
Potent and Selective Inhibition of CYP1A2 Enzyme by Obtusifolin and Its Chemopreventive Effects
 , , , and
, , , and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Enzymes
2.2. CYP1A2 Activity Assays
2.3. Molecular Docking Simulation
2.4. Time-Dependent Inhibition Assays
2.5. Kinetic Characterization of CYP1A2 Inhibition by Obtusifolin in Human Liver Microsomes and Recombinant CYP1A2
2.6. Inhibitory Effects of Obtusifolin against Human Cytochrome P450s Activity
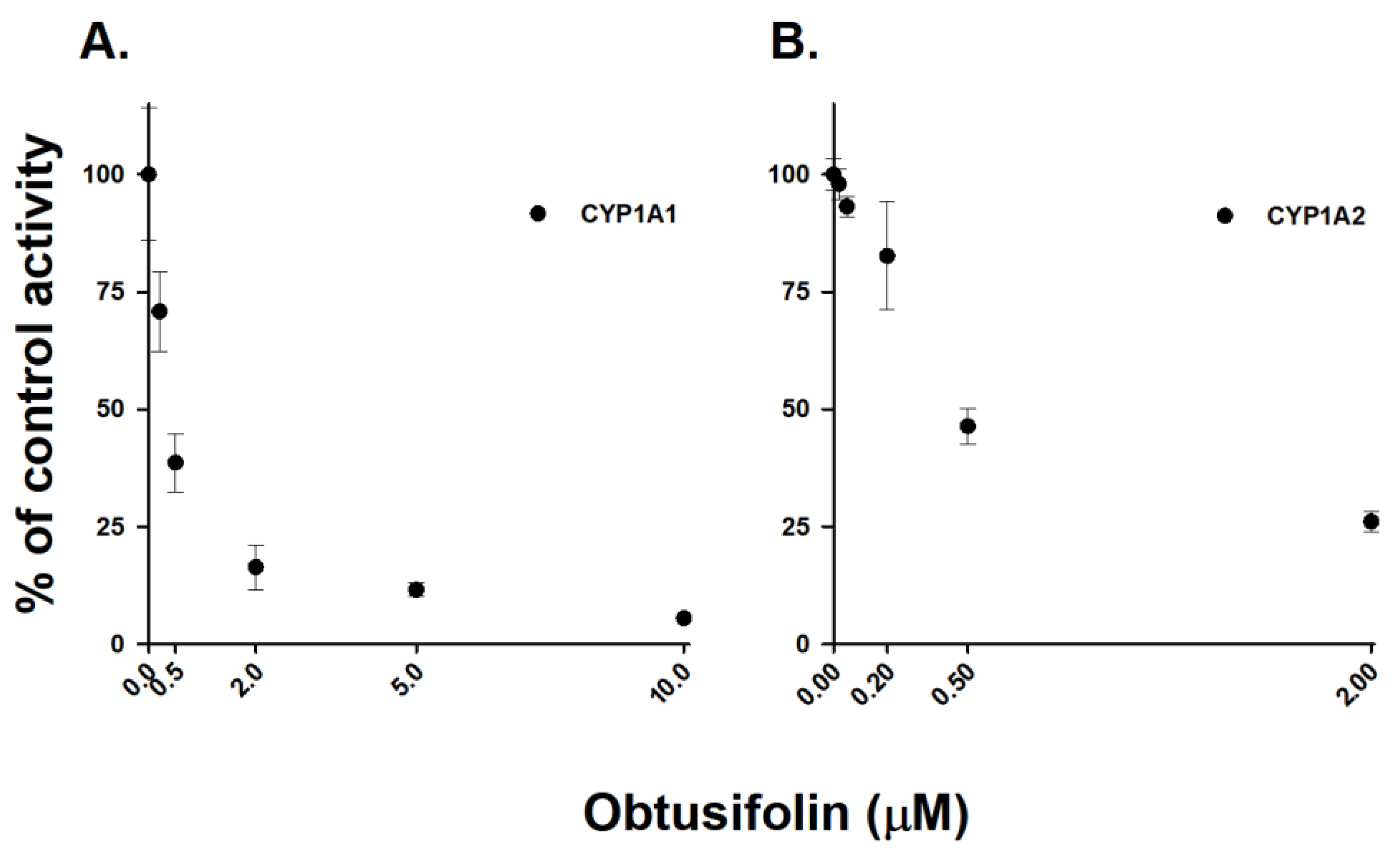
2.7. Inhibitory Effects of Obtusifolin on Human Recombinant CYP1A1 and CYP1A2 Enzymes
2.8. LC-MS/MS Analysis
2.9. Data Analysis
3. Results and Discussion
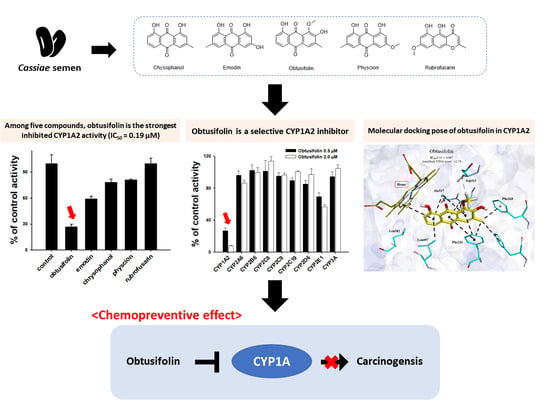
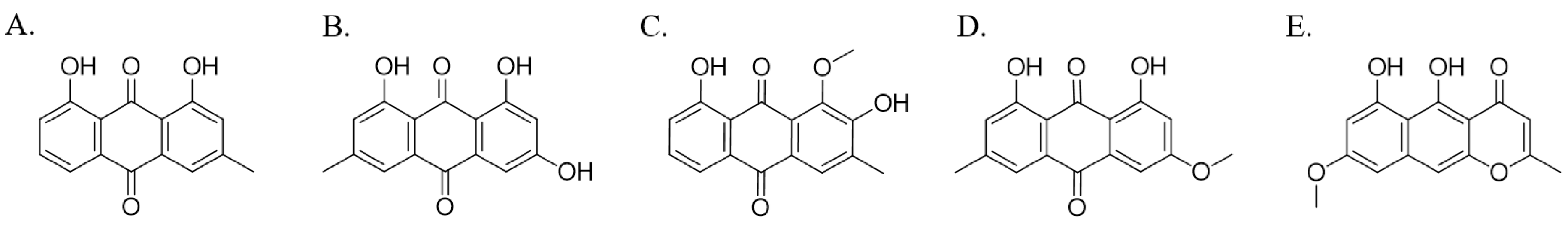
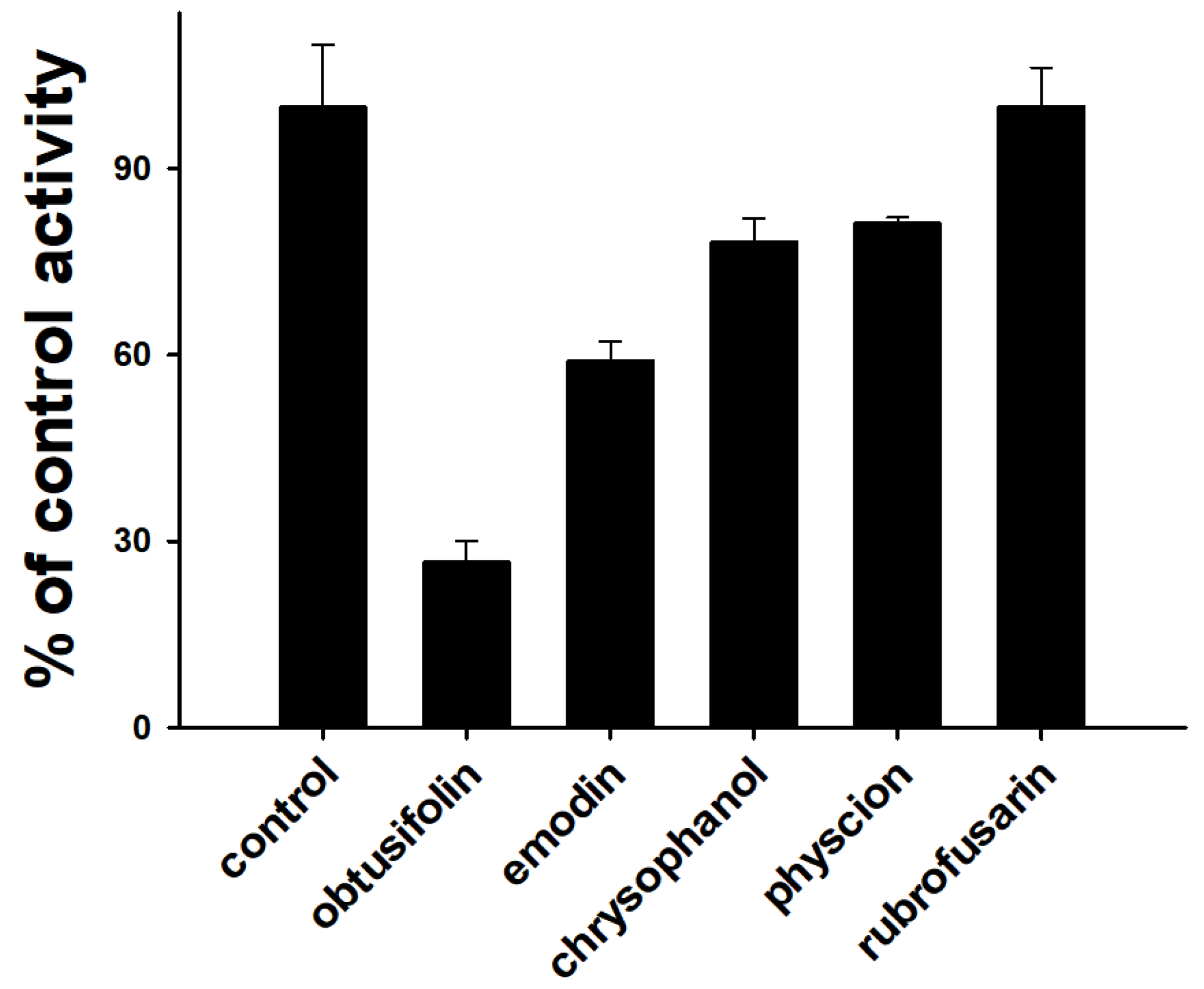
3.1. Inhibition of CYP1A2 Activity by Five Major Phytochemicals from Cassiae Semen
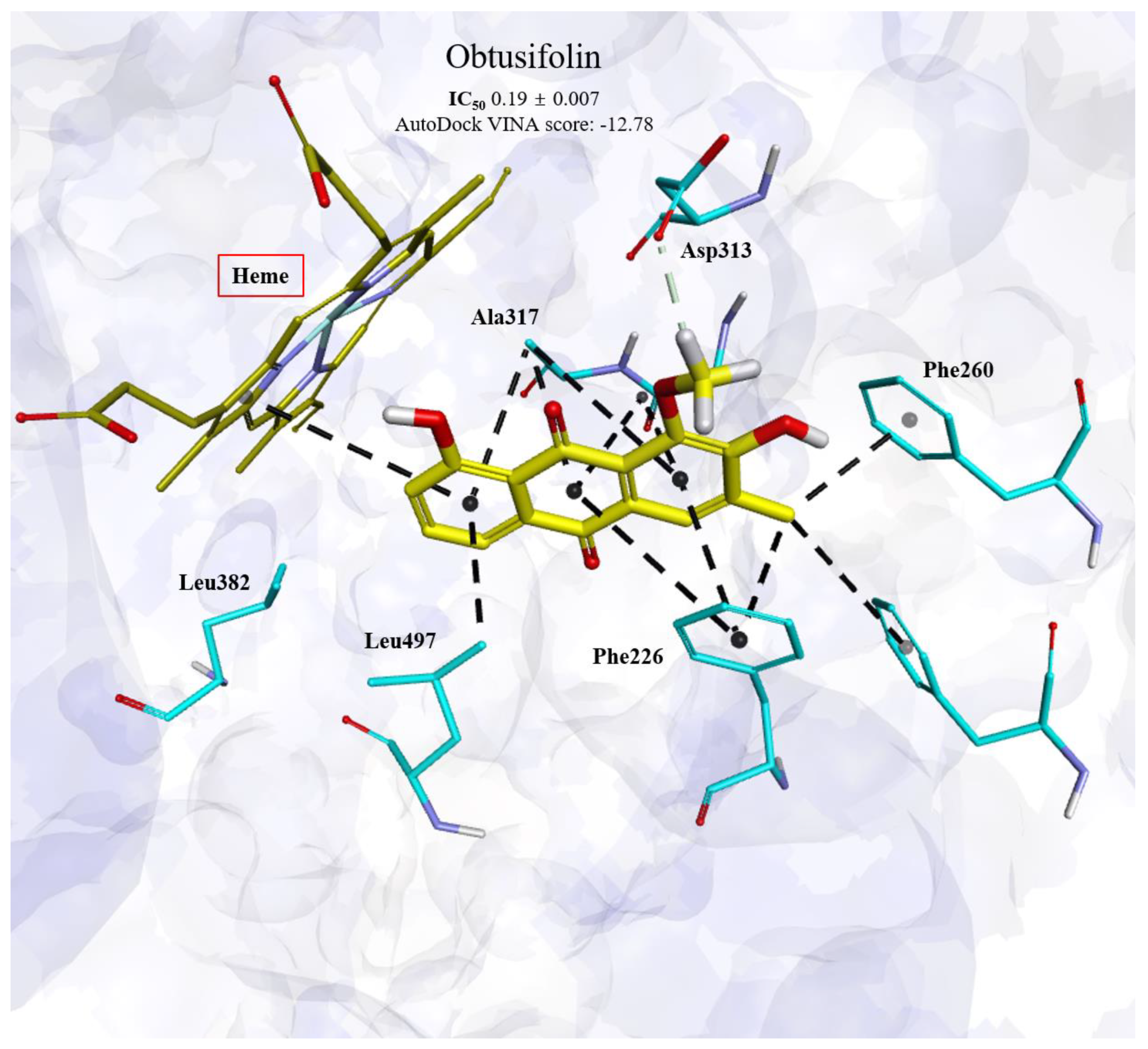
3.2. Binding Modes of Obtusifolin Assessed by Molecular Docking Simulation
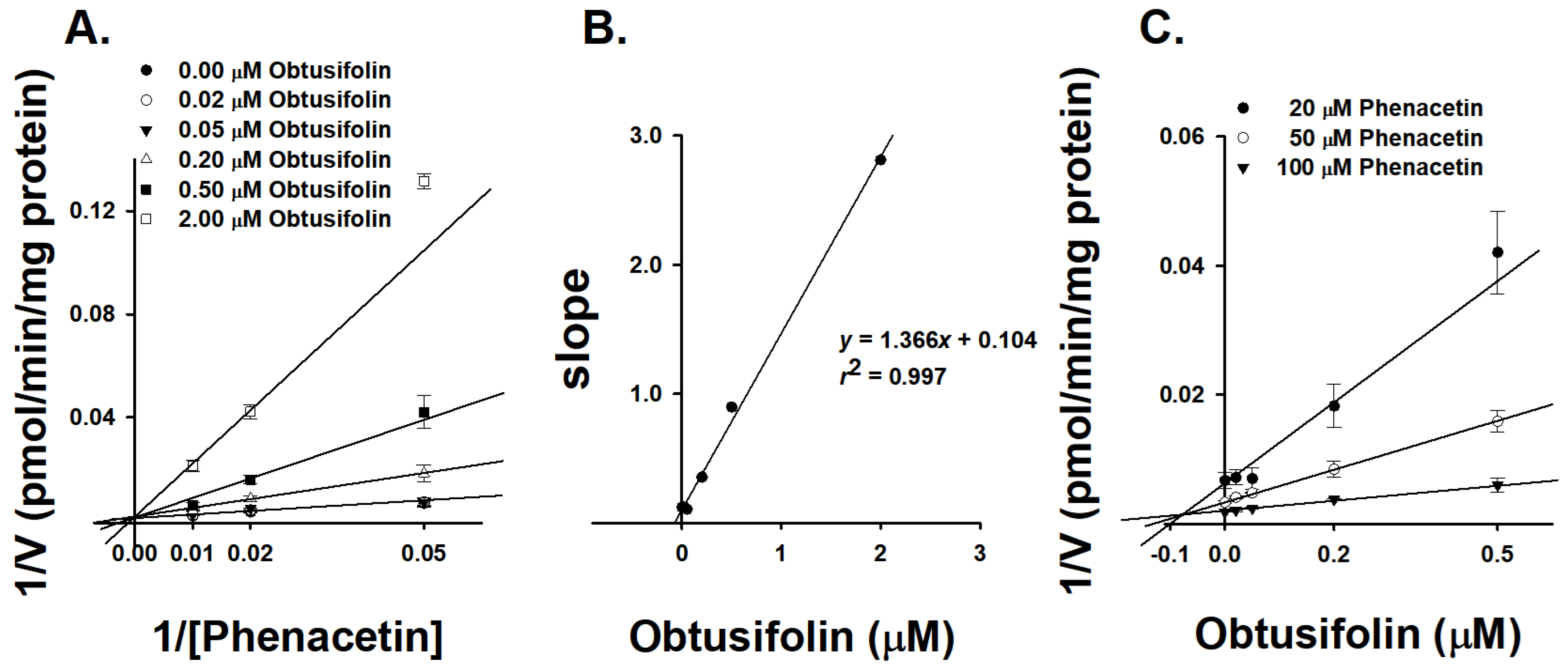
3.3. Characterizatin of Inhibition Kinetics of Obtusifolin against CYP1A2
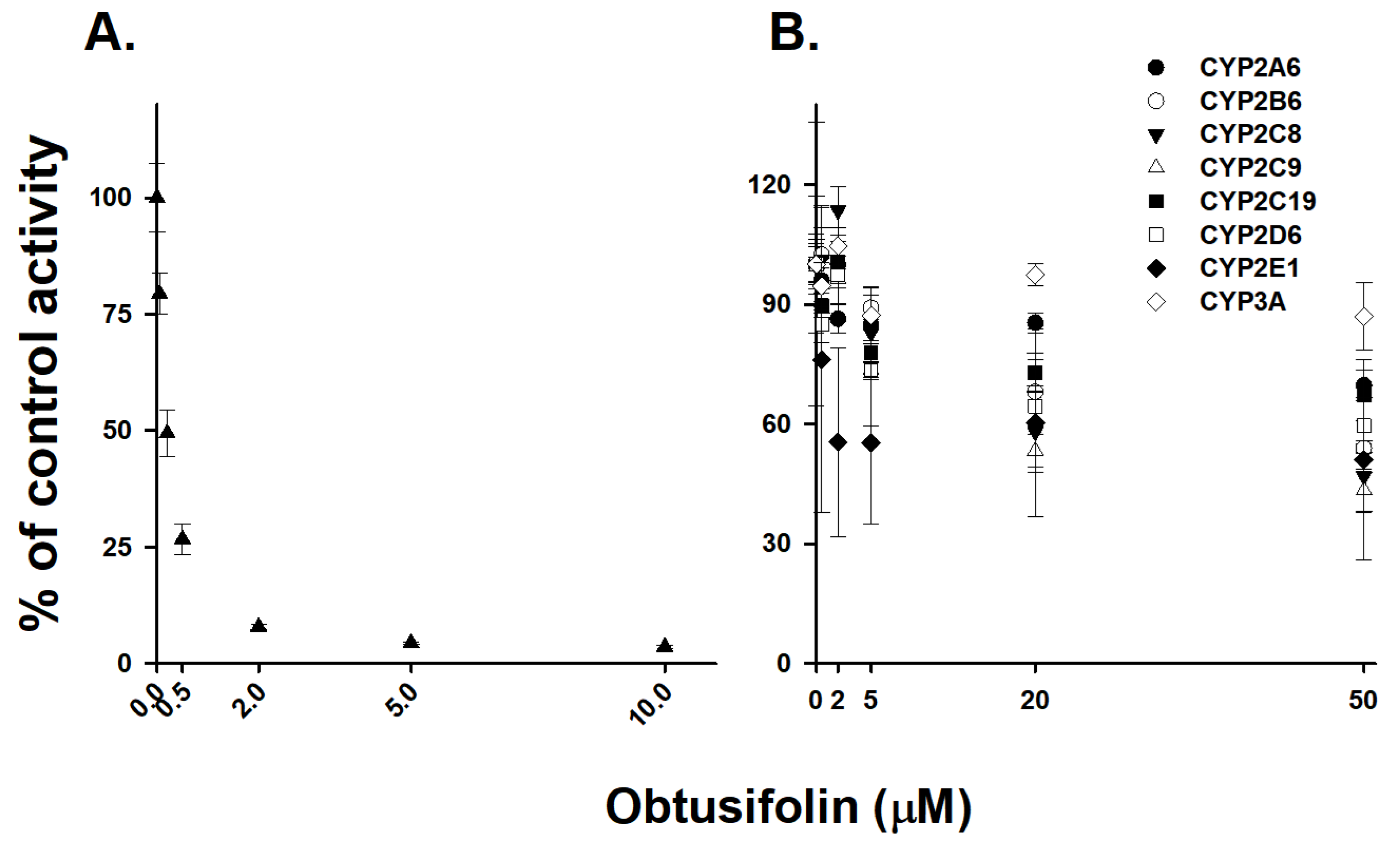
3.4. Selective Inhibition of CYP1A2 Activity by Obtusifolin
3.5. Chemopreventive Effects of Obtusifolin
3.6. Evaluation of Drug Interaction Potential of Obtusifolin
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, B.; Xie, L.; Peng, S.; Sun, K.; Jin, J.; Zhen, Y.; Qin, K.; Cai, B. Nine components pharmacokinetic study of rat plasma after oral administration raw and prepared Semen Cassiae in normal and acute liver injury rats. J. Sep. Sci. 2019, 42, 2341–2350. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Fu, J.; Yin, X.; Yang, C.; Zhang, X.; Wang, W.; Du, X.; Wang, Q.; Ni, J. Cassiae semen: A review of its phytochemistry and pharmacology (Review). Mol. Med. Rep. 2017, 16, 2331–2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, D.S.; Lee, G.Y.; Kim, Y.S.; Lee, Y.M.; Kim, C.S.; Yoo, J.L.; Kim, J.S. Anthraquinones from the seeds of Cassia tora with inhibitory activity on protein glycation and aldose reductase. Biol. Pharm. Bull. 2007, 30, 2207–2210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatano, T.; Uebayashi, H.; Ito, H.; Shiota, S.; Tsuchiya, T.; Yoshida, T. Phenolic constituents of Cassia seeds and antibacterial effect of some naphthalenes and anthraquinones on methicillin-resistant Staphylococcus aureus. Chem. Pharm. Bull. 1999, 47, 1121–1127. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Hyun, S.K.; Yoon, B.H.; Seo, J.H.; Lee, K.T.; Cheong, J.H.; Jung, S.Y.; Jin, C.; Choi, J.S.; Ryu, J.H. Gluco-obtusifolin and its aglycon, obtusifolin, attenuate scopolamine-induced memory impairment. J. Pharmacol. Sci. 2009, 111, 110–116. [Google Scholar] [CrossRef] [Green Version]
- Hsu, Y.L.; Tsai, E.M.; Hou, M.F.; Wang, T.N.; Hung, J.Y.; Kuo, P.L. Obtusifolin suppresses phthalate esters-induced breast cancer bone metastasis by targeting parathyroid hormone-related protein. J. Agric. Food Chem. 2014, 62, 11933–11940. [Google Scholar] [CrossRef]
- Nam, J.; Seol, D.W.; Lee, C.G.; Wee, G.; Yang, S.; Pan, C.H. Obtusifolin, an Anthraquinone Extracted from Senna obtusifolia (L.) H.S.Irwin & Barneby, Reduces Inflammation in a Mouse Osteoarthritis Model. Pharmaceuticals 2021, 14, 249. [Google Scholar]
- Liu, Y.; Mapa, M.S.T.; Sprando, R.L. Anthraquinones inhibit cytochromes P450 enzyme activity in silico and in vitro. J. Appl. Toxicol. 2021, 41, 1438–1445. [Google Scholar] [CrossRef]
- Kaminsky, L.S.; Spivack, S.D. Cytochromes P450 and cancer. Mol. Aspects Med. 1999, 20, 70–84, 137. [Google Scholar]
- Androutsopoulos, V.P.; Tsatsakis, A.M.; Spandidos, D.A. Cytochrome P450 CYP1A1: Wider roles in cancer progression and prevention. BMC Cancer 2009, 9, 187. [Google Scholar] [CrossRef] [Green Version]
- Mary, V.S.; Valdehita, A.; Navas, J.M.; Rubinstein, H.R.; Fernandez-Cruz, M.L. Effects of aflatoxin B(1), fumonisin B(1) and their mixture on the aryl hydrocarbon receptor and cytochrome P450 1A induction. Food Chem. Toxicol. 2015, 75, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Hodek, P.; Koblihova, J.; Kizek, R.; Frei, E.; Arlt, V.M.; Stiborova, M. The relationship between DNA adduct formation by benzo[a]pyrene and expression of its activation enzyme cytochrome P450 1A1 in rat. Environ. Toxicol. Pharmacol. 2013, 36, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Chun, Y.J.; Kim, M.Y.; Guengerich, F.P. Resveratrol is a selective human cytochrome P450 1A1 inhibitor. Biochem. Biophys. Res. Commun. 1999, 262, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Skupinska, K.; Misiewicz-Krzeminska, I.; Lubelska, K.; Kasprzycka-Guttman, T. The effect of isothiocyanates on CYP1A1 and CYP1A2 activities induced by polycyclic aromatic hydrocarbons in Mcf7 cells. Toxicol. In Vitro 2009, 23, 763–771. [Google Scholar] [CrossRef]
- Wang, A.; Savas, U.; Stout, C.D.; Johnson, E.F. Structural characterization of the complex between alpha-naphthoflavone and human cytochrome P450 1B1. J. Biol. Chem. 2011, 286, 5736–5743. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Tian, X.; Wang, Y.; Sun, Z.; Dong, P.; Wang, C.; Huo, X.; Zhang, B.; Huang, S.; Deng, S.; et al. The natural anthraquinones from Rheum palmatum induced the metabolic disorder of melatonin by inhibiting human CYP and SULT enzymes. Toxicol. Lett. 2016, 262, 27–38. [Google Scholar] [CrossRef]
- Liu, J.; Nguyen, T.T.; Dupart, P.S.; Sridhar, J.; Zhang, X.; Zhu, N.; Stevens, C.L.; Foroozesh, M. 7-Ethynylcoumarins: Selective inhibitors of human cytochrome P450s 1A1 and 1A2. Chem. Res. Toxicol. 2012, 25, 1047–1057. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Nguyen, P.H.; Kim, G.; Jang, S.N.; Lee, G.H.; Phuc, N.M.; Wu, Z.; Liu, K.H. Strong and Selective Inhibitory Effects of the Biflavonoid Selamariscina A against CYP2C8 and CYP2C9 Enzyme Activities in Human Liver Microsomes. Pharmaceutics 2020, 12, 343. [Google Scholar] [CrossRef]
- Sansen, S.; Yano, J.K.; Reynald, R.L.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. Adaptations for the oxidation of polycyclic aromatic hydrocarbons exhibited by the structure of human P450 1A2. J. Biol. Chem. 2007, 282, 14348–14355. [Google Scholar] [CrossRef] [Green Version]
- McNutt, A.T.; Francoeur, P.; Aggarwal, R.; Masuda, T.; Meli, R.; Ragoza, M.; Sunseri, J.; Koes, D.R. GNINA 1.0: Molecular docking with deep learning. J. Cheminform. 2021, 13, 43. [Google Scholar] [CrossRef]
- Heo, J.K.; Nguyen, P.H.; Kim, W.C.; Phuc, N.M.; Liu, K.H. Inhibitory Effect of Selaginellins from Selaginella tamariscina (Beauv.) Spring against Cytochrome P450 and Uridine 5′-Diphosphoglucuronosyltransferase Isoforms on Human Liver Microsomes. Molecules 2017, 22, 1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Lee, H.; Ji, H.K.; Lee, T.; Liu, K.H. Screening of ten cytochrome P450 enzyme activities with 12 probe substrates in human liver microsomes using cocktail incubation and liquid chromatography-tandem mass spectrometry. Biopharm. Drug Dispos. 2019, 40, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.B.; Park, S.Y.; Bae, S.; Seo, H.J.; Kim, S.E.; Lee, G.M.; Wu, Z.; Liu, K.H. Comprehensive Investigation of Stereoselective Food Drug Interaction Potential of Resveratrol on Nine P450 and Six UGT Isoforms in Human Liver Microsomes. Pharmaceutics 2021, 13, 1419. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.J.; Ji, S.B.; Kim, S.E.; Lee, G.M.; Park, S.Y.; Wu, Z.; Jang, D.S.; Liu, K.H. Inhibitory Effects of Schisandra Lignans on Cytochrome P450s and Uridine 5′-Diphospho-Glucuronosyl Transferases in Human Liver Microsomes. Pharmaceutics 2021, 13, 371. [Google Scholar] [CrossRef]
- Tang, J.C.; Yang, H.; Song, X.Y.; Song, X.H.; Yan, S.L.; Shao, J.Q.; Zhang, T.L.; Zhang, J.N. Inhibition of cytochrome P450 enzymes by rhein in rat liver microsomes. Phytother. Res. 2009, 23, 159–164. [Google Scholar] [CrossRef]
- Yamamoto, T.; Suzuki, A.; Kohno, Y. Application of microtiter plate assay to evaluate inhibitory effects of various compounds on nine cytochrome P450 isoforms and to estimate their inhibition patterns. Drug Metab. Pharmacokinet. 2002, 17, 437–448. [Google Scholar] [CrossRef]
- Kim, H.; Choi, H.K.; Jeong, T.C.; Jahng, Y.; Kim, D.H.; Lee, S.H.; Lee, S. Selective inhibitory effects of mollugin on CYP1A2 in human liver microsomes. Food Chem. Toxicol. 2013, 51, 33–37. [Google Scholar] [CrossRef]
- Bourrie, M.; Meunier, V.; Berger, Y.; Fabre, G. Cytochrome P450 isoform inhibitors as a tool for the investigation of metabolic reactions catalyzed by human liver microsomes. J. Pharmacol. Exp. Ther. 1996, 277, 321–332. [Google Scholar]
- Seo, K.A.; Kim, H.; Ku, H.Y.; Ahn, H.J.; Park, S.J.; Bae, S.K.; Shin, J.G.; Liu, K.H. The monoterpenoids citral and geraniol are moderate inhibitors of CYP2B6 hydroxylase activity. Chem. Biol. Interact. 2008, 174, 141–146. [Google Scholar] [CrossRef]
- Kim, S.J.; You, J.; Choi, H.G.; Kim, J.A.; Jee, J.G.; Lee, S. Selective inhibitory effects of machilin A isolated from Machilus thunbergii on human cytochrome P450 1A and 2B6. Phytomedicine 2015, 22, 615–620. [Google Scholar] [CrossRef]
- Kang, A.Y.; Young, L.R.; Dingfelder, C.; Peterson, S. Effects of furanocoumarins from apiaceous vegetables on the catalytic activity of recombinant human cytochrome P-450 1A2. Protein J. 2011, 30, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Fairman, D.A.; Collins, C.; Chapple, S. Progress curve analysis of CYP1A2 inhibition: A more informative approach to the assessment of mechanism-based inactivation? Drug Metab. Dispos. 2007, 35, 2159–2165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awortwe, C.; Manda, V.K.; Avonto, C.; Khan, S.I.; Khan, I.A.; Walker, L.A.; Bouic, P.J.; Rosenkranz, B. In Vitro Evaluation of Reversible and Time-Dependent Inhibitory Effects of Kalanchoe crenata on CYP2C19 and CYP3A4 Activities. Drug Metab. Lett. 2015, 9, 48–62. [Google Scholar] [CrossRef]
- Perloff, E.S.; Mason, A.K.; Dehal, S.S.; Blanchard, A.P.; Morgan, L.; Ho, T.; Dandeneau, A.; Crocker, R.M.; Chandler, C.M.; Boily, N.; et al. Validation of cytochrome P450 time-dependent inhibition assays: A two-time point IC50 shift approach facilitates kinact assay design. Xenobiotica 2009, 39, 99–112. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, J.H.; Lee, T.; Bae, J.S.; Jeong, T.C.; Lee, E.S.; Lee, S. Selective inhibitory effects of HYIpro-3-1 on CYP1A2 in human liver microsomes. Biopharm. Drug Dispos. 2021, 42, 35–41. [Google Scholar] [CrossRef]
- Shimada, T.; Fujii-Kuriyama, Y. Metabolic activation of polycyclic aromatic hydrocarbons to carcinogens by cytochromes P450 1A1 and 1B1. Cancer Sci. 2004, 95, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.Y.; Qi, M.; Zhao, L.; Zhu, M.K.; Guo, J.; Liu, J.; Gu, C.Q.; Rajput, S.A.; Krumm, C.S.; Qi, D.S.; et al. Curcumin Prevents Aflatoxin B(1) Hepatoxicity by Inhibition of Cytochrome P450 Isozymes in Chick Liver. Toxins 2016, 8, 327. [Google Scholar] [CrossRef] [Green Version]
- Gerhauser, C.; Klimo, K.; Heiss, E.; Neumann, I.; Gamal-Eldeen, A.; Knauft, J.; Liu, G.Y.; Sitthimonchai, S.; Frank, N. Mechanism-based in vitro screening of potential cancer chemopreventive agents. Mutat. Res. 2003, 523–524, 163–172. [Google Scholar] [CrossRef]
- Joshi, P.; McCann, G.J.P.; Sonawane, V.R.; Vishwakarma, R.A.; Chaudhuri, B.; Bharate, S.B. Identification of Potent and Selective CYP1A1 Inhibitors via Combined Ligand and Structure-Based Virtual Screening and Their in Vitro Validation in Sacchrosomes and Live Human Cells. J. Chem. Inf. Model. 2017, 57, 1309–1320. [Google Scholar] [CrossRef]
- Zhai, S.; Dai, R.; Friedman, F.K.; Vestal, R.E. Comparative inhibition of human cytochromes P450 1A1 and 1A2 by flavonoids. Drug Metab. Dispos. 1998, 26, 989–992. [Google Scholar]
- Yang, C.; Wang, S.; Guo, X.; Sun, J.; Liu, L.; Wu, L. Simultaneous determination of seven anthraquinones in rat plasma by Ultra High Performance Liquid Chromatography-tandem Mass Spectrometry and pharmacokinetic study after oral administration of Semen Cassiae extract. J. Ethnopharmacol. 2015, 169, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Sun, Q.; Hao, W.; Zhao, J. Pharmacokinetics and tissue distribution study of obtusifolin in rats by liquid chromatography-tandem mass spectrometry. Biomed. Chromatogr. 2021, 35, e5009. [Google Scholar] [CrossRef]
- Jiang, J.Y.; Yang, M.W.; Qian, W.; Lin, H.; Geng, Y.; Zhou, Z.Q.; Xiao, D.W. Quantitative determination of rhein in human plasma by liquid chromatography-negative electrospray ionization tandem mass/mass spectrometry and the application in a pharmacokinetic study. J. Pharm. Biomed. Anal. 2012, 57, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Koyama, E.; Chiba, K.; Tani, M.; Ishizaki, T. Reappraisal of human CYP isoforms involved in imipramine N-demethylation and 2-hydroxylation: A study using microsomes obtained from putative extensive and poor metabolizers of S-mephenytoin and eleven recombinant human CYPs. J. Pharmacol. Exp. Ther. 1997, 281, 1199–1210. [Google Scholar] [PubMed]
- Chiu, C.C.; Lane, H.Y.; Huang, M.C.; Liu, H.C.; Jann, M.W.; Hon, Y.Y.; Chang, W.H.; Lu, M.L. Dose-dependent alternations in the pharmacokinetics of olanzapine during coadministration of fluvoxamine in patients with schizophrenia. J. Clin. Pharmacol. 2004, 44, 1385–1390. [Google Scholar] [CrossRef]
- Granfors, M.T.; Backman, J.T.; Neuvonen, M.; Neuvonen, P.J. Ciprofloxacin greatly increases concentrations and hypotensive effect of tizanidine by inhibiting its cytochrome P450 1A2-mediated presystemic metabolism. Clin. Pharmacol. Ther. 2004, 76, 598–606. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | IC50 (µM) (1) | ||||
|---|---|---|---|---|---|
| CYP1A2 | Obtusifolin | Emodin | Chrysophanol | Physcion | Rubrofusarin |
| 0.19 ± 0.01 | 0.79 ± 0.26 | 1.99 ± 0.23 | 2.16 ± 0.48 | 6.33 ± 1.38 | |
| Chemical | IC50 (µM) | Docking Score (kcal/mol) | Interactions |
|---|---|---|---|
| Obtusifolin | 0.19 ± 0.01 | −12.78 | Phe226, Phe256, Phe260, Ala317, Asp313, and Leu497 |
| Emodin | 0.79 ± 0.26 | −11.38 | Phe226, Phe260, Ala317, and Leu497 |
| Chrysophanol | 1.99 ± 0.23 | −11.66 | Phe226, Phe260, Ala317, and Leu497 |
| Physcion | 2.16 ± 0.48 | −8.34 | Phe226, Phe260, Ala317, and Leu497 |
| Rubrofusarin | 6.33 ± 1.38 | −7.56 | Thr124, Phe226, Phe256, Phe260, Ala317, and Leu497 |
| Inhibitor | HLM/rCYP1A2 | IC50 (μM) | Ki (μM) |
|---|---|---|---|
| Obtusifolin | HLMs | 0.19 ± 0.01 | 0.11 ± 0.02 |
| rCYP1A2 | 0.37 ± 0.11 | 0.21 ± 0.053 | |
| α-Naphthoflavone | HLMs | 0.0061 ± 0.0008 | 0.0075 ± 0.0010 |
| P450 Isoforms | IC50 (µM) |
|---|---|
| CYP1A2 | 0.19 ± 0.01 |
| CYP2A6 | >50 |
| CYP2B6 | >50 |
| CYP2C8 | 31.44 ± 9.85 |
| CYP2C9 | 28.64 ± 6.98 |
| CYP2C19 | >50 |
| CYP2D6 | >50 |
| CYP2E1 | >50 |
| CYP3A | >50 |
| Recombinant Cytochrome P450 | Substrate | IC50 (μM) |
|---|---|---|
| rCYP1A1 | Phenacetin | 0.06 ± 0.02 |
| 7-Ethoxyresorufin | 0.39 ± 0.06 | |
| 9-cis-Retinal | >10 | |
| rCYP1A2 | Phenacetin | 0.37 ± 0.11 |
| 7-Ethoxyresorufin | 0.57 ± 0.11 | |
| 9-cis-Retinal | 0.15 ± 0.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, E.-J.; Park, K.; Durai, P.; Kim, K.-Y.; Park, S.-Y.; Kwon, J.; Lee, H.J.; Pan, C.-H.; Liu, K.-H. Potent and Selective Inhibition of CYP1A2 Enzyme by Obtusifolin and Its Chemopreventive Effects. Pharmaceutics 2022, 14, 2683. https://doi.org/10.3390/pharmaceutics14122683
Park E-J, Park K, Durai P, Kim K-Y, Park S-Y, Kwon J, Lee HJ, Pan C-H, Liu K-H. Potent and Selective Inhibition of CYP1A2 Enzyme by Obtusifolin and Its Chemopreventive Effects. Pharmaceutics. 2022; 14(12):2683. https://doi.org/10.3390/pharmaceutics14122683
Chicago/Turabian StylePark, Eun-Ji, Keunwan Park, Prasannavenkatesh Durai, Ki-Young Kim, So-Young Park, Jaeyoung Kwon, Hee Ju Lee, Cheol-Ho Pan, and Kwang-Hyeon Liu. 2022. "Potent and Selective Inhibition of CYP1A2 Enzyme by Obtusifolin and Its Chemopreventive Effects" Pharmaceutics 14, no. 12: 2683. https://doi.org/10.3390/pharmaceutics14122683