.; Patel, C.; Khimani, M.; Neogi, S.; Sharma, P.; Siva Kumar, N.; Vekariya, R.L. A brief review on solid lipid nanoparticles: Part and parcel of contemporary drug delivery systems. RSC Adv. 2020, 10, 26777–26791. [Google Scholar] [CrossRef]
- Correia, A.C.; Monteiro, A.R.; Silva, R.; Moreira, J.N.; Sousa Lobo, J.M.; Silva, A.C. Lipid nanoparticles strategies to modify pharmacokinetics of central nervous system targeting drugs: Crossing or circumventing the blood-brain barrier (BBB) to manage neurological disorders. Adv. Drug Deliv. Rev. 2022, 189, 114485. [Google Scholar] [CrossRef]
- Diwan, R.; Ravi, P.R.; Pathare, N.S.; Aggarwal, V. Pharmacodynamic, pharmacokinetic and physical characterization of cilnidipine loaded solid lipid nanoparticles for oral delivery optimized using the principles of design of experiments. Colloids Surf. B Biointerfaces 2020, 193, 111073. [Google Scholar] [CrossRef]
- Scioli Montoto, S.; Muraca, G.; Ruiz, M.E. Solid lipid nanoparticles for drug delivery: Pharmacological and biopharmaceutical aspects. Front. Mol. Biosci. 2020, 7, 587997. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Minko, T. Nanotherapeutics for nose-to-brain drug delivery: An approach to bypass the blood brain barrier. Pharmaceutics 2021, 13, 2049. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.J.; Huang, Y.B.; Wu, P.C.; Fu, Y.S.; Kao, Y.R.; Fang, J.Y.; Tsai, Y.H. Oral apomorphine delivery from solid lipid nanoparticles with different monostearate emulsifiers: Pharmacokinetic and behavioral evaluations. J. Pharm. Sci. 2011, 100, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Uppuluri, C.T.; Ravi, P.R.; Dalvi, A.V. Design, optimization and pharmacokinetic evaluation of Piribedil loaded solid lipid nanoparticles dispersed in nasal in situ gelling system for effective management of Parkinson’s disease. Int. J. Pharm. 2021, 606, 120881. [Google Scholar] [CrossRef]
- Dudhipala, N.; Gorre, T. Neuroprotective effect of ropinirole lipid nanoparticles enriched hydrogel for Parkinson’s disease: In vitro, ex vivo, pharmacokinetic and pharmacodynamic evaluation. Pharmaceutics 2020, 12, 448. [Google Scholar] [CrossRef]
- Pardeshi, C.V.; Belgamwar, V.S. Improved brain pharmacokinetics following intranasal administration of N,N,N-trimethyl chitosan tailored mucoadhesive NLCs. Mater. Technol. 2020, 35, 249–266. [Google Scholar] [CrossRef]
- Calvo, P.; Gouritin, B.; Chacun, H.; Desmaele, D.; D’Angelo, J.; Noel, J.P.; Georgin, D.; Fattal, E.; Andreux, J.P.; Couvreur, P. Long-circulating PEGylated polycyanoacrylate nanoparticles as new drug carrier for brain delivery. Pharm. Res. 2001, 18, 1157–1166. [Google Scholar] [CrossRef]
- Alqahtani, M.S.; Kazi, M.; Alsenaidy, M.A.; Ahmad, M.Z. Advances in oral drug delivery. Front. Pharmacol. 2021, 12, 618411. [Google Scholar] [CrossRef]
- Azman, M.; Sabri, A.H.; Anjani, Q.K.; Mustaffa, M.F.; Hamid, K.A. Intestinal absorption study: Challenges and absorption enhancement strategies in improving oral drug delivery. Pharmaceuticals 2022, 15, 975. [Google Scholar] [CrossRef]
- Wang, Y.; Burgess, D.J. Microsphere technologies. In Long Acting Injections and Implants; Jeremy, C.W., Diane, J.B., Eds.; Springer: New York, NY, USA, 2012; pp. 167–194. [Google Scholar]
- Uhrich, K.E.; Cannizzaro, S.M.; Langer, R.S.; Shakesheff, K.M. Polymeric systems for controlled drug release. Chem. Rev. 1999, 99, 3181–3198. [Google Scholar] [CrossRef]
- Jain, R.; Shah, N.H.; Malick, A.W.; Rhodes, C.T. Controlled drug delivery by biodegradable poly(ester) devices: Different preparative approaches. Drug Dev. Ind. Pharm. 1998, 24, 703–727. [Google Scholar] [CrossRef]
- Abadi, S.S.; Moin, A.; Veerabhadrappa, G.H. Fabricated microparticles: An innovative method to minimize the side effects of NSAIDs in arthritis. Crit. Rev. Ther. Drug Carrier Syst. 2016, 33, 433–488. [Google Scholar] [CrossRef]
- Tran, V.T.; Benoit, J.P.; Venier-Julienne, M.C. Why and how to prepare biodegradable, monodispersed, polymeric microparticles in the field of pharmacy? Int. J. Pharm. 2011, 407, 1–11. [Google Scholar] [CrossRef]
- Su, Y.; Liu, J.; Tan, S.; Liu, W.; Wang, R.; Chen, C. PLGA sustained-release microspheres loaded with an insoluble small-molecule drug: Microfluidic-based preparation, optimization, characterization, and evaluation in vitro and in vivo. Drug Deliv. 2022, 29, 1437–1446. [Google Scholar] [CrossRef]
- Su, Y.; Zhang, B.; Sun, R.; Liu, W.; Zhu, Q.; Zhang, X.; Wang, R.; Chen, C. PLGA-based biodegradable microspheres in drug delivery: Recent advances in research and application. Drug Deliv. 2021, 28, 1397–1418. [Google Scholar] [CrossRef]
- O’Brien, M.N.; Jiang, W.; Wang, Y.; Loffredo, D.M. Challenges and opportunities in the development of complex generic long-acting injectable drug products. J. Control. Release 2021, 336, 144–158. [Google Scholar] [CrossRef]
- Lam, P.L.; Gambari, R. Advanced progress of microencapsulation technologies: In vivo and in vitro models for studying oral and transdermal drug deliveries. J. Control. Release 2014, 178, 25–45. [Google Scholar] [CrossRef]
- Sharifi, F.; Otte, A.; Yoon, G.; Park, K. Continuous in-line homogenization process for scale-up production of naltrexone-loaded PLGA microparticles. J. Control. Release 2020, 325, 347–358. [Google Scholar] [CrossRef]
- Chereddy, K.K.; Vandermeulen, G.; Preat, V. PLGA based drug delivery systems: Promising carriers for wound healing activity. Wound Repair Regen. 2016, 24, 223–236. [Google Scholar] [CrossRef]
- Freitas, S.; Merkle, H.P.; Gander, B. Microencapsulation by solvent extraction/evaporation: Reviewing the state of the art of microsphere preparation process technology. J. Control. Release 2005, 102, 313–332. [Google Scholar] [CrossRef]
- Lee, J.; Kwon, H.J.; Ji, H.; Cho, S.H.; Cho, E.H.; Han, H.D.; Shin, B.C. Marbofloxacin-encapsulated microparticles provide sustained drug release for treatment of veterinary diseases. Mater. Sci. Eng. C Mater. Biol. Appl. 2016, 60, 511–517. [Google Scholar] [CrossRef]
- Ramazani, F.; Chen, W.; van Nostrum, C.F.; Storm, G.; Kiessling, F.; Lammers, T.; Hennink, W.E.; Kok, R.J. Strategies for encapsulation of small hydrophilic and amphiphilic drugs in PLGA microspheres: State-of-the-art and challenges. Int. J. Pharm. 2016, 499, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Li, L.C. Current advances in sustained-release systems for parenteral drug delivery. Expert. Opin. Drug Deliv. 2005, 2, 1039–1058. [Google Scholar] [CrossRef] [PubMed]
- Van de Weert, M.; Hennink, W.E.; Jiskoot, W. Protein instability in poly(lactic-co-glycolic acid) microparticles. Pharm. Res. 2000, 17, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Wissing, T.B.; Bonito, V.; van Haaften, E.E.; van Doeselaar, M.; Brugmans, M.; Janssen, H.M.; Bouten, C.V.C.; Smits, A. Macrophage-driven biomaterial degradation depends on scaffold microarchitecture. Front. Bioeng. Biotechnol. 2019, 7, 87. [Google Scholar] [CrossRef]
- Foged, C.; Brodin, B.; Frokjaer, S.; Sundblad, A. Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model. Int. J. Pharm. 2005, 298, 315–322. [Google Scholar] [CrossRef]
- Sinha, V.R.; Trehan, A. Biodegradable microspheres for protein delivery. J. Control. Release 2003, 90, 261–280. [Google Scholar] [CrossRef]
- Gasmi, H.; Siepmann, F.; Hamoudi, M.C.; Danede, F.; Verin, J.; Willart, J.F.; Siepmann, J. Towards a better understanding of the different release phases from PLGA microparticles: Dexamethasone-loaded systems. Int. J. Pharm. 2016, 514, 189–199. [Google Scholar] [CrossRef]
- Tamani, F.; Bassand, C.; Hamoudi, M.C.; Siepmann, F.; Siepmann, J. Mechanistic explanation of the (up to) 3 release phases of PLGA microparticles: Monolithic dispersions studied at lower temperatures. Int. J. Pharm. 2021, 596, 120220. [Google Scholar] [CrossRef]
- Shi, Z.L.; Fan, Z.Y.; Zhang, H.; Li, S.T.; Yuan, H.; Tong, J.H. Localized delivery of brain-derived neurotrophic factor from PLGA microspheres promotes peripheral nerve regeneration in rats. J. Orthop. Surg. Res. 2022, 17, 172. [Google Scholar] [CrossRef]
- Herran, E.; Ruiz-Ortega, J.A.; Aristieta, A.; Igartua, M.; Requejo, C.; Lafuente, J.V.; Ugedo, L.; Pedraz, J.L.; Hernandez, R.M. In vivo administration of VEGF- and GDNF-releasing biodegradable polymeric microspheres in a severe lesion model of Parkinson’s disease. Eur. J. Pharm. Biopharm. 2013, 85, 1183–1190. [Google Scholar] [CrossRef]
- Li, S.; Liu, J.; Li, G.; Zhang, X.; Xu, F.; Fu, Z.; Teng, L.; Li, Y.; Sun, F. Near-infrared light-responsive, pramipexole-loaded biodegradable PLGA microspheres for therapeutic use in Parkinson’s disease. Eur. J. Pharm. Biopharm. 2019, 141, 1–11. [Google Scholar] [CrossRef]
- Kanwar, N.; Bhandari, R.; Kuhad, A.; Sinha, V.R. Polycaprolactone-based neurotherapeutic delivery of rasagiline targeting behavioral and biochemical deficits in Parkinson’s disease. Drug Deliv. Transl. Res. 2019, 9, 891–905. [Google Scholar] [CrossRef]
- Agbay, A.; Mohtaram, N.K.; Willerth, S.M. Controlled release of glial cell line-derived neurotrophic factor from poly(epsilon-caprolactone) microspheres. Drug Deliv. Transl. Res. 2014, 4, 159–170. [Google Scholar] [CrossRef]
- Parthipan, A.K.; Gupta, N.; Pandey, K.; Sharma, B.; Jacob, J.; Saha, S. One-step fabrication of bicompartmental microparticles as a dual drug delivery system for Parkinson’s disease management. J. Mater. Sci 2019, 54, 730–744. [Google Scholar] [CrossRef]
- Kashif, P.M.; Madni, A.; Ashfaq, M.; Rehman, M.; Mahmood, M.A.; Khan, M.I.; Tahir, N. Development of eudragit RS 100 microparticles loaded with ropinirole: Optimization and in vitro evaluation studies. AAPS PharmSciTech 2017, 18, 1810–1822. [Google Scholar] [CrossRef]
- Garbayo, E.; Ansorena, E.; Lana, H.; Carmona-Abellan, M.D.; Marcilla, I.; Lanciego, J.L.; Luquin, M.R.; Blanco-Prieto, M.J. Brain delivery of microencapsulated GDNF induces functional and structural recovery in parkinsonian monkeys. Biomaterials 2016, 110, 11–23. [Google Scholar] [CrossRef]
- Fernandez, M.; Negro, S.; Slowing, K.; Fernandez-Carballido, A.; Barcia, E. An effective novel delivery strategy of rasagiline for Parkinson’s disease. Int. J. Pharm. 2011, 419, 271–280. [Google Scholar] [CrossRef]
- Fabbri, M.; Barbosa, R.; Rascol, O. Off-time treatment options for Parkinson’s disease. Neurol. Ther. 2023, 12, 391–424. [Google Scholar] [CrossRef]
- D’Aurizio, E.; van Nostrum, C.F.; van Steenbergen, M.J.; Sozio, P.; Siepmann, F.; Siepmann, J.; Hennink, W.E.; Di Stefano, A. Preparation and characterization of poly(lactic-co-glycolic acid) microspheres loaded with a labile antiparkinson prodrug. Int. J. Pharm. 2011, 409, 289–296. [Google Scholar] [CrossRef]
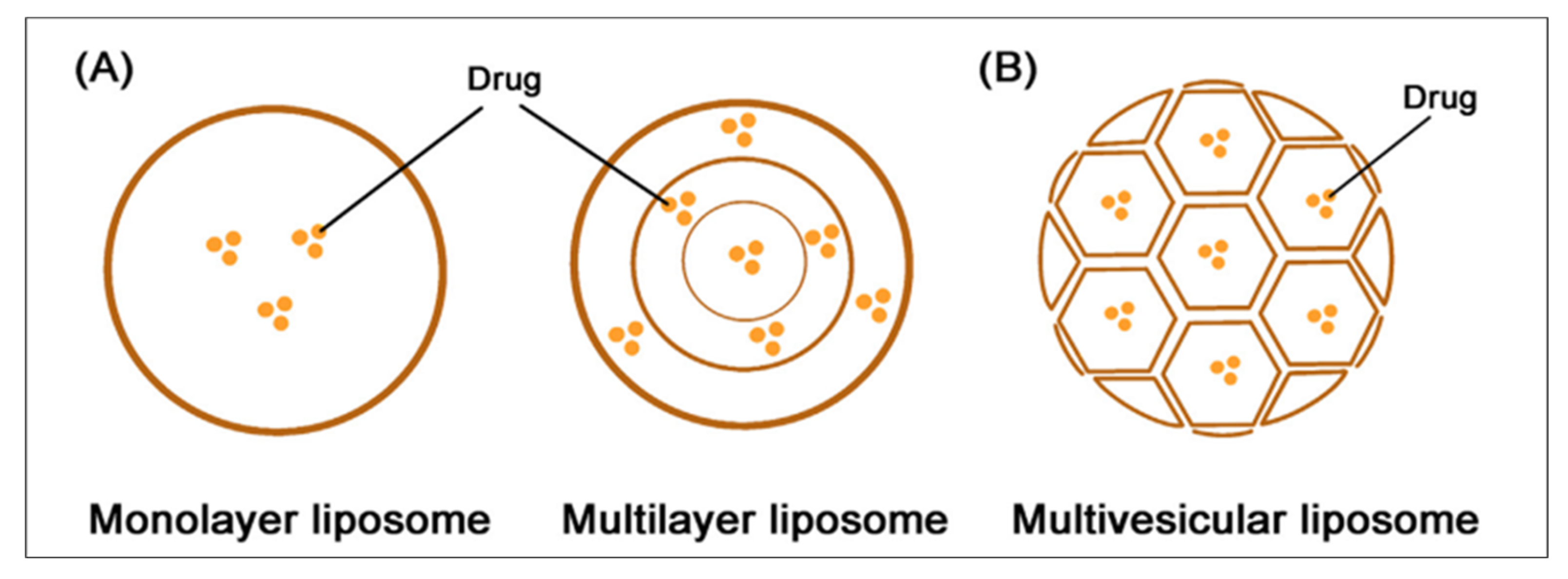
- Subramani, T.; Ganapathyswamy, H. An overview of liposomal nano-encapsulation techniques and its applications in food and nutraceutical. J. Food Sci. Technol. 2020, 57, 3545–3555. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lu, A.; Wang, X.; Belhadj, Z.; Wang, J.; Zhang, Q. A review of existing strategies for designing long-acting parenteral formulations: Focus on underlying mechanisms, and future perspectives. Acta Pharm. Sin. B 2021, 11, 2396–2415. [Google Scholar] [CrossRef] [PubMed]
- Grassin-Delyle, S.; Buenestado, A.; Naline, E.; Faisy, C.; Blouquit-Laye, S.; Couderc, L.J.; Le Guen, M.; Fischler, M.; Devillier, P. Intranasal drug delivery: An efficient and non-invasive route for systemic administration: Focus on opioids. Pharmacol. Ther. 2012, 134, 366–379. [Google Scholar] [CrossRef] [PubMed]
- Migliore, M.M.; Ortiz, R.; Dye, S.; Campbell, R.B.; Amiji, M.M.; Waszczak, B.L. Neurotrophic and neuroprotective efficacy of intranasal GDNF in a rat model of Parkinson’s disease. Neuroscience 2014, 274, 11–23. [Google Scholar] [CrossRef]
- Xiang, Y.; Wu, Q.; Liang, L.; Wang, X.; Wang, J.; Zhang, X.; Pu, X.; Zhang, Q. Chlorotoxin-modified stealth liposomes encapsulating levodopa for the targeting delivery against Parkinson’s disease in the MPTP-induced mice model. J. Drug Target 2012, 20, 67–75. [Google Scholar] [CrossRef]
- Trivedi, R.; Umekar, M.; Kotagale, N.; Bonde, S.; Taksande, J. Design, evaluation and in vivo pharmacokinetic study of a cationic flexible liposomes for enhanced transdermal delivery of pramipexole. J. Drug Deliv. Sci. Technol. 2021, 61, 102313. [Google Scholar] [CrossRef]
- Bjorklund, A.; Dunnett, S.B. Dopamine neuron systems in the brain: An update. Trends Neurosci. 2007, 30, 194–202. [Google Scholar] [CrossRef]
- Cassano, T.; Lopalco, A.; de Candia, M.; Laquintana, V.; Lopedota, A.; Cutrignelli, A.; Perrone, M.; Iacobazzi, R.M.; Bedse, G.; Franco, M.; et al. Oxazepam-dopamine conjugates increase dopamine delivery into striatum of intact rats. Mol. Pharm. 2017, 14, 3178–3187. [Google Scholar] [CrossRef]
- Denora, N.; Cassano, T.; Laquintana, V.; Lopalco, A.; Trapani, A.; Cimmino, C.S.; Laconca, L.; Giuffrida, A.; Trapani, G. Novel codrugs with GABAergic activity for dopamine delivery in the brain. Int. J. Pharm. 2012, 437, 221–231. [Google Scholar] [CrossRef]
- Lopalco, A.; Cutrignelli, A.; Denora, N.; Lopedota, A.; Franco, M.; Laquintana, V. Transferrin functionalized liposomes loading dopamine HCl: Development and permeability studies across an in vitro model of human blood-brain barrier. Nanomaterials 2018, 8, 178. [Google Scholar] [CrossRef]
- Day, C.M.; Sweetman, M.J.; Garg, S. Nanocarriers for the Delivery of Combination Drugs; Baboota, S., Ali, J., Eds.; Elsevier: New York, NY, USA, 2021; Chapter 4; p. 119. [Google Scholar]
- Venkataramana, N.K.; Kumar, S.K.V.; Balaraju, S.; Radhakrishnan, R.C.; Bansal, A.; Dixit, A.; Rao, D.K.; Das, M.; Jan, M.; Gupta, P.K.; et al. Open-labeled study of unilateral autologous bone-marrow-derived mesenchymal stem cell transplantation in Parkinson’s disease. Transl. Res. 2010, 155, 62–70. [Google Scholar] [CrossRef]
- Ribovski, L.; Hamelmann, N.M.; Paulusse, J.M.J. Polymeric nanoparticles properties and brain delivery. Pharmaceutics 2021, 13, 2045. [Google Scholar] [CrossRef]
- Park, K.; Kim, J.H.; Nam, Y.S.; Lee, S.; Nam, H.Y.; Kim, K.; Park, J.H.; Kim, I.S.; Choi, K.; Kim, S.Y.; et al. Effect of polymer molecular weight on the tumor targeting characteristics of self-assembled glycol chitosan nanoparticles. J. Control. Release 2007, 122, 305–314. [Google Scholar] [CrossRef]
- Priya James, H.; John, R.; Alex, A.; Anoop, K.R. Smart polymers for the controlled delivery of drugs—A concise overview. Acta Pharm. Sin. B 2014, 4, 120–127. [Google Scholar] [CrossRef]
- Saalwächter, K. Grand challenges in polymers. Front. Soft. Matter 2022, 2, 18. [Google Scholar] [CrossRef]
- Panchal, S.S.; Vasava, D.V. Biodegradable polymeric materials: Synthetic approach. ACS Omega 2020, 5, 4370–4379. [Google Scholar] [CrossRef]
- Sharma, S.; Parveen, R.; Chatterji, B.P. Toxicology of nanoparticles in drug delivery. Curr. Pathobiol. Rep. 2021, 9, 133–144. [Google Scholar] [CrossRef]
- Niaounakis, M. Biopolymers Reuse, Recycling, and Disposal; Niaounakis, M., Ed.; William Andrew Publishing: Oxford, UK, 2013; Chapter 7; pp. 193–241. [Google Scholar]








| Brand Name | Composition | Dosage Form | Drug-Release Mechanism | Limitation | Development Stage | Ref. |
|---|---|---|---|---|---|---|
| Sinemet CR® Merck Sharpe and Dohme corp | L-DOPA, CD, hydroxypropyl cellulose, polyvinyl acetate-crotonic copolymer, magnesium stearate, and red ferric oxide | Controlled-release (CR) tablet | The drug is released through a combination mechanism of erosion and surface dissolution | Bioavailability is 30% less than IR tablet, which increases the dosage requirement | Approved/ Marketed | [32] |
| Parcopa® Schwarz Pharma | L-DOPA, CD, aspartame, citric acid, cross povidone, magnesium stearate, mannitol, microcrystalline cellulose, natural and artificial mint flavor and sodium bicarbonate | Orally disintegrating | Tablet rapidly disintegrates on the tongue, releasing L-DOPA and CD | No substantial variations were observed in its motor advantages when compared to LD/CD IR | Approved/ Marketed | [33] |
| Stalevo® Novartis Pharmaceutical | L-DOPA, CD, entacapone, corn starch, croscarmellose sodium, glycerol 85%, hypromellose, magnesium stearate, mannitol, polysorbate 80, povidone, sucrose, red iron oxide, and titanium dioxide | Immediate-release (IR), CR | The tablet is designed to release the three drugs at different rates and locations in the GIT, CD is immediately released followed by extended the release of L-DOPA in the small intestine, and finally delayed release of entacapone portion in the small intestine | Requires administration four times a day. Evokes a few motor complications | Approved/ Marketed | [34] |
| Rytary® Impax pharmaceuticals | L-DOPA, CD, microcrystalline cellulose, mannitol, tartaric acid, ethyl cellulose, hypromellose, sodium starch glycolate, sodium lauryl sulfate, povidone, talc, methacrylic copolymers, triethyl citrate, croscarmellose sodium, and magnesium stearate | IR layer with delayed-release beads | The outermost layer disintegrates immediately providing the initial dose by dissolution, whereas the remaining layer releases the drug by diffusion | PD-induced gastroparesis might impact the effect of rotary movement | Approved/ Marketed | [35] |
| Duopa® Abbvie Inc | L-DOPA, CD, carmellose sodium | Intestinal gel | The drug is released by diffusion and dissolution from the gel | Recommended for only 16 h. Adverse events related to the device are more likely to occur within the initial two weeks and are in line with the recognized complications of the PEG-J procedure [36] | Approved/ Marketed | [37] |
| Requip® GlaxoSmithKline | Ropinirole, hypromellose, hydrogenated castor oil, carmellose sodium, povidone K29–32, maltodextrin, magnesium stearate, lactose monohydrate, anhydrous colloidal silica, mannitol, ferric oxide yellow (E172) and glycerol dibehenate | Prolonged-release tablet | The drug is released by diffusion and erosion where hypromellose acts as rate-controlling matrix | NA | Approved/ Marketed | [38] |
| Permax® Eli Lilly and Amarin | Pergolide mesylate, croscarmellose sodium, iron oxide, lactose, magnesium stearate, and povidone | IR tablet | Once administered, the tablet disintegrates rapidly due to croscarmellose sodium after encountering gastric fluid | Adverse effects on heart valve | Withdrawn from market | [39] |
| Mirapex ER® Boehringer Ingelheim, Pfizer | Pramipexole, hypromellose, corn starch, carbomer homopolymer, colloidal silicon dioxide, and magnesium stearate | ER tablet | Extended release was achieved by combining three polymers such as hypromellose, corn starch, and carbomer homopolymer, after encountering digestive fluid. Drug first dissolves from the surface followed by matrix swelling, slow diffusion of the drug, along with the erosion of the tablet | NA | Approved/ Marketed | [40] |
| Dostinex® Pfizer | Cabergoline, leucine, and lactose | IR tablet | Tablet disintegrates rapidly once administered, releasing the drug, which is absorbed quickly into the blood circulation | NA | Approved/ Marketed | [41] |
| Eldepryl® Somerset | Selegiline, citric acid, lactose, magnesium stearate, and microcrystalline cellulose | IR capsule | NA | NA | Approved/ Marketed | [42] |
| Zelapar® Valeant pharmaceutical | Selegiline, gelatin, mannitol, glycine, aspartame, citric acid, yellow iron oxide, and grapefruit flavor | Orally disintegrating tablet | When the tablet is kept on the tongue, it disintegrates into small particles, which then dissolve in the saliva and are absorbed through the mucous membranes of the mouth | Can irritate the buccal mucosa | Approved/ Marketed | [43] |
| Apokyn Mylan Bertek | Apomorphine hydrochloride, sodium metabisulphite, sodium hydroxide, benzyl alcohol, hydrochloric acid | Subcutaneous injection | The drug is immediately absorbed into the bloodstream after administration of subcutaneous solution formulation (peak time 10–60 min) | Skin nodules at the site of injection | Approved/ Marketed | [34] |
| APO-go® Pen Britannia Pharmaceuticals | Apomorphine, sodium bisulfite, hydrochloric acid | Subcutaneous injection | The drug is immediately absorbed into the bloodstream after administration of subcutaneous solution formulation | Skin nodules at the site of injection | Approved/ Marketed | [34] |
| Kynmobi Sunovion Pharmaceuticals Inc | Apomorphine, disodium EDTA, dihydrate, FDandC Blue #1, glycerol, glyceryl monostearate, hydroxyethyl cellulose, hypromellose, maltodextrin, (-)-menthol, pyridoxine hydrochloride, sodium hydroxide, sodium metabisulfite, sucralose, and white ink | Sublingual film | When kept under the tongue, the film disintegrates in 3 min, releasing the drug, which is rapidly absorbed through a thin mucous membrane under the tongue. Maximum concentration reached within 0.5 to 1 h | The problem of stomatitis at higher dose | Approved/ Marketed | [34] |
| Neupro® UCB Pharma Limited | Backing film—aluminized polyester Drug matrix—rotigotine, ascorbyl palmitate, povidone, silicone adhesive, sodium metabisulfite, and dl-α-tocopherol Protective liner—transparent fluoropolymer-coated polyester film | Transcutaneous patch | 45% of rotigotine is released within 24 h from the patch by diffusion | Requires replacement every 24 h | Approved/ Marketed | [34] |
| Treatments in Clinical Trials | ||||||
| AP09004 Accordin pill™ Intec Pharma Ltd. | L-DOPA, CD | Gastro- retentive capsule | NA | Further studies are required to study its effectiveness in delaying the onset of motor complications | Completed Phase 2 | [44] |
| DM-1992 Depomed Inc Acuform™ technology | L-DOPA, CD | Gastro- retentive bilayered tablet | NA | Not statistically significant | Completed Phase 2 | [45] |
| XP21279 Xenoport Inc | Prodrug of L-DOPA, CD | Sustained-release bilayered tablet | NA | No difference in the reduction of off period | Completed Phase 2 | [46] |
| V1512 Vernalis/Chiesi Farmaceutici SpA | L-DOPA methyl ester, CD | Effervescent tablet | NA | No significant difference in pharmacokinetics compared to that of a conventional tablet | Completed Phase 2 | [47] |
| TRIGEL LobSor Pharmaceuticals AB | L-DOPA, CD and Entacapone | Intestinal gel | NA | A patient needs to be hospitalized for 3 days (expected), and it cannot be given at home | Completed Phase 1 | [48] |
| CVT-301 Acorda Therapeutics Inc | L-DOPA | Dry powder inhaler | NA | Cough induction due to the inhalation of powder | Completed Phase 3 | [49] |
| LY03003 Luye Pharma | Rotigotine | Sustained-release microsphere | NA | NA | Completed Phase 1 | [50] |
| IPX203 Impax pharma | L-DOPA, CD | IR and ER capsule | NA | NA | Completed Phase 3 | [51] |
| ABBV-951 AbbVie | L-DOPA phosphate, CD phosphate | Subcutaneous infusion system | NA | NA | Completed Phase 3 | [52] |
| ND0612 Neuroderm Ltd. | L-DOPA, CD | Subcutaneous infusion | NA | Small transient papules at infusion sites | Phase 3 ongoing | [53] |
| Infudopa SubC Vastra Gotaland Region | L-DOPA | IV, SC | NA | NA | Completed Phase 1 | [54] |
| ProNeura™ Titan Pharmaceuticals | Ropinirole | Subcutaneous implant | NA | NA | Terminated Phase 2 | [55] |
| SPM 952 UCB Pharma Limited | Rotigotine | Intranasal | NA | NA | Completed Phase 2 | [56] |
| Prestwick Pharmaceuticals, Schering AG | Lisuride | Transcutaneous patch | NA | Increased incidence of psychotic events | Completed Phase 2 | [34] |
| Drug/API | Method | Polymer | Advantages | Route of Administration | References |
|---|---|---|---|---|---|
| Pramipexole | W/O/W emulsion solvent evaporation | D,L- PLGA 50:50 (M.w 10,000 Da) | Plasma concentration revealed drug release up to 2 weeks | Intramuscular | [158] |
| Rasagiline | W/O/W emulsion solvent evaporation | Polycaprolactone | In-vitro prolonged release up to 45 days was observed, whereas improved pharmacodynamics up to 30 days was observed after single-dose administration | Subcutaneous | [159] |
| GDNF | W/O/W emulsion solvent evaporation | Poly(ε-caprolactone) (M.w 45,000 Da) | In in-vitro release studies, GDNF was released from the microsphere for up to 25 days | - | [160] |
| L-DOPA, CD | Electrohydrodynamic co-jetting | D,L-PLGA 50:50 (Intrinsic viscosity-0.67 g/mol), PLA | More than 90% of the drugs were released within 24 h | Oral | [161] |
| Ropinirole | O/O emulsion solvent evaporation | Eudragit RS | 50% of the drug was released in 12 h following zero-order kinetics | Oral | [162] |
| GDNF | W/O/W emulsion solvent evaporative | D,L-PLGA 50:50 (M.w 34,000 Da) | Single administration provided sustained motor improvement and dopaminergic function restoration | Stereotaxic implantation | [163] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakmode, D.D.; Day, C.M.; Song, Y.; Garg, S. The Management of Parkinson’s Disease: An Overview of the Current Advancements in Drug Delivery Systems. Pharmaceutics 2023, 15, 1503. https://doi.org/10.3390/pharmaceutics15051503
Nakmode DD, Day CM, Song Y, Garg S. The Management of Parkinson’s Disease: An Overview of the Current Advancements in Drug Delivery Systems. Pharmaceutics. 2023; 15(5):1503. https://doi.org/10.3390/pharmaceutics15051503
Chicago/Turabian StyleNakmode, Deepa D., Candace M. Day, Yunmei Song, and Sanjay Garg. 2023. "The Management of Parkinson’s Disease: An Overview of the Current Advancements in Drug Delivery Systems" Pharmaceutics 15, no. 5: 1503. https://doi.org/10.3390/pharmaceutics15051503