1. Introduction
Influenza infections affect 20–30% of the child population and up to 10% adult population globally and cause from 290,000 to 650,000 deaths every year [
1], placing a burden on global health and the economy. Although anti-influenza drugs, such as oseltamivir, zanamivir, peramivir and baloxavir marboxil, have been approved for clinical use, the effectiveness of these drugs relies heavily on timely treatment, usually up to 48 h after the onset of symptoms [
2]. Moreover, these drugs face challenges with drug resistance [
3,
4,
5] due to the high mutation rate and frequent genetic reassortments of influenza viruses. Thus, developing novel influenza antivirals with longer therapeutical time windows and higher resistance to mutations is important to help in the treatment of this disease.
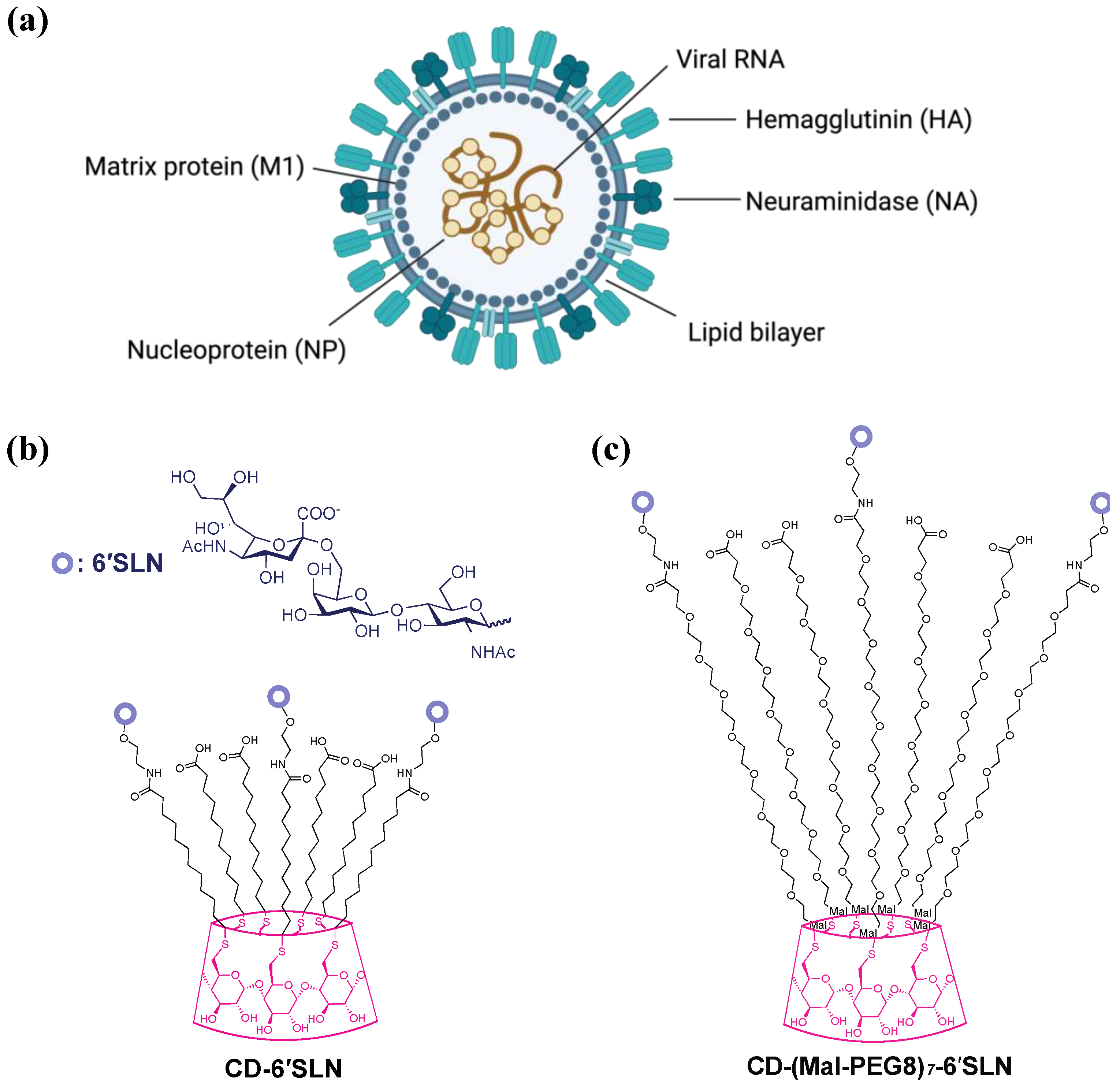
The influenza virion consists of a viral envelope and ribonucleoprotein complexes containing segmented, single-stranded RNA bound and encapsulated by nucleoproteins (
Figure 1a). The outer envelope is a lipid bilayer with integrated two-spike glycoproteins, hemagglutinin (HA) and neuraminidase (NA), which are vital for mediating viral attachment and release, respectively. Under the lipid bilayer is the Matrix protein 1 (M1), which forms a capsid enclosing the genome. Matrix protein 2 (M2), a proton-selective ion channel necessary for viral genome release during virus entry, is integrated into the influenza outer envelope.
In general, influenza antiviral drugs target various stages of the viral replication cycle. Small-molecule viral enzyme inhibitors [
2], such as the M2 proton channel blocker amantadine, endonuclease inhibitor baloxavir marboxil, and neuraminidase inhibitor oseltamivir, target the intracellular viral replication processes of viral fusion, genome replication, and viral budding, respectively. Large molecules, such as therapeutical antibodies [
6], interact directly with extracellular virions, in particular viral membrane proteins, to prevent viral attachment or fusion to host cells. Notably, such an extracellular mechanism avoids many side effects, often associated with the impact on the intracellular physiology of host cells.
Influenza HA has been studied as a target of antiviral drugs for decades, though no drug candidate with an HA inhibition mechanism has been licensed for clinical use. HA binds to sialic acids of glycoproteins and glycolipids on the cell surface to allow viral entry into the host cell. Therefore, sialic acid-based glycomimetics could be a potential HA inhibitor. Various scaffolds such as glyco-nanoparticle [
7], DNA [
8], polyglycerol [
9] and polyamidoamine dendrimer [
10] modified with multiple sialic acid units showed enhanced hemagglutination inhibition or influenza inhibition compared with the monovalent sialic acids due to the multivalency effect [
11,
12]. Most of these studies explored how the valency and spacing affected the antiviral effect, while few focused on the role of the linker between the binding motif and the scaffold.
In our previous study [
13], we reported CD-6′SLN [6-sialyllactosamine (6′SLN)-modified β-cyclodextrin (CD)], a virucidal macromolecule made of β-cyclodextrin core covalently grafted with the sialyl trisaccharide Neu5Acα2-6Galβ1-4GlcNAc (6′SLN) via hydrophobic undecyl linkers (
Figure 1b). β-cyclodextrin was selected as a scaffold because it’s a safe natural cyclic oligosaccharide with a well-defined structure and high biocompatibility [
14,
15,
16]. We found CD-6′SLN showed nanomolar to micromolar inhibition against seasonal influenza strains H1N1, H3N2 and influenza B, while the analogous molecule with hydrophilic polyethylene glycol linkers CD-(Mal-PEG8)
7-6′SLN (
Figure 1c) only shows reversible inhibition with lower potency. In the BALB/c H1N1 mouse model, the groups treated with CD-6′SLN showed a much higher survival rate than the group treated with oseltamivir 24 h after infection. The mechanisms of viral inhibition and more specifically the mechanism of virucidal action of CD-6′SLN are not fully understood.
Here, we aim to elucidate the antiviral mechanism of CD-6′SLN. HA is most likely the main target of CD-6′SLN because 6′SLN is the natural binding moiety for HA in human strains such as H1N1 and H3N2 [
17,
18,
19]. Moreover, HA is the most abundant envelope protein, with around 500 spikes per virion [
20]. Although NA accounts for only 1/5th to 1/4th of the total spike proteins, it could also be a potential target as it also bears sialic acid binding pockets [
21,
22]. It is possible that the catalytic activity of NA is blocked by CD-6′SLN, causing inefficient viral entry [
22,
23], endocytosis and release [
24]. Another potential target could be the viral envelope, as CD-6′SLN contains long hydrophobic chains that may interact with the viral envelope. CD-MUS [
25] is a compound structurally similar to CD-6′SLN but carries sulfonate groups instead of 6′SLN at the end of the undecyl chains. It irreversibly inhibits many viruses that depend on heparan sulfate proteoglycan for cell entry. Hence, both CD-6′SLN and CD-MUS are effective virucidal antivirals. While CD-MUS was designed to target the viral attachment ligands (VALs), it has been shown to disrupt the structural integrity of herpes simplex virus-2, forcing the release of the viral genomic DNA. A similar molecular mechanism may also contribute to the virucidal activity of CD-6′SLN.
We investigated the molecular targets of CD-6′SLN by applying the compound during various stages of infection. In addition, we performed assays to confirm whether viral membrane proteins and envelope interact with the compound and if the compound was able to break down the virus to release its genome. Our findings demonstrate that CD-6′SLN targets hemagglutinin and affects the viral lipid membrane. Unlike CD-MUS, CD-6′SLN did not cause the release of the viral genome, suggesting a somewhat different mechanism from that reported for CD-MUS [
25].
2. Materials and Methods
2.1. Cells, Virus and Chemicals
MDCK (Madin-Darby Canine Kidney Cells) were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Thermo Fisher Scientific, Waltham, MA, USA) with 1% Penicillin/Streptomycin and 10% FBS (Thermo Fisher Scientific, Waltham, MA, USA) in the humidified atmosphere in 5% CO2 at 37 °C.
H1N1 (A/Netherlands/2009) was a kind gift from Prof. M Schmolke (University of Geneva) and it was reproduced in MDCK cells and titrated by immunocytochemistry assay.
Heptakis-(6-deoxy-6-iodo)-β-Cyclodextrin was purchased from AraChem (Tilburg, Netherlands) and dried under high vacuum for 48 h prior to use. 12-mercaptododecanoic acid was purchased from Sigma-Aldrich (St. Louis, MO, USA) and dried in the same way. Neu5Acα2-6Galβ1-4GlcNAc-β-ethylamine was purchased from Asparia Glycomics (Donostia, Spain). Maleimide-PEG8-CH2CH2COOH was purchased from PurePEG (San Diego, CA, USA). All the other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) as reagent grade and used without further purification.
2.2. Chemical Synthesis and Characterization
CD-(S-C11-COOH)
7, CD-6′SLN, and CD-(Mal-PEG8)
7-6′SLN were synthesized based on a previously published procedure [
13], with modifications as described below. The results of NMR and mass spec of the molecules are shown in
Figures S3–S6 in Supplemental Materials.
2.2.1. CD-(S-C11-COOH)7
Potassium tert-butoxide (48.51 mmol, 5.443 g) and 12-mercaptododecanoic acid (22.05 mmol: 5.123 g) were dissolved in 50 mL of DMF under an argon atmosphere. The viscous mixture was stirred at room temperature for 30 min using a mechanical stirrer. Heptakis-(6-deoxy-6-iodo)-β-Cyclodextrin (2.625 mmol, 5 g) was dissolved separately in dry DMF and added to the mixture. The mixture was placed in an oil bath at 70 °C and allowed to react for 12 h. The crude was precipitated directly into 400 mL diethyl ether (Et2O) and vacuum filtered with a fritted disk funnel (Por 3). The solid on the filter was collected (5 g), dried under vacuum for 24 h, and then dissolved in a solution of 95 mL distilled water. This solution was precipitated into 190 mL Acetonitrile and filtered on a fritted disk (Por 4). The material was collected, dissolved in MilliQ water, and freeze-dried to obtain 1.05 g CD-(S-C11-COOH)7. 1H NMR (400 MHz, TFA-d) δ 5.37 (d, J = 3.6 Hz, 1H, glucose H-1), 4.35 (t, J = 9.0 Hz, 1H, glucose H-3), 4.28 (t, J = 7.4 Hz, 1H, glucose H-5), 4.10 (dd, J = 9.9, 3.4 Hz, 1H, glucose H-2), 3.88 (t, J = 9.3 Hz, 1H, glucose H-4), 3.48 (d, J = 12.8 Hz, 1H, glucose H-6a), 3.24 (dd, J = 13.5, 7.6 Hz, 1H, glucose H-6b), 3.00–2.84 (m, 2H, S-CH2-CH2), 2.64 (t, J = 7.6 Hz, 2H, CH2-COOH), 1.86 (q, J = 7.4 Hz, 4H, CH2-CH2-COOH, S-CH2-CH2), 1.53 (m, 14H, S-C-C-CH2-CH2-CH2-CH2-CH2-CH2-CH2-C-C-COO). HRMS (nanochip-ESI/LTQ-Orbitrap) m/z: [M–3H]3− Calcd for C126H221O42S73− 877.5133; Found 877.1148.
2.2.2. CD-6′SLN
1.05 g CD-(S-C11-COOH)7 was dissolved in 15 mL DMSO with the addition of N-hydroxysuccinimide (5.51 mmol, 634 mg), 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (10.2 mmol, 2 g), and 4-Dimethylaminopyridine (0.486 mmol, 60 mg) and stirred overnight. The crude sample was washed with 200 µL acidic water (1 M HCl in H2O) and centrifuged at 5500× g for 1 min. This step was repeated two more times followed by centrifugations at 5500× g for 10 min and the pellet was collected. A solution of 50:50 ACN: Et2O (15 mL) was added to the pellet and centrifuged again at 5500× g for 5 min after the resuspension of the pellet. The new pellet was collected, followed by the addition of Et2O (15 mL) and centrifugation at 5500× g for 5 min. The final pellet was collected and vacuum dried. A portion of the collected activated cyclodextrin derivative (0.28 mmol: 935 mg) and the Neu5Acα2-6Galβ1-4GlcNAc-β-ethylamine (6′SLN, 0.63 mmol: 467 mg) were dissolved in 16 mL DMSO. 100 µL of a triethylamine solution (200 µL TEA in 1 mL DMSO) was added to the DMSO solution and the mixture was stirred overnight. The next day, the resulting material was collected and was dialysed against MilliQ water for three days (the dialysis water was changed twice per day) in a cellulose membrane (MWCO: 2kDa). The material left in the membrane was collected at the end of three days and lyophilized. 1H NMR (400 MHz, D2O) δ 5.13 (s, 2.8H, β-CD Glc H-1) 4.58 (s, 1H, GlcNAc H-1), 4.46 (d, J = 7.8 Hz, 1H, Gal H-1), 4.16–3.29 (m, 31H, other H on 6′SLN and Glc H, S-CH2-CH2), 2.68 (d, J = 11.9 Hz, 1H, Neu5Ac H-2), 2.29 (m, 2.2H, CH2COO), 2.06 (d, J = 9.8 Hz, 6H, COCH3), 1.82–1.11 (m, 19H, S-C-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-C-COO, Neu5Ac H-2′).
2.2.3. CD-(Mal-PEG8)7-6′SLN
Heptakis-(6-deoxy-6-mercapto)-β-Cyclodextrin (0.024 mmol, 30 mg) was dissolved into 1.5 mL DMSO and mixed with 8 mL 0.1 M phosphate buffer (pH 6.8) containing Maleimide-PEG8-CH2CH2COOH (0.29 mmol, 150 mg). The reaction mixture was stirred overnight at room temperature and then dialysed against MiliQ water with cellulose membrane (MWCO: 1kDa) for three days and dried by lyophilization to yield pink powder CD-(Mal-PEG8)7-COOH. HRMS (nanochip-ESI/LTQ-Orbitrap) m/z: [M–4H]4− Calcd for C203H339N7O112S74−1223.5775; Found 1223.2257. CD-(Mal-PEG8)7-COOH (15.6 µmol, 76.4 mg), N-hydroxysuccinimide (0.42 mmol, 48 mg), 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (0.18 mmol, 36 mg) and 4-Dimethylaminopyridine (0.0097 mmol, 1.2 mg) were dissolved in 5 mL DMSO and stirred overnight. After reaction, the product was precipitated and washed by 500 mL dichloromethane/diethyl ether (1/50) mixture twice and dried under vacuum. 10 mg of obtained cyclodextrin intermediate was dissolved in 2 mL DMSO. Neu5Acα2-6Galβ1-4GlcNAc-β-ethylamine (4.6 µmol, 3.3 mg) and triethylamine (0.049 mmol, 5 mg) were then added in. The mixture was stirred overnight and the product CD-(Mal-PEG8)7-6′SLN was purified by three-day dialysis against MiliQ water with cellulose membrane (MWCO: 2kDa) and dried by lyophilization.
2.3. H1N1 Inhibition Assay
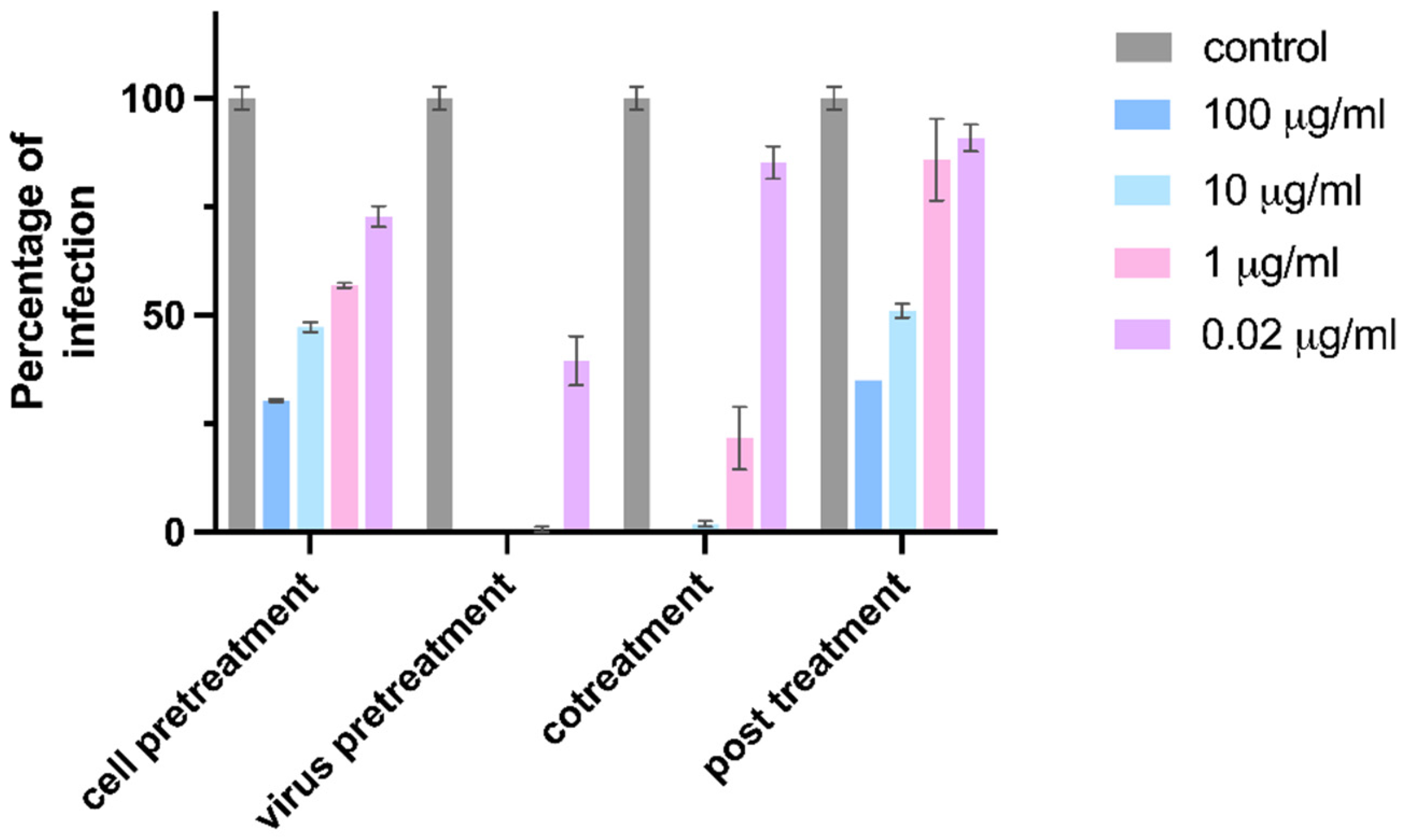
MDCK cells were pre-seeded on a 96-well plate at the density of 2 × 104 cells per well 24 h in advance. 0.005 MOI of H1N1 was used for all inhibition experiments. For the virus pretreatment groups, 10 μg/mL CD-6′SLN were incubated with H1N1 at 37 °C for 1 h and the mixture was added to the cell cultures. For the cell pretreatment groups, the cells were first treated with 10 μg/mL CD-6′SLN for 1 h and then infected with the virus. For the virus co-treatment groups, 10 μg/mL CD-6′SLN were added simultaneously with the virus to the cells. For the post-infection treatment groups, the virus was added to the cells, after 1 h incubation for viral attachment, the inoculum was removed and a medium containing 10 μg/mL CD-6′SLN was added to the cells. After 24 h incubation, the medium was removed and cells were fixed with methanol for 1 h. Then the LIGHT DIAGNOSTICS Flu A monoclonal antibody (1:100 dilution, Merck Millipore, Burlington, MA, USA) was added and incubated for 1 h at 37 °C. The cells were washed with wash buffer (PBS + Tween 0.05%) three times, followed by adding anti-mouse lgG, HRP-linked antibody (1:500 dilution, Cell Signaling Technology, Danvers, MA, USA). After 1 h, the cells were washed and the DAB solution was added. Infected cells were counted and percentages of infection were calculated by comparing the number of infected cells in treated and untreated conditions.
The EC
50 measurements of CD-6′SLN, CD-(S-C11-COOH)
7, and CD-(Mal-PEG8)7-6′SLN were performed using the virus pretreatment method. Prism 9 (GraphPad, San Diego, CA, USA) was used to estimate the EC
50 from the dose-response curve (
Figure S1).
2.4. Biolayer Interferometry
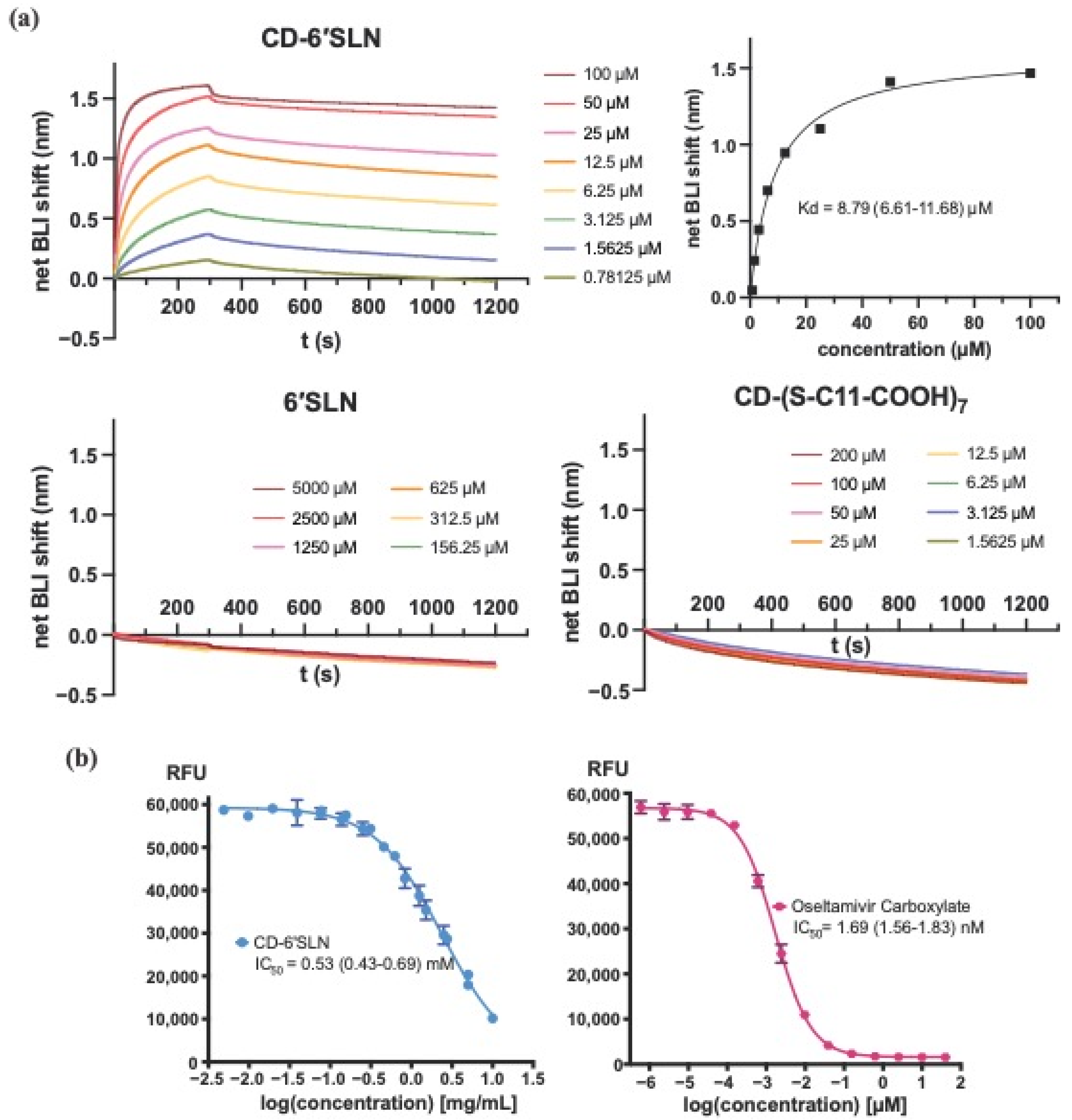
GatorTM Anti-His probes (No. 160009, Gator bio, Palo Alto, CA, USA) were first prewetted in 10 mM PBS buffer (75 mM NaCl, pH 7.6), and then immobilized with 10 μg/mL H1N1 Hemagglutinin (A/California/04/2009, GSC-Z03181, GenScript, Rijswijk, The Netherlands) for 300 s to reach a shift of 1.8 nm. After balancing the probes in the same buffer for 60 s, the probes were dipped into different concentrations of the test materials for 300 s, and then into the buffer for 900 s. Reference probes for each concentration were undergoing the same procedure without HA immobilization. The association and dissociation shift (nm) were plotted by subtracting the signal of reference probes from experimental probes, and the equilibrium dissociation constant KD was calculated by steady-state analysis using Prism 9 (GraphPad, San Diego, CA, USA).
2.5. Neuraminidase Inhibition Assay
The activities of Influenza H1N1 (A/California/04/2009) Neuraminidase (11058-VNAHC, Sino Biological, Bejing, China) were tested using a fluorogenic substrate MUNANA (2′-(4-Methylumbelliferyl)-alpha-D-N-acetylneuraminic acid sodium salt hydrate, Sigma Aldrich, 69587). The optimal final concentration of NA (1.75 unit/mL) and MUNANA (100 μM) were chosen after pretests to ensure the linear relationship between RFU readout and the fluorescence product concentration. In the assay, NA was incubated with different concentrations of tested materials in 33 mM MES buffer (4 mM CaCl2, pH 6.5) for 2 h. MUNANA was then added to the mixture and the fluorescence was measured after 1 h incubation at 37 °C using a Tecan Spark plate reader (excitation wavelength 355 ± 30 nm, emission wavelength 460 ± 25 nm). Prism 9 (GraphPad, San Diego, CA, USA) was used to determine the IC50 from the dose-response curve.
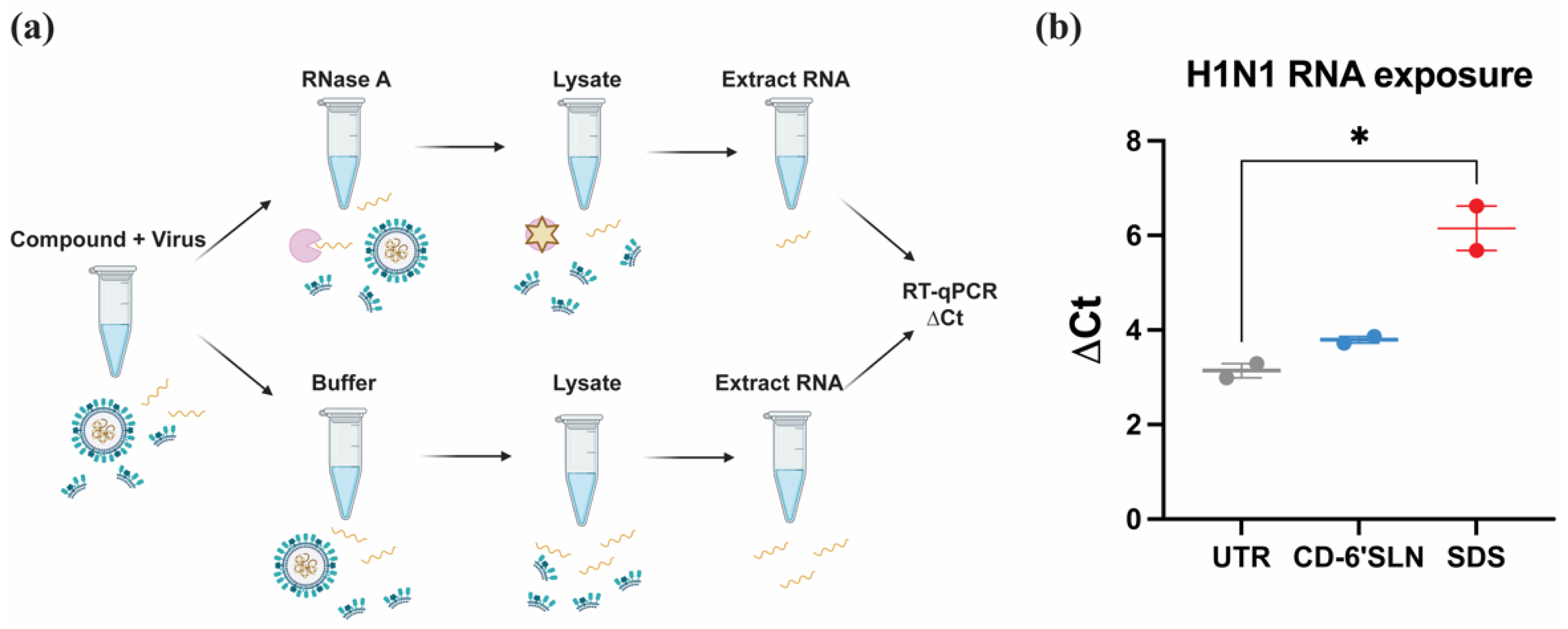
2.6. RNA Exposure Assay
Influenza H1N1 (A/Netherlands/2009, 1.3 × 105 PFU/mL) was incubated with 100 μg/mL tested material in DMEM medium containing 1% P/S at 37 °C for 3 h. 1.1 μL 10 mg/mL RNAse A (EN0531, Thermo Fisher Scientific, Waltham, MA, USA) or 1.1 μL MiliQ water was added into 10 μL 1× PBS buffer, 100 μL 1:20 diluted viral material solution in two Eppendorf tubes. After 30 min incubation at 37 °C, 500 μL GTC lysis buffer (Omega bio-tek, Norcross, GA, USA) was added into each tube and RNA was extracted using E.Z.N.A. DNA/RNA/isolation kit (Omega Bio-tek, Norcross, GA, USA) and quantified by RP-qPCR. The RT-qPCR reaction (final volume 10 μL) consisted of 5 μL QuantiTech Probe RT-PCR Master Mix 2×, 0.1 μL QuantiTect RT Mix, 2.5 μL extracted viral RNA and 2.4 μL primers (4 μM each, 5’-AAG ACC AAT CYT GTC ACC TCT GA-3’, 5’-CAA AGC GTC TAC GCT GCA GTC C-3’, Microsynth AG, Balgach, Switzerland) and probe (2 μM, FAM5’-TTT GTG TTC ACG CTC ACC GTG CC-3’TAMRA, Microsynth AG, Balgach, Switzerland). The RT-qPCR was executed on QuantiStudio 7 (Thermo Fisher Scientific, Waltham, MA, USA), starting at 50 °C for 30 min and 95 °C for 15 min, followed by 45 cycles of 15 s at 94 °C and 1 min at 60 °C. Cycle threshold (Ct) was analysed by QuantiStudio software v1.7.2(Thermo Fisher Scientific, Waltham, MA, USA) and ∆Ct was plotted as the difference between the RNAse-treated sample and MilliQ water-treated sample.
2.7. R18-Labeled H1N1 Preparation
2.33 μL of 3.12 μmol/mL ethanolic R18 (octadecyl rhodamine B chloride) solution was injected into 255 μL H1N1 suspension containing 2 mg/mL of viral protein in total, quantified by modified Lowry protein assay Thermo Scientific™ Pierce™ Modified Lowry Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA) under vortex mixing. The mixture was incubated in the dark at room temperature for 1 h, and the non-inserted fluorophore was removed by chromatography on Zeba™ Spin Desalting column (7K MWCO, Thermo Fisher Scientific, Waltham, MA, USA) using 10 mM TES, 150 mM NaCl (pH 7.4) as elution buffer. The protein concentration of the R18-labeled virus was determined again by the modified Lowry assay.
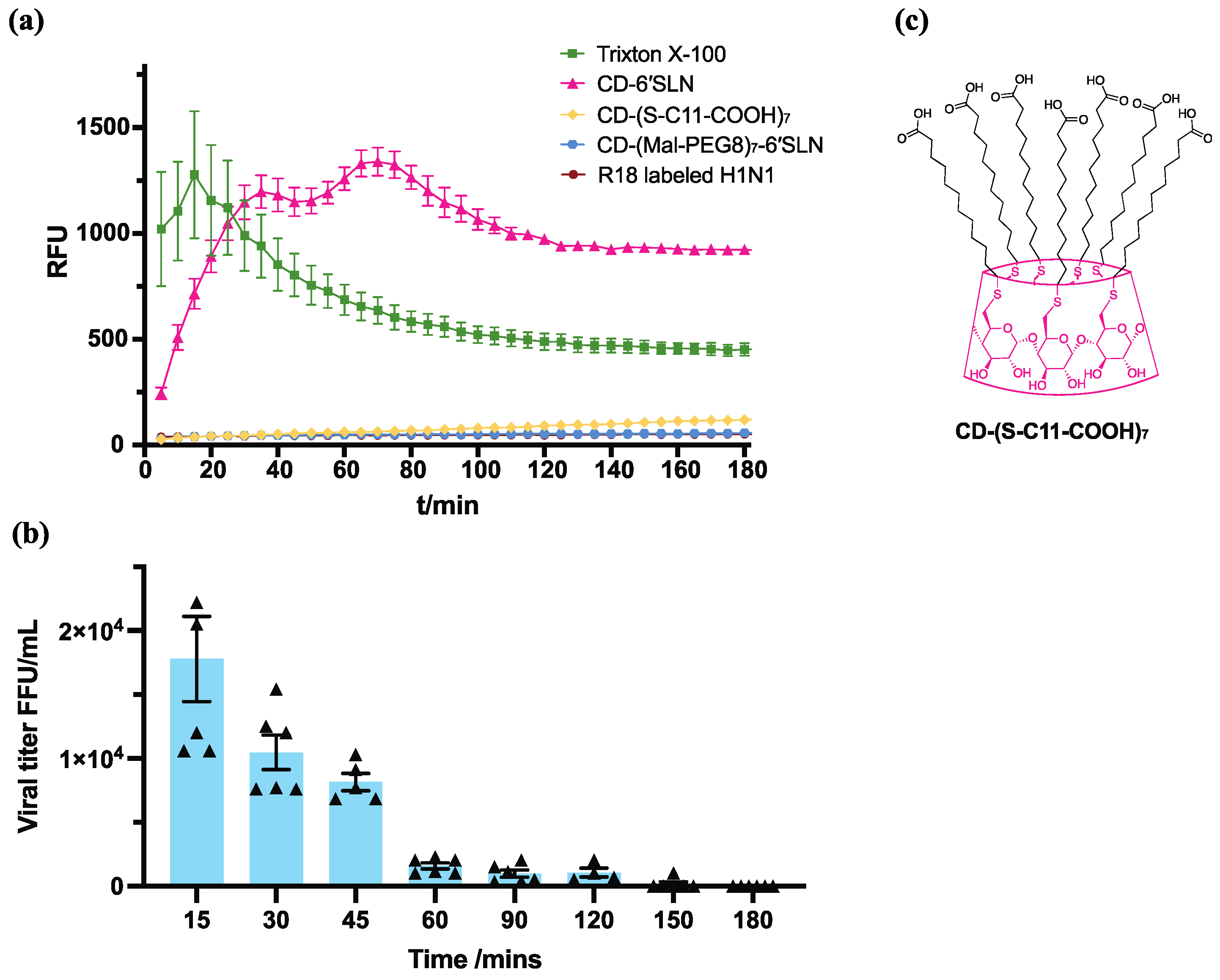
2.8. R18 Release Assay
R18-labelled H1N1 (final viral protein concentration 2 μg/mL) was added into 1% P/S DMEM medium containing triton X-100, CD-6′SLN, CD-(S-C11-COOH)7 or CD-(Mal-PEG8)7-6′SLN. Fluorescence was measured at different timepoints using Tecan infinite 200 plate reader with excitation at 560 nm and emission at 590 nm.
2.9. H1N1 Virucidal Assay
100 μg/mL test compound was incubated with 1.3 × 105 FFU/mL H1N1 at 37 °C. After 15 min, 30 min, 45 min, 1 h, 1.5 h, 2 h, 2.5 h and 3 h, the virus compound mixture was titrated using the ICC assay mentioned in the H1N1 inhibition assay. Viral titre calculated the number of stained cells times the dilution factor.
4. Discussion and Conclusions
Most approved small-molecule antivirals inhibit the intracellular replication process, which may induce cell mutagenesis [
28], carcinogenesis [
29], and teratogenesis [
30] depending on the specific mechanism. Therefore, developing new safe antivirals, focusing on novel mechanisms is essential to, not only increase the antiviral toolkit but also be ready for emerging viral diseases. Here we showed that CD-6′SLN inhibited influenza A H1N1 with an extracellular mechanism by interacting with the viral envelope protein hemagglutinin and viral envelope lipids that ultimately blocked viral entry. In this case, both the hemagglutinin ligand 6′SLN and aliphatic linker contribute to the antiviral effect. The 6′SLN group directly binds to HA on the viral envelope and CD-6′SLN interacts with the viral envelope thanks to its hydrophobic linker, likely causing envelope destruction.
The prevalence and accessibility of hemagglutinin make it an important drug discovery target. Nevertheless, small sialic acid-containing inhibitors are not effective in this case due to the low affinity and mutational variability of the sialic acid binding site [
31]. It seems that CD-6′SLN overcomes this limitation due to the multivalent interaction and irreversible damage mediated by the hydrophobic linker. Meanwhile, the natural ligand mimic provides a simple specificity mechanism, and the modular construction of CD-6′SLN enables further modifications in response to mutational variations of the sialic acid binding site.
CD-6′SLN is not the only antiviral that targets lipid bilayers. N-docosanol, which has a similar saturated fatty structure of 22 carbon atoms, is a commercial anti-herpes simplex virus (HSV-2) drug and has been reported to inhibit enveloped viruses including HSV-1, HSV-2, respiratory syncytial virus, cytomegalovirus, varicella-zoster virus, human herpesvirus 6 and human immunodeficiency virus-1 with EC
50 ranging from 3 mM to 12 mM [
32]. Unlike CD-6′SLN, n-docosanol is not directly virucidal and the optimal viral inhibition occurs when the compound is applied to host cells prior to viral infection. The underlying mechanism is that n-docosanol incorporates into cell membranes and is metabolized into phosphatidylcholine- and phosphatidylethanolamine-like species [
33], thus, inhibiting viral fusion in the early stage of HSV-2 replication [
34]. The inhibition activity of CD-6′SLN based on its action on the virus instead of the host cells, and the resulting virucidal effect, could probably be explained by the enrichment of CD-6′SLN on the viral envelope induced by specific 6SLN-hemagglutinin interactions.
In summary, the influenza inhibition and virucidal activity of CD-6′SLN result from the compound binding to the viral protein hemagglutinin and interacting with the viral envelope. This result implies that combining a virus-specific epitope with a lipid-targeting aliphatic linker is likely a general strategy for developing virucidal yet non-toxic antivirals. Based on the dual action of the viral protein and the envelope, we hypothesize that such compounds may be also effective against emergent viral strains in the future.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




