1. Introduction
Lambert–Eaton myasthenic syndrome (LEMS) is a rare autoimmune disorder of the neuromuscular junction. In patients with LEMS, antibodies against voltage-gated calcium channels decrease the release of acetylcholine into neuromuscular synapses, resulting in muscle weakness [
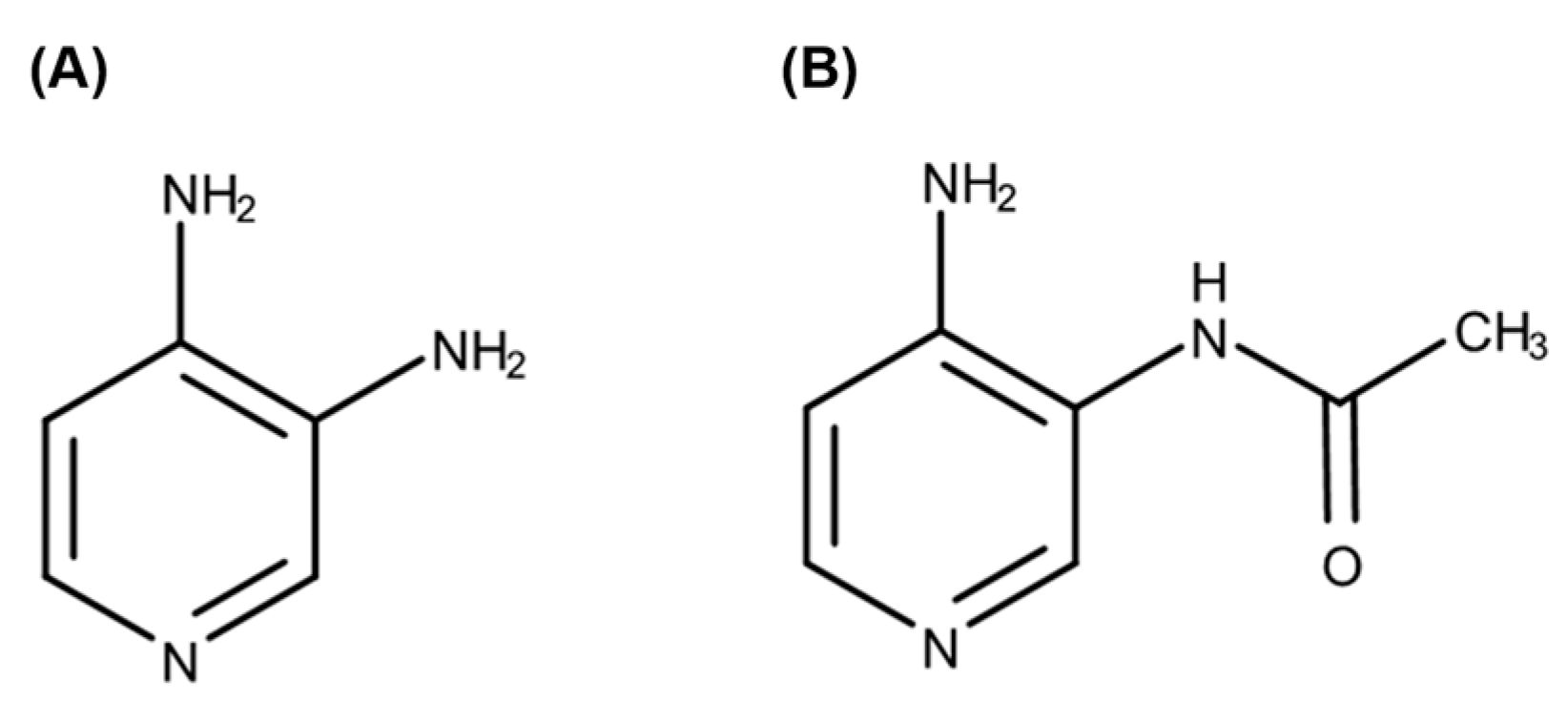
1]. Amifampridine (
Figure 1A), a potassium channel blocker, increases calcium influx, resulting in exocytosis of synaptic vesicles containing acetylcholine [
2]. Clinical trials have confirmed that amifampridine effectively controls fatigability and weakness in patients with LEMS, and its adverse effects are generally tolerable, with mild to moderate severity [
3]. The Food and Drug Administration (FDA) of the United States (US) approved amifampridine tablets for the treatment of LEMS in adults (Firdapse
®) as well as in patients 6 to 17 years of age (Ruzurgi
®) in 2018 [
4]. Amifampridine remains the only FDA-approved evidence-based treatment option for LEMS.
In humans, amifampridine is rapidly absorbed after oral administration and reaches its maximum plasma concentration (C
max) within 1 h, which is delayed by food intake [
5]. It is extensively metabolized to 3-
N-acetylamifampridine (
Figure 1B), a pharmacologically inactive metabolite mainly formed by
N-acetyltransferase 2 (NAT2). NAT1 is also involved in the metabolism of amifampridine to 3-
N-acetylamifampridine at a much slower rate [
6]. NAT2 genes are highly polymorphic in humans, and slow acetylators exhibit significantly higher blood levels of amifampridine, with more frequent adverse events [
4,
7], suggesting a significant contribution of NAT2 to the pharmacokinetics and safety of amifampridine. More than 90% of the dose of amifampridine (parent drug, 19%) and 3-
N-acetylamifampridine (metabolite, 74.0–81.7%) is excreted via the urine within 24 h after administration. The elimination half-life is approximately 2.5 h for amifampridine and 4 h for 3-
N-cetylamifampridine in humans [
4].
Acetylation is one of the pathways involved in the detoxification and biotransformation of many drugs [
8]. NATs are phase II metabolizing enzymes that transfer an acetyl group from acetyl coenzyme A (acetyl-CoA) to a xenobiotic acceptor substrate [
9,
10] and are responsible for the acetylation of aromatic and heterocyclic amines as well as hydrazines. The mechanism is a ping-pong bi-bi reaction involving two successive steps. First, catalytic Cys68 is acetylated by binding to acetyl-CoA. Acetyl-CoA is then released, followed by substrate binding and transfer of an acetyl group to the exocyclic amine or the oxygen of the hydroxylated amine in the second reaction [
11,
12]. NAT2 is an enzyme found in many organs, such as the liver, lungs, breast, colon, ureter, and prostate [
13], and catalyzes numerous clinically used drugs such as dapsone, procainamide, isoniazid, phenelzine, and sulfonamide [
14,
15]. Additionally, several NAT2 inhibitors, including acetaminophen, apocynin, and cimetidine, have been reported [
16,
17,
18]. Especially, the inhibitory effects of acetaminophen on NAT2 have been reported both in vitro and in vivo [
8,
16]. Acetaminophen inhibited sulfamethazine acetylation in human liver cytosol isolated from fast and slow acetylators (
Ki value of 2144 and 712 µM in fast and slow acetylators, respectively) and also the acetylation of sulfamethazine decreased in humans when acetaminophen was co-administered. These previous findings indicate that acetylation inhibition by acetaminophen is likely to induce clinically relevant drug interactions. However, drug interaction studies of acetaminophen with drugs mainly metabolized by the
N-acetylation process, such as amifampridine, are still quite limited. Considering its widespread use and high therapeutic dose (usual dose of 1000–2000 mg/day and maximum recommended therapeutic dose of 4000 mg/day), the potential for drug interactions between acetaminophen and NAT2 substrate drugs should be thoroughly investigated.
Unexpected alterations in drug pharmacokinetics may lead to decreased efficacy and increased toxicity, eventually resulting in treatment failure. In the case of amifampridine, increased systemic exposure may induce nervous system disorders based on its mechanism of action [
19]. However, investigations on NAT2-mediated drug interactions with amifampridine have rarely been reported. Therefore, this study aimed to investigate the effects of NAT2 inhibitors, including acetaminophen, on the pharmacokinetics of amifampridine, a NAT2 substrate, using in vitro and in vivo systems.
2. Materials and Methods
2.1. Materials
Acetaminophen, acetyl-CoA, amifampridine, apocynin, cimetidine, dimethyl sulfoxide (DMSO), formic acid, phosphate-buffered saline (PBS), Rompun 2% injection, and sodium chloride were purchased from Sigma Aldrich (St Louis, MO, USA). 3-N-acetylamifampridine and ondansetron were obtained from Toronto Research Chemicals (North York, ON, Canada) and Cadila (Ahmedabad, India), respectively. Heparin was purchased from Huons (Seongnam, Korea) and Zoletil was obtained from Vibrac SA (Carros, France). High-performance liquid chromatography (HPLC) grade methanol and acetonitrile from Honeywell Burdick and Jackson Co. (Ulsan, Korea) were used in this study. Liver S9 fractions of rats, humans, mice, and rat plasma were purchased from Sekisui XenoTech (Kansas City, KS, USA). Water was purified using an AquaMAX ultrapure water purification system (YL Instruments, Anyang, Korea). All other chemicals and solvents were of reagent or HPLC grade and were used as received without further purification.
2.2. In Vitro Metabolic Profiles of Amifampridine in Rat Liver S9 Fraction
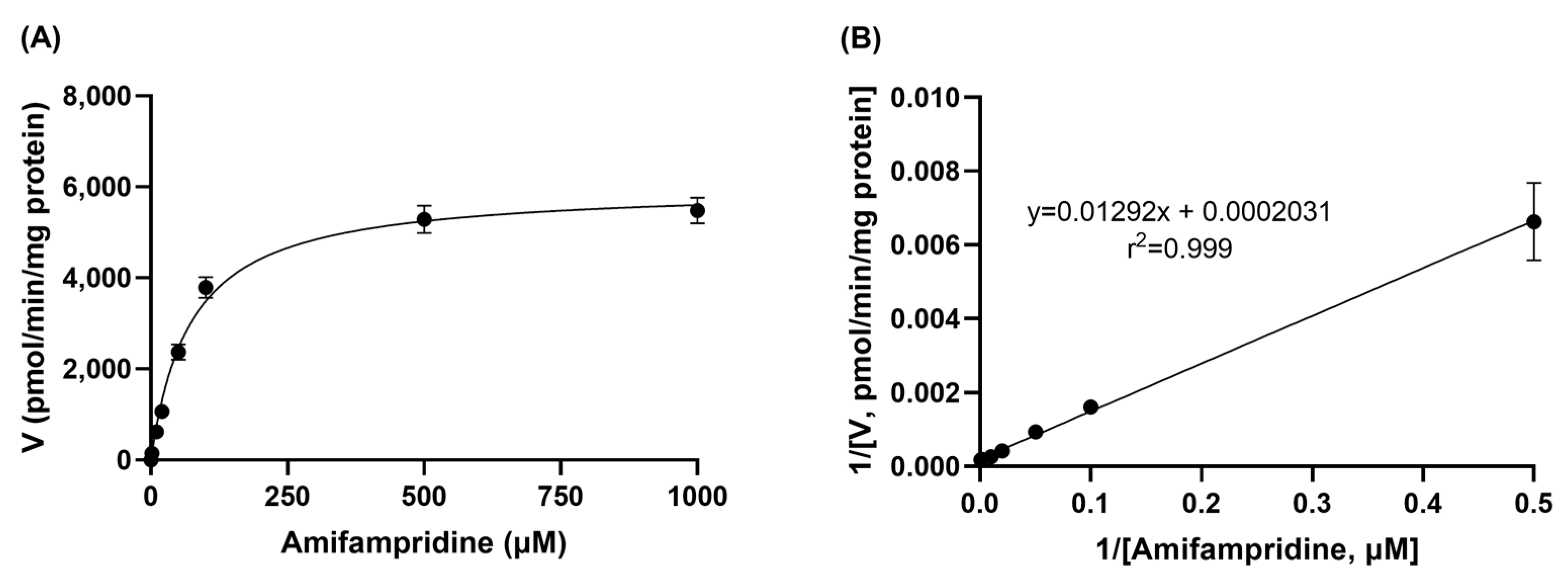
An in vitro metabolic study was performed to investigate the metabolic profile of amifampridine in the rat liver S9 fraction. The reaction mixture, which consisted of rat liver S9 fraction (2 mg/mL protein) and 2 mM acetyl-CoA in potassium phosphate buffer, was prepared and incubated for 5 min in a thermal mixer (200 oscillations/min) at 37 °C. Then, amifampridine was added to the reaction mixture to make the final concentrations of amifampridine at 2, 10, 20, 50, 100, 500, and 1000 μM. After further incubation for 15 min at 37 °C, 50 μL of the sample was transferred into the tube containing 200 μL of ice-cold potassium phosphate buffer and mixed by vortexing. An amount of 50 μL of the mixture was then transferred into the tube containing 100 μL of internal standard (IS; 5 ng/mL ondansetron in acetonitrile), followed by vortexing and centrifugation at 14,000 rpm for 15 min at 4 °C. The supernatant was transferred into vials for HPLC-tandem mass spectrometry (MS/MS) analysis and the concentration of 3-N-acetylamifampridine was determined.
2.3. In Vitro Inhibition of Amifampridine Acetylation in the Liver and Intestinal S9 Fractions
To determine the half maximal inhibitory concentration (IC50) value of NAT2 inhibitors (acetaminophen, apocynin, and cimetidine) on the metabolism of amifampridine to 3-N-acetylamifampridine, the reaction mixture consisted of a liver S9 fraction (2 mg/mL protein) and 2 mM acetyl-CoA in potassium phosphate buffer was pre-incubated for 5 min in a thermomixer at 37 °C. Then, the NAT2 inhibitors (final concentrations of 0, 1, 5, 10, 50, 100, 500, 1000, and 5000 μM) and amifampridine (final concentration of 50 μM) were added to the reaction mixture and incubated for 15 min at 37 °C. An amount of 50 μL of the mixture was then transferred into the tube and mixed with 100 μL of IS (5 ng/mL ondansetron in acetonitrile). The mixture was vortexed and centrifuged at 14,000 rpm for 15 min at 4 °C. The supernatant was transferred into vials for HPLC-MS/MS analysis and the concentration of 3-N-acetylamifampridine was determined.
The inhibitory effects of acetaminophen on amifampridine acetylation were compared in rat liver and intestine S9 fractions. The rat S9 fraction was prepared following a method previously reported [
20]. The reaction mixture consisted of rat liver or intestinal S9 fraction (2 mg/mL protein), and 2 mM acetyl-CoA in potassium phosphate buffer was pre-incubated for 5 min in a thermomixer at 37 °C. Then, acetaminophen (final concentration of 300 μM) and amifampridine (final concentration of 50 μM) were added to the reaction mixture and incubated for 15 min at 37 °C. The reaction mixture was then pre-treated and analyzed as mentioned above.
2.4. Mechanism of Metabolism Inhibition of Amifampridine by Acetaminophen
To determine the inhibition mechanism of amifampridine metabolism by acetaminophen, various concentrations of amifampridine (40, 80, 160, and 320 μM) were incubated with the rat liver S9 fraction in the presence of different concentrations of acetaminophen (0, 10, 50, 100, and 500 μM). After pre-incubating the reaction mixture containing 2 mM acetyl-CoA for 5 min in a thermomixer at 37 °C, acetaminophen and amifampridine were added and further incubated for 15 min at 37 °C. The samples were diluted five-fold using potassium phosphate buffer, then 50 μL of the diluted samples was transferred into the tube containing 100 μL of IS (5 ng/mL ondansetron in acetonitrile). The mixture was vortexed and centrifuged at 14,000 rpm for 15 min at 4 °C. The supernatant was transferred into vials for HPLC-MS/MS analysis and the concentration of 3-N-acetylamifampridine was determined.
To examine whether acetaminophen could inhibit the metabolic activity of amifampridine in a time-dependent manner, the experiment was conducted by using a high concentration of amifampridine (approximately four-fold Km value) for different pre-incubation times (0, 10, 30, and 60 min) in the presence of different concentrations of acetaminophen (0, 10, 50, 100, and 500 μM). The reaction mixture contained the liver S9 fraction (2 mg/mL protein) and 2 mM acetyl-CoA in potassium phosphate buffer. Then, acetaminophen was added and pre-incubated at different times in a thermomixer at 37 °C. The reaction was then initiated by adding amifampridine (240 μM as a final concentration) and vortex mixing followed by incubation at 37 °C for 15 min. After incubation, the 50 μL aliquot was transferred into a tube containing 200 μL of ice-cold potassium phosphate to dilute it five-fold. Then, the 50 μL of the mixture was transferred into the tube containing 100 μL of IS (5 ng/mL ondansetron in acetonitrile) to terminate the reaction, followed by vortexing and centrifugation at 14,000 rpm for 15 min at 4 °C. Finally, the supernatant was transferred to liquid chromatography (LC) vials for analysis, and the concentration of 3-N-acetylamifampridine was determined by LC-MS/MS.
2.5. In Vivo Pharmacokinetic Interaction Study in Rats
Male Sprague Dawley (SD) rats (7–8 weeks old, 260–330 g) were purchased from Orient Bio, Inc. (Seongnam, Korea). The rats were housed under a light/dark cycle for 12 h and were left for at least a week before the experiment to adapt to the laboratory environment. Water and food were provided ad libitum. The rats were fasted overnight with free access to water prior to pharmacokinetic studies. The protocols for animal experiments were approved in accordance with the Guidelines for Animal Care and Use of Gachon University (approval no.: GIACUC-R2021006; 13 May 2021).
The rats were anesthetized with a mixture of Rompun and Zoletil by intramuscular injection and received an intraperitoneal injection of vehicle (40:60 PEG400:normal saline; n = 8) or 100 mg/kg acetaminophen (n = 8). After 10 min, 2 mg/kg amifampridine (dissolved in saline) was administered orally. Blood samples were collected at 0, 5, 15, 30, 60, 90, 120, 240, and 360 min and centrifuged at 14,000 rpm for 15 min at 4 °C. For analysis of amifampridine and 3-N-acetylamifampridine, 50 μL of plasma samples was mixed with 100 μL IS solution (5 ng/mL ondansetron in acetonitrile) and the mixture was vortexed followed by centrifugation at 14,000 rpm for 15 min at 4 °C. For the analysis of acetaminophen, 200 μL of IS (tolbutamide 200 ng/mL in methanol) was added to the plasma samples which were diluted 10-fold using the blank plasma and then vortexed for one minute. Further, the samples were centrifuged at 14,000 rpm for 15 min at 4 °C. The supernatants were transferred to LC vials for quantification.
Urine samples were collected at intervals of 0–4, 4–8, and 8–24 h after the administration of amifampridine to control and acetaminophen-treated rats (n = 6), as described above, and were diluted 500-fold with potassium phosphate buffer. An amount of 200 μL of IS was then added to the samples and vortexed for a minute. After centrifugation at 14,000 rpm for 15 min at 4 °C, the supernatant was transferred into vials for quantification of amifampridine and 3-N-acetylamifampridine.
To investigate the effects of acetaminophen on the tissue distribution of amifampridine and 3-N-acetylamifampridine, rats were sacrificed at 60 min after administration of amifampridine in control and acetaminophen-treated rats (n = 6) as mentioned above, and the tissues, including liver, kidney, heart, lungs, spleen, brain, and muscle tissues, were obtained. Tissue samples were homogenized using a WheatonTM Dounce tissue grinder (Wheaton Millipore, Billerica, MA, USA) after adding a three-fold volume of PBS. An amount of 100 μL of tissue homogenates was transferred to the tube and 200 μL of IS solution was then added and vortexed followed by centrifugation at 14,000 rpm for 15 min at 4 °C. The supernatants were transferred to LC vials for quantification. For muscle samples, the supernatant was concentrated using a desiccator and reconstituted with the mobile phase solvent.
2.6. Quantification of In Vitro and In Vivo Samples Using LC-MS/MS
An Agilent 6490 Triple Quadrupole MS coupled with an Agilent Technologies 1260 HPLC system (LC-MS/MS) was used to quantify amifampridine and 3-N-acetylamifampridine. The mass spectrometer was operated in multiple reaction monitoring (MRM) modes with positive electrospray ionization (ESI+). The MRM transitions for amifampridine, 3-N-acetylamifampridine, and IS (ondansetron) were m/z 110.1→93.0, 152.1→110.1, and 294.1→170.0, respectively. The collision energy was 20, 16, and 28 V for amifampridine, 3-N-acetylamifampridine, and IS, respectively. Simultaneous chromatographic separation of amifampridine and 3-N-acetylamifampridine was performed using a reversed-phase HPLC column (Synergi Polar—RP column 80 Å, 150 × 2.0 mm, 4 µm; Phenomenex (Torrance, CA, USA)) with a linear gradient of 0.1% formic acid in water and acetonitrile (95%:5%→30%:70%) at a flow rate of 0.2 mL/min for 17 min. The column oven was set to 25 °C and the injection volume was 2 µL.
For the analysis of acetaminophen, MRM mode with ESI+ was used with the MRM transitions of m/z 152.1→110 and 271.1→155 for acetaminophen and IS (tolbutamide), respectively. The collision energy for both acetaminophen and IS was 14 V. The chromatographic separation was performed with Synergi 4 μM and a Polar-RP column 80 Å (150 × 2.0 mm; Phenomenex) with a linear gradient of 0.1% formic acid in water and acetonitrile (80%:20%→20%:80%) at a flow rate of 0.2 mL/min for 20 min. The column oven was set to 25 °C and the injection volume was 2 µL.
Calibration curves were constructed by plotting the peak area ratios of the analytes to IS on the y-axis against their concentration on the x-axis using weighted (1/x) least-squares linear regression analysis.
2.7. Data Analysis
The
Vm and
Km values, Michaelis–Menten parameters, were determined by linear regression using Lineweaver–Burk equation, as follows:
where
v is the rate of amifampridine metabolism to 3-
N-acetylamifampridine in the S9 fraction, [
S] is the concentration of amifampridine,
Vmax is the maximum metabolic rate of amifampridine, and
Km is the concentration of amifampridine at half
Vmax. The intrinsic clearance (CL
int) was calculated using
Vmax/Km, with the assumption that
Km is much greater than [
S], as is usual in clinical practice.
The inhibition mode was determined using Lineweaver–Burk plots, and
Ki was calculated using the following equation, assuming mixed inhibition:
where
v is the rate of metabolism to 3-
N-acetylamifampridine at each concentration of amifampridine,
Vmax is the maximum rate of metabolism,
Km is the Michaelis constant, I is the concentration of acetaminophen,
Ki is the inhibition constant, and [
S] is the amifampridine concentration. The α value is the degree to which the binding of acetaminophen changes the affinity of the enzyme for amifampridine.
Kobs, the observed rate of enzyme loss, was estimated from the negative slopes of the lines using a linear regression analysis of the natural logarithm of metabolite formation as a function of incubation time [
21]. The first-order rate constant,
Kinact, which represents the maximum rate of enzyme inactivation, was calculated using a nonlinear regression analysis as follows:
Non-compartment analysis (NCA) was performed to calculate the pharmacokinetic parameters for the in vivo study using WinNonlin (Version 8.3. (Pharsight Corporation, Mountain View, CA, USA)). The area under the plasma concentration–time curve from time 0 to infinity (AUCinf) was calculated by the linear trapezoidal method and standard extrapolation using λ, where λ represents the slope of the terminal phase in a semi-log graph for plasma concentration–time profiles. Renal clearance (CLr) was determined by dividing the accumulated amount of drug excreted in the urine by AUCinf.
A two-tailed unpaired Student’s t-test was performed to determine statistically significant differences between groups and p-values less than 0.05 were considered significant. Data are presented as means ± SD.
4. Discussion
Amifampridine is primarily eliminated via metabolism to 3-
N-acetylamifampridine, which is formed mainly by NAT2 and NAT1, and the NAT1 has a much slower metabolic rate than NAT2 [
5]. The C
max and AUC of amifampridine are approximately 3.5-fold and 5–10-fold higher in slow acetylators than in fast acetylators for NAT2 [
19]. In addition, a more than 10-fold higher frequency of drug-related treatment-emergent adverse events has been reported with slow acetylators than with fast acetylators for NAT2 [
19]. Thus, NAT2 activity is critical for maintaining treatment efficacy with minimal adverse effects. However, to the best of our knowledge, information regarding the drug interactions of amifampridine mediated by NAT2 has not yet been reported, which possibly limits the effective treatment of LEMS with amifampridine. Therefore, we investigated the effects of NAT2-mediated interactions with amifampridine using in vitro and in vivo systems.
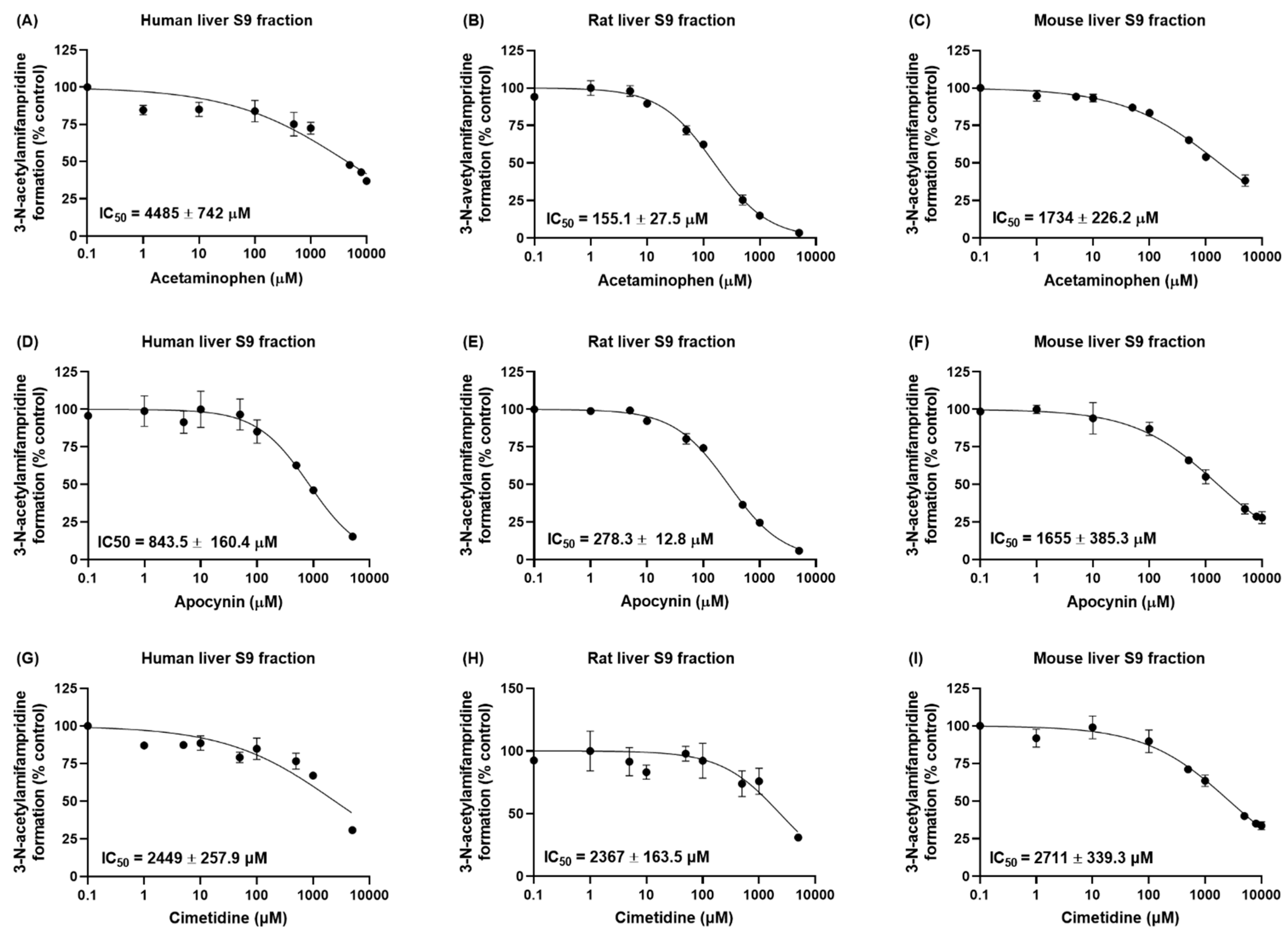
In this study, we confirmed that amifampridine was metabolized through the acetylation pathway in the rat liver S9 fraction. We then evaluated the in vitro inhibitory effects of the well-known NAT2 inhibitors acetaminophen [
16], apocynin [
17], and cimetidine [
18,
22] in liver S9 fractions obtained from humans, rats, and mice (
Figure 3). The IC
50 value of cimetidine on NAT2 was calculated as 2367 µM in rat liver S9 fraction, which is similar to the value in a previous report (IC
50 = 2060 µM [
22]). In contrast, the IC
50 value of apocynin on NAT2 obtained in this study (278.3 µM in rat liver S9 fractions) was less than the previous report (690 µM [
17]). However, considering the dependency of IC
50 values on experimental conditions, such as substrate or enzyme concentrations, the IC
50 value obtained in this study seems within a reasonable range. The inhibitory effects of cimetidine on the acetylation of amifampridine were similar in human, rat, and mouse liver S9 fractions; acetaminophen and apocynin showed substantial species differences in their inhibitory effects. In particular, an approximately 29-fold stronger inhibitory effect of acetaminophen was observed in rats compared to that in humans (IC
50 values of 155.1 μM and 4485 μM in rat and human liver S9 fractions, respectively). These results indicate that meticulous care is required in the interpretation of NAT2 inhibitory study results obtained in species other than humans.
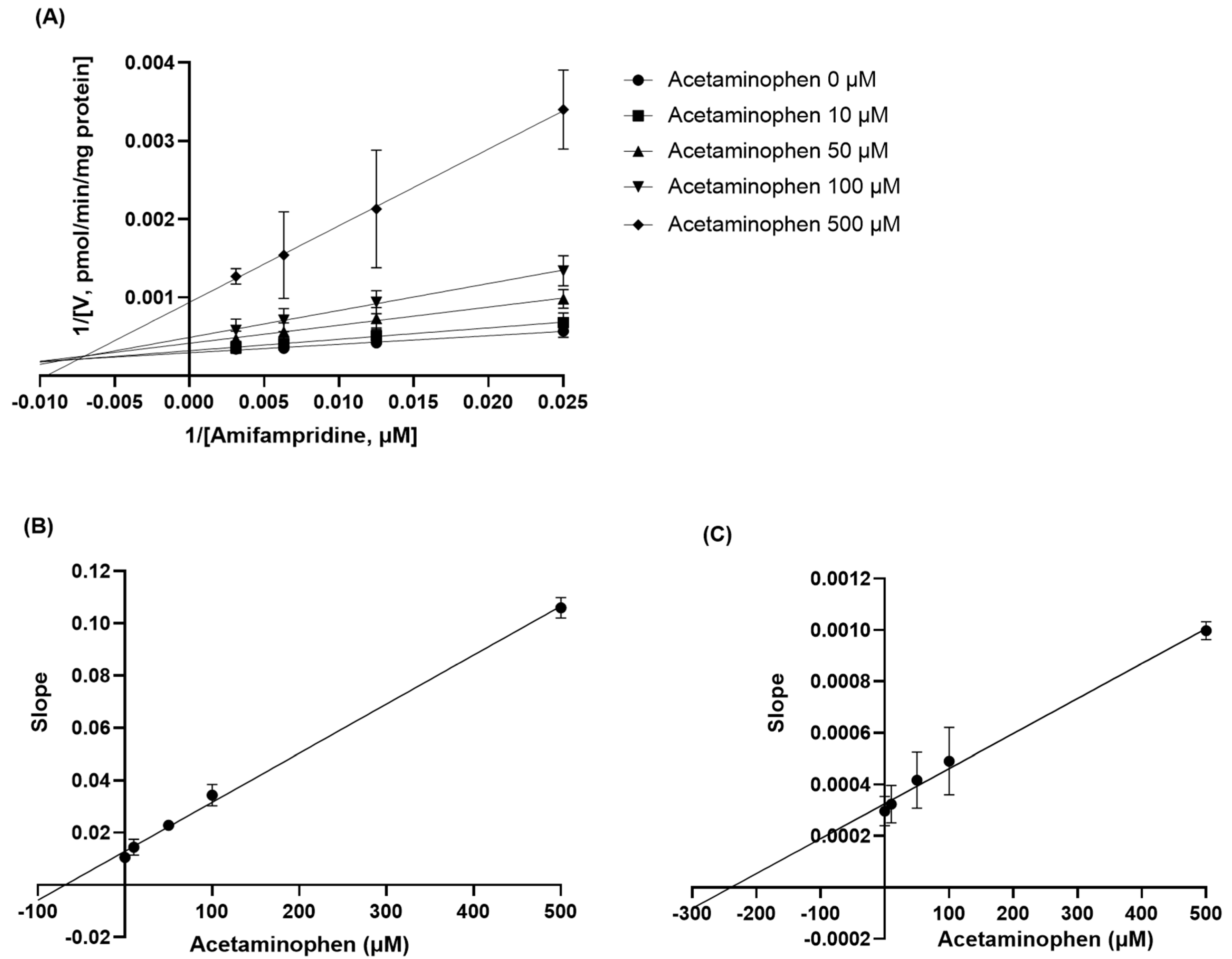
To further investigate the inhibitory mechanisms of acetaminophen on
N-acetylation of amifampridine, rat liver S9 fraction was treated with various concentrations of amifampridine and acetaminophen and the formation rate of 3-
N-acetylamifampridine was determined (
Figure 4). We observed that acetaminophen inhibits
N-acetylation of amifampridine in a mixed inhibition manner, which implies that acetaminophen can bind not only with unbound NAT2 but also bound NAT2 with amifampridine, resulting in altered binding affinities of substrates to the enzyme and a maximum metabolism capacity. Acetaminophen also inhibited the
N-acetylation of amifampridine in a time-dependent manner, with a
Kinact of 0.0031 min
−1 (
Figure 5), indicating that the metabolic activity of NAT2 on amifampridine decreased by approximately 0.31% per minute in the presence of acetaminophen. The underlying mechanisms of the acetaminophen-mediated inhibition of amifampridine acetylation determined in this study will help understand the interactions between acetaminophen and amifampridine.
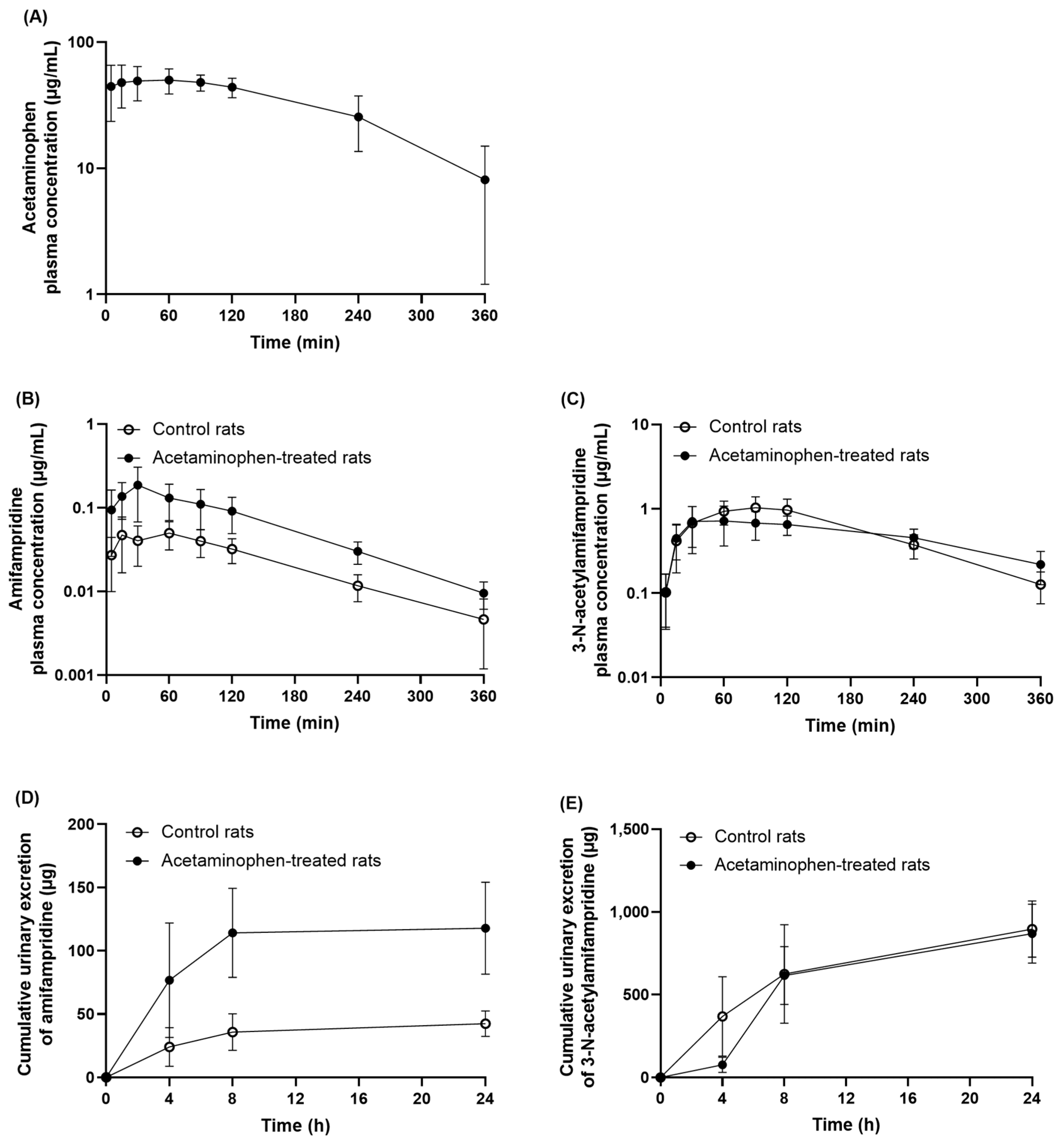
Since acetaminophen strongly inhibited the metabolism of amifampridine in the rat liver and intestinal S9 fractions in vitro, the potential of in vivo pharmacokinetic interactions between amifampridine and acetaminophen was investigated in rats (
Figure 6). The dose of amifampridine (2 mg/kg) was determined based on the clinical dose and several pharmacokinetic studies of amifampridine in rats [
4,
23]. The dose of acetaminophen (100 mg/kg) was selected based on clinical doses and pharmacokinetic interaction studies with acetaminophen in rats [
24,
25]. The systemic exposure to amifampridine was significantly increased and the total clearance (CL/F) decreased when acetaminophen was administered to rats. However, t
1/2 remained unchanged after treatment with acetaminophen, likely due to both a decreased CL/F and volume of distribution (V
d/F) (
Table 1), which affect t
1/2. The reasons behind a decreased V
d/F remain unclear; however, the increased F, possibly due to inhibition of first-pass intestinal and hepatic metabolism of amifampridine by acetaminophen before reaching systemic circulation, and the decreased distribution of amifampridine into several tissues by acetaminophen may be considered as the relevant mechanisms. Similarly, the increased F can affect the CL/F of amifampridine in the presence of acetaminophen. To confirm the effect of acetaminophen on the systemic CL and V
d of amifampridine, an additional in vivo PK study with intravenous administration of amifampridine would be necessary. The AUC ratio of 3-
N-acetylamifampridine to amifampridine (AUC
m/AUC
p) significantly decreased by 65% after pretreatment with acetaminophen (
Table 1), suggesting that the pharmacokinetic profiles of amifampridine and its metabolite were strongly affected by acetaminophen in vivo. Furthermore, a highly significant increase was observed in the cumulative urinary recovery of amifampridine in acetaminophen-treated rats compared to control rats. The renal clearance of amifampridine did not differ between the groups, which may be due to the proportional increase in cumulative urinary recovery to the increase in AUC
inf in the plasma of acetaminophen-treated rats. In addition, the accumulated amount excreted in the urine or renal clearance of 3-
N-acetylamifamrpdine was not altered by treatment with acetaminophen. Taken together, the increased systemic exposure to amifampridine and the decreased AUC
m/AUC
p are unlikely to be attributable to the interaction with acetaminophen via the urinary excretion pathway of amifampridine or its metabolite.
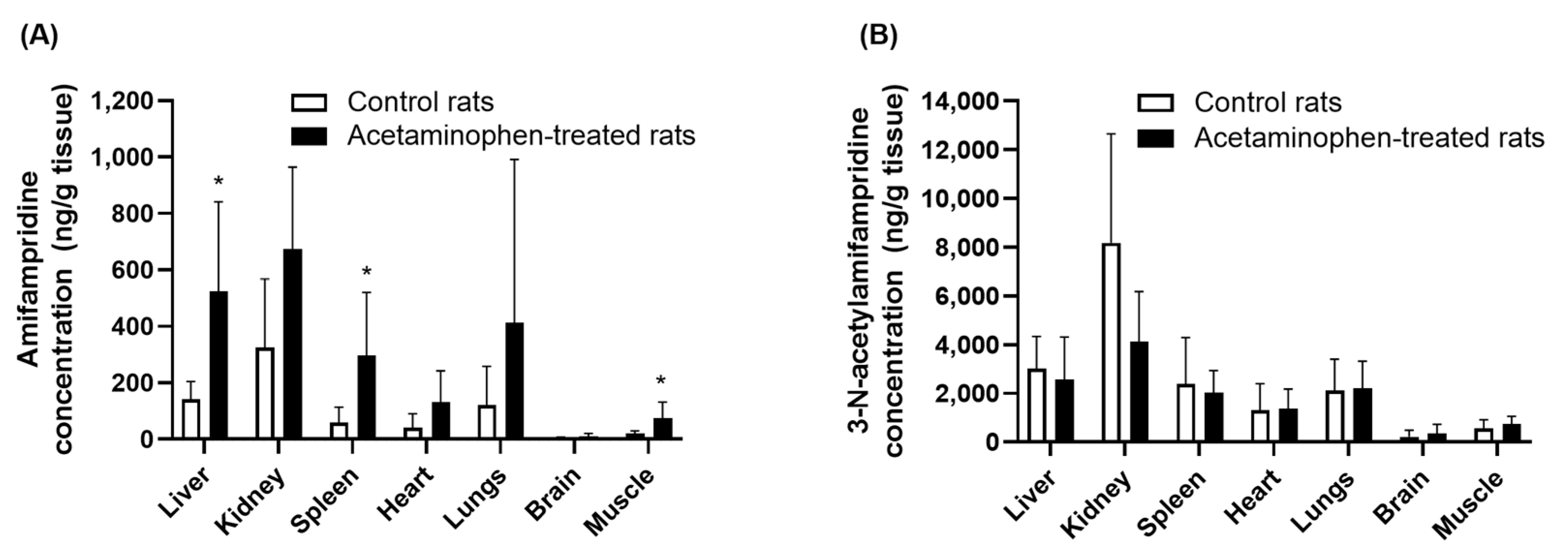
A tissue distribution analysis showed that the amount of amifampridine distributed in the tissue was higher in acetaminophen-treated rats than in control rats (
Figure 7). However, the K
p values, which were calculated by dividing the concentration of amifampridine in the tissue by that in the plasma, were not significantly altered by treatment with acetaminophen, except in the kidneys (
Table 2). The K
p value of 3-
N-acetylamifampridile was not significantly altered by acetaminophen in any tissue studied. These results suggested that the mechanisms governing the tissue distribution of amifampridine and 3-
N-acetylamifampridile were not significantly associated with acetaminophen, except in the kidneys. The K
p value of amifampridine in the kidneys decreased by 51% in acetaminophen-treated rats. This was likely a result of the increased urinary excretion of amifampridine in acetaminophen-treated rats. The mechanism underlying this finding remains unclear, and further studies are necessary to elucidate it. However, the altered distribution of amifampridine only to the kidneys after treatment with acetaminophen is less likely to affect the plasma concentrations of amifampridine, considering the volume of the kidney and total plasma volume. Collectively, the inhibitory effects of acetaminophen on the
N-acetylation of amifampridine seem to be the most plausible mechanism to explain the in vivo pharmacokinetic interactions between acetaminophen and amifampridine, based on the in vitro and in vivo results (urinary excretion and tissue distribution studies).
It is generally known that acetaminophen is safe and less likely to induce clinically important drug interactions; thus, this drug is available over the counter as well as by prescription in most countries. However, several recent studies have provided evidence that acetaminophen may induce drug interactions by altering the pharmacokinetics of victim drugs. For example, systemic exposure to sorafenib, a tyrosine kinase inhibitor used for the treatment of advanced renal cell carcinoma, and its metabolite N-oxide sorafenib increased in the presence of acetaminophen in rats [
26], which may be due to P-glycoprotein (P-gp) inhibition by acetaminophen [
27]. In addition, acetaminophen increases the clearance of lamotrigine glucuronide conjugates, resulting in decreased systemic exposure to lamotrigine in humans [
28]. Our study also highlights the importance of acetaminophen-mediated drug interactions; thus, drug interactions with acetaminophen should receive more attention regarding their clinical implications. However, in our study, acetaminophen inhibited the amifampridine metabolism much more strongly in the rat liver S9 fraction than in the human liver S9 fraction, and species differences should be carefully considered when interpreting the in vivo interaction results in rats and humans. The observed C
max values after oral or intravenous administration of acetaminophen are ~200 µM at most in humans [
29,
30]. Considering the IC
50 of acetaminophen on NAT2 inhibition in human liver S9 fractions and the pharmacokinetic profiles of acetaminophen reported in humans, the potency of drug interactions might be lower than those observed in rats. Further translational research between different species and clinical studies are warranted to obtain more confirmative evidence on the drug interactions between amifampridine and acetaminophen in humans. Since the NAT2 gene is expressed in the tissues other than the liver, such as the intestine [
31], the tissue distribution of the NAT2 gene in different species should be also considered.
This study was originally designed to investigate the systemic drug interactions between acetaminophen and amifampridine via hepatic NATs, and thus acetaminophen was administered by the intraperitoneal route. However, acetaminophen administered via the intraperitoneal route was absorbed rapidly and possibly affected the intestinal and hepatic first-pass metabolism of amifampridine, which was administered orally. Additionally, acetaminophen distributed into the tissues is likely to affect metabolism of amifampridine during systemic circulation not only in the liver but also in the intestine. Thus, diverse processes may be involved in the observed in vivo interactions between amifampridine and acetaminophen in rats, even though the level of contribution in each process cannot be separated quantitatively from this study. We confirmed that the inhibitory effects of acetaminophen on amifampridine acetylation are comparable between rat liver and intestinal S9 fractions; however, more comprehensive and systematic information on the interaction between amifampridine and acetaminophen, including a comparison of the inhibitory effects of acetaminophen in human and rat intestinal S9 fractions, is required to facilitate translation of the interaction effect into humans.
To the best of our knowledge, this is the first study to investigate the NAT2-mediated drug interaction potential of amifampridine with acetaminophen in vitro and in vivo. This study reported that acetaminophen inhibited the metabolism of amifampridine, leading to altered pharmacokinetics in rats. Therefore, care should be taken when amifampridine is co-administered with acetaminophen.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}