
Commercially Available Cell-Free Permeability Tests for Industrial Drug Development: Increased Sustainability through Reduction of In Vivo Studies
 , and
, and
Abstract
:1. Introduction
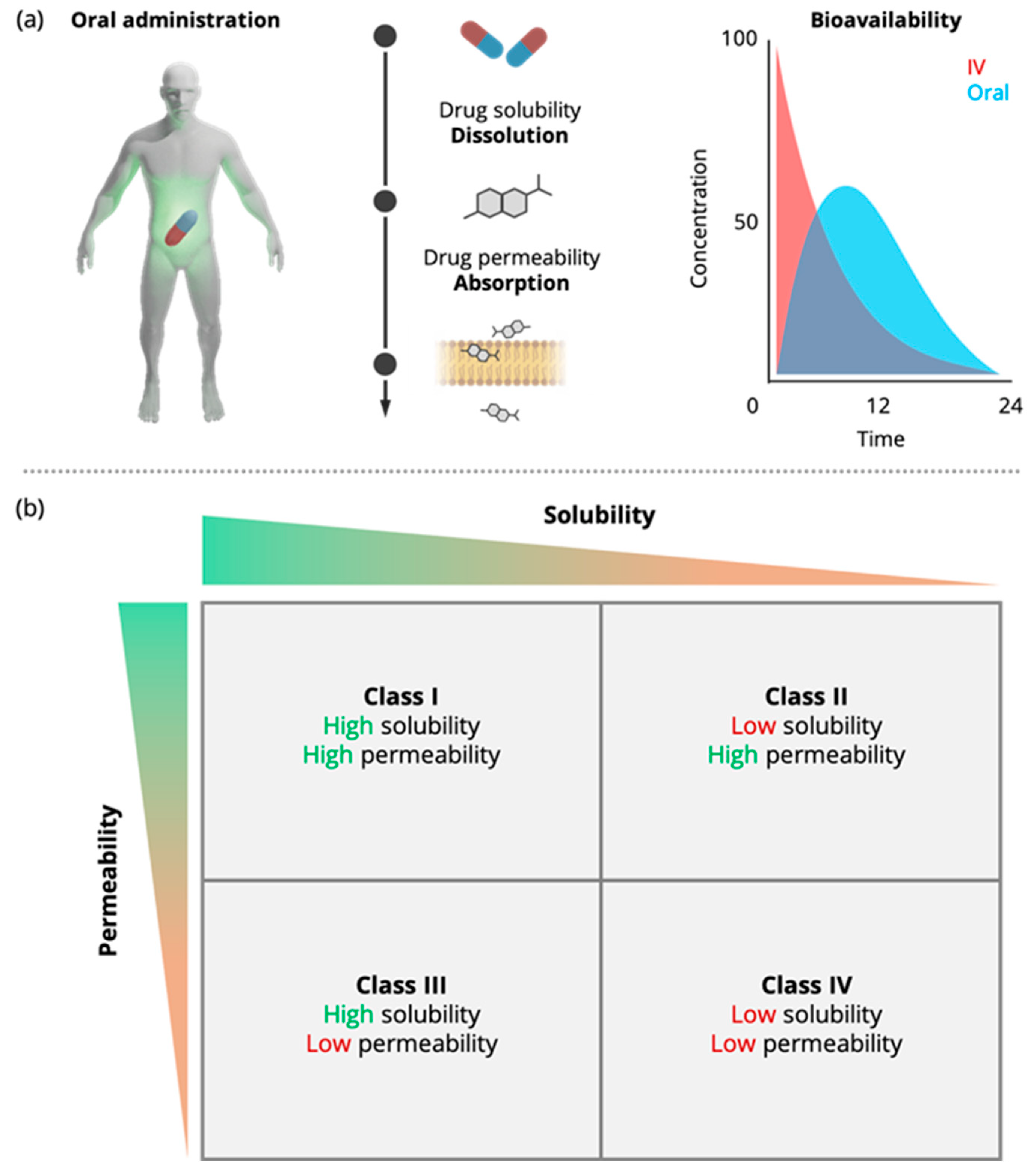
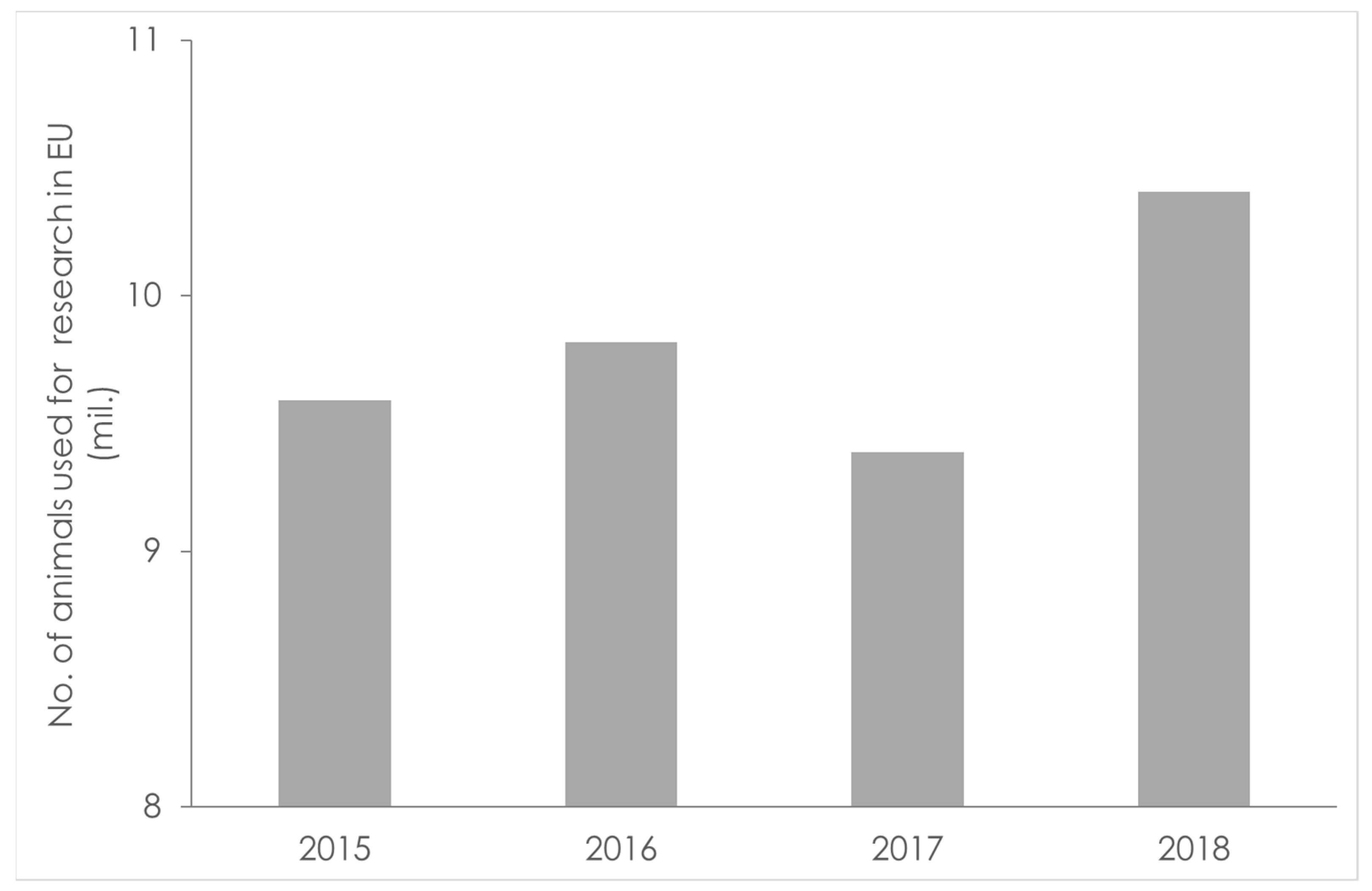
1.1. In Vivo Studies in Humans and Animals—A Challenge for Sustainability
1.2. Replacing In Vivo Studies with In Vitro Studies
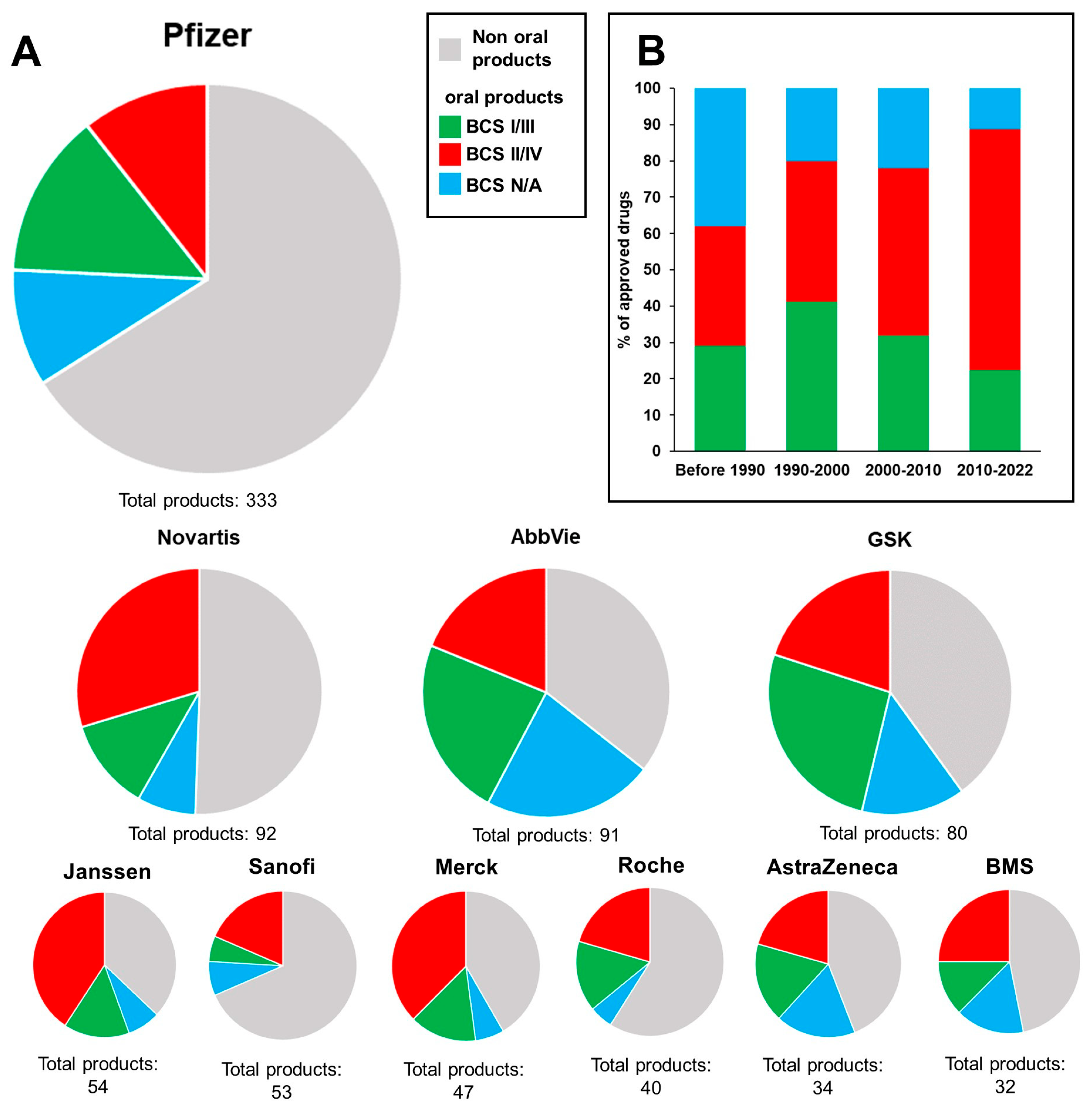
2. Industrial Implementation of In Vitro Permeability Testing
2.1. Methodology of the Search
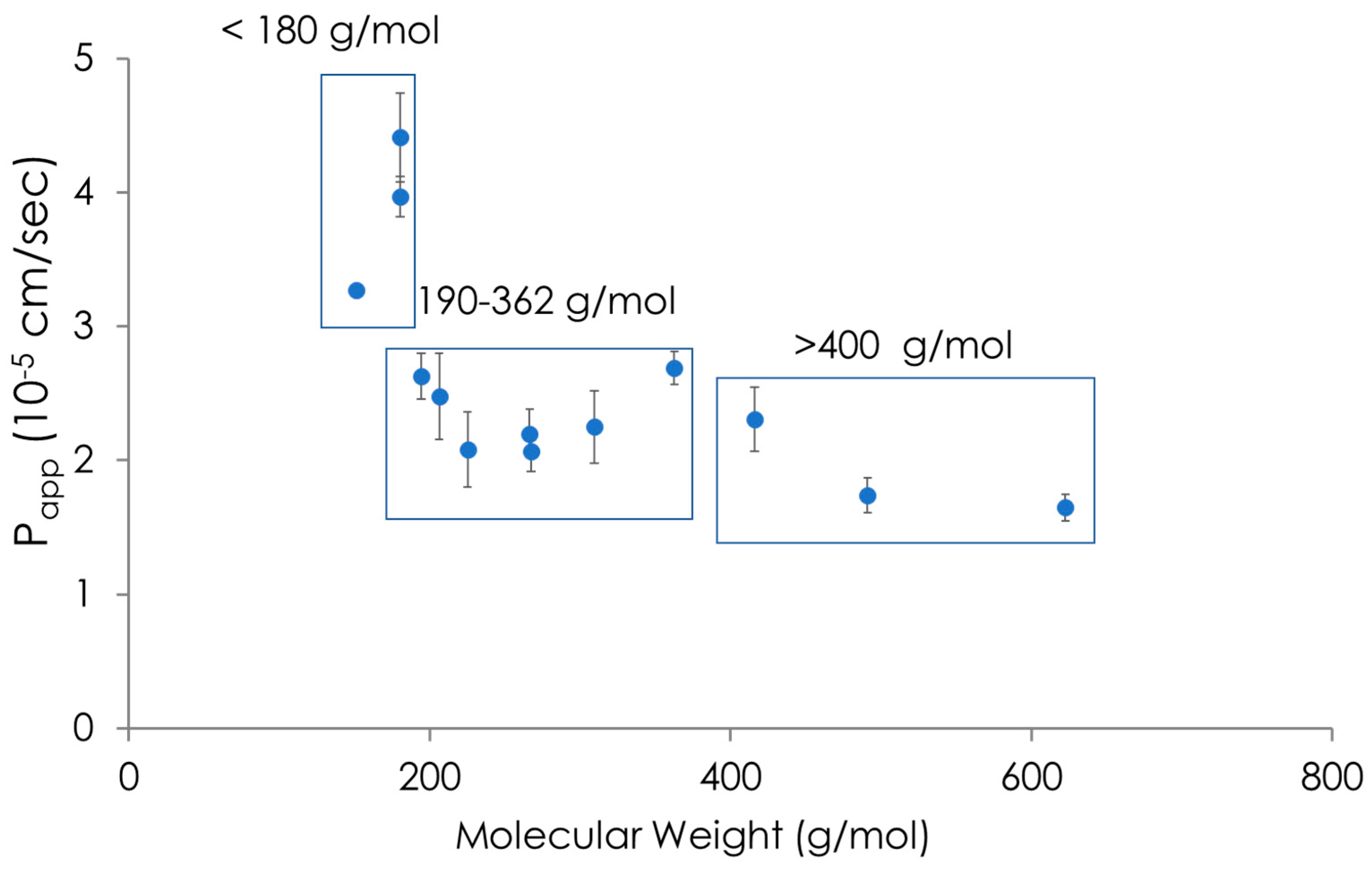
2.2. Results of the Search
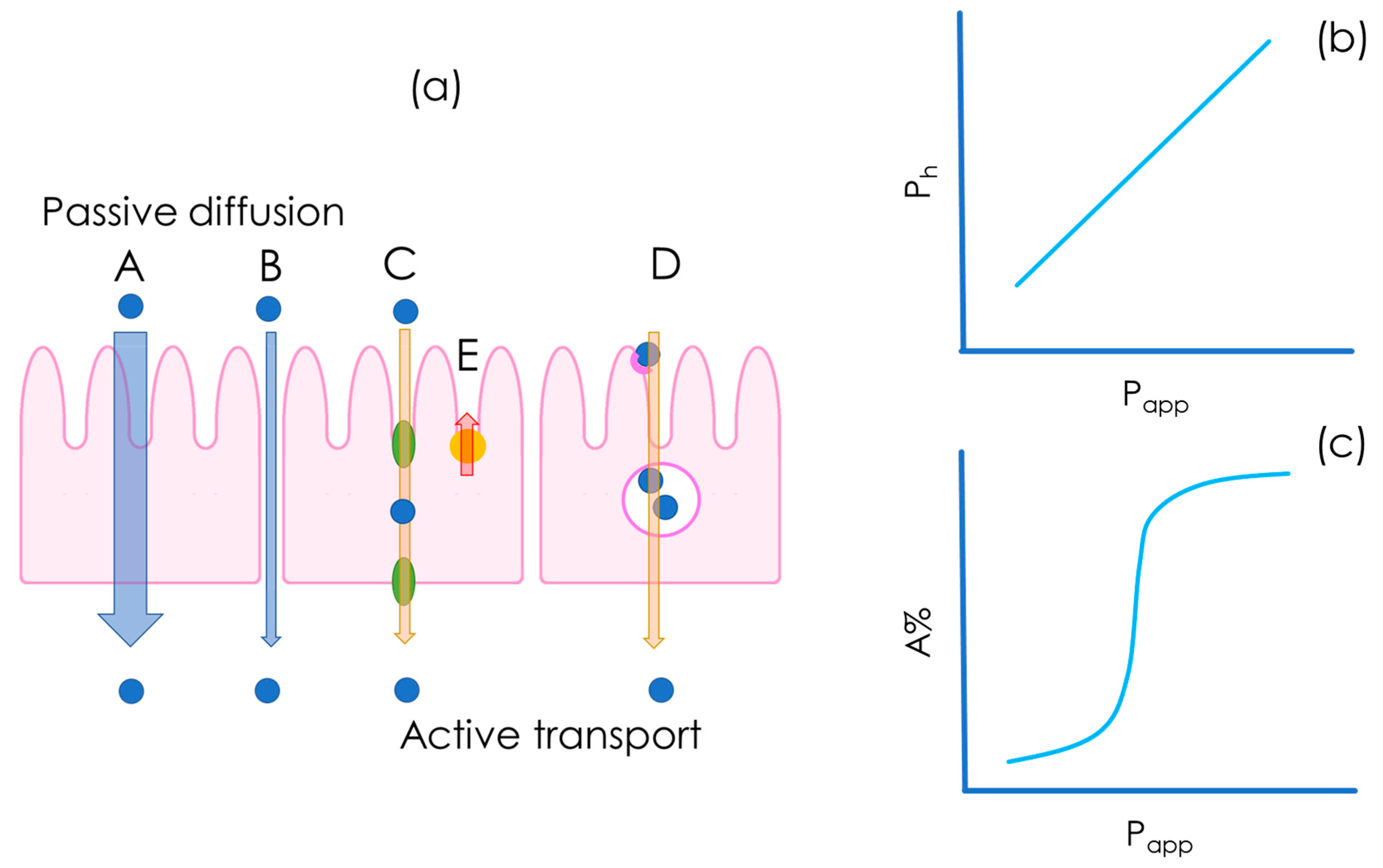
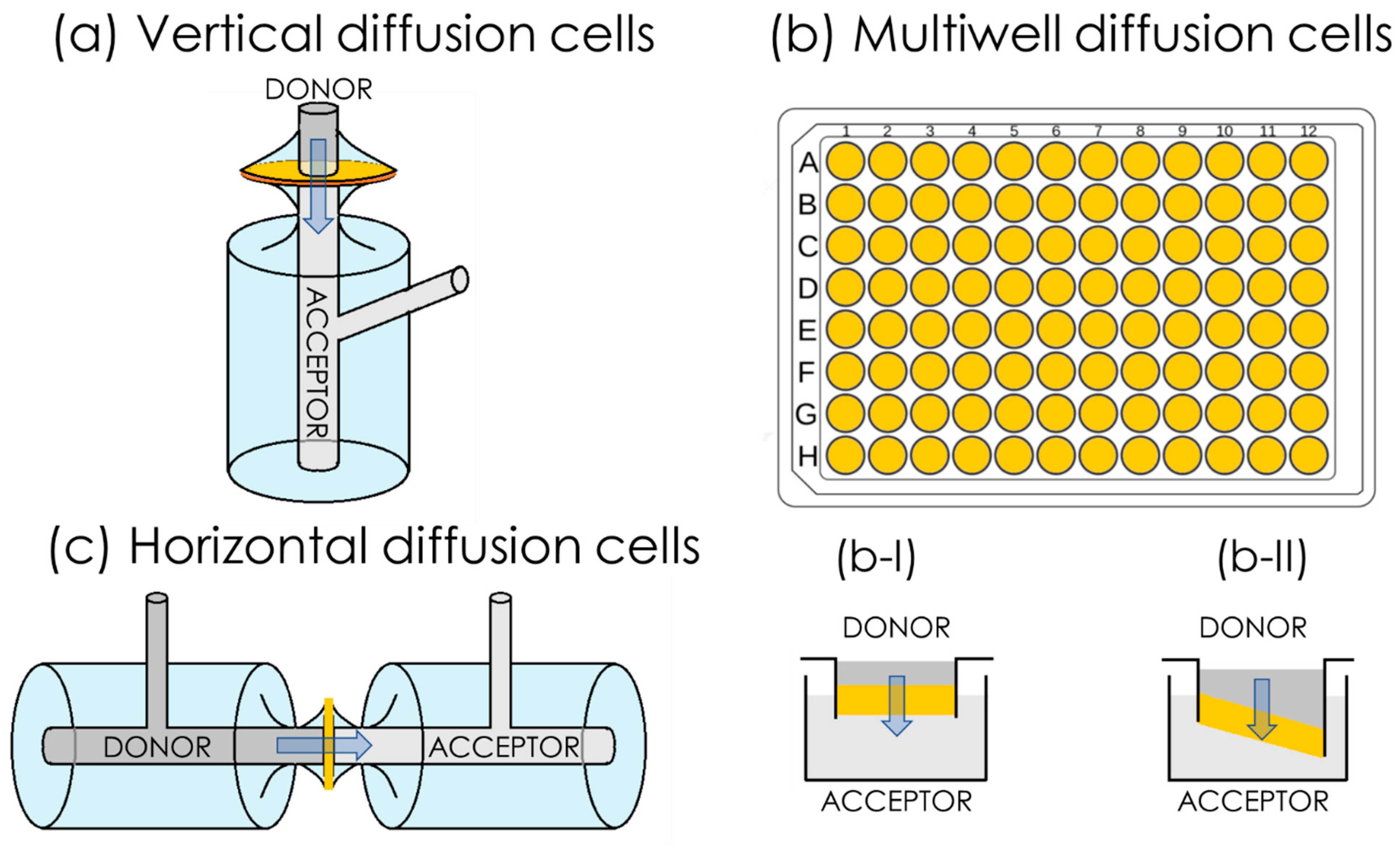
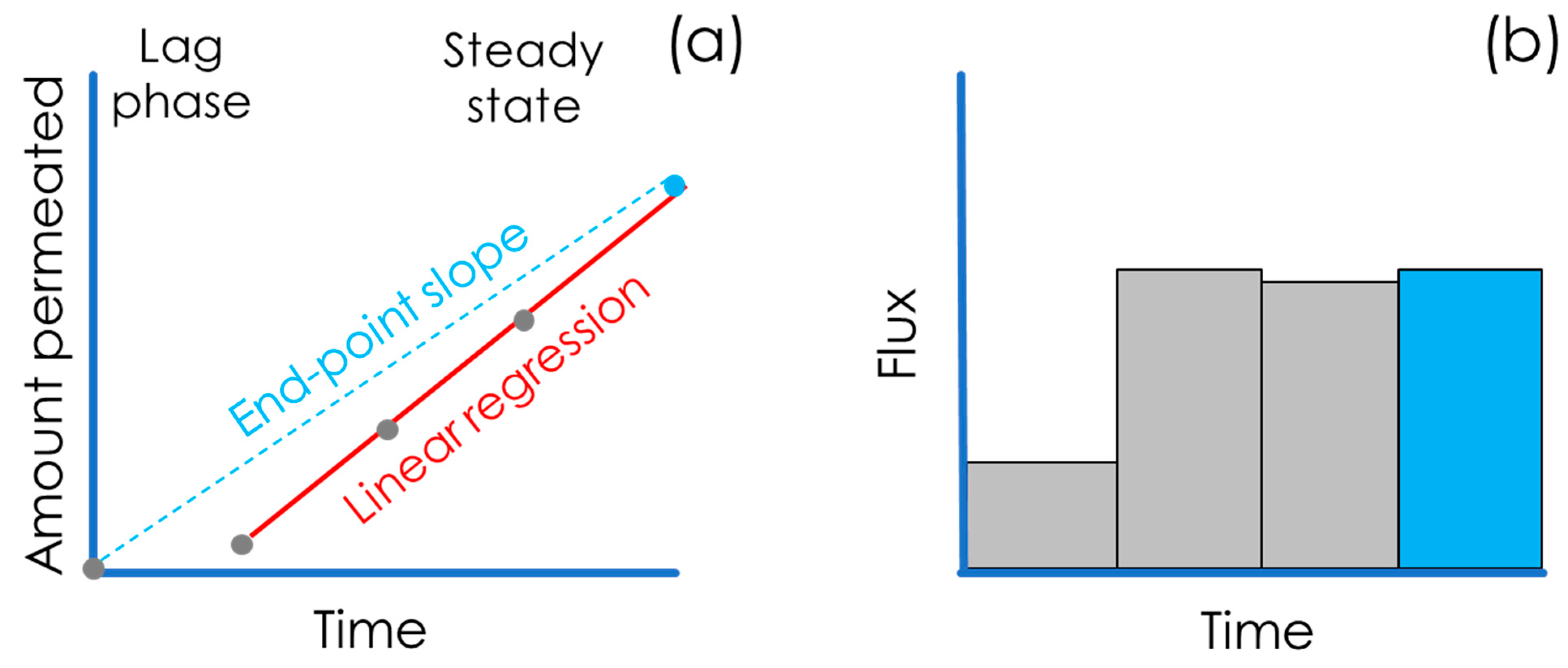
3. In Vitro Flux Studies and Permeability Quantification
3.1. Conventionally Accepted Physical Models of Passive Permeability
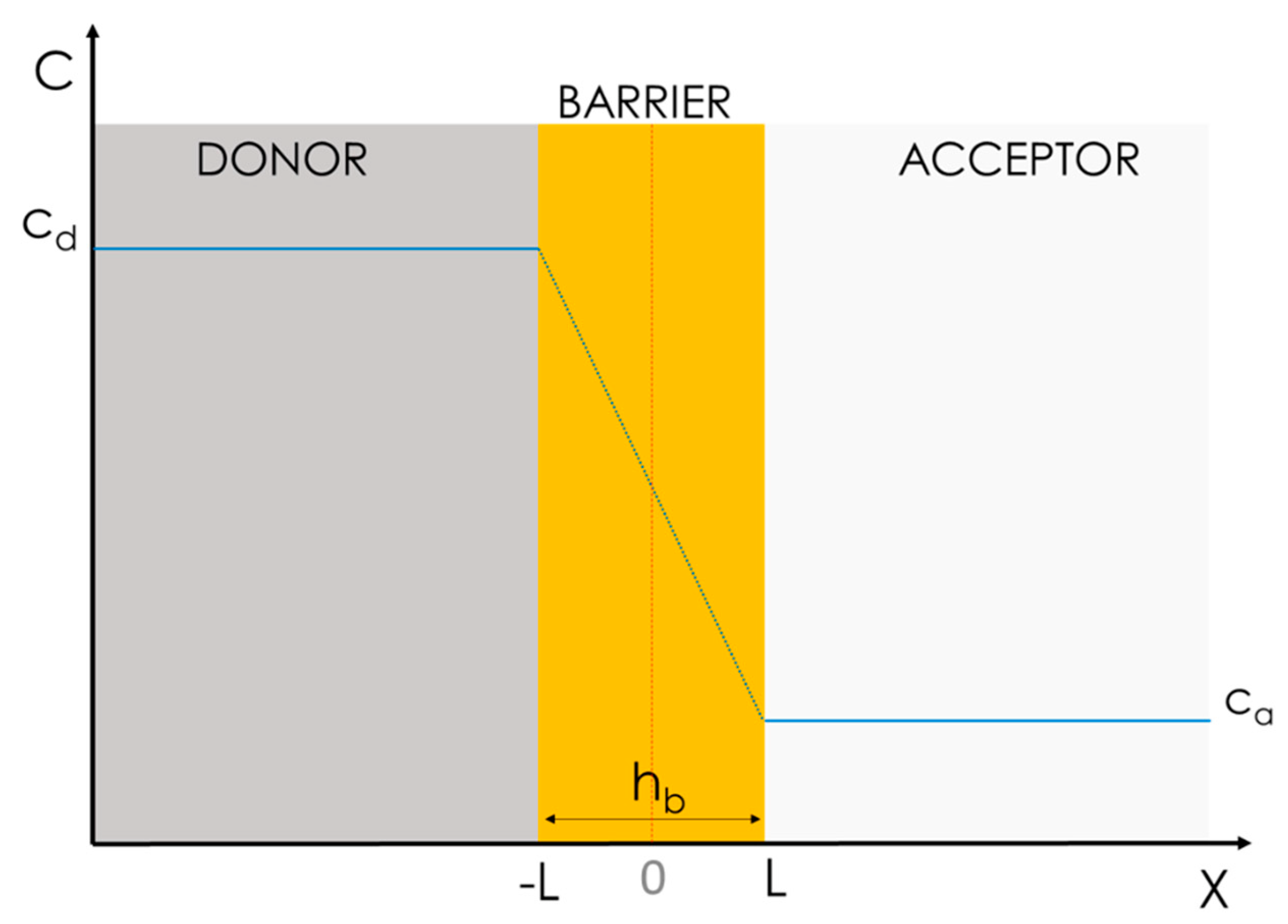
3.1.1. Simplified Homogeneous One-Phase Model
- The drug must have homogeneous and constant cd and ca values, i.e., a constant concentration gradient;
- The interface transition kinetic within the water phase and the barrier should be irrelevant; and
- D must be a constant all over space x.
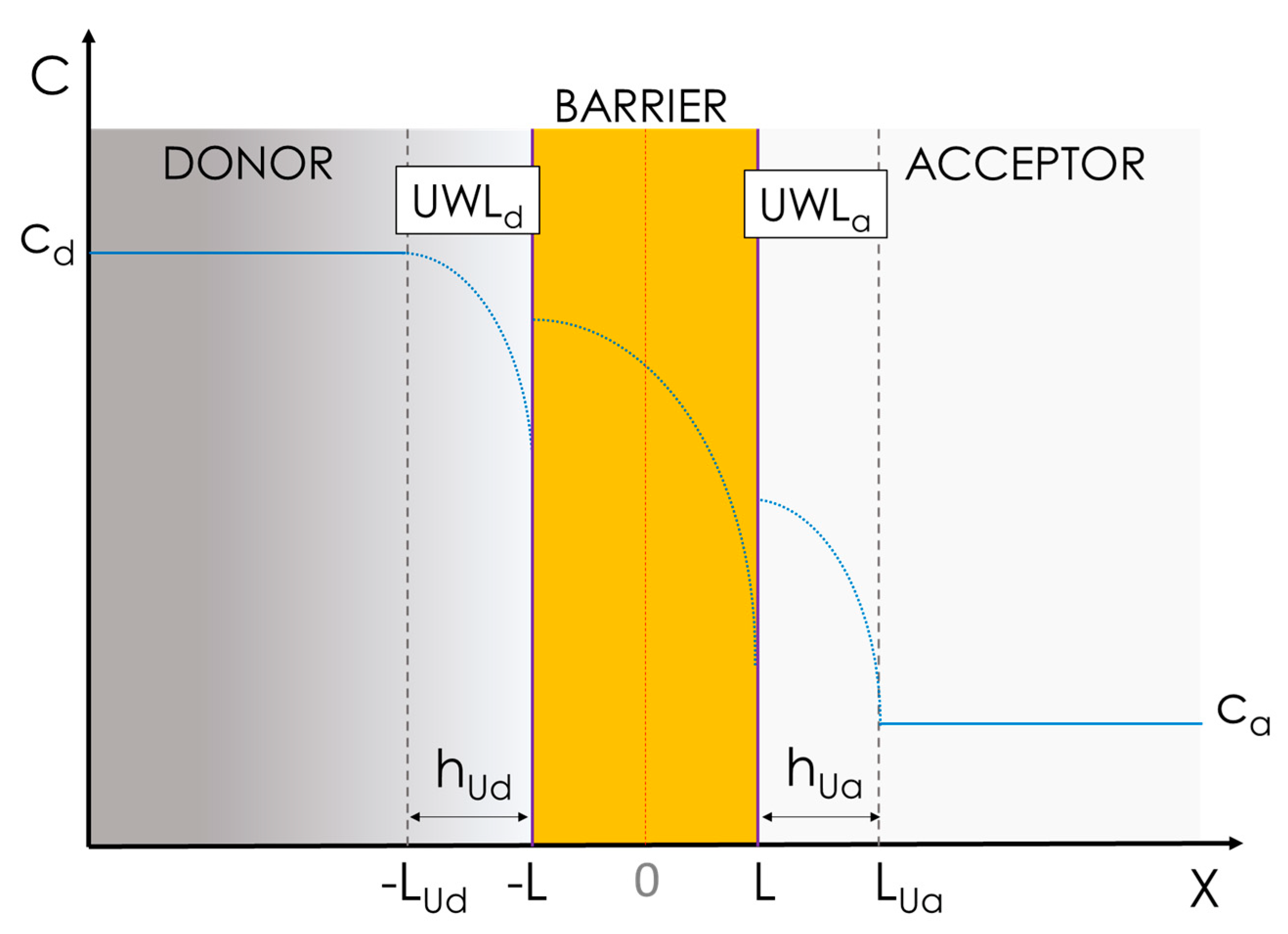
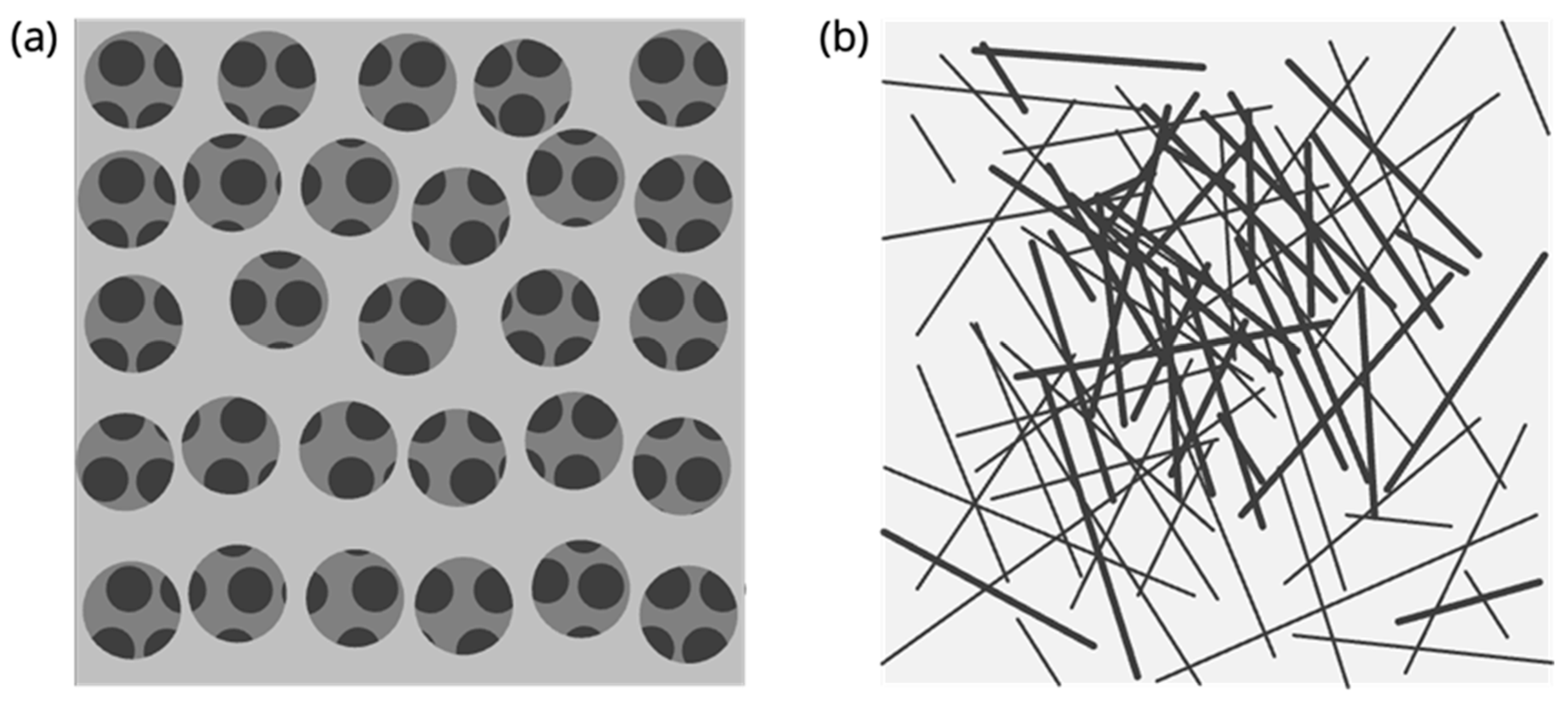
3.1.2. Advanced Multiphasic Model with Interfaces
4. Commercially Available Biomimetic Cell-Free In Vitro Permeability Assays
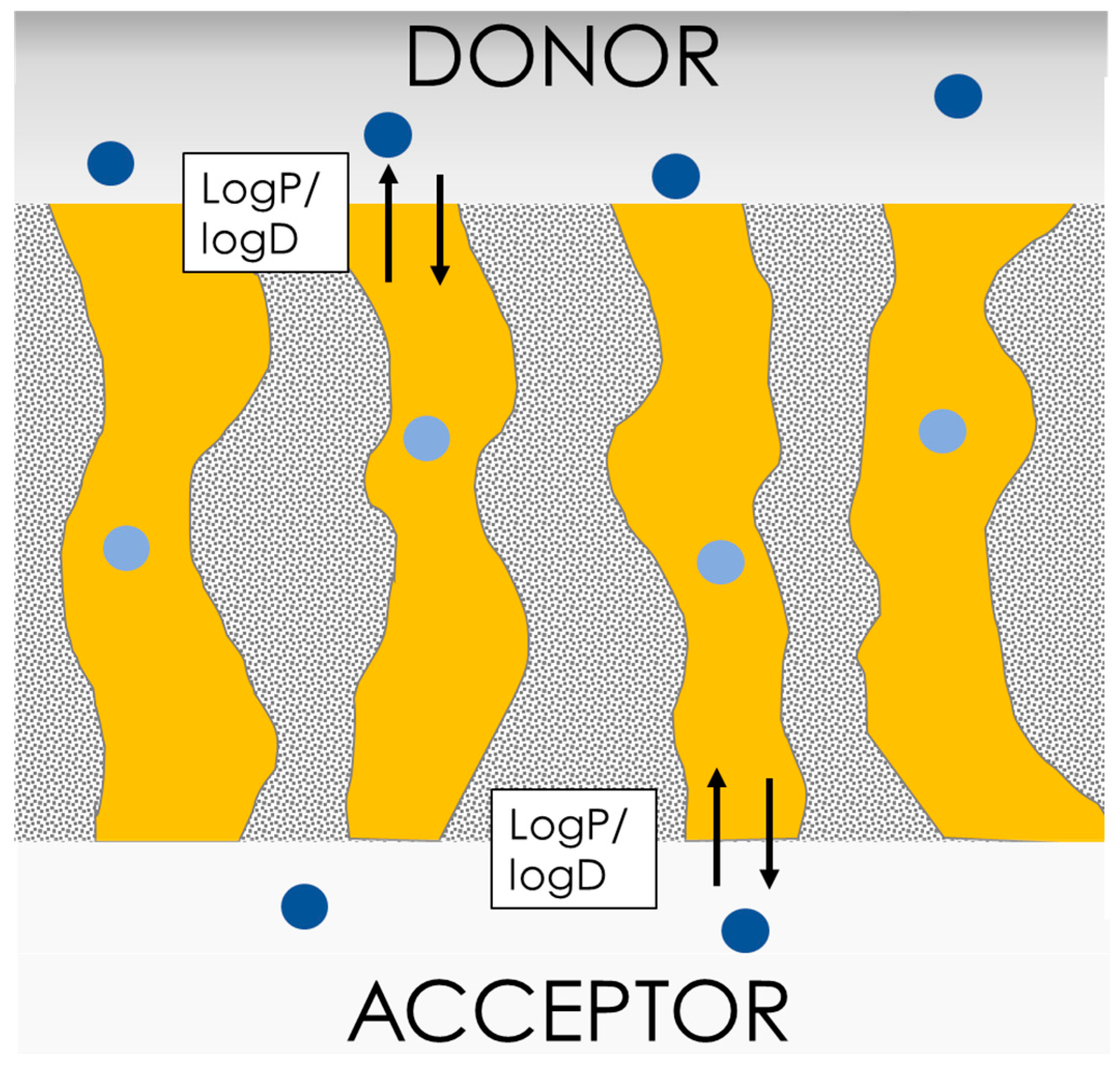
4.1. The Parallel Artificial Membrane Permeation Assay (PAMPA)
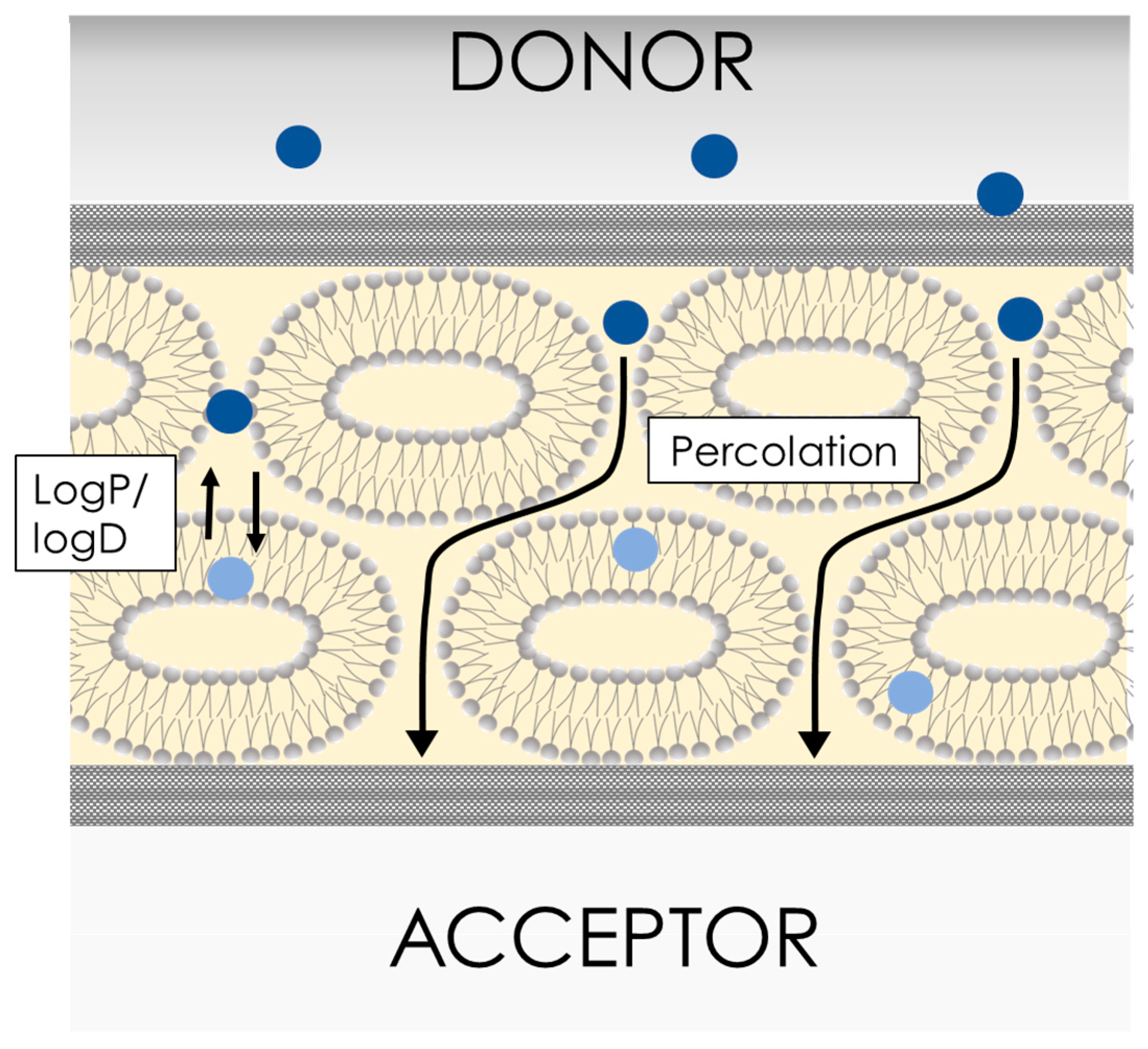
4.2. PermeaPad
5. Lipid-Free Membranes for In Vitro Permeability/Absorption Profiling
5.1. Regenerated Cellulose
5.2. Strat-M®
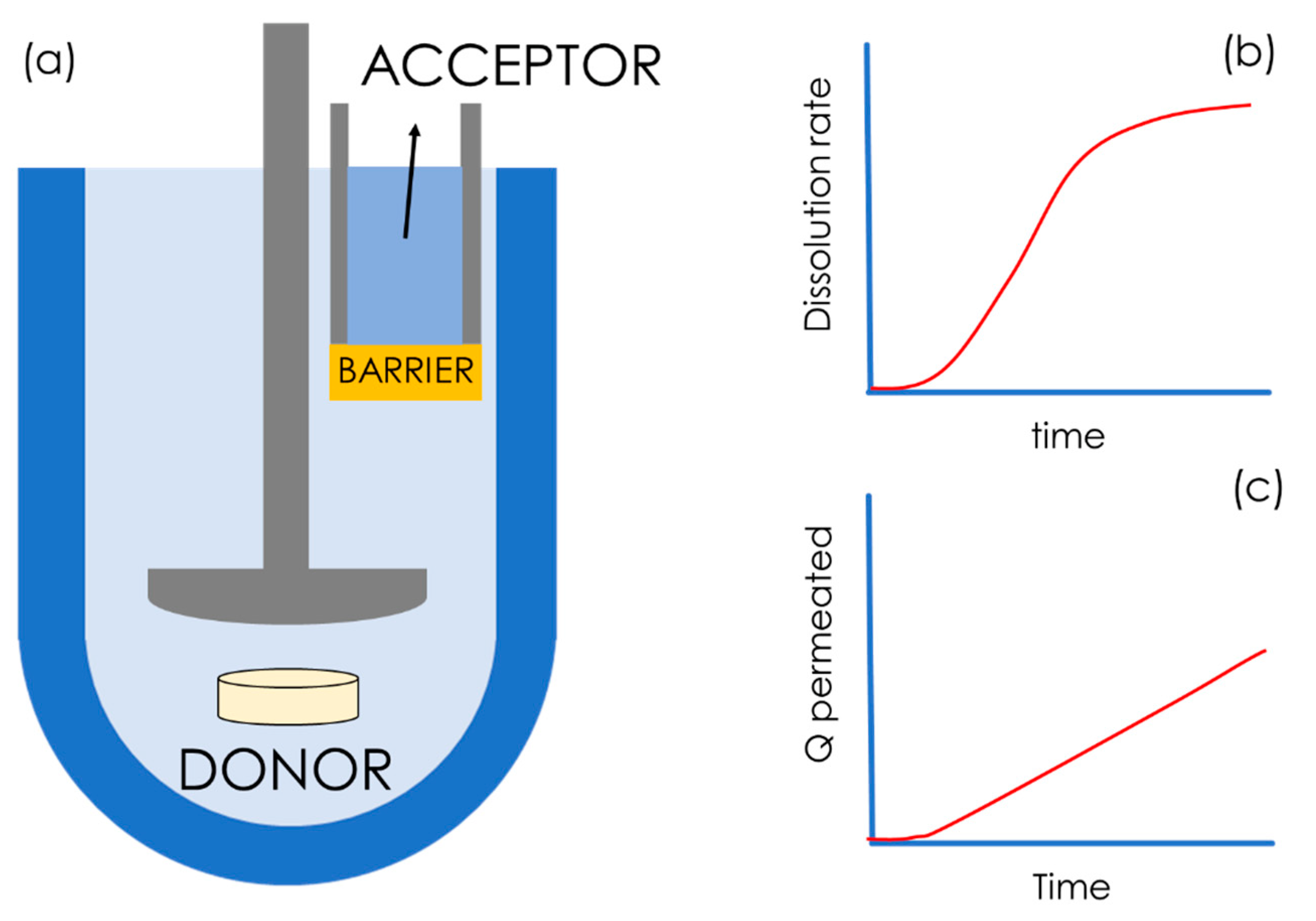
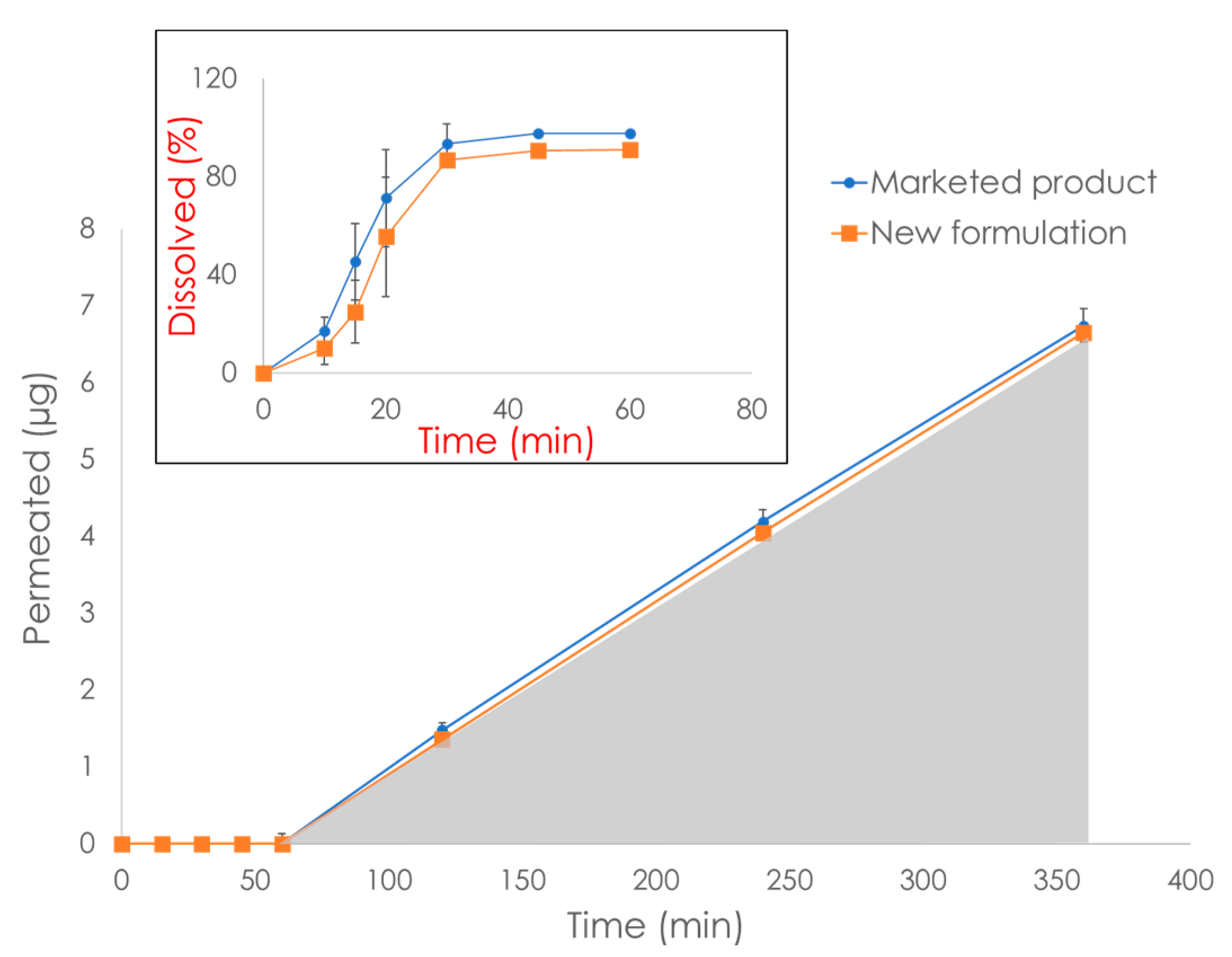
6. Commercially Available In Vitro Dissolution/Permeation Systems
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Belkhir, L.; Elmeligi, A. Carbon footprint of the global pharmaceutical industry and relative impact of its major players. J. Clean. Prod. 2019, 214, 185–194. [Google Scholar] [CrossRef]
- Forin, S.; Scholz, R. Are medicines more greenhouse gas intensive than cars? Comment to Belkhir, L., Elmeligi, A., 2019: Carbon footprint of the global pharmaceutical industry and relative impact of its major players. J. Clean. Prod. 2022, 331, 129963. [Google Scholar] [CrossRef]
- Milanesi, M.; Runfola, A.; Guercini, S. Pharmaceutical industry riding the wave of sustainability: Review and opportunities for future research. J. Clean. Prod. 2020, 261, 121204. [Google Scholar] [CrossRef]
- Adshead, F.; Salman, R.A.-S.; Aumonier, S.; Collins, M.; Hood, K.; McNamara, C.; Moore, K.; Smith, R.; Sydes, M.R.; Williamson, P.R. A strategy to reduce the carbon footprint of clinical trials Comment. Lancet 2021, 398, 281–282. [Google Scholar] [CrossRef]
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A Theoretical Basis for a Biopharmaceutic Drug Classification: The Correlation of in Vitro Drug Product Dissolution and in Vivo Bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef]
- European Medicines Agency. ICH M9 Guideline on Biopharmaceutics Classification System-Based Biowaivers; European Medicines Agency: Brussels, Belgium, 2020. [Google Scholar]
- Hayeshi, R.; Hilgendorf, C.; Artursson, P.; Augustijns, P.; Brodin, B.; Dehertogh, P.; Fisher, K.; Fossati, L.; Hovenkamp, E.; Korjamo, T.; et al. Comparison of drug transporter gene expression and functionality in Caco-2 cells from 10 different laboratories. Eur. J. Pharm. Sci. 2008, 35, 383–396. [Google Scholar] [CrossRef]
- Uchida, M.; Fukazawa, T.; Yamazaki, Y.; Hashimoto, H.; Miyamoto, Y. A modified fast (4 day) 96-well plate Caco-2 permeability assay. J. Pharmacol. Toxicol. Methods 2009, 59, 39–43. [Google Scholar] [CrossRef]
- MacLeod, M.; Leinonen, I.; Wall, E.; Houdijk, J.; Eory, V.; Burns, J.; Ahmadi, B.V.; Gómez-Barbero, M. Impact of Animal Breeding on GHG Emissions and Farm Economics; European Union: Maastricht, The Netherlands, 2019; Available online: https://publications.jrc.ec.europa.eu/ (accessed on 1 October 2022).
- Groff, K.; Bachli, E.; Lansdowne, M.; Capaldo, T. Review of Evidence of Environmental Impacts of Animal Research and Testing. Environments 2014, 1, 14–30. [Google Scholar] [CrossRef]
- Commission, E. 2019 Report on the Statistics on the Use of Animals for Scientific Purposes in the Member States of the European Union in 2015–2017. 2020.
- Hobson, H. EU-Wide Animal Research Statistics, 2018. 2021. Available online: https://www.understandinganimalresearch.org.uk/news/eu-wide-animals-in-research-statistics-for-2018-released#:~:text=EU%2Dwide%20animal%20research%20statistics%2C%202018,-Posted%3A%20by%20Hannah&text=In%202018%2C%2012%2C093%2C096%20animals%20were,breeding%20of%20genetically%20altered%20animals (accessed on 9 September 2022).
- Taylor, K.; Alvarez, L.R. An Estimate of the Number of Animals Used for Scientific Purposes Worldwide in 2015. Altern. Lab. Anim. 2019, 47, 196–213. [Google Scholar] [CrossRef]
- Akhtar, A. The Flaws and Human Harms of Animal Experimentation. Camb. Q. Healthc. Ethics 2015, 24, 407–419. [Google Scholar] [CrossRef] [Green Version]
- Goh, J.-Y.; Weaver, R.J.; Dixon, L.; Platt, N.J.; Roberts, R.A. Development and use of in vitro alternatives to animal testing by the pharmaceutical industry 1980–2013. Toxicol. Res. 2015, 4, 1297–1307. [Google Scholar] [CrossRef]
- Vinarov, Z.; Abrahamsson, B.; Artursson, P.; Batchelor, H.; Berben, P.; Bernkop-Schnürch, A.; Butler, J.; Ceulemans, J.; Davies, N.; Dupont, D.; et al. Current challenges and future perspectives in oral absorption research: An opinion of the UNGAP network. Adv. Drug Deliv. Rev. 2021, 171, 289–331. [Google Scholar] [CrossRef]
- Berben, P.; Bauer-Brandl, A.; Brandl, M.; Faller, B.; Flaten, G.E.; Jacobsen, A.-C.; Brouwers, J.; Augustijns, P. Drug permeability profiling using cell-free permeation tools: Overview and applications. Eur. J. Pharm. Sci. 2018, 119, 219–233. [Google Scholar] [CrossRef]
- Pharma 50: The 50 largest Pharmaceutical Companies in the World. 2022. Available online: https://www.drugdiscoverytrends.com/pharma-50-the-50-largest-pharmaceutical-companies-in-the-world-for-2022/ (accessed on 19 August 2022).
- Papich, M.G.; Martinez, M.N. Applying Biopharmaceutical Classification System (BCS) Criteria to Predict Oral Absorption of Drugs in Dogs: Challenges and Pitfalls. AAPS J. 2015, 17, 948–964. [Google Scholar] [CrossRef]
- Brodin, B. Passive diffusion of drug substances: The concept of flux and permeabilty. In Molecular Biopharmaceutics; Bente Steffansen, B.B., Nielsen, C.U., Eds.; Pharmaceutical Press: London, UK, 2010; pp. 135–152. [Google Scholar]
- Di, L.; Artursson, P.; Avdeef, A.; Benet, L.Z.; Houston, J.B.; Kansy, M.; Kerns, E.H.; Lennernäs, H.; Smith, D.A.; Sugano, K. The Critical Role of Passive Permeability in Designing Successful Drugs. Chemmedchem 2020, 15, 1862–1874. [Google Scholar] [CrossRef]
- Artursson, P.; Karlsson, J. Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem. Biophys. Res. Commun. 1991, 175, 880–885. [Google Scholar] [CrossRef]
- Artursson, P.; Palm, K.; Luthman, K. Caco-2 monolayers in experimental and theoretical predictions of drug transport1PII. Adv. Drug Deliv. Rev. 2001, 46, 27–43. [Google Scholar] [CrossRef]
- Carsten, U.; Nielsen, B.S.; Brodin, B. Carrier-mediated transport kinetics. In Molecular Biopharmaceutics; Bente Steffansen, B.B., Nielsen, C.U., Eds.; Pharmaceutical Press: London, UK, 2010; pp. 175–211. [Google Scholar]
- Di, L.; Artursson, P.; Avdeef, A.; Ecker, G.F.; Faller, B.; Fischer, H.; Houston, J.B.; Kansy, M.; Kerns, E.H.; Krämer, S.D.; et al. Evidence-based approach to assess passive diffusion and carrier-mediated drug transport. Drug Discov. Today 2012, 17, 905–912. [Google Scholar] [CrossRef]
- Sugano, K.; Kansy, M.; Artursson, P.; Avdeef, A.; Bendels, S.; Di, L.; Ecker, G.F.; Faller, B.; Fischer, H.; Gerebtzoff, G.; et al. Coexistence of passive and carrier-mediated processes in drug transport. Nat. Rev. Drug Discov. 2010, 9, 597–614. [Google Scholar] [CrossRef]
- Kell, D.B.; Dobson, P.D.; Oliver, S.G. Pharmaceutical drug transport: The issues and the implications that it is essentially carrier-mediated only. Drug Discov. Today 2011, 16, 704–714. [Google Scholar] [CrossRef]
- Sawada, G.A.; Barsuhn, C.L.; Lutzke, B.S.; Houghton, M.E.; Padbury, G.E.; Ho, N.F.; Raub, T.J. Increased lipophilicity and subsequent cell partitioning decrease passive transcellular diffusion of novel, highly lipophilic antioxidants. Experiment 1999, 288, 1317–1326. [Google Scholar]
- Crank, J. Infinite and semi-infinite media. In The Mathematics of Diffusion; Crank, J., Ed.; Oxford University Press: London, UK, 1975; pp. 28–44. [Google Scholar]
- Diamond, J.M.; Katz, Y. Interpretation of nonelectrolyte partition coefficients between dimyristoyl lecithin and water. J. Membr. Biol. 1974, 17, 121–154. [Google Scholar] [CrossRef] [PubMed]
- Parisio, G.; Stocchero, M.; Ferrarini, A. Passive Membrane Permeability: Beyond the Standard Solubility-Diffusion Model. J. Chem. Theory Comput. 2013, 9, 5236–5246. [Google Scholar] [CrossRef] [PubMed]
- Barry, P.H.; Diamond, J.M. Effects of unstirred layers on membrane phenomena. Physiol. Rev. 1984, 64, 763–872. [Google Scholar] [CrossRef]
- Katneni, K.; Charman, S.A.; Porter, C.J. An evaluation of the relative roles of the unstirred water layer and receptor sink in limiting the in-vitro intestinal permeability of drug compounds of varying lipophilicity. J. Pharm. Pharmacol. 2008, 60, 1311–1319. [Google Scholar] [CrossRef]
- Avdeef, A.; Nielsen, P.E.; Tsinman, O. PAMPA—A drug absorption in vitro model 11. Matching the in vivo unstirred water layer thickness by individual-well stirring in microtitre plates. Eur. J. Pharm. Sci. 2004, 22, 365–374. [Google Scholar]
- Brewster, M.E.; Noppe, M.; Peeters, J.; Loftsson, T. Effect of the unstirred water layer on permeability enhancement by hydrophilic cyclodextrins. Int. J. Pharm. 2007, 342, 250–253. [Google Scholar] [CrossRef]
- Korjamo, T.; Heikkinen, A.; Mönkkönen, J. Analysis of Unstirred Water Layer in In Vitro Permeability Experiments. J. Pharm. Sci. 2009, 98, 4469–4479. [Google Scholar] [CrossRef]
- Eriksen, J.B.; Jacobsen, A.-C.; Christensen, K.T.; Bauer-Brandl, A.; Brandl, M. ‘Stirred not Shaken!’ Comparing Agitation Methods for Permeability Studies Using a Novel Type of 96-Well Sandwich-Plates. J. Pharm. Sci. 2022, 111, 32–40. [Google Scholar] [CrossRef]
- Tzanova, M.M.; Randelov, E.; Stein, P.C.; Hiorth, M.; di Cagno, M.P. Towards a better mechanistic comprehension of drug permeation and absorption: Introducing the diffusion-partitioning interplay. Int. J. Pharm. 2021, 608, 121116. [Google Scholar] [CrossRef]
- Kansy, M.; Senner, F.; Gubernator, K. Physicochemical High Throughput Screening: Parallel Artificial Membrane Permeation Assay in the Description of Passive Absorption Processes. J. Med. Chem. 1998, 41, 1007–1010. [Google Scholar] [CrossRef]
- Bermejo, M.; Avdeef, A.; Ruiz, A.; Nalda, R.; Ruell, J.; Tsinman, O.; González, I.; Fernández, C.; Sánchez, G.; Garrigues, T.; et al. PAMPA—A drug absorption in vitro model 7. Comparing rat in situ, Caco-2, and PAMPA permeability of fluoroquinolones. Eur. J. Pharm. Sci. 2004, 21, 429–441. [Google Scholar] [CrossRef]
- Sugano, K.; Hamada, H.; Machida, M.; Ushio, H.; Saitoh, K.; Terada, K. Optimized conditions of bio-mimetic artificial membrane permeation assay. Int. J. Pharm. 2001, 228, 181–188. [Google Scholar] [CrossRef]
- Avdeef, A.; Strafford, M.; Block, E.; Balogh, M.P.; Chambliss, W.; Khan, I. Drug absorption in vitro model: Filter-immobilized artificial membranes: 2. Studies of the permeability properties of lactones in Piper methysticum Forst. Eur. J. Pharm. Sci. 2001, 14, 271–280. [Google Scholar] [CrossRef]
- Seo, P.R.; Teksin, Z.S.; Kao, J.P.; Polli, J.E. Lipid composition effect on permeability across PAMPA. Eur. J. Pharm. Sci. 2006, 29, 259–268. [Google Scholar] [CrossRef]
- Ottaviani, G.; Martel, S.; Carrupt, P.-A. Parallel Artificial Membrane Permeability Assay: A New Membrane for the Fast Prediction of Passive Human Skin Permeability. J. Med. Chem. 2006, 49, 3948–3954. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H.; Fan, K.; McConnell, O.J.; Carter, G.T. High throughput artificial membrane permeability assay for blood–brain barrier. Eur. J. Med. Chem. 2003, 38, 223–232. [Google Scholar] [CrossRef]
- Dahan, A.; Miller, J.M.; Hoffman, A.; Amidon, G.E.; Amidon, G.L. The Solubility–Permeability Interplay in Using Cyclodextrins as Pharmaceutical Solubilizers: Mechanistic Modeling and Application to Progesterone. J. Pharm. Sci. 2010, 99, 2739–2749. [Google Scholar] [CrossRef]
- Beig, A.; Fine-Shamir, N.; Porat, D.; Lindley, D.; Miller, J.M.; Dahan, A. Concomitant solubility-permeability increase: Vitamin E TPGS vs. amorphous solid dispersion as oral delivery systems for etoposide. Eur. J. Pharm. Biopharm. 2017, 121, 97–103. [Google Scholar] [CrossRef]
- Chen, X.; Murawski, A.; Patel, K.; Crespi, C.L.; Balimane, P.V. A Novel Design of Artificial Membrane for Improving the PAMPA Model. Pharm. Res. 2008, 25, 1511–1520. [Google Scholar] [CrossRef]
- Kuhl, N.; Lang, J.; Leuthold, M.; Klein, C.D. Discovery of potent benzoxaborole inhibitors against SARS-CoV-2 main and dengue virus proteases. Eur. J. Med. Chem. 2022, 240, 114585. [Google Scholar] [CrossRef] [PubMed]
- Abram, M.; Jakubiec, M.; Reeb, K.; Cheng, M.H.; Gedschold, R.; Rapacz, A.; Mogilski, S.; Socała, K.; Nieoczym, D.; Nieoczym, D.; et al. Discovery of (R)-N-Benzyl-2-(2,5-dioxopyrrolidin-1-yl)propanamide [(R)-AS-1], a Novel Orally Bioavailable EAAT2 Modulator with Drug- like Properties and Potent Antiseizure Activity In Vivo. J. Med. Chem. 2022, 6, 11703–11725. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.; Soto-Acosta, R.; Xie, J.; Wilson, D.J.; Dreis, C.D.; Majima, R.; Edwards, T.C.; Geraghty, R.J.; Chen, L. Bisubstrate Inhibitors of Severe Acute Respiratory Syndrome Coronavirus-2 Nsp14 Methyltransferase. ACS Med. Chem. Lett. 2022, 13, 1477–1484. [Google Scholar] [CrossRef] [PubMed]
- Zareba, P.; Śliwa, P.; Satała, G.; Zajdel, P.; Latacz, G.; Jaśkowska, J. New N-aryl-N’-aryl-/(thio)ureido-/sulfamoylamino-derivatives of alkyl/alkylcarbamoyl piperazines: Effect of structural modifications on selectivity over 5-HT1A receptor. Eur. J. Med. Chem. 2022, 235, 114319. [Google Scholar] [CrossRef]
- Wang, C.; Northfield, S.; Swedberg, J.; Colless, B.; Chaousis, S.; Price, D.A.; Liras, S.; Craik, D.J. Exploring experimental and computational markers of cyclic peptides: Charting islands of permeability. Eur. J. Med. Chem. 2015, 97, 202–213. [Google Scholar] [CrossRef]
- Bennion, B.J.; Be, N.A.; McNerney, M.W.; Lao, V.; Carlson, E.M.; Valdez, C.A.; Malfatti, M.A.; Enright, H.A.; Nguyen, T.H.; Lightstone, F.C.; et al. Predicting a Drug’s Membrane Permeability: A Computational Model Validated With in Vitro Permeability Assay Data. J. Phys. Chem. B 2017, 121, 5228–5237. [Google Scholar] [CrossRef]
- Butnarasu, C.; Caron, G.; Pacheco, D.P.; Petrini, P.; Visentin, S. Cystic Fibrosis Mucus Model to Design More Efficient Drug Therapies. Mol. Pharm. 2021, 19, 520–531. [Google Scholar] [CrossRef]
- Yamauchi, S.; Inoue, D.; Sugano, K. Permeation characteristics of tetracyclines in parallel artificial membrane permeation assay II: Effect of divalent metal ions and mucin. ADMET DMPK 2020, 8, 129–138. [Google Scholar] [CrossRef]
- Yamauchi, S.; Sugano, K. Permeation characteristics of tetracyclines in parallel artificial membrane permeation assay. ADMET DMPK 2019, 7, 151–160. [Google Scholar] [CrossRef]
- Wexler, D.S.; Gao, L.; Anderson, F.; Ow, A.; Nadasdi, L.; McAlorum, A.; Urfer, R.; Huang, S.-G. Linking Solubility and Permeability Assays for Maximum Throughput and Reproducibility. SLAS Discov. Adv. Sci. Drug Discov. 2005, 10, 383–390. [Google Scholar] [CrossRef]
- Blanchard, J.; Sawers, S.J.A. The absolute bioavailability of caffeine in man. Eur. J. Clin. Pharmacol. 1983, 24, 93–98. [Google Scholar] [CrossRef]
- Teixeira, L.D.S.; Chagas, T.V.; Alonso, A.; Gonzalez-Alvarez, I.; Bermejo, M.; Polli, J.; Rezende, K.R. Biomimetic Artificial Membrane Permeability Assay over Franz Cell Apparatus Using BCS Model Drugs. Pharmaceutics 2020, 12, 988. [Google Scholar] [CrossRef]
- Soriano-Meseguer, S.; Fuguet, E.; Port, A.; Rosés, M. Optimization of experimental conditions for skin-PAMPA measurements. ADMET DMPK 2020, 8, 16–28. [Google Scholar] [CrossRef]
- Azman, M.; Sabri, A.H.; Anjani, Q.K.; Mustaffa, M.F.; Hamid, K.A. Intestinal Absorption Study: Challenges and Absorption Enhancement Strategies in Improving Oral Drug Delivery. Pharmaceuticals 2022, 15, 975. [Google Scholar] [CrossRef]
- Sun, H.; Nguyen, K.; Kerns, E.; Yan, Z.; Yu, K.R.; Shah, P.; Jadhav, A.; Xu, X. Highly predictive and interpretable models for PAMPA permeability. Bioorg. Med. Chem. 2017, 25, 1266–1276. [Google Scholar] [CrossRef]
- Leung, S.S.F.; Mijalkovic, J.; Borrelli, K.; Jacobson, M.P. Testing Physical Models of Passive Membrane Permeation. J. Chem. Inf. Model. 2012, 52, 1621–1636. [Google Scholar] [CrossRef]
- Di Cagno, A.B.-B. Assembly for Assessing Drug Permeability with Adjustable Biomimetic Properties; Southern Denmak University: Odense, Denmark, 2015; p. 27. [Google Scholar]
- Di Cagno, M.; Bibi, H.A.; Bauer-Brandl, A. New biomimetic barrier Permeapad (TM) for efficient investigation of passive permeability of drugs. Eur. J. Pharm. Sci. 2015, 73, 29–34. [Google Scholar] [CrossRef]
- Jacobsen, A.-C.; Elvang, P.A.; Bauer-Brandl, A.; Brandl, M. A dynamic in vitro permeation study on solid mono- and diacyl-phospholipid dispersions of celecoxib. Eur. J. Pharm. Sci. 2019, 127, 199–207. [Google Scholar] [CrossRef]
- Flaten, G.E.; Luthman, K.; Vasskog, T.; Brandl, M. Drug permeability across a phospholipid vesicle-based barrier: 4. The effect of tensides, co-solvents and pH changes on barrier integrity and on drug permeability. Eur. J. Pharm. Sci. 2008, 34, 173–180. [Google Scholar] [CrossRef]
- Flaten, G.E.; Bunjes, H.; Luthman, K.; Brandl, M. Drug permeability across a phospholipid vesicle-based barrier 2. Characterization of barrier structure, storage stability and stability towards pH changes. Eur. J. Pharm. Sci. 2006, 28, 336–343. [Google Scholar] [CrossRef]
- Flaten, G.E.; Dhanikula, A.B.; Luthman, K.; Brandl, M. Drug permeability across a phospholipid vesicle based barrier: A novel approach for studying passive diffusion. Eur. J. Pharm. Sci. 2006, 27, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Flaten, G.E.; Skar, M.; Luthman, K.; Brandl, M. Drug permeability across a phospholipid vesicle based barrier: 3. Characterization of drug-membrane interactions and the effect of agitation on the barrier integrity and on the permeability. Eur. J. Pharm. Sci. 2007, 30, 324–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falavigna, M.; Brurok, S.; Klitgaard, M.; Flaten, G.E. Simultaneous assessment of in vitro lipolysis and permeation in the mucus-PVPA model to predict oral absorption of a poorly water soluble drug in SNEDDSs. Int. J. Pharm. 2021, 596, 120258. [Google Scholar] [CrossRef] [PubMed]
- Falavigna, M.; Klitgaard, M.; Berthelsen, R.; Müllertz, A.; Flaten, G.E. Predicting Oral Absorption of fenofibrate in Lipid-Based Drug Delivery Systems by Combining In Vitro Lipolysis with the Mucus-PVPA Permeability Model. J. Pharm. Sci. 2021, 110, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Falavigna, M.; Klitgaard, M.; Brase, C.; Ternullo, S.; Škalko-Basnet, N.; Flaten, G.E. Mucus-PVPA (mucus Phospholipid Vesicle-based Permeation Assay): An artificial permeability tool for drug screening and formulation development. Int. J. Pharm. 2018, 537, 213–222. [Google Scholar] [CrossRef]
- Falavigna, M.; Klitgaard, M.; Steene, E.; Flaten, G.E. Mimicking regional and fasted/fed state conditions in the intestine with the mucus-PVPA in vitro model: The impact of pH and simulated intestinal fluids on drug permeability. Eur. J. Pharm. Sci. 2019, 132, 44–54. [Google Scholar] [CrossRef]
- Brandl, M.; Tardi, C.; Drechsler, M.; Bachmann, D.; Reszka, R.; Bauer, K.; Schubert, R. Three-dimensional liposome networks: Freeze fracture electron microscopical evaluation of their structure and in vitro analysis of release of hydrophilic markers. Adv. Drug Deliv. Rev. 1997, 24, 161–164. [Google Scholar] [CrossRef]
- Jacobsen, A.C.; Nielsen, S.; Brandl, M.; Bauer-Brandl, A. Drug Permeability Profiling Using the Novel Permeapad (R) 96-Well Plate. Pharm. Res. 2020, 37, 93. [Google Scholar] [CrossRef]
- Eriksen, J.B.; Barakat, H.; Luppi, B.; Brandl, M.; Bauer-Brandl, A. Modulation of Paracellular-like Drug Transport across an Artificial Biomimetic Barrier by Osmotic Stress-Induced Liposome Shrinking. Pharmaceutics 2022, 14, 721. [Google Scholar] [CrossRef]
- Maher, S.; Mrsny, R.J.; Brayden, D.J. Intestinal permeation enhancers for oral peptide delivery. Adv. Drug Deliv. Rev. 2016, 106, 277–319. [Google Scholar] [CrossRef]
- Bibi, H.A.; di Cagno, M.; Holm, R.; Bauer-Brandl, A. Permeapad (TM) for investigation of passive drug permeability: The effect of surfactants, co-solvents and simulated intestinal fluids (FaSSIF and FeSSIF). Int. J. Pharm. 2015, 493, 192–197. [Google Scholar] [CrossRef]
- Magnano, G.C.; Sut, S.; Dall’Acqua, S.; Di Cagno, M.P.; Lee, L.; Lee, M.; Filon, F.L.; Perissutti, B.; Hasa, D.; Voinovich, D. Validation and testing of a new artificial biomimetic barrier for estimation of transdermal drug absorption. Int. J. Pharm. 2022, 628, 122266. [Google Scholar] [CrossRef]
- Sripetch, S.; Prajapati, M.; Loftsson, T. Cyclodextrins and Drug Membrane Permeation: Thermodynamic Considerations. J. Pharm. Sci. 2022, 111, 2571–2580. [Google Scholar] [CrossRef]
- Eriksen, J.B.; Christensen, S.B.; Bauer-Brandl, A.; Brandl, M. Dissolution/Permeation of Albendazole in the Presence of Cyclodextrin and Bile Salts: A Mechanistic In Vitro Study into Factors Governing Oral Bioavailability. J. Pharm. Sci. 2022, 111, 1667–1673. [Google Scholar] [CrossRef]
- Volkova, T.V.; Simonova, O.R.; Perlovich, G.L. Permeability of diverse drugs through a lipid barrier: Impact of pH and cyclodextrin. J. Mol. Liq. 2022, 357, 119135. [Google Scholar] [CrossRef]
- Volkova, T.V.; Simonova, O.R.; Perlovich, G.L. Thiazolidine-2,4-dione derivative in 2-hydroxypropyl-beta-cyclodextrin solutions: Complexation/solubilization, distribution and permeability. J. Mol. Liq. 2021, 333, 115931. [Google Scholar] [CrossRef]
- Agafonov, M.; Volkova, T.; Kumeev, R.; Chibunova, E.; Terekhova, I. Impact of pluronic F127 on aqueous solubility and membrane permeability of antirheumatic compounds of different structure and polarity. J. Mol. Liq. 2019, 274, 770–777. [Google Scholar] [CrossRef]
- Agafonov, M.; Volkova, T.; Kumeev, R.; Delyagina, E.; Terekhova, I. Experimental study on interactions occurring between Pluronics and leflunomide in solution. J. Mol. Liq. 2020, 302, 112289. [Google Scholar] [CrossRef]
- Klitgaard, M.; Müllertz, A.; Berthelsen, R. Estimating the Oral Absorption from Self-Nanoemulsifying Drug Delivery Systems Using an In Vitro Lipolysis-Permeation Method. Pharmaceutics 2021, 13, 489. [Google Scholar] [CrossRef]
- Jacobsen, A.-C.; Ejskjær, L.; Brandl, M.; Holm, R.; Bauer-Brandl, A. Do Phospholipids Boost or Attenuate Drug Absorption? In Vitro and In Vivo Evaluation of Mono- and Diacyl Phospholipid-Based Solid Dispersions of Celecoxib. J. Pharm. Sci. 2021, 110, 198–207. [Google Scholar] [CrossRef]
- Bibi, H.A.; Holm, R.; Bauer-Brandl, A. Simultaneous lipolysis/permeation in vitro model, for the estimation of bioavailability of lipid based drug delivery systems. Eur. J. Pharm. Biopharm. 2017, 117, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.M.; Beig, A.; Krieg, B.J.; Carr, R.A.; Borchardt, T.B.; Amidon, G.E.; Amidon, G.L.; Dahan, A. The Solubility–Permeability Interplay: Mechanistic Modeling and Predictive Application of the Impact of Micellar Solubilization on Intestinal Permeation. Mol. Pharm. 2011, 8, 1848–1856. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.M.; Beig, A.; Carr, R.A.; Webster, G.K.; Dahan, A. The Solubility–Permeability Interplay When Using Cosolvents for Solubilization: Revising the Way We Use Solubility-Enabling Formulations. Mol. Pharm. 2012, 9, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Wu, I.Y.; Bala, S.; Škalko-Basnet, N.; di Cagno, M.P. Interpreting non-linear drug diffusion data: Utilizing Korsmeyer-Peppas model to study drug release from liposomes. Eur. J. Pharm. Sci. 2019, 138, 105026. [Google Scholar] [CrossRef]
- Tzanova, M.M.; Moretti, F.; Grassi, G.; Stein, P.C.; Hiorth, M.; Abrami, M.; Grassi, M.; di Cagno, M.P. Modelling drug diffusion through unstirred water layers allows real-time quantification of free/loaded drug fractions and release kinetics from colloidal-based formulations. Eur. J. Pharm. Biopharm. 2022, 178, 168–178. [Google Scholar] [CrossRef]
- Holzem, F.L.; Weck, A.; Schaffland, J.P.; Stillhart, C.; Klein, S.; Bauer-Brandl, A.; Brandl, M. Biopredictive capability assessment of two dissolution/permeation assays, µFLUX™ and PermeaLoop™, using supersaturating formulations of Posaconazole. Eur. J. Pharm. Sci. 2022, 176, 106260. [Google Scholar] [CrossRef]
- Eriksen, J.B.; Messerschmid, R.; Andersen, M.L.; Wada, K.; Bauer-Brandl, A.; Brandl, M. Dissolution/permeation with PermeaLoop (TM): Experience and IVIVC exemplified by dipyridamole enabling formulations. Eur. J. Pharm. Sci. 2020, 154, 105532. [Google Scholar] [CrossRef]
- Ilie, A.R.; Griffin, B.; Brandl, M.; Bauer-Brandl, A.; Jacobsen, A.-C.; Vertzoni, M.; Kuentz, M.; Kolakovic, R.; Holm, R. Exploring impact of supersaturated lipid-based drug delivery systems of celecoxib on in vitro permeation across Permeapad (R) membrane and in vivo absorption. Eur. J. Pharm. Sci. 2020, 152, 105452. [Google Scholar] [CrossRef]
- Palmelund, H.; Eriksen, J.B.; Bauer-Brandl, A.; Rantanen, J.; Löbmann, K. Enabling formulations of aprepitant: In vitro and in vivo comparison of nanocrystalline, amorphous and deep eutectic solvent based formulations. Int. J. Pharm. X 2021, 3, 100083. [Google Scholar] [CrossRef]
- Bibi, H.A.; Holm, R.; Bauer-Brandl, A. Use of Permeapad (R) for prediction of buccal absorption: A comparison to in vitro, ex vivo and in vivo method. Eur. J. Pharm. Sci. 2016, 93, 399–404. [Google Scholar] [CrossRef]
- Corazza, E.; Abruzzo, A.; Giordani, B.; Cerchiara, T.; Bigucci, F.; Vitali, B.; di Cagno, M.P.; Luppi, B. Human Lactobacillus Biosurfactants as Natural Excipients for Nasal Drug Delivery of Hydrocortisone. Pharmaceutics 2022, 14, 524. [Google Scholar] [CrossRef]
- Corazza, E.; di Cagno, M.P.; Bauer-Brandl, A.; Abruzzo, A.; Cerchiara, T.; Bigucci, F.; Luppi, B. Drug delivery to the brain: In situ gelling formulation enhances carbamazepine diffusion through nasal mucosa models with mucin. Eur. J. Pharm. Sci. 2022, 179, 106294. [Google Scholar] [CrossRef]
- Sinko, P.D.; Gidley, D.; Vallery, R.; Lamoureux, A.; Amidon, G.L.; Amidon, G.E. In Vitro Characterization of the Biomimetic Properties of Poly(dimethylsiloxane) To Simulate Oral Drug Absorption. Mol. Pharm. 2017, 14, 4661–4674. [Google Scholar] [CrossRef]
- Garrett, E.; Chemburkar, P.B. Evaluation, Control, and Prediction of Drug Diffusion Through Polymeric Membranes III: Diffusion of Barbiturates, Phenylalkylamines, Dextromethorphan, Progesterone, and Other Drugs. J. Pharm. Sci. 1968, 57, 1401–1409. [Google Scholar] [CrossRef]
- Wasdo, S.C.; Juntunen, J.; Devarajan, H.; Sloan, K. A correlation of flux through a silicone membrane with flux through hairless mouse skin and human skin in vitro. Int. J. Pharm. 2009, 373, 62–67. [Google Scholar] [CrossRef]
- Geinoz, S.; Rey, S.; Boss, G.; Bunge, A.L.; Guy, R.H.; Carrupt, P.-A.; Reist, M.; Testa, B. Quantitative structure-permeation relationships for solute transport across silicone membranes. Pharm. Res. 2002, 19, 1622–1629. [Google Scholar] [CrossRef]
- Wei, D.Q.S.; Hossain, M.; Saleh, Z.S. Separation of Polyphenolics and Sugar by Ultrafiltration: Effects of Operating Conditions on Fouling and Diafiltration. Proceedings of World Academy of Science. Eng. Technol. 2007, 23, 349. [Google Scholar]
- Sironi, D.; Rosenberg, J.; Bauer-Brandl, A.; Brandl, M. PermeaLoop™, a novel in vitro tool for small-scale drug-dissolution/permeation studies. J. Pharm. Biomed. Anal. 2018, 156, 247–251. [Google Scholar] [CrossRef]
- Wilson, V.; Lou, X.; Osterling, D.J.; Stolarik, D.F.; Jenkins, G.; Gao, W.; Zhang, G.G.; Taylor, L.S. Relationship between amorphous solid dispersion in vivo absorption and in vitro dissolution: Phase behavior during dissolution, speciation, and membrane mass transport. J. Control. Release 2018, 292, 172–182. [Google Scholar] [CrossRef]
- Jacobsen, A.-C.; Krupa, A.; Brandl, M.; Bauer-Brandl, A. High-Throughput Dissolution/Permeation Screening—A 96-Well Two-Compartment Microplate Approach. Pharmaceutics 2019, 11, 227. [Google Scholar] [CrossRef]
- Di Cagno, M.P.; Luppi, B. Drug ‘‘supersaturation’’ states induced by polymeric micelles and liposomes: A mechanistic investigation into permeability enhancements. Eur. J. Pharm. Sci. 2013, 48, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Braeckmans, M.; Augustijns, P.; Mols, R.; Servais, C.; Brouwers, J. Investigating the Mechanisms behind the Positive Food Effect of Abiraterone Acetate: In Vitro and Rat In Situ Studies. Pharmaceutics 2022, 14, 952. [Google Scholar] [CrossRef] [PubMed]
- Pellett, M.A.; Watkinson, A.C.; Hadgraft, J.; Brain, K.R. Comparison of permeability data from traditional diffusion cells and ATR-FTIR spectroscopy. Part II. Determination of diffusional pathlengths in synthetic membranes and human stratum corneum. Int. J. Pharm. 1997, 154, 217–227. [Google Scholar] [CrossRef]
- Inc., M.C. Pioneering Skin Testing, Experience the Unmatched Predicabiltiy of Strat-M(R) Membrane. 2018, p. 8. Available online: https://dicsa.es/assets/downloads/2018-13609_Strat-M_membrane_product_brochure_MRK_Web_DP.pdf (accessed on 1 October 2022).
- Haq, A.; Goodyear, B.; Ameen, D.; Joshi, V.; Michniak-Kohn, B. Strat-M® synthetic membrane: Permeability comparison to human cadaver skin. Int. J. Pharm. 2018, 547, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Arce, F.J.; Asano, N.; See, G.L.; Itakura, S.; Todo, H.; Sugibayashi, K. Usefulness of Artificial Membrane, Strat-M®, in the Assessment of Drug Permeation from Complex Vehicles in Finite Dose Conditions. Pharmaceutics 2020, 12, 173. [Google Scholar] [CrossRef]
- O’Shea, J.P.; Augustijns, P.; Brandl, M.; Brayden, D.J.; Brouwers, J.; Griffin, B.T.; Holm, R.; Jacobsen, A.-C.; Lennernäs, H.; Vinarov, Z.; et al. Best practices in current models mimicking drug permeability in the gastrointestinal tract—An UNGAP review. Eur. J. Pharm. Sci. 2022, 170, 106098. [Google Scholar] [CrossRef]
- Pion. MicroFLUX. Available online: https://pion-inc.com/scientific-instruments/in-vivo-predictive-tools/absorption/microflux (accessed on 9 September 2022).
- Alvebratt, C.; Keemink, J.; Edueng, K.; Cheung, O.; Strømme, M.; Bergström, C.A. An in vitro dissolution–digestion–permeation assay for the study of advanced drug delivery systems. Eur. J. Pharm. Biopharm. 2020, 149, 21–29. [Google Scholar] [CrossRef]
- Eliasen, J.N.; Berthelsen, R.; Slot, A.L.; Müllertz, A. Evaluating side-by-side diffusion models for studying drug supersaturation in an absorptive environment: A case example of fenofibrate and felodipine. J. Pharm. Pharmacol. 2019, 72, 371–384. [Google Scholar] [CrossRef]
- Tsinman, K.; Tsinman, O.; Lingamaneni, R.; Zhu, S.; Riebesehl, B.; Grandeury, A.; Juhnke, M.; Van Eerdenbrugh, B. Ranking Itraconazole Formulations Based on the Flux through Artificial Lipophilic Membrane. Pharm. Res. 2018, 35, 161. [Google Scholar] [CrossRef]
- Borbas, E.; Balogh, A.; Bocz, K.; Müller, J.; Kiserdei, É.; Vigh, T.; Sinkó, B.; Marosi, A.; Halász, A.; Dohányos, Z.; et al. In vitro dissolution-permeation evaluation of an electrospun cyclodextrin-based formulation of aripiprazole using mu Flux (TM). Int. J. Pharm. 2015, 491, 180–189. [Google Scholar] [CrossRef]
- Borbas, E.; Kádár, S.; Tsinman, K.; Tsinman, O.; Csicsák, D.; Takacs-Novak, K.; Völgyi, G.; Sinko, B.; Pataki, H. Prediction of Bioequivalence and Food Effect Using Flux- and Solubility-Based Methods. Mol. Pharm. 2019, 16, 4121–4130. [Google Scholar] [CrossRef] [Green Version]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Company Name | Location of Headquarters | Revenue 2021 (Bill. USD) [18] | No. Results | Articles Published by Affiliation (%) |
|---|---|---|---|---|
| Pfizer Inc. | New York, USA | 81.3 | 232 | 0.4 |
| Sinopharm | Shanghai, CN | 60.5 | 1 | 0.1 |
| AbbVie | Chicago, USA | 56.2 | 58 | 1.4 |
| Johnson & Johnson | New Brunswick, USA | 52.1 | 50 | 0.9 |
| Novartis | Basel, CH | 51.6 | 92 | 0.4 |
| F. Hoffmann–La Roche SA | Basel, CH | 49.3 | 74 | 0.5 |
| Merck & Co. Inc. | Kenilworth, USA | 48.7 | 149 | 0.3 |
| GlaxoSmithKline | Brentford, UK | 47.9 | 114 | 0.4 |
| Bristol Myers Squibb | New York, USA | 46.4 | 96 | 0.6 |
| Sanofi S.A. | Paris, FR | 44.6 | 61 | 0.3 |
| AstraZeneca | Cambridge, UK | 37.4 | 144 | 0.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jacobsen, A.-C.; Visentin, S.; Butnarasu, C.; Stein, P.C.; di Cagno, M.P. Commercially Available Cell-Free Permeability Tests for Industrial Drug Development: Increased Sustainability through Reduction of In Vivo Studies. Pharmaceutics 2023, 15, 592. https://doi.org/10.3390/pharmaceutics15020592
Jacobsen A-C, Visentin S, Butnarasu C, Stein PC, di Cagno MP. Commercially Available Cell-Free Permeability Tests for Industrial Drug Development: Increased Sustainability through Reduction of In Vivo Studies. Pharmaceutics. 2023; 15(2):592. https://doi.org/10.3390/pharmaceutics15020592
Chicago/Turabian StyleJacobsen, Ann-Christin, Sonja Visentin, Cosmin Butnarasu, Paul C. Stein, and Massimiliano Pio di Cagno. 2023. "Commercially Available Cell-Free Permeability Tests for Industrial Drug Development: Increased Sustainability through Reduction of In Vivo Studies" Pharmaceutics 15, no. 2: 592. https://doi.org/10.3390/pharmaceutics15020592