
Structure–Activity Relationship Studies of 9-Alkylamino-1,2,3,4-tetrahydroacridines against Leishmania (Leishmania) infantum Promastigotes
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. General
2.2. Instrumentalization
2.3. Computational Studies
2.3.1. Virtual Screening
2.3.2. Molecular Docking
2.4. Synthesis
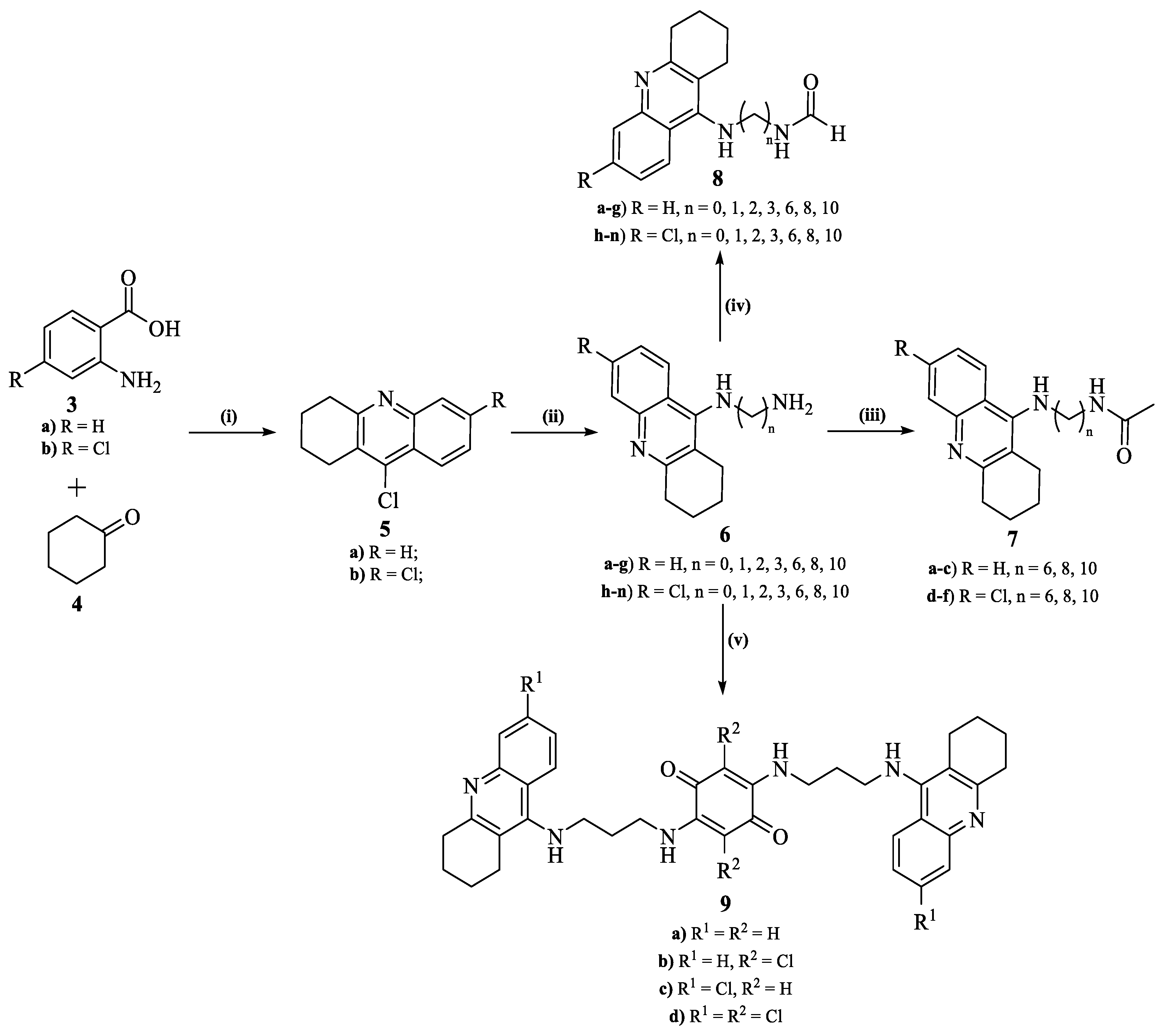
2.4.1. General Procedure for the Synthesis of 9-Chloroacridines (5)
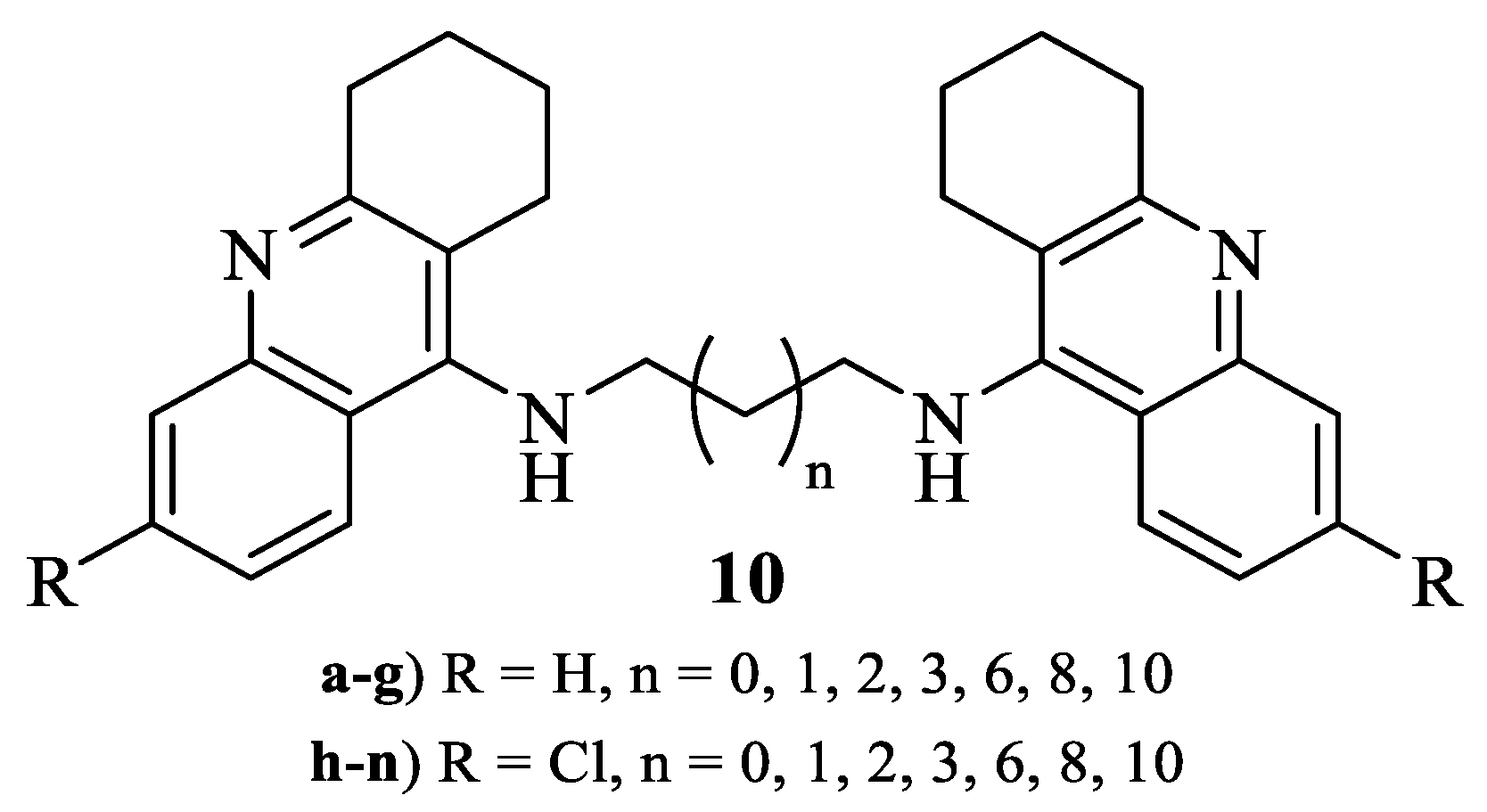
2.4.2. General Procedure for the Synthesis of 9-Alkylamino-1,2,3,4-tetrahydroacridines (6.a–n)
2.4.3. General Procedure for the Synthesis of N-Acetylated-9-alkylamino-1,2,3,4-tetrahydroacridines (7.a–n)
2.4.4. General Procedure for the Synthesis of N-Formylated-9-alkylamino-1,2,3,4-tetrahydroacridine (8.a–f)
2.4.5. General Procedure for the Synthesis of Compounds 9.a–d
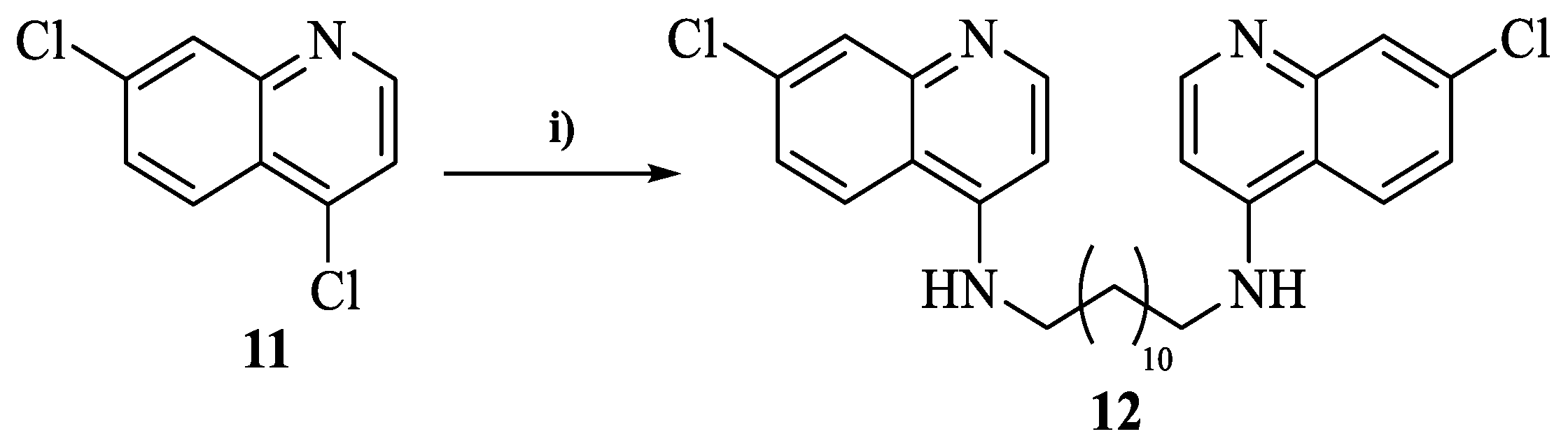
2.4.6. General Procedure for the Synthesis of N1,N12-bis(7-chloroquinolin-4-yl)dodecane-1,12-diamine (12)
2.5. Biological Evaluation
2.5.1. Promastigote Stage of the Parasite
2.5.2. Toxicity against Mammalian Macrophages
3. Results and Discussion
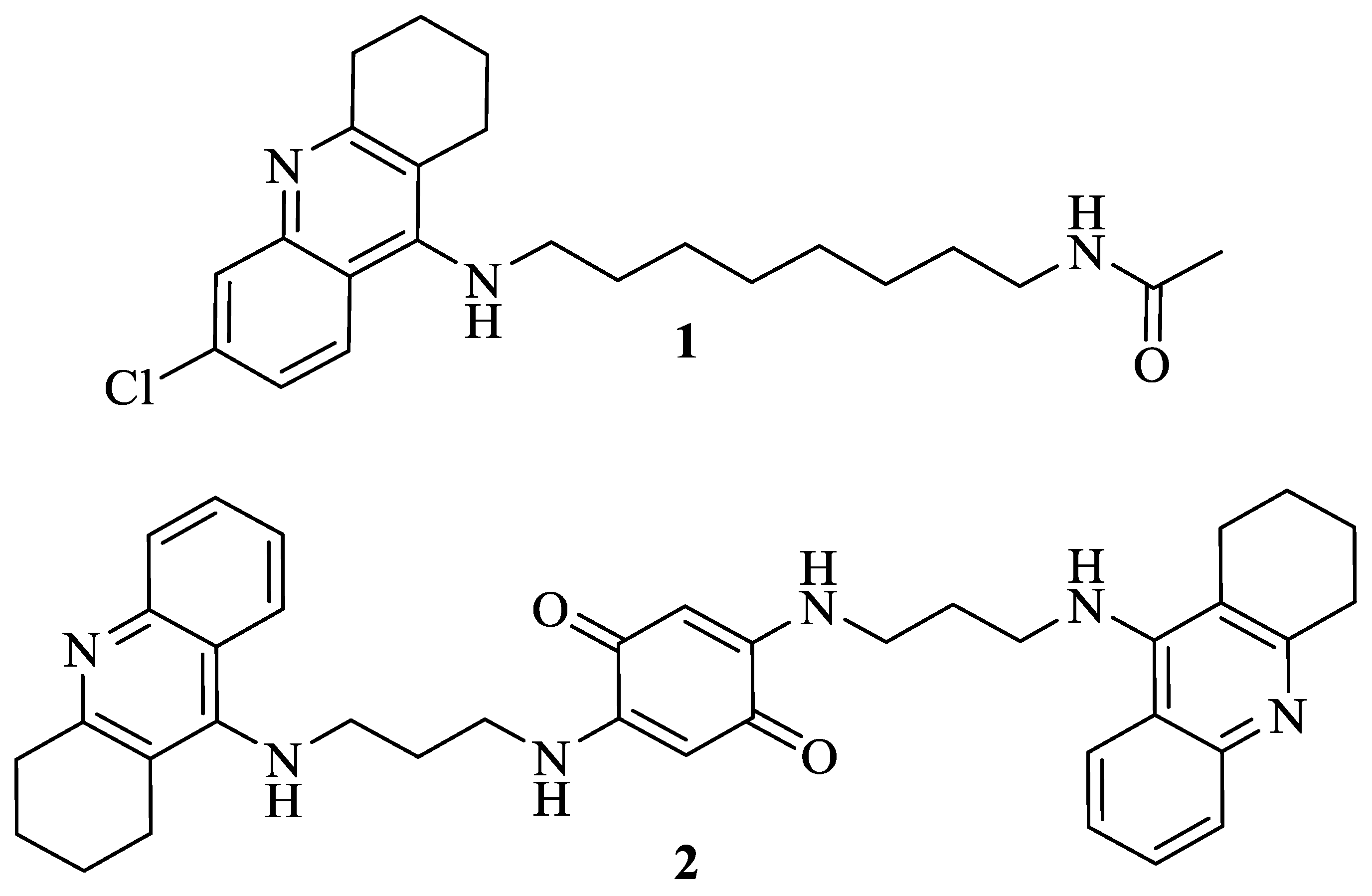
3.1. Virtual Screening against AdoMet DC
3.2. Chemistry
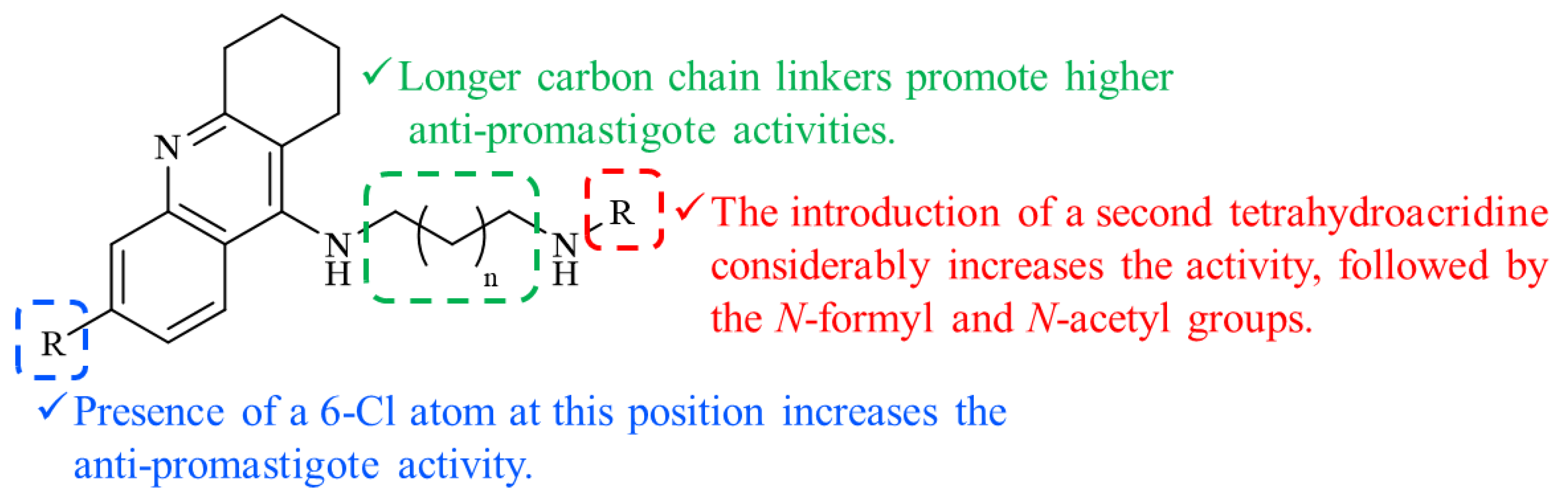
3.3. Antileishmanial Evaluation against L. infantum Promastigotes
3.4. Toxicity against Mammalian Macrophages
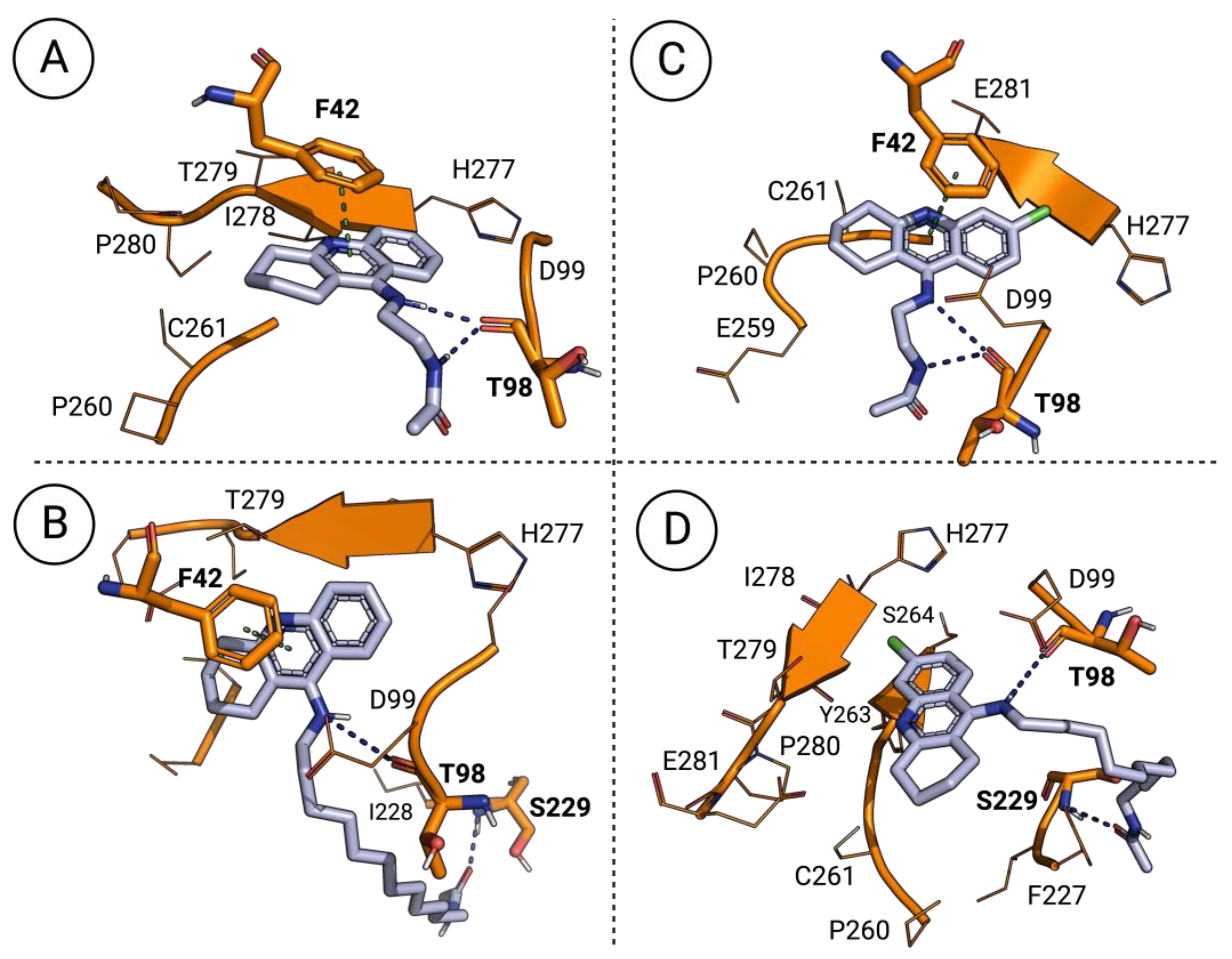
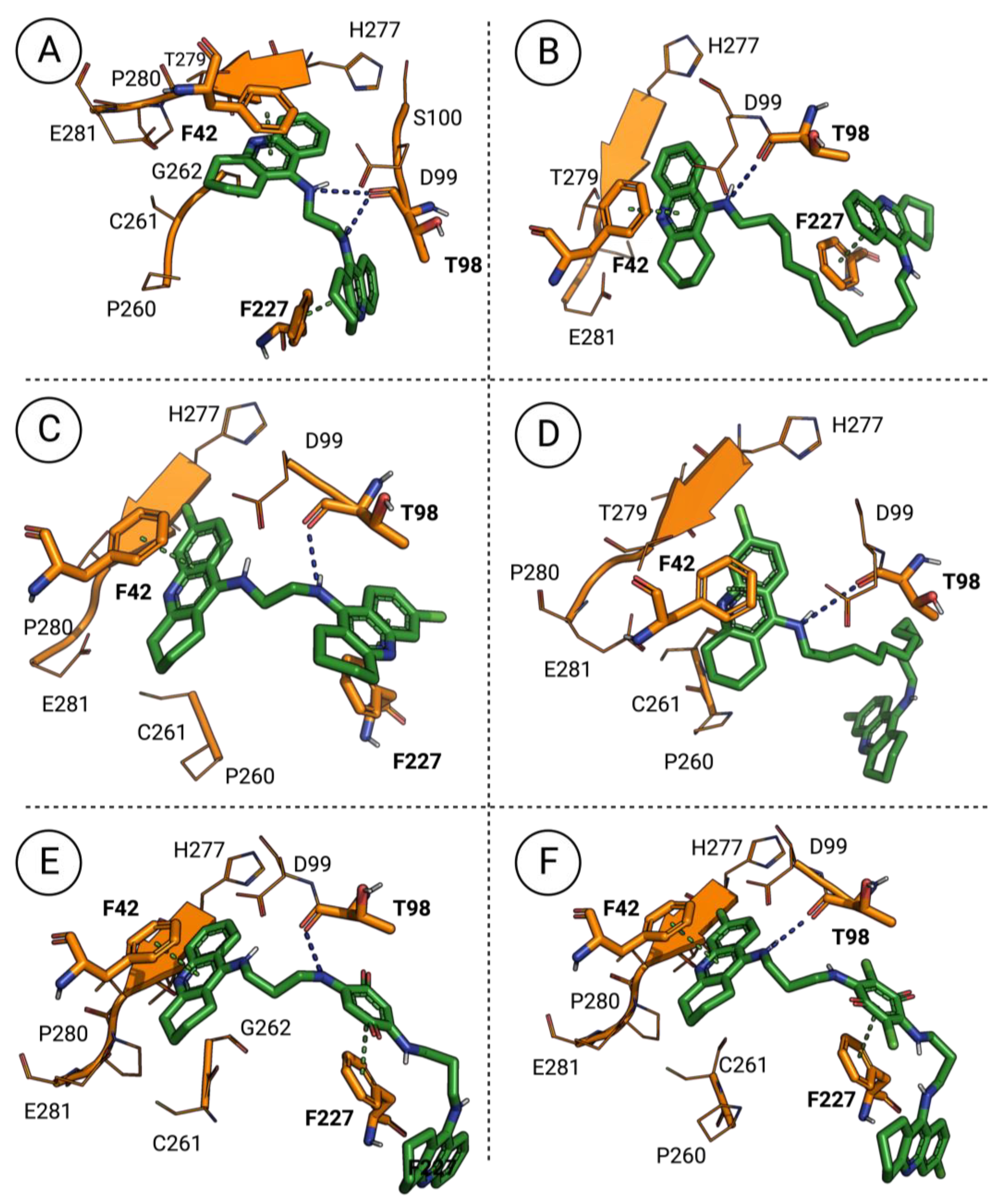
3.5. Molecular Docking of the Most Promising Antileishmanial Agents
4. Comments and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). First WHO Report on Neglected Tropical Diseases: Working to Overcome the Global Impact of Neglected Tropical Diseases; WHO: Geneva, Switzerland, 2010. [Google Scholar]
- Bodimeade, C.; Marks, M.; Mabey, D. Neglected Tropical Diseases: Elimination and Eradication. Clin. Med. 2019, 19, 157–160. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). The Control of Leishmaniases; WHO: Geneva, Switzerland, 2010; Volume 949. [Google Scholar]
- Oryan, A.; Akbari, M. Worldwide Risk Factors in Leishmaniasis. Asian Pac. J. Trop. Med. 2016, 9, 925–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pace, D. Leishmaniasis. J. Infect. 2014, 69, S10–S18. [Google Scholar] [CrossRef] [PubMed]
- Sen, R.; Chatterjee, M. Plant Derived Therapeutics for the Treatment of Leishmaniasis. Phytomedicine 2011, 18, 1056–1069. [Google Scholar] [CrossRef] [PubMed]
- Ogungbe, I.V.; Singh, M.; Setzer, W.N. Antileishmanial Natural Products from Plants. In Studies in Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 2012; Volume 36, pp. 331–382. ISBN 9780444538369. [Google Scholar]
- Chawla, B.; Madhubala, R. Drug Targets in Leishmania. J. Parasit. Dis. 2010, 34, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willert, E.K.; Fitzpatrick, R.; Phillips, M.A. Allosteric Regulation of an Essential Trypanosome Polyamine Biosynthetic Enzyme by a Catalytically Dead Homolog. Proc. Natl. Acad. Sci. USA 2007, 104, 8275–8280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willert, E.K.; Phillips, M.A. Cross-Species Activation of Trypanosome S-Adenosylmethionine Decarboxylase by the Regulatory Subunit Prozyme. Mol. Biochem. Parasitol. 2009, 168, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heby, O.; Persson, L.; Rentala, M. Targeting the Polyamine Biosynthetic Enzymes: A Promising Approach to Therapy of African Sleeping Sickness, Chagas’ Disease, and Leishmaniasis. Amino Acids 2007, 33, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, X.; Li, B.; Gao, C.; Jiang, Y. Acridine and Its Derivatives: A Patent Review (2009–2013). Expert Opin. Ther. Pat. 2014, 24, 647–664. [Google Scholar] [CrossRef] [PubMed]
- Gensicka-Kowalewska, M.; Cholewiński, G.; Dzierzbicka, K. Recent Developments in the Synthesis and Biological Activity of Acridine/Acridone Analogues. RSC Adv. 2017, 7, 15776–15804. [Google Scholar] [CrossRef] [Green Version]
- Silva, C.F.M.; Pinto, D.C.G.A.; Fernandes, P.A.; Silva, A.M.S. Evolution of Acridines and Xanthenes as a Core Structure for the Development of Antileishmanial Agents. Pharmaceuticals 2022, 15, 148. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.-K.; Wu, L.-J.; Hsiao, G.; Yen, M.-H. Homodimeric Tacrine Congeners as Acetylcholinesterase Inhibitors. J. Med. Chem. 2002, 45, 2277–2282. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Alves, A.G.; Duarte, M.; Cruz, T.; Castro, H.; Lopes, F.; Moreira, R.; Ressurreição, A.S.; Tomás, A.M. Biological Evaluation and Mechanistic Studies of Quinolin-(1H)-Imines as a New Chemotype against Leishmaniasis. Antimicrob. Agents Chemother. 2021, 65, e01513-20. [Google Scholar] [CrossRef] [PubMed]
- Heravi, M.M.; Ghavidel, M.; Mohammadkhani, L. Beyond a Solvent: Triple Roles of Dimethylformamide in Organic Chemistry. RSC Adv. 2018, 8, 27832–27862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bongarzone, S.; Tran, H.N.A.; Cavalli, A.; Roberti, M.; Carloni, P.; Legname, G.; Bolognesi, M.L. Parallel Synthesis, Evaluation, and Preliminary Structure−Activity Relationship of 2,5-Diamino-1,4-Benzoquinones as a Novel Class of Bivalent Anti-Prion Compound. J. Med. Chem. 2010, 53, 8197–8201. [Google Scholar] [CrossRef] [PubMed]
- Robertson, P.A.; Villani, L.; Dissanayake, U.L.M.; Duncan, L.F.; Abbott, B.M.; Wilson, D.J.D.; Robertson, E.G. Halocarbons as Hydrogen Bond Acceptors: A Spectroscopic Study of Haloethylbenzenes (PhCH2CH2X, X=F, Cl, Br) and Their Hydrate Clusters. Phys. Chem. Chem. Phys. 2018, 20, 8218–8227. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Anti-Promastigote Activity (IC50) a | Cytotoxicity (% of Death) a | Therapeutic Index |
|---|---|---|---|
| 5.a | >100 µM | N.t. b | N.t. b |
| 5.b | >100 µM | N.t. b | N.t. b |
| 7.a | >100 µM | N.t. b | N.t. b |
| 7.b | >100 µM | N.t. b | N.t. b |
| 7.c | >100 µM | N.t. b | N.t. b |
| 7.d | >100 µM | N.t. b | N.t. b |
| 7.e | >100 µM | N.t. b | N.t. b |
| 7.f | 11.20 ± 0.66 µM | 77.5% (10 μM) | <10 |
| 7.g | 1.86 ± 0.48 µM | 98.8% (10 μM) | <10 |
| 7.h | >100 µM | N.t. b | N.t. b |
| 7.i | >100 µM | N.t. b | N.t. b |
| 7.j | >100 µM | N.t. b | N.t. b |
| 7.k | >100 µM | N.t. b | N.t. b |
| 7.l | >50 µM | N.t. b | N.t. b |
| 7.m | 5.78 ± 0.65 µM | 94.2% (10 μM) | <10 |
| 7.n | 4.47 ± 0.79 µM | 97.3% (10 μM) | <10 |
| 8.a | >50 µM | N.t. b | N.t. b |
| 8.b | 5.64 ± 0.65 µM | 88.1% (10 μM) | <10 |
| 8.c | 1.69 ± 0.22 µM | 99.5% (10 μM) | <10 |
| 8.d | 14.17 ± 0.98 µM | 100.0% (50 μM) | <10 |
| 8.e | 3.23 ± 0.41 µM | 94.7% (10 μM) | <10 |
| 8.f | 1.55 ± 0.38 µM | 91.2% (10 μM) | <10 |
| 10.a | >100 µM | N.t. b | N.t. b |
| 10.b | >100 µM | N.t. b | N.t. b |
| 10.c | >50 µM | N.t. b | N.t. b |
| 10.d | >50 µM | N.t. b | N.t. b |
| 10.e | 2.99 ± 0.98 µM | 99.5% (10 μM) | <10 |
| 10.f | 0.94 ± 0.33 µM | 99.5% (10 µM) | <10 |
| 10.g | 0.37 ± 0.06 µM | 99.7% (10 μM) | <10 |
| 10.h | 9.50 ± 0.51 µM | N.t. b | N.t. b |
| 10.i | 8.58 ± 0.47 µM | 90.4% (10 μM) | <10 |
| 10.j | 8.56 ± 1.56 µM | 9.02 ± 1.79 µM | <10 |
| 10.k | 5.26 ± 4.89 µM | 87.5% (10 μM) | <10 |
| 10.l | 1.42 ± 0.22 µM | 95.0% (10 μM) | <10 |
| 10.m | 1.92 ± 0.21 µM | 99.0% (10 μM) | <10 |
| 10.n | 3.50 ± 0.63 µM | 92.5% (50 μM) | N.t. b |
| 9.a | >100 µM | N.t. b | N.t. b |
| 9.b | >100 µM | N.t. b | N.t. b |
| 9.c | >50 µM | N.t. b | N.t. b |
| 9.d | 2.17 ± 0.45 µM | N.t. b | N.t. b |
| 12 | 0.60 ± 0.11 µM | 11.69 ± 3.96 μM | 19.48 |
| Amphotericin B | 0.17 ± 0.09 µM | 3.1 ± 0.6 µM | 18.24 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, C.F.M.; Leão, T.; Dias, F.; Tomás, A.M.; Pinto, D.C.G.A.; Oliveira, E.F.T.; Oliveira, A.; Fernandes, P.A.; Silva, A.M.S. Structure–Activity Relationship Studies of 9-Alkylamino-1,2,3,4-tetrahydroacridines against Leishmania (Leishmania) infantum Promastigotes. Pharmaceutics 2023, 15, 669. https://doi.org/10.3390/pharmaceutics15020669
Silva CFM, Leão T, Dias F, Tomás AM, Pinto DCGA, Oliveira EFT, Oliveira A, Fernandes PA, Silva AMS. Structure–Activity Relationship Studies of 9-Alkylamino-1,2,3,4-tetrahydroacridines against Leishmania (Leishmania) infantum Promastigotes. Pharmaceutics. 2023; 15(2):669. https://doi.org/10.3390/pharmaceutics15020669
Chicago/Turabian StyleSilva, Carlos F. M., Teresa Leão, Filipa Dias, Ana M. Tomás, Diana C. G. A. Pinto, Eduardo F. T. Oliveira, Ana Oliveira, Pedro A. Fernandes, and Artur M. S. Silva. 2023. "Structure–Activity Relationship Studies of 9-Alkylamino-1,2,3,4-tetrahydroacridines against Leishmania (Leishmania) infantum Promastigotes" Pharmaceutics 15, no. 2: 669. https://doi.org/10.3390/pharmaceutics15020669