
Antiviral Mechanisms of N-Phenyl Benzamides on Coxsackie Virus A9
 , , ,
, , ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemistry
2.2. Cells, Viruses, and Molecules
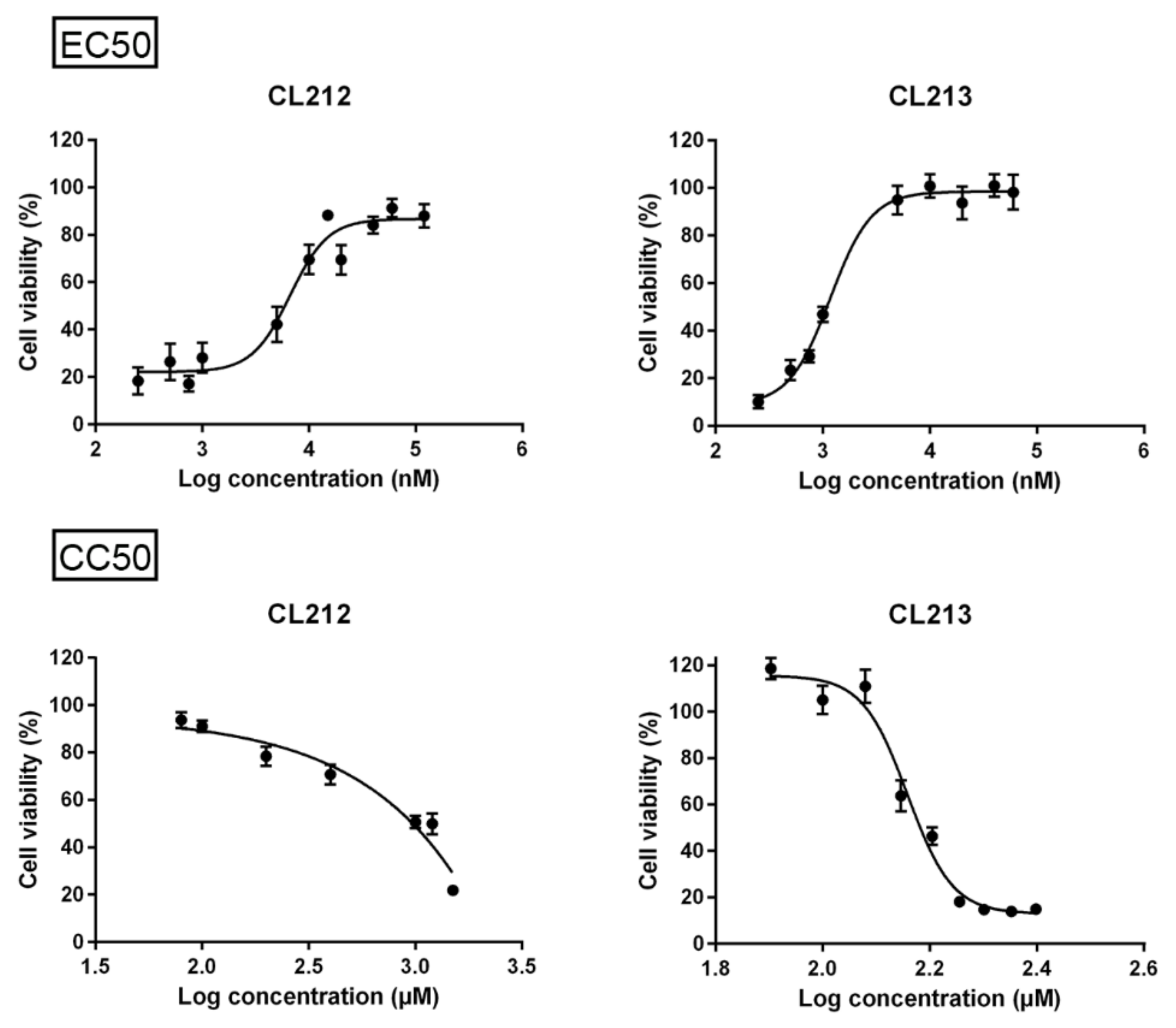
2.3. Concentration Series, EC50 and CC50
2.4. Time-of-Addition Assay
2.5. Real-Time Fluorescence Uncoating Assay
2.6. Radioactive Gradient Assay
2.7. Transmission Electron Microscope (TEM) Imaging
2.8. Docking Imaging
2.9. Statistical Analysis
3. Results
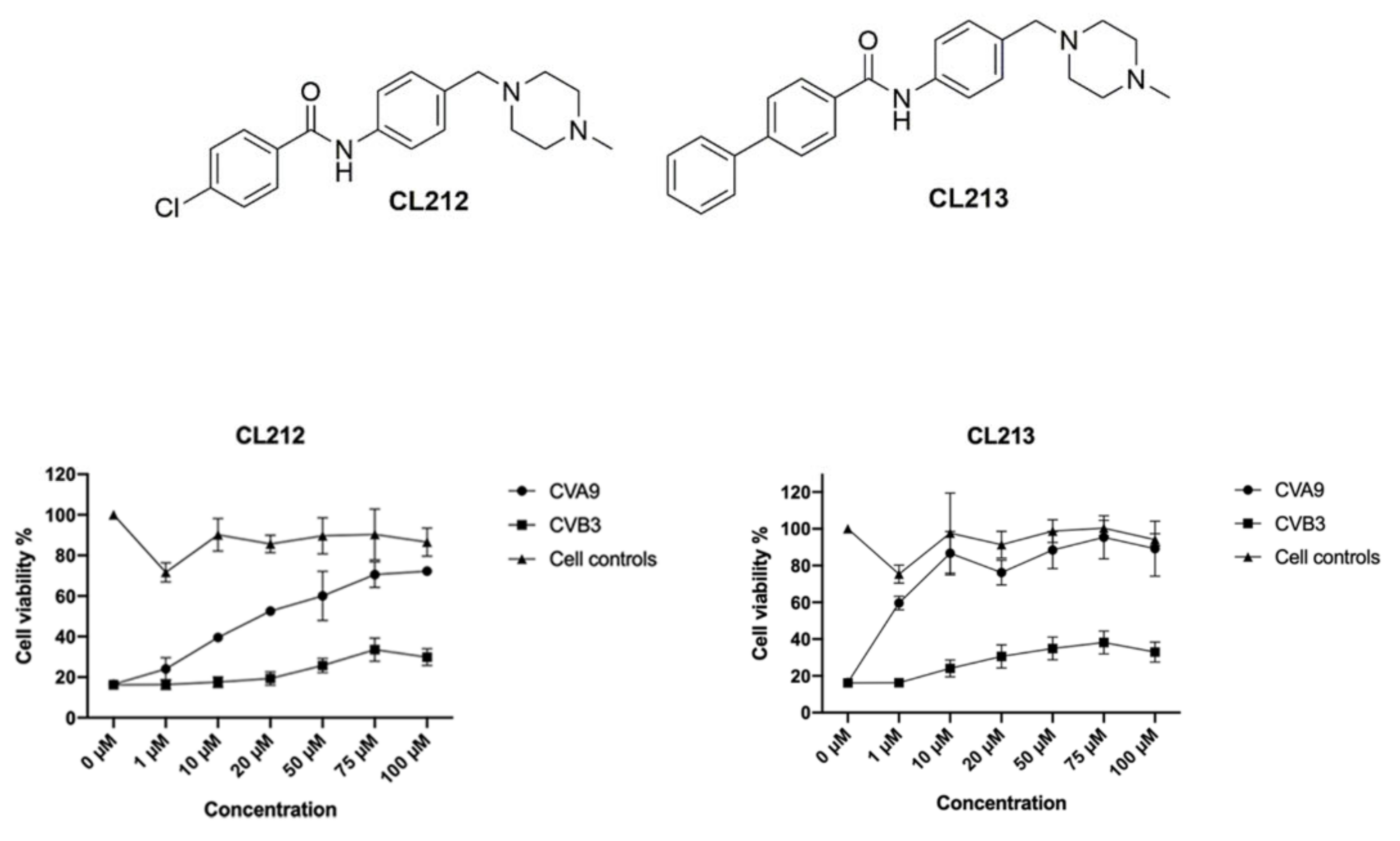
3.1. Potential Antiviral Identification
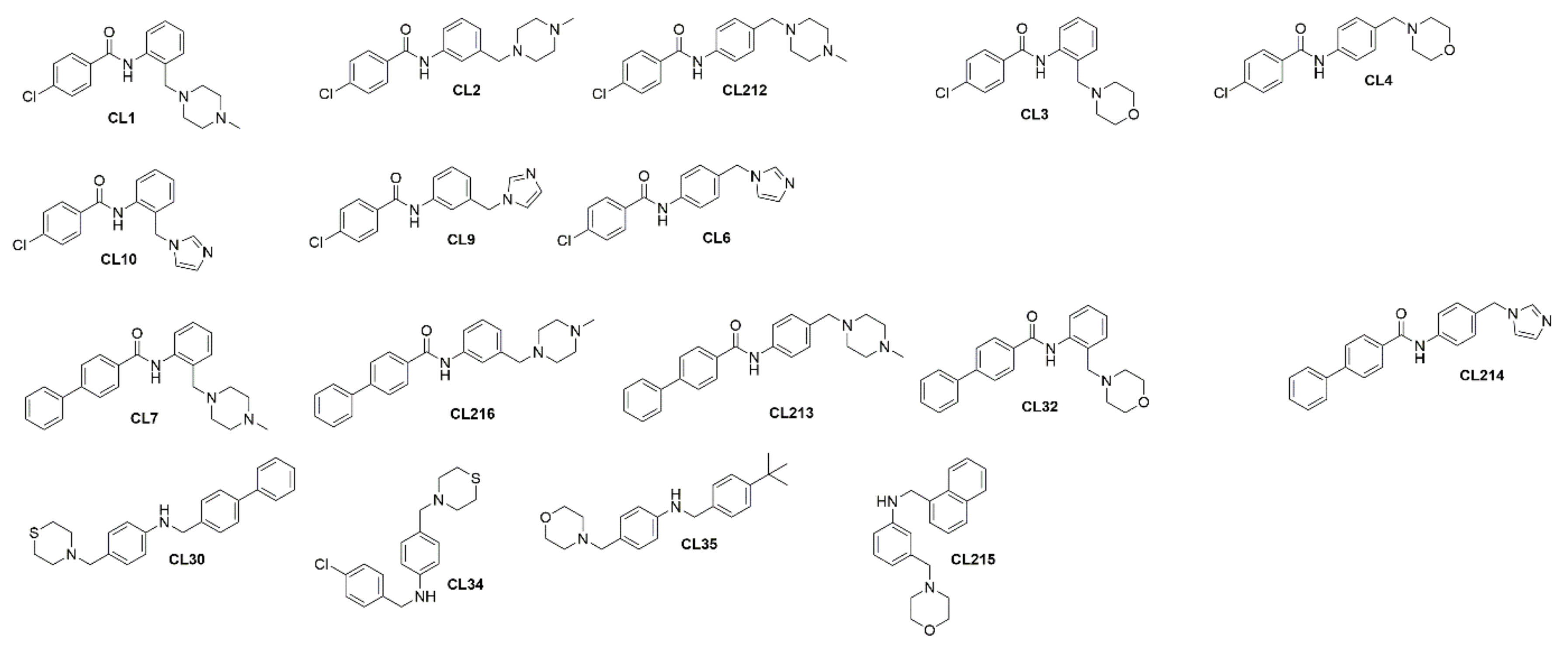
3.2. Initial Structure Activity Relationship (SAR) Studies
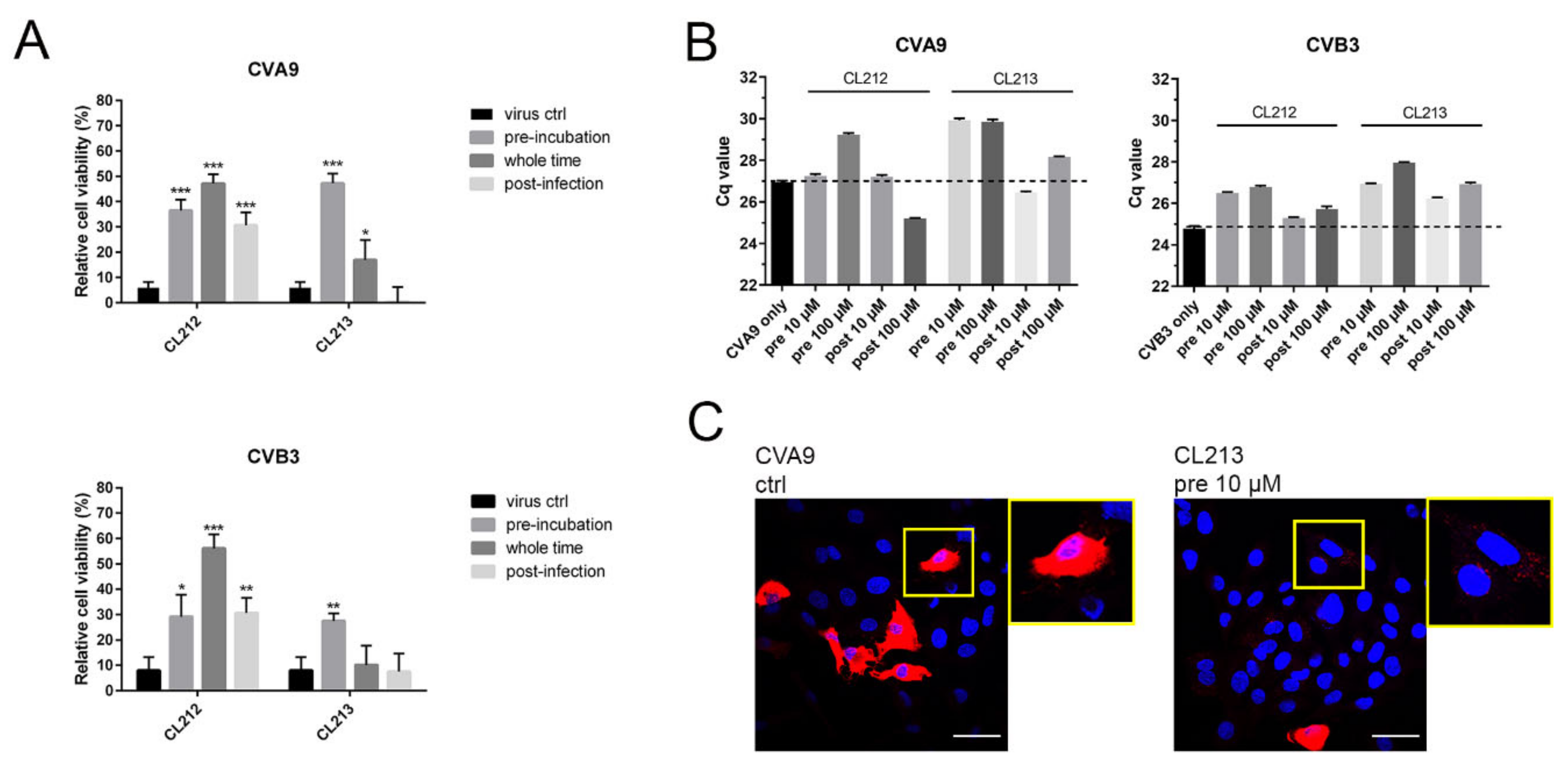
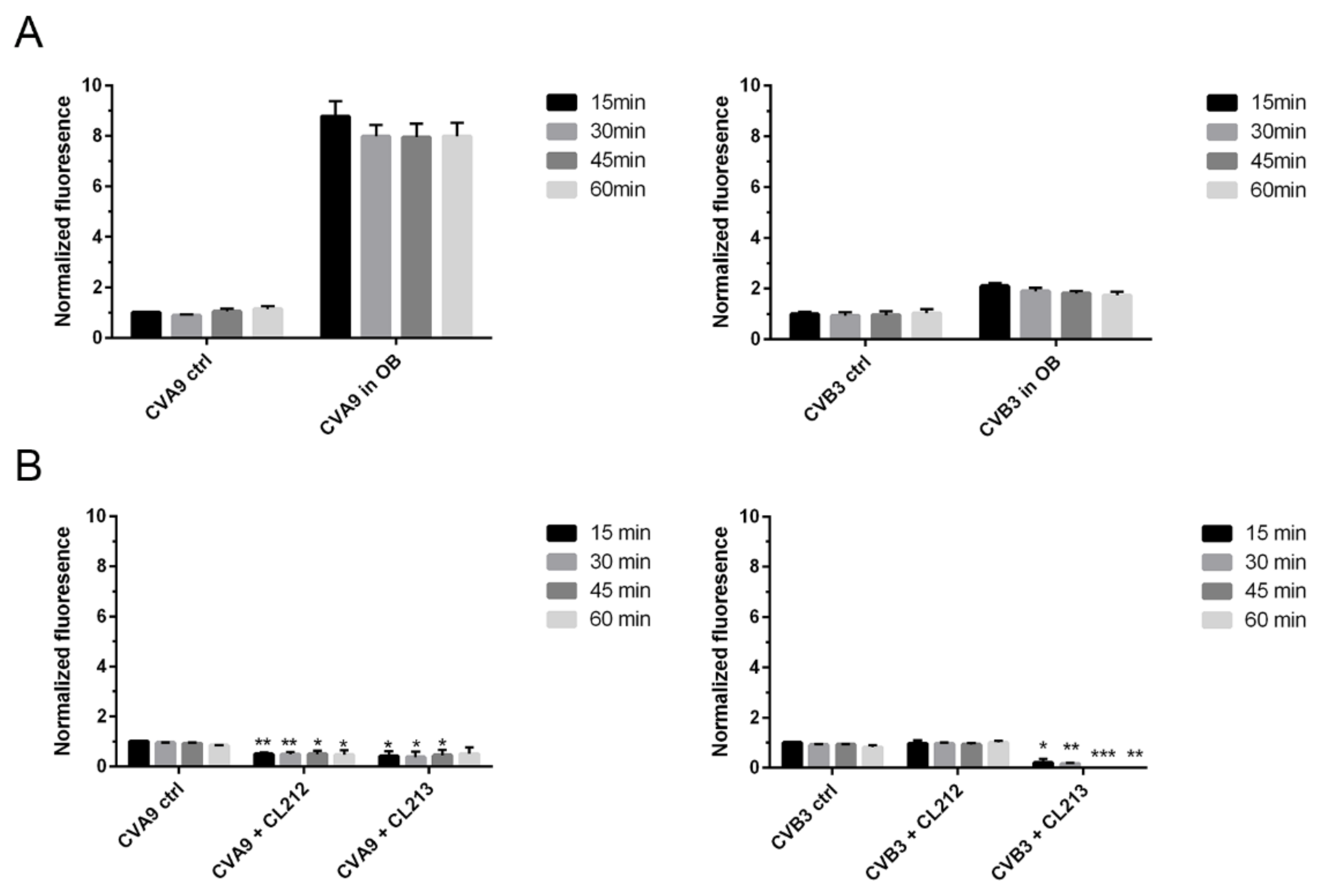
3.3. CL212 and CL213 Prevent the Infection Already after Pre-Incubation
3.4. CL212 and CL213 Stabilize the Virus Capsid
3.4.1. Uncoating
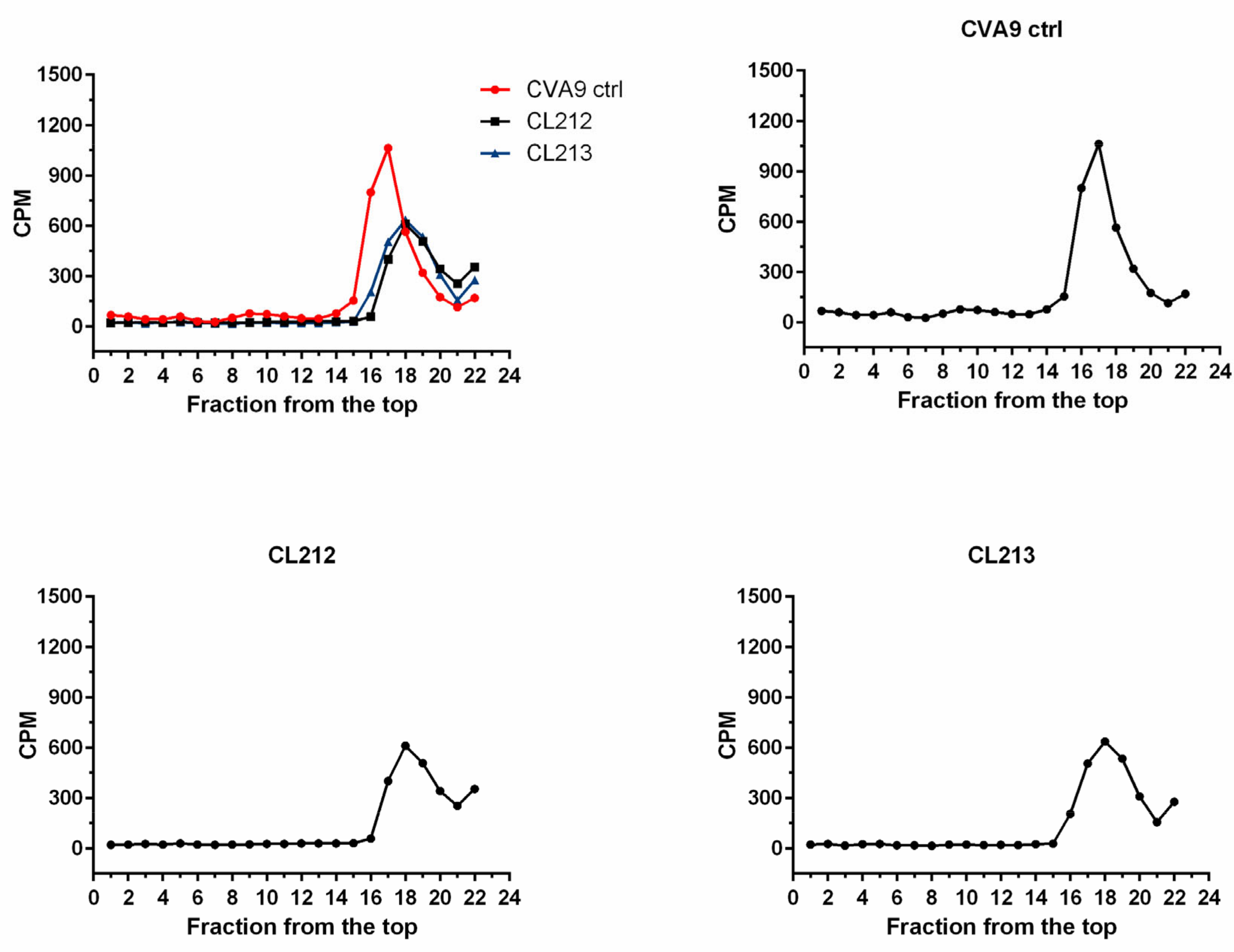
3.4.2. Virus Stays Intact Detected by Sucrose Gradient Fractionation
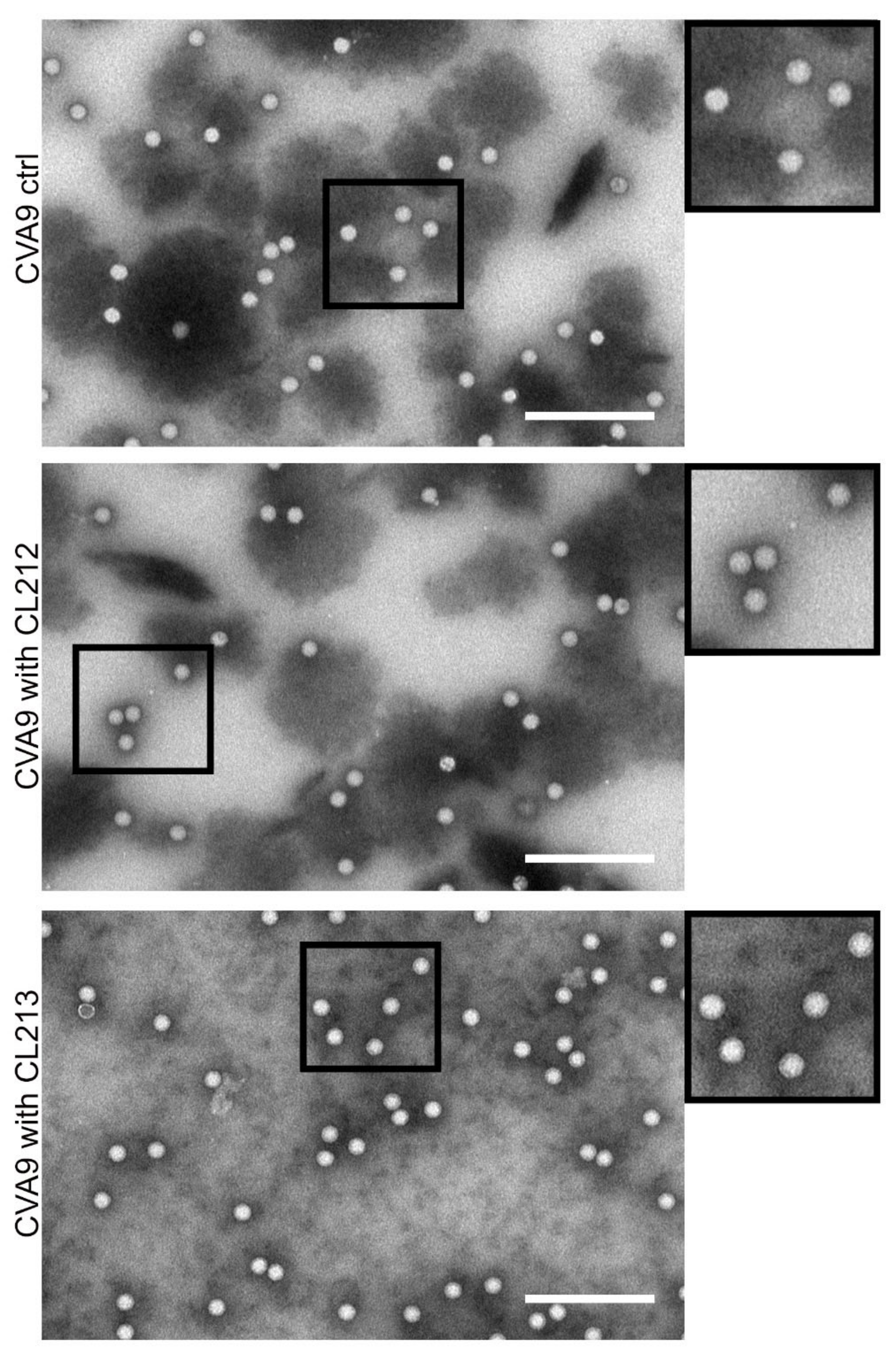
3.4.3. Virus Stays Intact Detected by Electron Microscopy
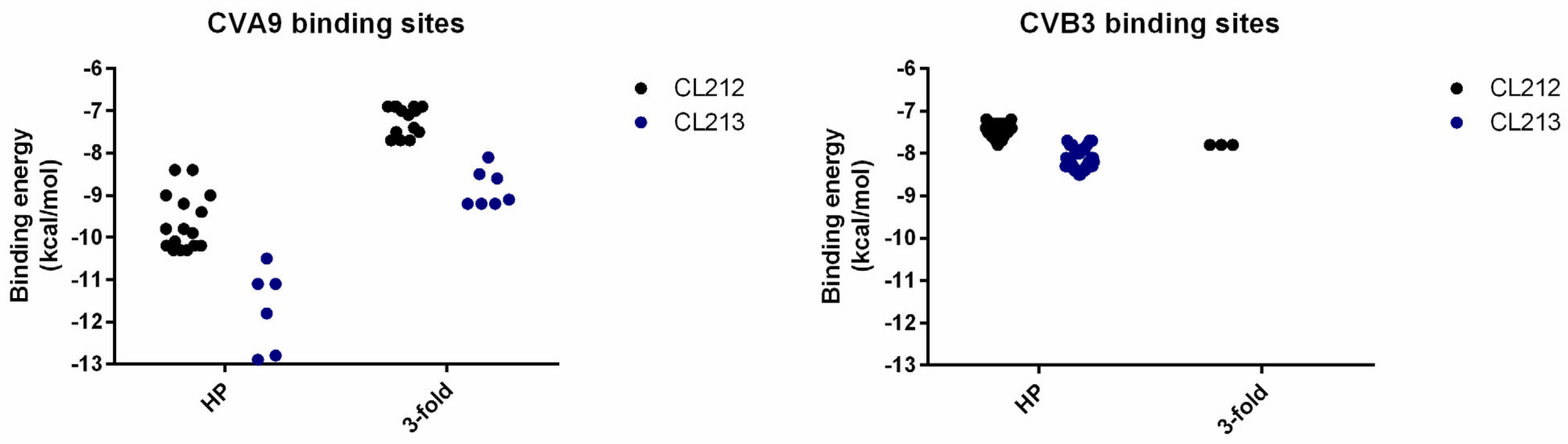
3.5. CL212 and CL213 Bind to the Hydrophobic Pocket and 3-Fold Axis Pore
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Laitinen, O.H.; Honkanen, H.; Pakkanen, O.; Oikarinen, S.; Hankaniemi, M.M.; Huhtala, H.; Ruokoranta, T.; Lecouturier, V.; André, P.; Harju, R.; et al. Coxsackievirus B1 Is Associated with Induction of β-Cells Autoimmunity That Portends Type 1 Diabetes. Diabetes 2014, 63, 446–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roivainen, M.; Alfthan, G.; Jousilahti, P.; Kimpimäki, M.; Hovi, T.; Tuomilehto, J. Enterovirus Infections as a Possible Risk Factor for Myocardial Infarction. Circulation 1998, 98, 2534–2537. [Google Scholar] [CrossRef] [Green Version]
- Andriushkova, N.G.; Turchyna, N.S.; Poniatowski, V.A.; Dolinchuk, L.V.; Melnyk, V.V.; Shyrobokov, V.P.; Zakharchenko, N.V. The Role of the Persistent Enterovirus Infection in Development of Acute Stroke. Wiad. Lek. 2017, 70, 187–191. [Google Scholar]
- Cui, A.; Yu, D.; Zhu, Z.; Meng, L.; Li, H.; Liu, J.; Liu, G.; Mao, N.; Xu, W. An Outbreak of Aseptic Meningitis Caused by Coxsackievirus A9 in Gansu, the People’s Republic of China. Virol. J. 2010, 7, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreau, B.; Bastedo, C.; Michel, R.P.; Ghali, P. Hepatitis and Encephalitis Due to Coxsackie Virus A9 in an Adult. Case Rep. Gastroenterol. 2011, 5, 617–622. [Google Scholar] [CrossRef]
- Cheung, P.K.M.; Yuan, J.; Zhang, H.M.; Chau, D.; Yanagawa, B.; Suarez, A.; McManus, B.; Yang, D. Specific Interactions of Mouse Organ Proteins with the 5′untranslated Region of Coxsackievirus B3: Potential Determinants of Viral Tissue Tropism. J. Med. Virol. 2005, 77, 414–424. [Google Scholar] [CrossRef]
- Oberste, M.S.; Maher, K.; Schnurr, D.; Flemister, M.R.; Lovchik, J.C.; Peters, H.; Sessions, W.; Kirk, C.; Chatterjee, N.; Fuller, S.; et al. Enterovirus 68 Is Associated with Respiratory Illness and Shares Biological Features with Both the Enteroviruses and the Rhinoviruses. J. Gen. Virol. 2004, 85, 2577–2584. [Google Scholar] [CrossRef]
- Muehlenbachs, A.; Bhatnagar, J.; Zaki, S.R. Tissue Tropism, Pathology and Pathogenesis of Enterovirus Infection. J. Pathol. 2015, 235, 217–228. [Google Scholar] [CrossRef] [Green Version]
- Egorova, A.; Ekins, S.; Schmidtke, M.; Makarov, V. Back to the Future: Advances in Development of Broad-Spectrum Capsid-Binding Inhibitors of Enteroviruses. Eur. J. Med. Chem. 2019, 178, 606–622. [Google Scholar] [CrossRef]
- Turkki, P.; Laajala, M.; Flodström-Tullberg, M.; Marjomäki, V. Human Enterovirus Group B Viruses Rely on Vimentin Dynamics for Efficient Processing of Viral Nonstructural Proteins. J. Virol. 2020, 94, e01393-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruokolainen, V.; Laajala, M.; Marjomäki, V. Real-Time Fluorescence Measurement of Enterovirus Uncoating. Bio. Protoc. 2020, 10, e3582. [Google Scholar] [CrossRef]
- Myllynen, M.; Kazmertsuk, A.; Marjomäki, V. A Novel Open and Infectious Form of Echovirus 1. J. Virol. 2016, 90, 6759–6770. [Google Scholar] [CrossRef] [Green Version]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeerschd, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.; Huey, R.; Linkstrom, W.; Sanner, M.; Belew, R.; Goodsell, D.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2010, 31, 2785–2791. [Google Scholar]
- Hassan, N.M.; Alhossary, A.A.; Mu, Y.; Kwoh, C.K. Protein-Ligand Blind Docking Using QuickVina-W with Inter-Process Spatio-Temporal Integration. Sci. Rep. 2017, 7, 15451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reshamwala, D.; Shroff, S.; Sheik Amamuddy, O.; Laquintana, V.; Denora, N.; Zacheo, A.; Lampinen, V.; Hytonen, V.P.; Tastan Bishop, Ö.; Krol, S.; et al. Polyphenols Epigallocatechin Gallate and Resveratrol, and Polyphenol-Functionalized Nanoparticles Prevent Enterovirus Infection through Clustering and Stabilization of the Viruses. Pharmaceutics 2021, 13, 1182. [Google Scholar] [CrossRef]
- Seeliger, D.; de Groot, B.L. Ligand Docking and Binding Site Analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des. 2010, 24, 417–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruokolainen, V.; Domanska, A.; Laajala, M.; Pelliccia, M.; Butcher, S.J.; Marjomäki, V. Extracellular Albumin and Endosomal Ions Prime Enterovirus Particles for Uncoating That Can Be Prevented by Fatty Acid Saturation. J. Virol. 2019, 93, e00599-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laajala, M.; Reshamwala, D.; Marjomäki, V. Therapeutic Targets for Enterovirus Infections. Expert Opin. Ther. Targets 2020, 24, 745–757. [Google Scholar] [CrossRef]
- Huttunen, M.; Waris, M.; Kajander, R.; Hyypiä, T.; Marjomäki, V. Coxsackievirus A9 Infects Cells via Nonacidic Multivesicular Bodies. J. Virol. 2014, 88, 5138–5151. [Google Scholar] [CrossRef] [Green Version]
- Pevear, D.C.; Tull, T.M.; Seipel, M.E.; Groarke, J.M. Activity of Pleconaril against Enteroviruses. Antimicrob. Agents Chemother. 1999, 43, 2109–2115. [Google Scholar] [CrossRef] [Green Version]
- Watson, K.G.; Brown, R.N.; Cameron, R.; Chalmers, D.K.; Hamilton, S.; Jin, B.; Krippner, G.Y.; Luttick, A.; McConnell, D.B.; Reece, P.A.; et al. An Orally Bioavailable Oxime Ether Capsid Binder with Potent Activity against Human Rhinovirus. J. Med. Chem. 2003, 46, 3181–3184. [Google Scholar] [CrossRef] [PubMed]
- Andries, K.; Dewindt, B.; Snoeks, J.; Willebrords, R.; van Eemeren, K.; Stokbroekx, R.; Janssen, P.A.J. In Vitro Activity of Pirodavir (R 77975), a Substituted Phenoxy- Pyridazinamine with Broad-Spectrum Antipicornaviral Activity. Antimicrob. Agents Chemother. 1992, 36, 100–107. [Google Scholar] [CrossRef] [Green Version]
- Buontempo, P.J.; Cox, S.; Wright-Minogue, J.; DeMartino, J.L.; Skelton, A.M.; Ferrari, E.; Albin, R.; Rozhon, E.J.; Girijavallabhan, V.; Modlin, J.F.; et al. SCH 48973: A Potent, Broad-Spectrum, Antienterovirus Compound. Antimicrob. Agents Chemother. 1997, 41, 1220–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberste, M.S.; Moore, D.; Anderson, B.; Pallansch, M.A.; Pevear, D.C.; Collett, M.S. In Vitro Antiviral Activity of V-073 against Polioviruses. Antimicrob. Agents Chemother. 2009, 53, 4501–4503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarov, V.A.; Braun, H.; Richter, M.; Riabova, O.B.; Kirchmair, J.; Kazakova, E.S.; Seidel, N.; Wutzler, P.; Schmidtke, M. Pyrazolopyrimidines: Potent Inhibitors Targeting the Capsid of Rhino- and Enteroviruses. ChemMedChem 2015, 10, 1629–1634. [Google Scholar] [CrossRef] [Green Version]
- Abdelnabi, R.; Geraets, J.A.; Ma, Y.; Mirabelli, C.; Flatt, J.W.; Domanska, A.; Delang, L.; Jochmans, D.; Kumar, T.A.; Jayaprakash, V.; et al. A Novel Druggable Interprotomer Pocket in the Capsid of Rhino-and Enteroviruses. PLoS Biol. 2019, 17, e3000281. [Google Scholar] [CrossRef] [Green Version]
- Schmidtke, M.; Hammerschmidt, E.; Schüler, S.; Zell, R.; Birch-Hirschfeld, E.; Makarov, V.A.; Riabova, O.B.; Wutzler, P. Susceptibility of Coxsackievirus B3 Laboratory Strains and Clinical Isolates to the Capsid Function Inhibitor Pleconaril: Antiviral Studies with Virus Chimeras Demonstrate the Crucial Role of Amino Acid 1092 in Treatment. J. Antimicrob. Chemother. 2005, 56, 648–656. [Google Scholar] [CrossRef] [Green Version]
- Groarke, J.M.; Pevear, D.C. Attenuated Virulence of Pleconaril-Resistant Coxsackievirus B3 Variants. J. Infect. Dis. 1999, 179, 1538–1541. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | EC50 | CC50 | SI |
|---|---|---|---|
| CL212 | 5.92 µM | >1000 µM 1 | >168.92 |
| CL213 | 1.09 µM | 152.05 µM | 139.50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laajala, M.; Kalander, K.; Consalvi, S.; Amamuddy, O.S.; Bishop, Ö.T.; Biava, M.; Poce, G.; Marjomäki, V. Antiviral Mechanisms of N-Phenyl Benzamides on Coxsackie Virus A9. Pharmaceutics 2023, 15, 1028. https://doi.org/10.3390/pharmaceutics15031028
Laajala M, Kalander K, Consalvi S, Amamuddy OS, Bishop ÖT, Biava M, Poce G, Marjomäki V. Antiviral Mechanisms of N-Phenyl Benzamides on Coxsackie Virus A9. Pharmaceutics. 2023; 15(3):1028. https://doi.org/10.3390/pharmaceutics15031028
Chicago/Turabian StyleLaajala, Mira, Kerttu Kalander, Sara Consalvi, Olivier Sheik Amamuddy, Özlem Tastan Bishop, Mariangela Biava, Giovanna Poce, and Varpu Marjomäki. 2023. "Antiviral Mechanisms of N-Phenyl Benzamides on Coxsackie Virus A9" Pharmaceutics 15, no. 3: 1028. https://doi.org/10.3390/pharmaceutics15031028