
Nanotechnology-Based Drug Delivery Systems to Control Bacterial-Biofilm-Associated Lung Infections
 and
and
Abstract
:1. Introduction
2. Respiratory Tract Anatomy and Mucosal Barriers
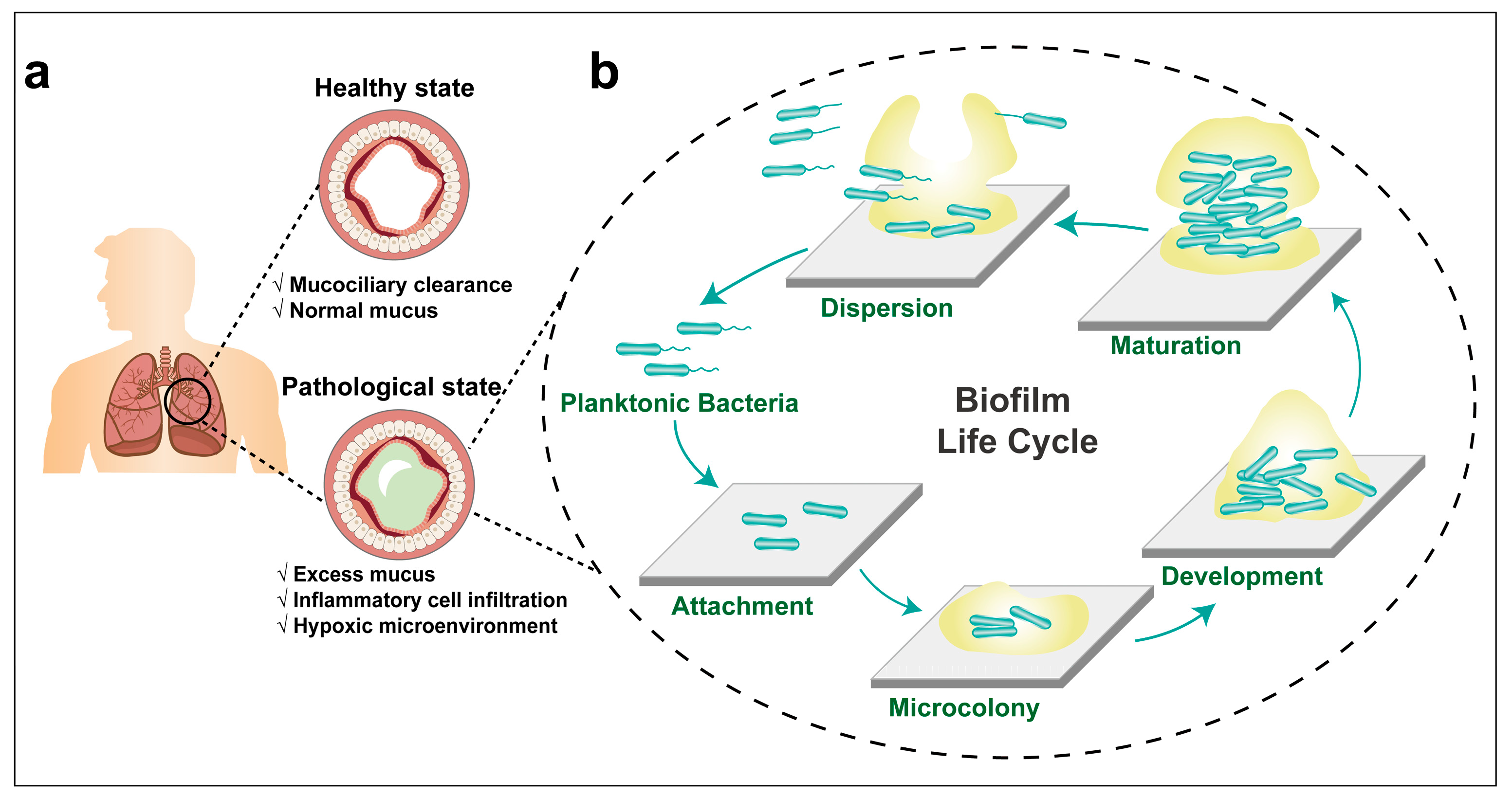
3. Lung Infections and Bacterial Biofilm Formation
3.1. The Formation of P. aeruginosa Biofilm
3.2. The Important Roles of EPS in the Development of Bacterial Biofilm
3.3. P. aeruginosa Biofilm-Associated Lung Infections in Cystic Fibrosis Patients
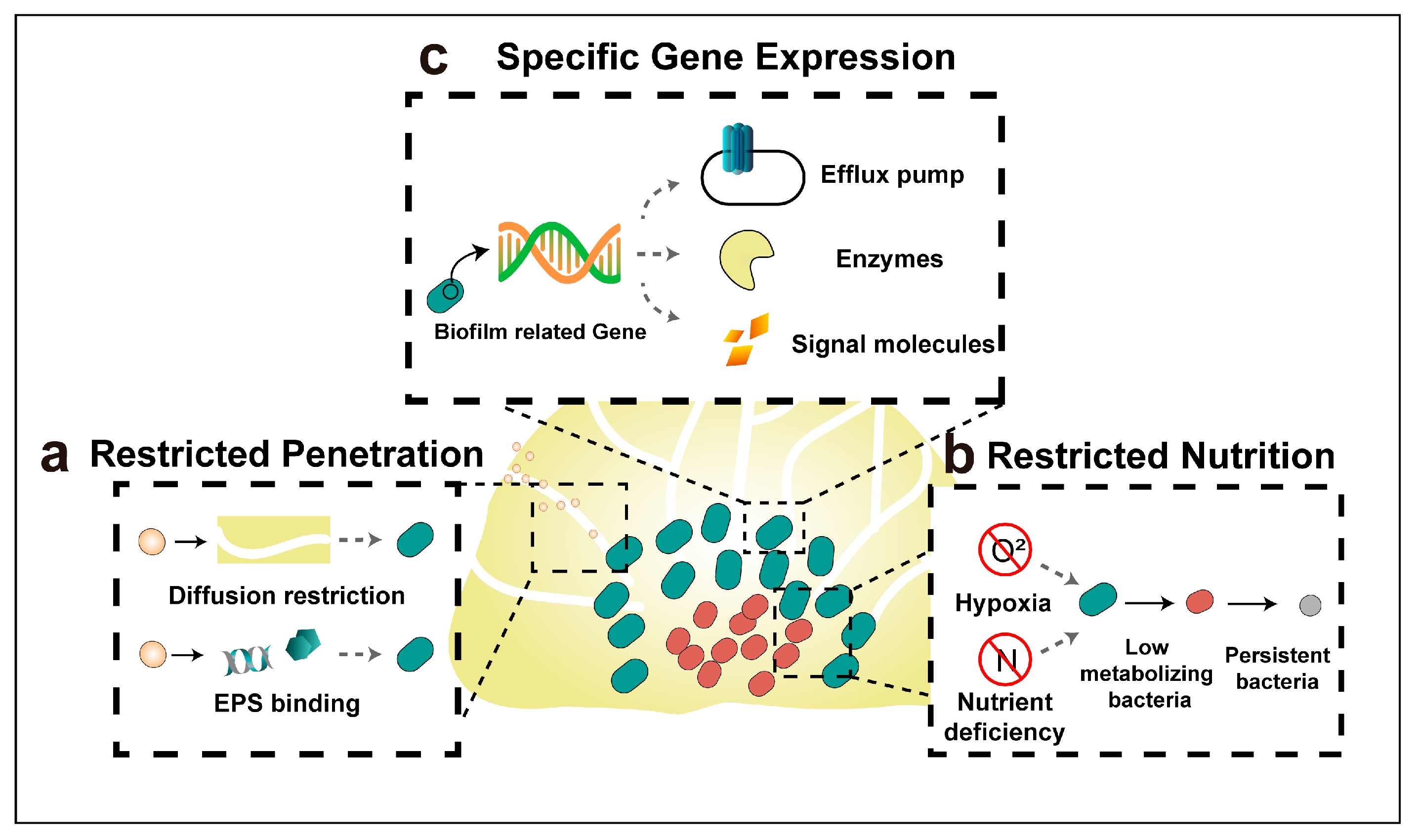
4. Mechanisms of Antibiotic Resistance of Biofilm Bacteria
4.1. Restricted Penetration by The Biofilm Matrix
4.2. Restricted Nutrition and Emerging of Persister Bacteria
4.3. Specific Gene Expression
5. Administration Routes of Nanomedicines for the Treatment of Lung Infections
5.1. Systemic Administration
5.2. Inhalation Administration
5.2.1. Strategies to Overcome Airway Mucus
5.2.2. Strategies to Avoid Macrophage Phagocytosis
5.2.3. Interaction with Pulmonary Surfactants
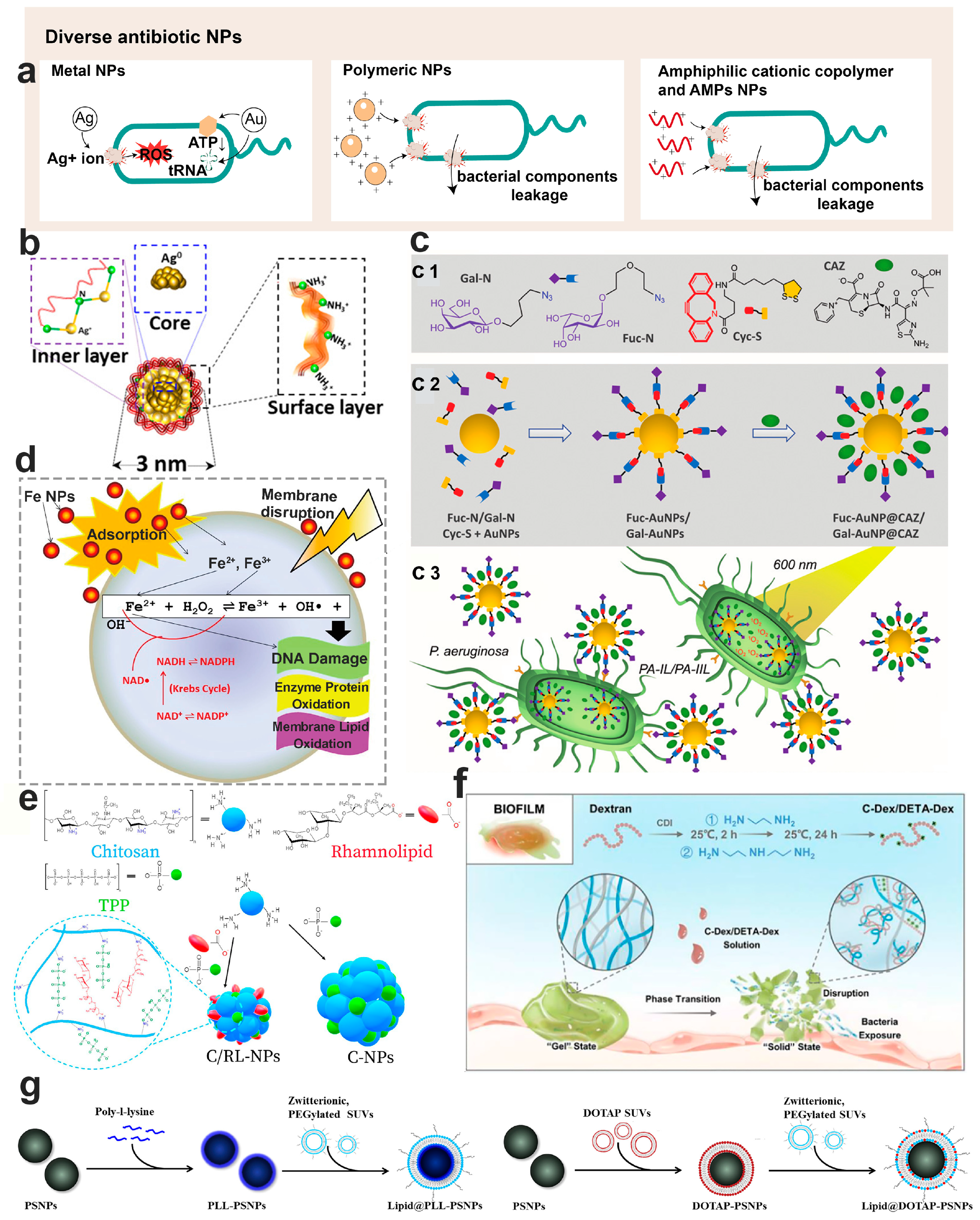
6. Nanotechnology-Based Diverse Antimicrobials for Bacterial Biofilm Control
6.1. Physicochemical Properties of Anti-Biofilm Nanoparticles
6.2. Inorganic Antimicrobial Nanoparticles
6.2.1. Ag Antimicrobial Nanoparticles
6.2.2. Au Antimicrobial Nanoparticles
6.2.3. Metal Oxide Antimicrobial Nanoparticles
6.3. Polymeric Antimicrobial Nanomaterials
6.3.1. Lipid-Based Antimicrobial Nanoparticles
6.3.2. Chitosan Antimicrobial Nanoparticles
6.3.3. Dextran Antimicrobial Nanoparticles

6.3.4. Amphiphilic Cationic Copolymer Antimicrobial Nanoparticles
6.3.5. Antimicrobial Peptides Loaded Nanoparticles
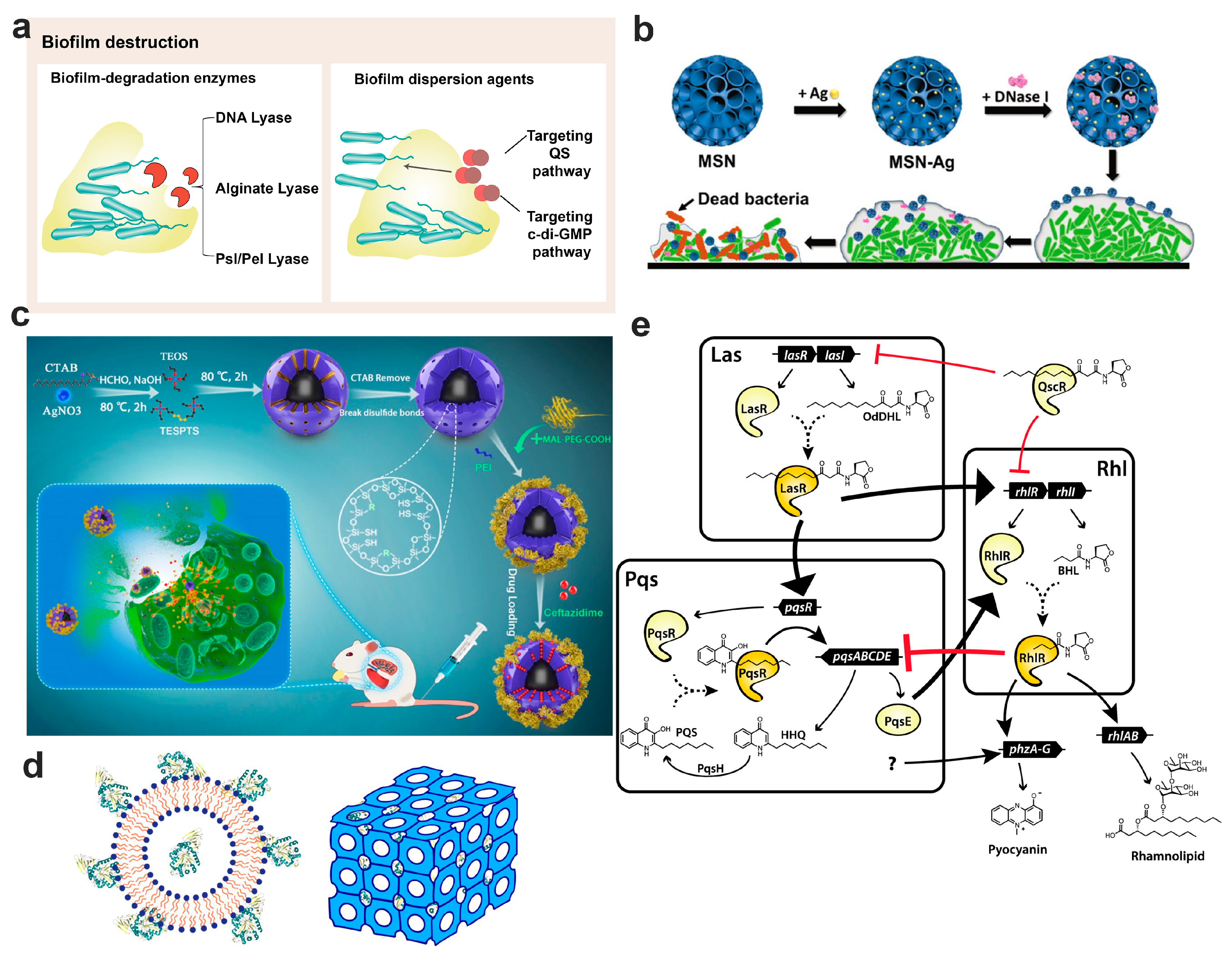
7. Nanotechnology-Based Bacterial Biofilm Matrix Degradation Strategy
7.1. Disruption by Biofilm Degradation Enzymes
7.2. Dispersion the Biofilm through Signaling Pathway
7.2.1. Biofilm Dispersion Mediated by the Quorum Sensing Pathway

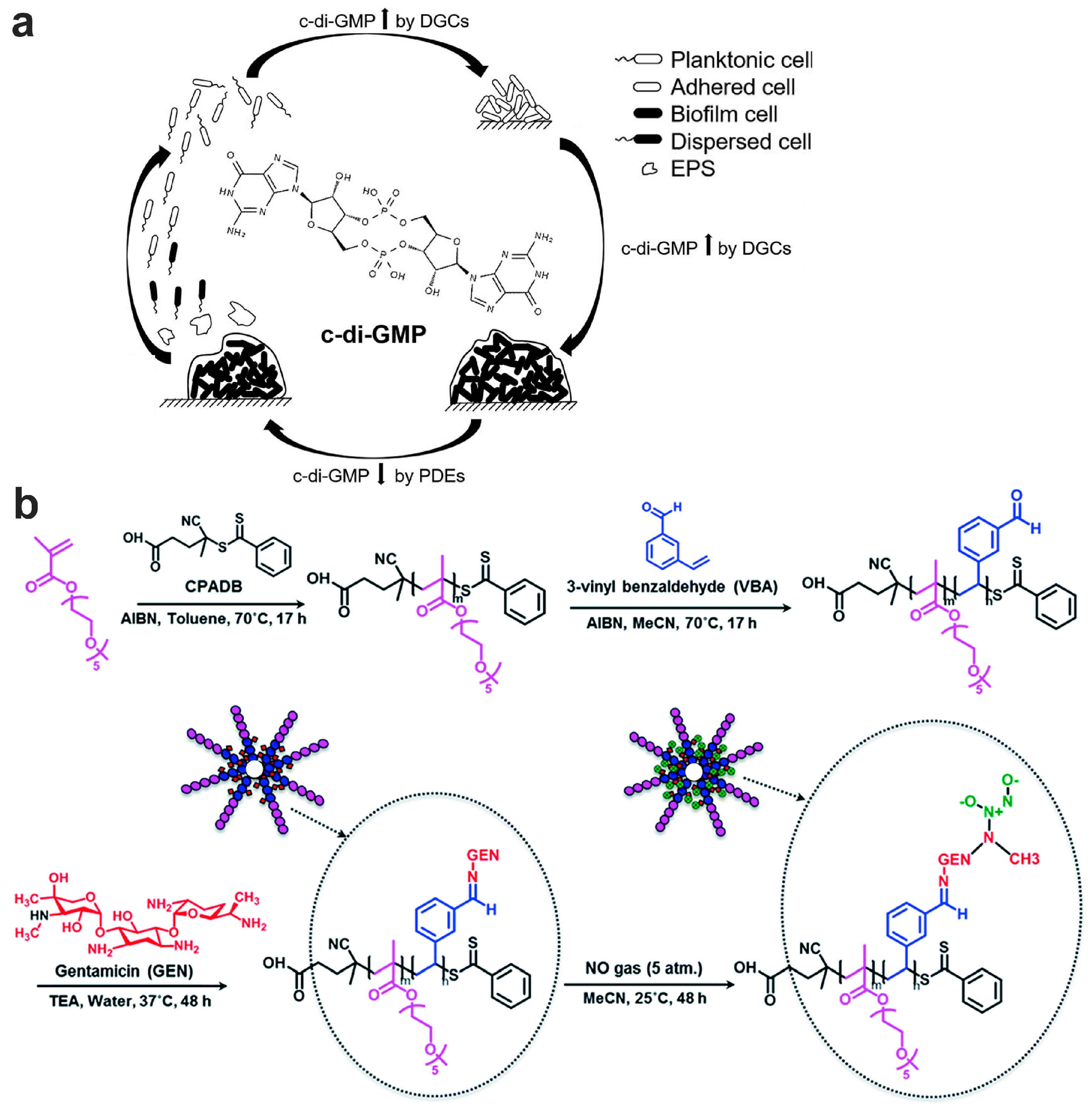
7.2.2. Biofilm Dispersion Mediated by c-di-GMP Pathway

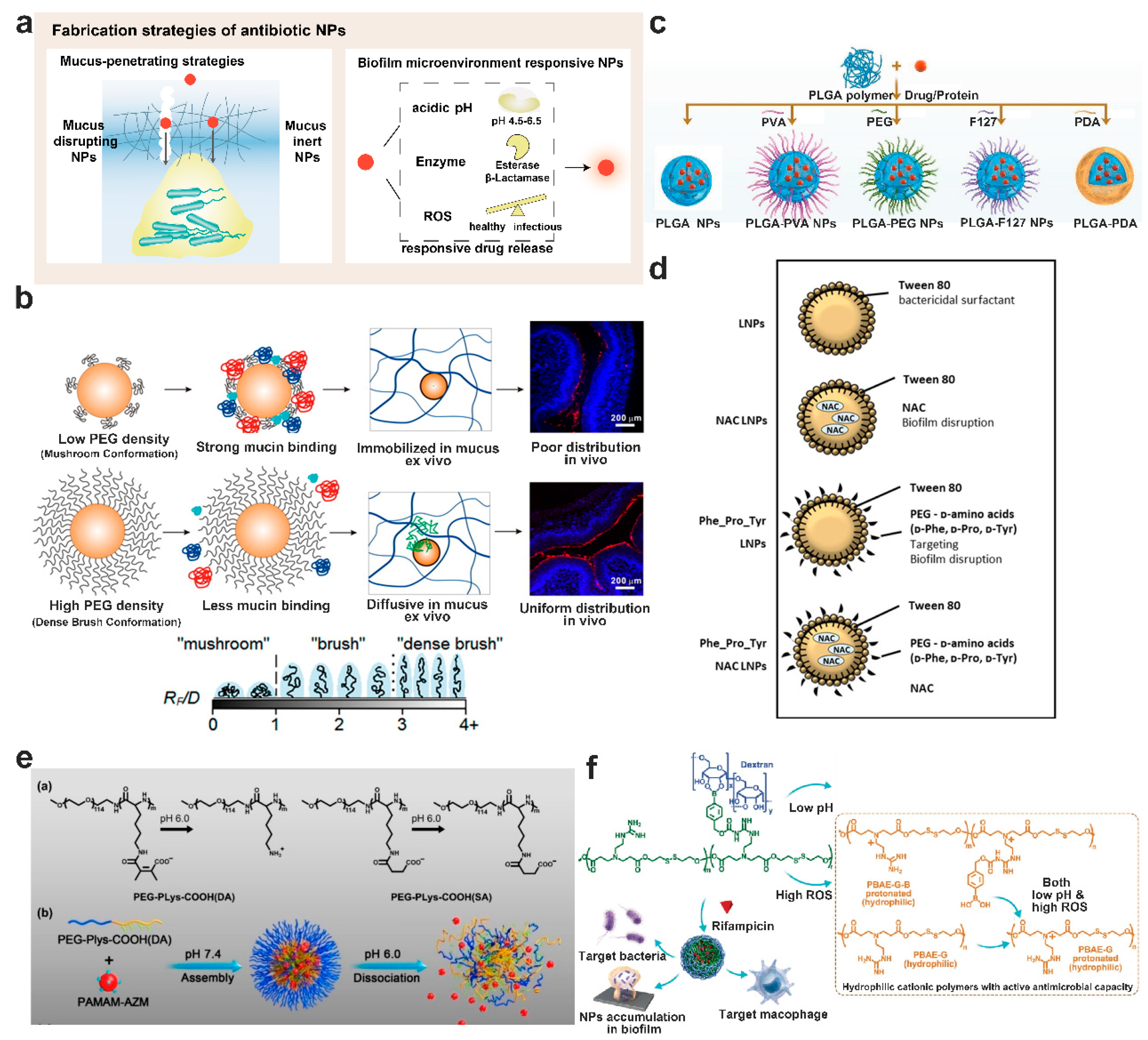
8. Nanotechnology-Based Nanoparticle Fabrication Strategies for Bacterial Biofilm Control
8.1. Mucus-Penetrating Particles or Muco-Inert Particles
8.2. Enhance Mucus Penetration of Particles by Mucus Disrupting Agents
8.3. Biofilm Microenvironment Responsive Nanoparticulate Systems

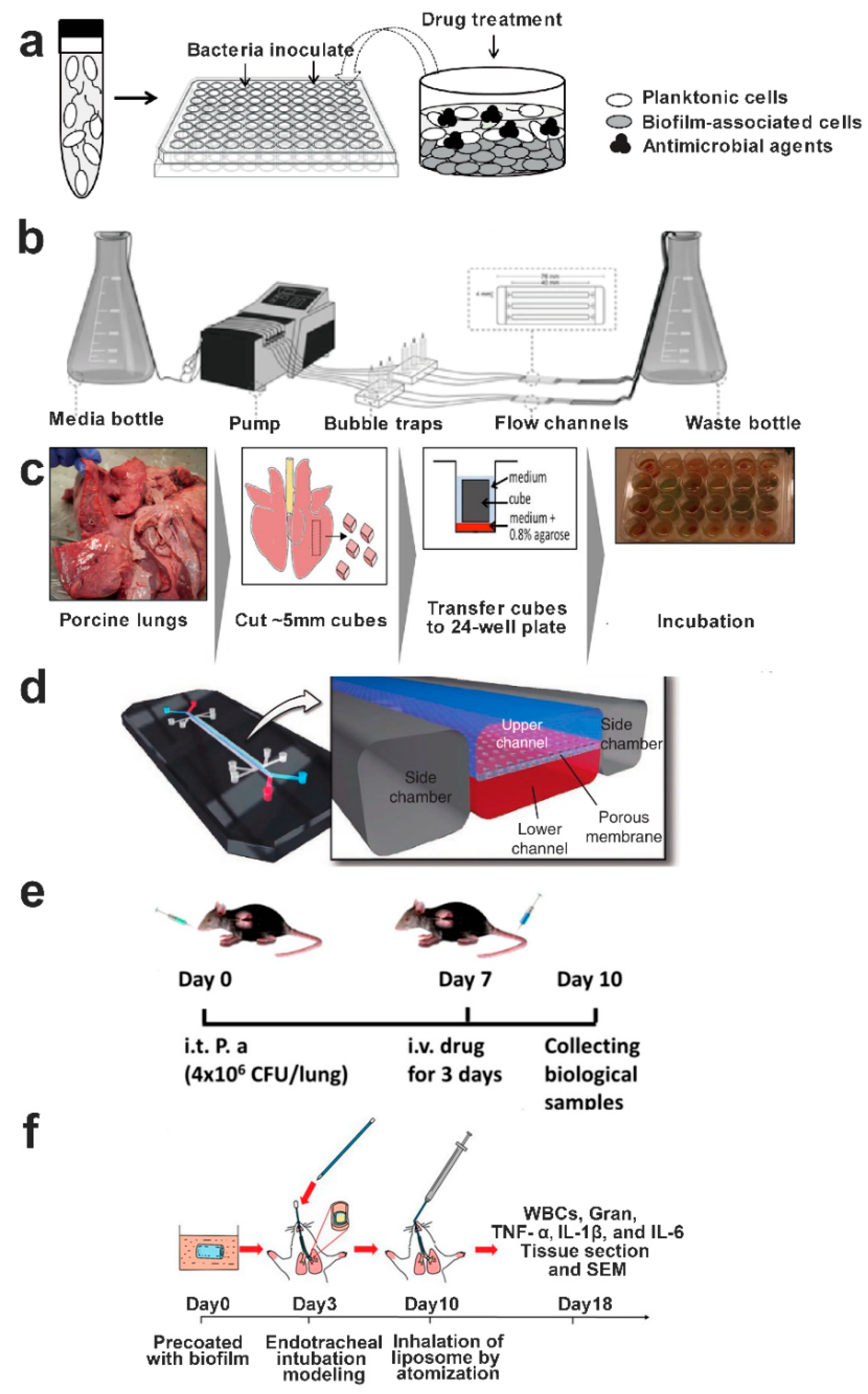
9. Biofilm Infectious Models for Evaluation of Antibiofilm Activity
9.1. In Vitro Biofilm Models

9.2. In Vivo Biofilm Models
10. Summary and Outlook
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Azoulay, E.; Russell, L.; Van de Louw, A.; Metaxa, V.; Bauer, P.; Povoa, P.; Montero, J.G.; Loeches, I.M.; Mehta, S.; Puxty, K.; et al. Diagnosis of severe respiratory infections in immunocompromised patients. Intensive Care Med. 2020, 46, 298–314. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, E.C. Influenza Virus. Trends Microbiol. 2018, 26, 809–810. [Google Scholar] [CrossRef] [PubMed]
- Langedijk, A.C.; Bont, L.J. Respiratory syncytial virus infection and novel interventions. Nat. Rev. Microbiol. 2023, 21, 734–749. [Google Scholar] [CrossRef]
- Borczuk, A.C.; Yantiss, R.K. The pathogenesis of coronavirus-19 disease. J. Biomed. Sci. 2022, 29, 87. [Google Scholar] [CrossRef] [PubMed]
- Rostami, A.; Liu, M.-C.; Xu, Q.; Li, T.-T.; Wang, T.; Jiang, B.-G.; Lv, C.-L.; Zhang, X.-A.; Liu, W.; Fang, L.-Q. Prevalence of human infection with respiratory adenovirus in China: A systematic review and meta-analysis. PLoS Neglected Trop. Dis. 2023, 17, e0011151. [Google Scholar] [CrossRef]
- Shang, L.; Xu, J.; Cao, B. Viral pneumonia in China: From surveillance to response. Lancet Public. Health 2020, 5, e633–e634. [Google Scholar] [CrossRef] [PubMed]
- Cone, R.A. Barrier properties of mucus. Adv. Drug Deliv. Rev. 2009, 61, 75–85. [Google Scholar] [CrossRef]
- Vanaki, S.M.; Holmes, D.; Saha, S.C.; Chen, J.; Brown, R.J.; Jayathilake, P.G. Muco-ciliary clearance: A review of modelling techniques. J. Biomech. 2020, 99, 109578. [Google Scholar] [CrossRef]
- Fahy, J.V.; Dickey, B.F. Airway mucus function and dysfunction. N. Engl. J. Med. 2010, 363, 2233–2247. [Google Scholar] [CrossRef]
- Ding, L.; Wang, J.L.; Cai, S.H.; Smyth, H.; Cui, Z.R. Pulmonary biofilm-based chronic infections and inhaled treatment strategies. Int. J. Pharm. 2021, 604, 120768. [Google Scholar] [CrossRef]
- Ciofu, O.; Tolker-Nielsen, T.; Jensen, P.O.; Wang, H.; Hoiby, N. Antimicrobial resistance, respiratory tract infections and role of biofilms in lung infections in cystic fibrosis patients. Adv. Drug Deliv. Rev. 2015, 85, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Potera, C. Microbiology—Forging a link between biofilms and disease. Science 1999, 283, 1837–1839. [Google Scholar] [CrossRef] [PubMed]
- Costerton, J.W.; Stewart, P.S.; Greenberg, E.P. Bacterial biofilms: A common cause of persistent infections. Science 1999, 284, 1318–1322. [Google Scholar] [CrossRef] [PubMed]
- Ciofu, O.; Moser, C.; Jensen, P.O.; Hoiby, N. Tolerance and resistance of microbial biofilms. Nat. Rev. Microbiol. 2022, 20, 621–635. [Google Scholar] [CrossRef]
- Sharma, A.; Kumar, D.; Dahiya, K.; Hawthorne, S.; Jha, S.K.; Jha, N.K.; Nand, P.; Girgis, S.; Raj, S.; Srivastava, R.; et al. Advances in pulmonary drug delivery targeting microbial biofilms in respiratory diseases. Nanomedicine 2021, 16, 1905–1923. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Bera, H.; Wang, H.; Wang, J.; Guo, Y.; Shi, C.; Cun, D.; Moser, C.; Hoiby, N.; Yang, M. Combination and nanotechnology based pharmaceutical strategies for combating respiratory bacterial biofilm infections. Int. J. Pharm. 2022, 616, 121507. [Google Scholar] [CrossRef]
- Selmani, A.; Kovačević, D.; Bohinc, K. Nanoparticles: From synthesis to applications and beyond. Adv. Colloid. Interface Sci. 2022, 303, 102640. [Google Scholar] [CrossRef]
- Wu, L.; Shan, W.; Zhang, Z.; Huang, Y. Engineering nanomaterials to overcome the mucosal barrier by modulating surface properties. Adv. Drug Deliv. Rev. 2018, 124, 150–163. [Google Scholar] [CrossRef]
- Murgia, X.; Loretz, B.; Hartwig, O.; Hittinger, M.; Lehr, C.M. The role of mucus on drug transport and its potential to affect therapeutic outcomes. Adv. Drug Deliv. Rev. 2018, 124, 82–97. [Google Scholar] [CrossRef]
- Leal, J.; Smyth, H.D.C.; Ghosh, D. Physicochemical properties of mucus and their impact on transmucosal drug delivery. Int. J. Pharm. 2017, 532, 555–572. [Google Scholar] [CrossRef]
- Button, B.; Cai, L.H.; Ehre, C.; Kesimer, M.; Hill, D.B.; Sheehan, J.K.; Boucher, R.C.; Rubinstein, M. A periciliary brush promotes the lung health by separating the mucus layer from airway epithelia. Science 2012, 337, 937–941. [Google Scholar] [CrossRef] [PubMed]
- Sears, P.R.; Davis, C.W.; Chua, M.; Sheehan, J.K. Mucociliary interactions and mucus dynamics in ciliated human bronchial epithelial cell cultures. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 301, L181–L186. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhang, J.; Shan, W.; Huang, Y. Developments of mucus penetrating nanoparticles. Asian J. Pharm. Sci. 2015, 10, 275–282. [Google Scholar] [CrossRef]
- Song, D.; Iverson, E.; Kaler, L.; Bader, S.; Scull, M.A.; Duncan, G.A. Modeling Airway Dysfunction in Asthma Using Synthetic Mucus Biomaterials. ACS Biomater. Sci. Eng. 2021, 7, 2723–2733. [Google Scholar] [CrossRef]
- Patarin, J.; Ghiringhelli, E.; Darsy, G.; Obamba, M.; Bochu, P.; Camara, B.; Quetant, S.; Cracowski, J.L.; Cracowski, C.; Robert de Saint Vincent, M. Rheological analysis of sputum from patients with chronic bronchial diseases. Sci. Rep. 2020, 10, 15685. [Google Scholar] [CrossRef]
- Duncan, G.A.; Jung, J.; Joseph, A.; Thaxton, A.L.; West, N.E.; Boyle, M.P.; Hanes, J.; Suk, J.S. Microstructural alterations of sputum in cystic fibrosis lung disease. JCI Insight 2016, 1, e88198. [Google Scholar] [CrossRef]
- Linssen, R.S.; Chai, G.; Ma, J.; Kummarapurugu, A.B.; van Woensel, J.B.M.; Bem, R.A.; Kaler, L.; Duncan, G.A.; Zhou, L.; Rubin, B.K.; et al. Neutrophil Extracellular Traps Increase Airway Mucus Viscoelasticity and Slow Mucus Particle Transit. Am. J. Respir. Cell Mol. Biol. 2021, 64, 69–78. [Google Scholar] [CrossRef]
- Lai, S.K.; Wang, Y.Y.; Hanes, J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv. Drug Deliv. Rev. 2009, 61, 158–171. [Google Scholar] [CrossRef]
- Schneider, C.S.; Xu, Q.; Boylan, N.J.; Chisholm, J.; Tang, B.C.; Schuster, B.S.; Henning, A.; Ensign, L.M.; Lee, E.; Adstamongkonkul, P.; et al. Nanoparticles that do not adhere to mucus provide uniform and long-lasting drug delivery to airways following inhalation. Sci. Adv. 2017, 3, e1601556. [Google Scholar] [CrossRef]
- Xu, Q.; Ensign, L.M.; Boylan, N.J.; Schon, A.; Gong, X.; Yang, J.C.; Lamb, N.W.; Cai, S.; Yu, T.; Freire, E.; et al. Impact of Surface Polyethylene Glycol (PEG) Density on Biodegradable Nanoparticle Transport in Mucus ex Vivo and Distribution in Vivo. ACS Nano 2015, 9, 9217–9227. [Google Scholar] [CrossRef]
- Seiler, F.; Lepper, P.M.; Bals, R.; Beisswenger, C. Regulation and function of antimicrobial peptides in immunity and diseases of the lung. Protein Pept. Lett. 2014, 21, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Lecaille, F.; Lalmanach, G.; Andrault, P.M. Antimicrobial proteins and peptides in human lung diseases: A friend and foe partnership with host proteases. Biochimie 2016, 122, 151–168. [Google Scholar] [CrossRef] [PubMed]
- James, G.A.; Swogger, E.; Wolcott, R.; Pulcini, E.; Secor, P.; Sestrich, J.; Costerton, J.W.; Stewart, P.S. Biofilms in chronic wounds. Wound Repair. Regen. 2008, 16, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Haesler, E.; Swanson, T.; Ousey, K.; Carville, K. Clinical indicators of wound infection and biofilm: Reaching international consensus. J. Wound Care 2019, 28, s4–s12. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Meng, Z.; Han, X.; Li, S.; Ma, X.; Chen, X.; Liang, Y.; Deng, X.; Xia, K.; Zhang, Y.; et al. Cholesterol Microdomain Enhances the Biofilm Eradication of Antibiotic Liposomes. Adv. Healthc. Mater. 2022, 11, 2101745. [Google Scholar] [CrossRef]
- Jean-Pierre, V.; Boudet, A.; Sorlin, P.; Menetrey, Q.; Chiron, R.; Lavigne, J.-P.; Marchandin, H. Biofilm Formation by Staphylococcus aureus in the Specific Context of Cystic Fibrosis. Int. J. Mol. Sci. 2022, 24, 597. [Google Scholar] [CrossRef]
- Weiser, J.N.; Ferreira, D.M.; Paton, J.C. Streptococcus pneumoniae: Transmission, colonization and invasion. Nat. Rev. Microbiol. 2018, 16, 355–367. [Google Scholar] [CrossRef]
- Margalit, I.; Goldberg, E.; Ben Ari, Y.; Ben-Zvi, H.; Shostak, Y.; Krause, I.; Muhsen, K. Clinical correlates of nocardiosis. Sci. Rep. 2020, 10, 14272. [Google Scholar] [CrossRef]
- Xiu, W.; Ren, L.; Xiao, H.; Zhang, Y.; Wang, D.; Yang, K.; Wang, S.; Yuwen, L.; Li, X.; Dong, H.; et al. Ultrasound-responsive catalytic microbubbles enhance biofilm elimination and immune activation to treat chronic lung infections. Sci. Adv. 2023, 9, eade5446. [Google Scholar] [CrossRef]
- Bajire, S.K.; Prabhu, A.; Bhandary, Y.P.; Irfan, K.M.; Shastry, R.P. 7-Ethoxycoumarin rescued Caenorhabditis elegans from infection of COPD derived clinical isolate Pseudomonas aeruginosa through virulence and biofilm inhibition via targeting Rhl and Pqs quorum sensing systems. World J. Microbiol. Biotechnol. 2023, 39, 208. [Google Scholar] [CrossRef]
- Phillips-Houlbracq, M.; Ricard, J.-D.; Foucrier, A.; Yoder-Himes, D.; Gaudry, S.; Bex, J.; Messika, J.; Margetis, D.; Chatel, J.; Dobrindt, U.; et al. Pathophysiology of Escherichia coli pneumonia: Respective contribution of pathogenicity islands to virulence. Int. J. Med. Microbiol. 2018, 308, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Kharga, K.; Dhar, I.; Kashyap, S.; Sengupta, S.; Kumar, D.; Kumar, L. Zingerone inhibits biofilm formation and enhances antibiotic efficacy against Salmonella biofilm. World J. Microbiol. Biotechnol. 2023, 39, 268. [Google Scholar] [CrossRef]
- Li, Y.; Ni, M. Regulation of biofilm formation in Klebsiella pneumoniae. Front. Microbiol. 2023, 14, 1238482. [Google Scholar] [CrossRef] [PubMed]
- Marienborg, K.; Ambur, O.H.; Økstad, O.A.L.; Skaare, D. The alginate polymer OligoG alters susceptibility of biofilm-embedded non-typeable Haemophilus influenzae to ampicillin and ciprofloxacin. JAC-Antimicrob. Resist. 2023, 5, dlad046. [Google Scholar] [CrossRef] [PubMed]
- Earle, K.; Valero, C.; Conn, D.P.; Vere, G.; Cook, P.C.; Bromley, M.J.; Bowyer, P.; Gago, S. Pathogenicity and virulence of Aspergillus fumigatus. Virulence 2023, 14, 2172264. [Google Scholar] [CrossRef]
- Bouyssi, A.; Déméautis, T.; Trecourt, A.; Delles, M.; Agostini, F.; Monneret, G.; Glehen, O.; Wallon, M.; Persat, F.; Devouassoux, G.; et al. Characterization of Lung Inflammatory Response to Aspergillus fumigatus Spores. J. Fungi 2023, 9, 682. [Google Scholar] [CrossRef]
- Linder, K.A.; Kauffman, C.A.; Miceli, M.H. Blastomycosis: A Review of Mycological and Clinical Aspects. J. Fungi 2023, 9, 117. [Google Scholar] [CrossRef]
- Rather, M.A.; Saha, D.; Bhuyan, S.; Jha, A.N.; Mandal, M. Quorum Quenching: A Drug Discovery Approach Against Pseudomonas aeruginosa. Microbiol. Res. 2022, 264, 127173. [Google Scholar] [CrossRef]
- Hansen, C.R.; Pressler, T.; Hoiby, N. Early aggressive eradication therapy for intermittent Pseudomonas aeruginosa airway colonization in cystic fibrosis patients: 15 years experience. J. Cyst. Fibros. 2008, 7, 523–530. [Google Scholar] [CrossRef]
- Rumbaugh, K.P.; Sauer, K. Biofilm dispersion. Nat. Rev. Microbiol. 2020, 18, 571–586. [Google Scholar] [CrossRef]
- Arora, D.P.; Hossain, S.; Xu, Y.; Boon, E.M. Nitric Oxide Regulation of Bacterial Biofilms. Biochemistry 2015, 54, 3717–3728. [Google Scholar] [CrossRef] [PubMed]
- Bansil, R.; Turner, B.S. The biology of mucus: Composition, synthesis and organization. Adv. Drug Deliv. Rev. 2018, 124, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Curran, C.S.; Bolig, T.; Torabi-Parizi, P. Mechanisms and Targeted Therapies for Pseudomonas aeruginosa Lung Infection. Am. J. Respir. Crit. Care Med. 2018, 197, 708–727. [Google Scholar] [CrossRef] [PubMed]
- Proft, T.; Baker, E.N. Pili in Gram-negative and Gram-positive bacteria—Structure, assembly and their role in disease. Cell Mol. Life Sci. 2009, 66, 613–635. [Google Scholar] [CrossRef]
- Whiteley, M.; Diggle, S.P.; Greenberg, E.P. Progress in and promise of bacterial quorum sensing research. Nature 2017, 551, 313–320. [Google Scholar] [CrossRef]
- Koo, H.; Allan, R.N.; Howlin, R.P.; Stoodley, P.; Hall-Stoodley, L. Targeting microbial biofilms: Current and prospective therapeutic strategies. Nat. Rev. Microbiol. 2017, 15, 740–755. [Google Scholar] [CrossRef]
- Yang, L.; Hu, Y.; Liu, Y.; Zhang, J.; Ulstrup, J.; Molin, S. Distinct roles of extracellular polymeric substances in Pseudomonas aeruginosa biofilm development. Environ. Microbiol. 2011, 13, 1705–1717. [Google Scholar] [CrossRef]
- van Schaik, E.J.; Giltner, C.L.; Audette, G.F.; Keizer, D.W.; Bautista, D.L.; Slupsky, C.M.; Sykes, B.D.; Irvin, R.T. DNA binding: A novel function of Pseudomonas aeruginosa type IV pili. J. Bacteriol. 2005, 187, 1455–1464. [Google Scholar] [CrossRef]
- Yang, L.; Nilsson, M.; Gjermansen, M.; Givskov, M.; Tolker-Nielsen, T. Pyoverdine and PQS mediated subpopulation interactions involved in Pseudomonas aeruginosa biofilm formation. Mol. Microbiol. 2009, 74, 1380–1392. [Google Scholar] [CrossRef]
- Bjarnsholt, T.; Ciofu, O.; Molin, S.; Givskov, M.; Hoiby, N. Applying insights from biofilm biology to drug development—Can a new approach be developed? Nat. Rev. Drug Discov. 2013, 12, 791–808. [Google Scholar] [CrossRef]
- Klausen, M.; Heydorn, A.; Ragas, P.; Lambertsen, L.; Aaes-Jorgensen, A.; Molin, S.; Tolker-Nielsen, T. Biofilm formation by Pseudomonas aeruginosa wild type, flagella and type IV pili mutants. Mol. Microbiol. 2003, 48, 1511–1524. [Google Scholar] [CrossRef] [PubMed]
- Purevdorj-Gage, B.; Costerton, W.J.; Stoodley, P. Phenotypic differentiation and seeding dispersal in non-mucoid and mucoid Pseudomonas aeruginosa biofilms. Microbiology 2005, 151, 1569–1576. [Google Scholar] [CrossRef] [PubMed]
- Flemming, H.C.; Wingender, J. The biofilm matrix. Nat. Rev. Microbiol. 2010, 8, 623–633. [Google Scholar] [CrossRef]
- Byrd, M.S.; Sadovskaya, I.; Vinogradov, E.; Lu, H.P.; Sprinkle, A.B.; Richardson, S.H.; Ma, L.Y.; Ralston, B.; Parsek, M.R.; Anderson, E.M.; et al. Genetic and biochemical analyses of the Pseudomonas aeruginosa Psl exopolysaccharide reveal overlapping roles for polysaccharide synthesis enzymes in Psl and LPS production. Mol. Microbiol. 2009, 73, 622–638. [Google Scholar] [CrossRef] [PubMed]
- Mann, E.E.; Wozniak, D.J. Pseudomonas biofilm matrix composition and niche biology. FEMS Microbiol. Rev. 2012, 36, 893–916. [Google Scholar] [CrossRef]
- Ma, L.; Wang, S.; Wang, D.; Parsek, M.R.; Wozniak, D.J. The roles of biofilm matrix polysaccharide Psl in mucoid Pseudomonas aeruginosa biofilms. FEMS Immunol. Med. Microbiol. 2012, 65, 377–380. [Google Scholar] [CrossRef]
- Ma, L.; Conover, M.; Lu, H.; Parsek, M.R.; Bayles, K.; Wozniak, D.J. Assembly and development of the Pseudomonas aeruginosa biofilm matrix. PLoS Pathog. 2009, 5, e1000354. [Google Scholar] [CrossRef]
- Colvin, K.M.; Gordon, V.D.; Murakami, K.; Borlee, B.R.; Wozniak, D.J.; Wong, G.C.L.; Parsek, M.R. The Pel Polysaccharide Can Serve a Structural and Protective Role in the Biofilm Matrix of Pseudomonas aeruginosa. PLoS Pathog. 2011, 7, e1001264. [Google Scholar] [CrossRef]
- Jennings, L.K.; Storek, K.M.; Ledvina, H.E.; Coulon, C.; Marmont, L.S.; Sadovskaya, I.; Secor, P.R.; Tseng, B.S.; Scian, M.; Filloux, A.; et al. Pel is a cationic exopolysaccharide that cross-links extracellular DNA in the Pseudomonas aeruginosa biofilm matrix. Proc. Natl. Acad. Sci. USA 2015, 112, 11353–11358. [Google Scholar] [CrossRef]
- Rossi, E.; La Rosa, R.; Bartell, J.A.; Marvig, R.L.; Haagensen, J.A.J.; Sommer, L.M.; Molin, S.; Johansen, H.K. Pseudomonas aeruginosa adaptation and evolution in patients with cystic fibrosis. Nat. Rev. Microbiol. 2021, 19, 331–342. [Google Scholar] [CrossRef]
- Maurice, N.M.; Bedi, B.; Sadikot, R.T. Pseudomonas aeruginosa Biofilms: Host Response and Clinical Implications in Lung Infections. Am. J. Respir. Cell Mol. Biol. 2018, 58, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Hoiby, N.; Bjarnsholt, T.; Moser, C.; Jensen, P.O.; Kolpen, M.; Qvist, T.; Aanaes, K.; Pressler, T.; Skov, M.; Ciofu, O. Diagnosis of biofilm infections in cystic fibrosis patients. APMIS 2017, 125, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Cabra, N.; Paetzold, B.; Ferrar, T.; Mazzolini, R.; Torrents, E.; Serrano, L.; Lluch-Senar, M. Characterization of different alginate lyases for dissolving Pseudomonas aeruginosa biofilms. Sci. Rep. 2020, 10, 9390. [Google Scholar] [CrossRef] [PubMed]
- Wan, B.; Zhu, Y.; Tao, J.; Zhu, F.; Chen, J.; Li, L.; Zhao, J.; Wang, L.; Sun, S.; Yang, Y.; et al. Alginate Lyase Guided Silver Nanocomposites for Eradicating Pseudomonas aeruginosa from Lungs. ACS Appl. Mater. Interfaces 2020, 12, 9050–9061. [Google Scholar] [CrossRef] [PubMed]
- Powell, L.C.; Pritchard, M.F.; Ferguson, E.L.; Powell, K.A.; Patel, S.U.; Rye, P.D.; Sakellakou, S.M.; Buurma, N.J.; Brilliant, C.D.; Copping, J.M.; et al. Targeted disruption of the extracellular polymeric network of Pseudomonas aeruginosa biofilms by alginate oligosaccharides. NPJ Biofilms Microbiomes 2018, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- Cendra, M.D.; Torrents, E. Pseudomonas aeruginosa biofilms and their partners in crime. Biotechnol. Adv. 2021, 49, 107734. [Google Scholar] [CrossRef]
- Sarkar, S. Release mechanisms and molecular interactions of Pseudomonas aeruginosa extracellular DNA. Appl. Microbiol. Biotechnol. 2020, 104, 6549–6564. [Google Scholar] [CrossRef]
- Das, T.; Manefield, M. Pyocyanin promotes extracellular DNA release in Pseudomonas aeruginosa. PLoS ONE 2012, 7, e46718. [Google Scholar] [CrossRef]
- Mulcahy, H.; Charron-Mazenod, L.; Lewenza, S. Extracellular DNA chelates cations and induces antibiotic resistance in Pseudomonas aeruginosa biofilms. PLoS Pathog. 2008, 4, e1000213. [Google Scholar] [CrossRef]
- Wang, S.; Liu, X.; Liu, H.; Zhang, L.; Guo, Y.; Yu, S.; Wozniak, D.J.; Ma, L.Z. The exopolysaccharide Psl-eDNA interaction enables the formation of a biofilm skeleton in Pseudomonas aeruginosa. Environ. Microbiol. Rep. 2015, 7, 330–340. [Google Scholar] [CrossRef]
- Saunders, S.H.; Tse, E.C.M.; Yates, M.D.; Otero, F.J.; Trammell, S.A.; Stemp, E.D.A.; Barton, J.K.; Tender, L.M.; Newman, D.K. Extracellular DNA Promotes Efficient Extracellular Electron Transfer by Pyocyanin in Pseudomonas aeruginosa Biofilms. Cell 2020, 182, 919–932.e919. [Google Scholar] [CrossRef] [PubMed]
- Chiang, W.C.; Nilsson, M.; Jensen, P.O.; Hoiby, N.; Nielsen, T.E.; Givskov, M.; Tolker-Nielsen, T. Extracellular DNA shields against aminoglycosides in Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 2013, 57, 2352–2361. [Google Scholar] [CrossRef] [PubMed]
- Fuxman Bass, J.I.; Russo, D.M.; Gabelloni, M.L.; Geffner, J.R.; Giordano, M.; Catalano, M.; Zorreguieta, A.; Trevani, A.S. Extracellular DNA: A major proinflammatory component of Pseudomonas aeruginosa biofilms. J. Immunol. 2010, 184, 6386–6395. [Google Scholar] [CrossRef] [PubMed]
- Caiazza, N.C.; Shanks, R.M.; O’Toole, G.A. Rhamnolipids modulate swarming motility patterns of Pseudomonas aeruginosa. J. Bacteriol. 2005, 187, 7351–7361. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Singh, N.; Shetye, G.S.; Jin, Y.; Li, D.; Luk, Y.Y. Synthetic analogs of rhamnolipids modulate structured biofilms formed by rhamnolipid-nonproducing mutant of Pseudomonas aeruginosa. Bioorg. Med. Chem. 2017, 25, 1830–1838. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yu, B.; Tian, D.; Ni, M. Rhamnolipid but not motility is associated with the initiation of biofilm seeding dispersal of Pseudomonas aeruginosa strain PA17. J. Biosci. 2013, 38, 149–156. [Google Scholar] [CrossRef]
- O’Sullivan, B.P.; Freedman, S.D. Cystic fibrosis. Lancet 2009, 373, 1891–1904. [Google Scholar] [CrossRef]
- Domingue, J.C.; Drewes, J.L.; Merlo, C.A.; Housseau, F.; Sears, C.L. Host responses to mucosal biofilms in the lung and gut. Mucosal Immunol. 2020, 13, 413–422. [Google Scholar] [CrossRef]
- Hoiby, N.; Ciofu, O.; Bjarnsholt, T. Pseudomonas aeruginosa biofilms in cystic fibrosis. Future Microbiol. 2010, 5, 1663–1674. [Google Scholar] [CrossRef]
- Parks, Q.M.; Young, R.L.; Poch, K.R.; Malcolm, K.C.; Vasil, M.L.; Nick, J.A. Neutrophil enhancement of Pseudomonas aeruginosa biofilm development: Human F-actin and DNA as targets for therapy. J. Med. Microbiol. 2009, 58, 492–502. [Google Scholar] [CrossRef]
- Pedraz, L.; Blanco-Cabra, N.; Torrents, E. Gradual adaptation of facultative anaerobic pathogens to microaerobic and anaerobic conditions. FASEB J. 2020, 34, 2912–2928. [Google Scholar] [CrossRef] [PubMed]
- Kamath, K.S.; Krisp, C.; Chick, J.; Pascovici, D.; Gygi, S.P.; Molloy, M.P. Pseudomonas aeruginosa Proteome under Hypoxic Stress Conditions Mimicking the Cystic Fibrosis Lung. J. Proteome Res. 2017, 16, 3917–3928. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Shi, R.; Ma, F.; Han, S.; Zhang, Y. Oxygen effects on rhamnolipids production by Pseudomonas aeruginosa. Microb. Cell Fact. 2018, 17, 39. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, C.M.; Pu, Q.; Wu, Q.; Tan, S.; Shao, X.; Zhang, W.; Xie, Y.; Li, R.; Yu, X.J.; et al. Pseudomonas aeruginosa Regulatory Protein AnvM Controls Pathogenicity in Anaerobic Environments and Impacts Host Defense. mBio 2019, 10, 10–128. [Google Scholar] [CrossRef] [PubMed]
- Baral, B.; Mozafari, M.R. Strategic Moves of “Superbugs” Against Available Chemical Scaffolds: Signaling, Regulation, and Challenges. ACS Pharmacol. Transl. 2020, 3, 373–400. [Google Scholar] [CrossRef]
- Olivares, E.; Badel-Berchoux, S.; Provot, C.; Prevost, G.; Bernardi, T.; Jehl, F. Clinical Impact of Antibiotics for the Treatment of Pseudomonas aeruginosa Biofilm Infections. Front. Microbiol. 2019, 10, 2894. [Google Scholar] [CrossRef]
- Uruen, C.; Chopo-Escuin, G.; Tommassen, J.; Mainar-Jaime, R.C.; Arenas, J. Biofilms as Promoters of Bacterial Antibiotic Resistance and Tolerance. Antibiotics 2021, 10, 3. [Google Scholar] [CrossRef]
- Paranjape, S.S.; Shashidhar, R. Comparison of Starvation-Induced Persister Cells with Antibiotic-Induced Persister Cells. Curr. Microbiol. 2019, 76, 1495–1502. [Google Scholar] [CrossRef]
- Moldoveanu, A.L.; Rycroft, J.A.; Helaine, S. Impact of bacterial persisters on their host. Curr. Opin. Microbiol. 2021, 59, 65–71. [Google Scholar] [CrossRef]
- Chattagul, S.; Khan, M.M.; Scott, A.J.; Nita-Lazar, A.; Ernst, R.K.; Goodlett, D.R.; Sermswan, R.W. Transcriptomics Analysis Uncovers Transient Ceftazidime Tolerance in Burkholderia Biofilms. ACS Infect. Dis. 2021, 7, 2324–2336. [Google Scholar] [CrossRef]
- Poudyal, B.; Sauer, K. The PA3177 Gene Encodes an Active Diguanylate Cyclase That Contributes to Biofilm Antimicrobial Tolerance but Not Biofilm Formation by Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2018, 62, 10–128. [Google Scholar] [CrossRef] [PubMed]
- Poudyal, B.; Sauer, K. The ABC of Biofilm Drug Tolerance: The MerR-Like Regulator BrlR Is an Activator of ABC Transport Systems, with PA1874-77 Contributing to the Tolerance of Pseudomonas aeruginosa Biofilms to Tobramycin. Antimicrob. Agents Chemother. 2018, 62, 10–128. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wang, J.; Chai, M.; Li, X.; Deng, Y.; Jin, Q.; Ji, J. Size and Charge Adaptive Clustered Nanoparticles Targeting the Biofilm Microenvironment for Chronic Lung Infection Management. ACS Nano 2020, 14, 5686–5699. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Dong, H.; Xiu, W.; Yuwen, L.; Mou, Y.; Yin, Z.; Liang, B.; Wang, L. Self-Adaptive Antibiofilm Effect and Immune Regulation by Hollow Cu2MoS4 Nanospheres for Treatment of Implant Infections. ACS Appl. Mater. Interfaces 2023, 15, 18720–18733. [Google Scholar] [CrossRef]
- Hu, H.; Hua, S.Y.; Lin, X.; Lu, F.; Zhang, W.; Zhou, L.; Cui, J.; Wang, R.; Xia, J.; Xu, F.; et al. Hybrid Biomimetic Membrane Coated Particles-Mediated Bacterial Ferroptosis for Acute MRSA Pneumonia. ACS Nano 2023, 17, 11692–11712. [Google Scholar] [CrossRef]
- Zhao, Y.; Yu, C.; Yu, Y.; Wei, X.; Duan, X.; Dai, X.; Zhang, X. Bioinspired Heteromultivalent Ligand-Decorated Nanotherapeutic for Enhanced Photothermal and Photodynamic Therapy of Antibiotic-Resistant Bacterial Pneumonia. ACS Appl. Mater. Interfaces 2019, 11, 39648–39661. [Google Scholar] [CrossRef]
- Liu, Q.Y.; Guan, J.; Qin, L.; Zhang, X.; Mao, S.R. Physicochemical properties affecting the fate of nanoparticles in pulmonary drug delivery. Drug Discov. Today 2020, 25, 150–159. [Google Scholar] [CrossRef]
- Yue, L.; Zhang, X.; Zhao, C.; Chen, R.; Chen, X.; Rao, L. Inhaled drug delivery: Past, present, and future. Nano Today 2023, 52, 101942. [Google Scholar] [CrossRef]
- Chen, D.Q.; Liu, J.H.; Wu, J.; Suk, J.S. Enhancing nanoparticle penetration through airway mucus to improve drug delivery efficacy in the lung. Expert. Opin. Drug Deliv. 2021, 18, 595–606. [Google Scholar] [CrossRef]
- Pangeni, R.; Meng, T.; Poudel, S.; Sharma, D.; Hutsell, H.; Ma, J.; Rubin, B.K.; Longest, W.; Hindle, M.; Xu, Q. Airway mucus in pulmonary diseases: Muco-adhesive and muco-penetrating particles to overcome the airway mucus barriers. Int. J. Pharm. 2023, 634, 122661. [Google Scholar] [CrossRef]
- Zhao, J.; Qin, L.; Song, R.X.; Su, J.; Yuan, Y.; Zhang, X.; Mao, S.R. Elucidating inhaled liposome surface charge on its interaction with biological barriers in the lung. Eur. J. Pharm. Biopharm. 2022, 172, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Guo, Y.; Liu, S.; Hu, Y.; Yang, C.; Cheng, G.; Xue, C.; Zuo, Y.Y.; Sun, B. pH-Mediated Mucus Penetration of Zwitterionic Polydopamine-Modified Silica Nanoparticles. Nano Lett. 2023, 23, 7552–7560. [Google Scholar] [CrossRef]
- Araújo, F.; Martins, C.; Azevedo, C.; Sarmento, B. Chemical modification of drug molecules as strategy to reduce interactions with mucus. Adv. Drug Deliv. Rev. 2018, 124, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Chono, S.; Tanino, T.; Seki, T.; Morimoto, K. Influence of particle size on drug delivery to rat alveolar macrophages following pulmonary administration of ciprofloxacin incorporated into liposomes. J. Drug Target. 2008, 14, 557–566. [Google Scholar] [CrossRef]
- Seydoux, E.; Rodriguez-Lorenzo, L.; Blom, R.A.M.; Stumbles, P.A.; Petri-Fink, A.; Rothen-Rutishauser, B.M.; Blank, F.; von Garnier, C. Pulmonary delivery of cationic gold nanoparticles boost antigen-specific CD4(+) T Cell Proliferation. Nanomed-Nanotechnol. 2016, 12, 1815–1826. [Google Scholar] [CrossRef] [PubMed]
- Falahati, M.; Hasan, A.; Zeinabad, H.A.; Serpooshan, V.; von der Thüsen, J.H.; ten Hagen, T.L.M. Engineering of pulmonary surfactant corona on inhaled nanoparticles to operate in the lung system. Nano Today 2023, 52, 101998. [Google Scholar] [CrossRef]
- Garcia-Mouton, C.; Hidalgo, A.; Cruz, A.; Perez-Gil, J. The Lord of the Lungs: The essential role of pulmonary surfactant upon inhalation of nanoparticles. Eur. J. Pharm. Biopharm. 2019, 144, 230–243. [Google Scholar] [CrossRef]
- Liu, Y.; Shi, L.; Su, L.; van der Mei, H.C.; Jutte, P.C.; Ren, Y.; Busscher, H.J. Nanotechnology-based antimicrobials and delivery systems for biofilm-infection control. Chem. Soc. Rev. 2019, 48, 428–446. [Google Scholar] [CrossRef]
- Peulen, T.-O.; Wilkinson, K.J. Diffusion of Nanoparticles in a Biofilm. Environ. Sci. Technol. 2011, 45, 3367–3373. [Google Scholar] [CrossRef]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Zou, X.; Zhang, L.; Wang, Z.; Luo, Y. Mechanisms of the Antimicrobial Activities of Graphene Materials. J. Am. Chem. Soc. 2016, 138, 2064–2077. [Google Scholar] [CrossRef] [PubMed]
- Elbourne, A.; Coyle, V.E.; Truong, V.K.; Sabri, Y.M.; Kandjani, A.E.; Bhargava, S.K.; Ivanova, E.P.; Crawford, R.J. Multi-directional electrodeposited gold nanospikes for antibacterial surface applications. Nanoscale Adv. 2019, 1, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Elahi, N.; Kamali, M.; Baghersad, M.H. Recent biomedical applications of gold nanoparticles: A review. Talanta 2018, 184, 537–556. [Google Scholar] [CrossRef] [PubMed]
- Nicolae-Maranciuc, A.; Chicea, D.; Chicea, L.M. Ag Nanoparticles for Biomedical Applications—Synthesis and Characterization—A Review. Int. J. Mol. Sci. 2022, 23, 5778. [Google Scholar] [CrossRef]
- Jamkhande, P.G.; Ghule, N.W.; Bamer, A.H.; Kalaskar, M.G. Metal nanoparticles synthesis: An overview on methods of preparation, advantages and disadvantages, and applications. J. Drug Deliv. Sci. Technol. 2019, 53, 101174. [Google Scholar] [CrossRef]
- Rani, N.; Singh, P.; Kumar, S.; Kumar, P.; Bhankar, V.; Kumar, K. Plant-mediated synthesis of nanoparticles and their applications: A review. Mater. Res. Bull. 2023, 163, 112233. [Google Scholar] [CrossRef]
- Jadoun, S.; Chauhan, N.P.S.; Zarrintaj, P.; Barani, M.; Varma, R.S.; Chinnam, S.; Rahdar, A. Synthesis of nanoparticles using microorganisms and their applications: A review. Environ. Chem. Lett. 2022, 20, 3153–3197. [Google Scholar] [CrossRef]
- Khan, F.; Shahid, A.; Zhu, H.; Wang, N.; Javed, M.R.; Ahmad, N.; Xu, J.; Alam, M.A.; Mehmood, M.A. Prospects of algae-based green synthesis of nanoparticles for environmental applications. Chemosphere 2022, 293, 133571. [Google Scholar] [CrossRef]
- Kashyap, M.; Samadhiya, K.; Ghosh, A.; Anand, V.; Lee, H.; Sawamoto, N.; Ogura, A.; Ohshita, Y.; Shirage, P.M.; Bala, K. Synthesis, characterization and application of intracellular Ag/AgCl nanohybrids biosynthesized in Scenedesmus sp. as neutral lipid inducer and antibacterial agent. Environ. Res. 2021, 201, 111499. [Google Scholar] [CrossRef]
- Zheng, K.; Setyawati, M.I.; Leong, D.T.; Xie, J. Antimicrobial silver nanomaterials. Coord. Chem. Rev. 2018, 357, 1–17. [Google Scholar] [CrossRef]
- Mozafari, M.R.; Torkaman, S.; Karamouzian, F.M.; Rasti, B.; Baral, B. Antimicrobial Applications of Nanoliposome Encapsulated Silver Nanoparticles: A Potential Strategy to Overcome Bacterial Resistance. Curr. Nanosci. 2021, 17, 26–40. [Google Scholar] [CrossRef]
- Mirzajani, F.; Ghassempour, A.; Aliahmadi, A.; Esmaeili, M.A. Antibacterial effect of silver nanoparticles on Staphylococcus aureus. Res. Microbiol. 2011, 162, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Khan, H.M.; Khan, A.A.; Ahmad, M.K.; Mahdi, A.A.; Pal, R.; Cameotra, S.S. Interaction of silver nanoparticles with Escherichia coli and their cell envelope biomolecules. J. Basic. Microbiol. 2014, 54, 905–915. [Google Scholar] [CrossRef]
- Azizi-Lalabadi, M.; Garavand, F.; Jafari, S.M. Incorporation of silver nanoparticles into active antimicrobial nanocomposites: Release behavior, analyzing techniques, applications and safety issues. Adv. Colloid. Interfac. 2021, 293, 102440. [Google Scholar] [CrossRef]
- Haidari, H.; Bright, R.; Kopecki, Z.; Zilm, P.S.; Garg, S.; Cowin, A.J.; Vasilev, K.; Goswami, N. Polycationic Silver Nanoclusters Comprising Nanoreservoirs of Ag(+) Ions with High Antimicrobial and Antibiofilm Activity. ACS Appl. Mater. Interfaces 2022, 14, 390–403. [Google Scholar] [CrossRef]
- Wang, Y.; Kadiyala, U.; Qu, Z.; Elvati, P.; Altheim, C.; Kotov, N.A.; Violi, A.; VanEpps, J.S. Anti-Biofilm Activity of Graphene Quantum Dots via Self-Assembly with Bacterial Amyloid Proteins. ACS Nano 2019, 13, 4278–4289. [Google Scholar] [CrossRef]
- Ma, J.; Cheng, X.; Xu, Z.; Zhang, Y.; Valle, J.; Fan, S.; Zuo, X.; Lasa, I.; Fang, X. Structural mechanism for modulation of functional amyloid and biofilm formation by Staphylococcal Bap protein switch. EMBO J. 2021, 40, e107500. [Google Scholar] [CrossRef]
- Nicastro, L.; Tukel, C. Bacterial Amyloids: The Link between Bacterial Infections and Autoimmunity. Trends Microbiol. 2019, 27, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Dizaj, S.M.; Lotfipour, F.; Barzegar-Jalali, M.; Zarrintan, M.H.; Adibkia, K. Antimicrobial activity of the metals and metal oxide nanoparticles. Mater. Sci. Eng. C Mater. Biol. Appl. 2014, 44, 278–284. [Google Scholar] [CrossRef]
- Cui, Y.; Zhao, Y.; Tian, Y.; Zhang, W.; Lu, X.; Jiang, X. The molecular mechanism of action of bactericidal gold nanoparticles on Escherichia coli. Biomaterials 2012, 33, 2327–2333. [Google Scholar] [CrossRef]
- Zhang, C.; Shi, D.T.; Yan, K.C.; Sedgwick, A.C.; Chen, G.R.; He, X.P.; James, T.D.; Ye, B.; Hu, X.L.; Chen, D. A glycoconjugate-based gold nanoparticle approach for the targeted treatment of Pseudomonas aeruginosa biofilms. Nanoscale 2020, 12, 23234–23240. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Jin, Y.Y.; Chen, W.; Wang, J.J.; Chen, H.; Sun, L.; Li, X.; Ji, J.; Yu, Q.; Shen, L.Y.; et al. Construction of nanomaterials with targeting phototherapy properties to inhibit resistant bacteria and biofilm infections. Chem. Eng. J. 2019, 358, 74–90. [Google Scholar] [CrossRef]
- Cui, T.; Wu, S.; Sun, Y.; Ren, J.; Qu, X. Self-Propelled Active Photothermal Nanoswimmer for Deep-Layered Elimination of Biofilm In Vivo. Nano Lett. 2020, 20, 7350–7358. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Martinez, J.C.; Martinez-Castanon, G.A.; Martinez-Gutierrez, F.; Zavala-Alonso, N.V.; Patino-Marin, N.; Nino-Martinez, N.; Zaragoza-Magana, V.; Cabral-Romero, C. Antibacterial and Antibiofilm Activities of the Photothermal Therapy Using Gold Nanorods against Seven Different Bacterial Strains. J. Nanomater. 2015, 2015, 783671. [Google Scholar] [CrossRef]
- Khantamat, O.; Li, C.H.; Yu, F.; Jamison, A.C.; Shih, W.C.; Cai, C.Z.; Lee, T.R. Gold Nanoshell-Decorated Silicone Surfaces for the Near-Infrared (NIR) Photothermal Destruction of the Pathogenic Bacterium E. faecalis. Acs Appl. Mater. Interfaces 2015, 7, 3981–3993. [Google Scholar] [CrossRef]
- Jo, W.; Kim, M.J. Influence of the photothermal effect of a gold nanorod cluster on biofilm disinfection. Nanotechnology 2013, 24, 195104. [Google Scholar] [CrossRef]
- Dong, X.W.; Ye, J.; Chen, Y.; Tanziela, T.; Jiang, H.; Wang, X.M. Intelligent peptide-nanorods against drug-resistant bacterial infection and promote wound healing by mild-temperature photothermal therapy. Chem. Eng. J. 2022, 432, 134061. [Google Scholar] [CrossRef]
- Fang, J.; Wan, Y.; Sun, Y.; Sun, X.L.; Qi, M.L.; Cheng, S.; Li, C.Y.; Zhou, Y.M.; Xu, L.; Dong, B.; et al. Near-infrared-activated nanohybrid coating with black phosphorus/zinc oxide for efficient biofilm eradication against implant-associated infections. Chem. Eng. J. 2022, 435, 134935. [Google Scholar] [CrossRef]
- Zhuang, Q.Q.; Deng, Q.; He, S.B.; Chen, Q.Q.; Peng, H.P.; Deng, H.H.; Xia, X.H.; Chen, W. Bifunctional cupric oxide nanoparticle-catalyzed self-cascade oxidation reactions of ascorbic acid for bacterial killing and wound disinfection. Compos. Part B Eng. 2021, 222, 109074. [Google Scholar] [CrossRef]
- Mao, Z.; Li, X.; Wang, P.; Yan, H. Iron oxide nanoparticles for biomedical applications: An updated patent review (2015–2021). Expert. Opin. Ther. Pat. 2022, 32, 939–952. [Google Scholar] [CrossRef]
- Armijo, L.M.; Wawrzyniec, S.J.; Kopciuch, M.; Brandt, Y.I.; Rivera, A.C.; Withers, N.J.; Cook, N.C.; Huber, D.L.; Monson, T.C.; Smyth, H.D.C.; et al. Antibacterial activity of iron oxide, iron nitride, and tobramycin conjugated nanoparticles against Pseudomonas aeruginosa biofilms. J. Nanobiotechnol. 2020, 18, 35. [Google Scholar] [CrossRef] [PubMed]
- Flores-Lopez, L.Z.; Espinoza-Gomez, H.; Somanathan, R. Silver nanoparticles: Electron transfer, reactive oxygen species, oxidative stress, beneficial and toxicological effects. Mini review. J. Appl. Toxicol. 2019, 39, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Mann, R.; Holmes, A.; McNeilly, O.; Cavaliere, R.; Sotiriou, G.A.; Rice, S.A.; Gunawan, C. Evolution of biofilm-forming pathogenic bacteria in the presence of nanoparticles and antibiotic: Adaptation phenomena and cross-resistance. J. Nanobiotechnol. 2021, 19, 291. [Google Scholar] [CrossRef] [PubMed]
- Musielak, E.; Feliczak-Guzik, A.; Nowak, I. Synthesis and Potential Applications of Lipid Nanoparticles in Medicine. Materials 2022, 15, 682. [Google Scholar] [CrossRef]
- Baek, J.S.; Tan, C.H.; Ng, N.K.J.; Yeo, Y.P.; Rice, S.A.; Loo, S.C.J. A programmable lipid-polymer hybrid nanoparticle system for localized, sustained antibiotic delivery to Gram-positive and Gram-negative bacterial biofilms. Nanoscale Horiz. 2018, 3, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Wan, F.; Nylander, T.; Klodzinska, S.N.; Foged, C.; Yang, M.; Baldursdottir, S.G.; Nielsen, H.M. Lipid Shell-Enveloped Polymeric Nanoparticles with High Integrity of Lipid Shells Improve Mucus Penetration and Interaction with Cystic Fibrosis-Related Bacterial Biofilms. ACS Appl. Mater. Interfaces 2018, 10, 10678–10687. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.X.; Garrido-Maestu, A.; Jeong, K.C. Application, mode of action, and in vivo activity of chitosan and its micro and nanoparticles as antimicrobial agents: A review. Carbohyd. Polym. 2017, 176, 257–265. [Google Scholar] [CrossRef]
- Jha, R.; Mayanovic, R.A. A Review of the Preparation, Characterization, and Applications of Chitosan Nanoparticles in Nanomedicine. Nanomaterials 2023, 13, 1302. [Google Scholar] [CrossRef]
- Ma, S.; Moser, D.; Han, F.; Leonhard, M.; Schneider-Stickler, B.; Tan, Y.L. Preparation and antibiofilm studies of curcumin loaded chitosan nanoparticles against polymicrobial biofilms of Candida albicans and Staphylococcus aureus. Carbohydr. Polym. 2020, 241, 116254. [Google Scholar] [CrossRef]
- Marangon, C.A.; Martins, V.C.A.; Ling, M.H.; Melo, C.C.; Plepis, A.M.G.; Meyer, R.L.; Nitschke, M. Combination of Rhamnolipid and Chitosan in Nanoparticles Boosts Their Antimicrobial Efficacy. Acs Appl. Mater. Interfaces 2020, 12, 5488–5499. [Google Scholar] [CrossRef]
- Varshosaz, J. Dextran conjugates in drug delivery. Expert Opin. Drug Deliv. 2012, 9, 509–523. [Google Scholar] [CrossRef] [PubMed]
- Naha, P.C.; Liu, Y.; Hwang, G.; Huang, Y.; Gubara, S.; Jonnakuti, V.; Simon-Soro, A.; Kim, D.; Gao, L.Z.; Koo, H.; et al. Dextran-Coated Iron Oxide Nanoparticles as Biomimetic Catalysts for Localized and pH-Activated Biofilm Disruption. ACS Nano 2019, 13, 4960–4971. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Lu, Y.; Luo, Y. Recent advances in dextran-based drug delivery systems: From fabrication strategies to applications. Carbohydr. Polym. 2021, 264, 117999. [Google Scholar] [CrossRef]
- Barros, C.H.N.; Hiebner, D.W.; Fulaz, S.; Vitale, S.; Quinn, L.; Casey, E. Synthesis and self-assembly of curcumin-modified amphiphilic polymeric micelles with antibacterial activity. J. Nanobiotechnol. 2021, 19, 104. [Google Scholar] [CrossRef]
- Li, Y.R.; Wang, S.C.; Xing, Z.; Niu, Y.M.; Liao, Z.C.; Lu, Y.; Qiu, J.N.; Zhang, J.F.; Wang, C.M.; Dong, L. Destructing biofilms by cationic dextran through phase transition. Carbohydr. Polym. 2022, 279, 118778. [Google Scholar] [CrossRef]
- Hasannia, M.; Aliabadi, A.; Abnous, K.; Taghdisi, S.M.; Ramezani, M.; Alibolandi, M. Synthesis of block copolymers used in polymersome fabrication: Application in drug delivery. J. Control Release 2022, 341, 95–117. [Google Scholar] [CrossRef] [PubMed]
- Konai, M.M.; Bhattacharjee, B.; Ghosh, S.; Haldar, J. Recent Progress in Polymer Research to Tackle Infections and Antimicrobial Resistance. Biomacromolecules 2018, 19, 1888–1917. [Google Scholar] [CrossRef]
- Takahashi, H.; Nadres, E.T.; Kuroda, K. Cationic Amphiphilic Polymers with Antimicrobial Activity for Oral Care Applications: Eradication of S. mutans Biofilm. Biomacromolecules 2016, 18, 257–265. [Google Scholar] [CrossRef]
- Zhao, S.; Huang, W.; Wang, C.; Wang, Y.; Zhang, Y.; Ye, Z.; Zhang, J.; Deng, L.; Dong, A. Screening and Matching Amphiphilic Cationic Polymers for Efficient Antibiosis. Biomacromolecules 2020, 21, 5269–5281. [Google Scholar] [CrossRef]
- Vishwakarma, A.; Dang, F.; Ferrell, A.; Barton, H.A.; Joy, A. Peptidomimetic Polyurethanes Inhibit Bacterial Biofilm Formation and Disrupt Surface Established Biofilms. J. Am. Chem. Soc. 2021, 143, 9440–9449. [Google Scholar] [CrossRef]
- Fadaka, A.O.; Sibuyi, N.R.S.; Madiehe, A.M.; Meyer, M. Nanotechnology-Based Delivery Systems for Antimicrobial Peptides. Pharmaceutics 2021, 13, 1795. [Google Scholar] [CrossRef] [PubMed]
- Hiemstra, P.S.; Amatngalim, G.D.; van der Does, A.M.; Taube, C. Antimicrobial Peptides and Innate Lung Defenses: Role in Infectious and Noninfectious Lung Diseases and Therapeutic Applications. Chest 2016, 149, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Travkova, O.G.; Moehwald, H.; Brezesinski, G. The interaction of antimicrobial peptides with membranes. Adv. Colloid. Interface Sci. 2017, 247, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chang, R.Y.K.; Britton, W.J.; Chan, H.K. Advances in the development of antimicrobial peptides and proteins for inhaled therapy. Adv. Drug Deliv. Rev. 2022, 180, 114066. [Google Scholar] [CrossRef]
- Nordstrom, R.; Malmsten, M. Delivery systems for antimicrobial peptides. Adv. Colloid. Interface Sci. 2017, 242, 17–34. [Google Scholar] [CrossRef]
- Wang, C.; Hong, T.; Cui, P.; Wang, J.; Xia, J. Antimicrobial peptides towards clinical application: Delivery and formulation. Adv. Drug Deliv. Rev. 2021, 175, 113818. [Google Scholar] [CrossRef]
- Casciaro, B.; d’Angelo, I.; Zhang, X.; Loffredo, M.R.; Conte, G.; Cappiello, F.; Quaglia, F.; Di, Y.P.; Ungaro, F.; Mangoni, M.L. Poly(lactide- co-glycolide) Nanoparticles for Prolonged Therapeutic Efficacy of Esculentin-1a-Derived Antimicrobial Peptides against Pseudomonas aeruginosa Lung Infection: In Vitro and in Vivo Studies. Biomacromolecules 2019, 20, 1876–1888. [Google Scholar] [CrossRef]
- Falciani, C.; Zevolini, F.; Brunetti, J.; Riolo, G.; Gracia, R.; Marradi, M.; Loinaz, I.; Ziemann, C.; Cossio, U.; Llop, J.; et al. Antimicrobial Peptide-Loaded Nanoparticles as Inhalation Therapy for Pseudomonas aeruginosa Infections. Int. J. Nanomed. 2020, 15, 1117–1128. [Google Scholar] [CrossRef]
- Yu, M.; Chua, S.L. Demolishing the great wall of biofilms in Gram-negative bacteria: To disrupt or disperse? Med. Res. Rev. 2020, 40, 1103–1116. [Google Scholar] [CrossRef]
- Rivero Berti, I.; Islan, G.A.; Castro, G.R. Enzymes and biopolymers. The opportunity for the smart design of molecular delivery systems. Bioresour. Technol. 2021, 322, 124546. [Google Scholar] [CrossRef]
- d’Angelo, I.; Conte, C.; La Rotonda, M.I.; Miro, A.; Quaglia, F.; Ungaro, F. Improving the efficacy of inhaled drugs in cystic fibrosis: Challenges and emerging drug delivery strategies. Adv. Drug Deliv. Rev. 2014, 75, 92–111. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Ma, S.; Leonhard, M.; Moser, D.; Haselmann, G.M.; Wang, J.; Eder, D.; Schneider-Stickler, B. Enhancing antibiofilm activity with functional chitosan nanoparticles targeting biofilm cells and biofilm matrix. Carbohydr. Polym. 2018, 200, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Tasia, W.; Lei, C.; Cao, Y.; Ye, Q.; He, Y.; Xu, C. Enhanced eradication of bacterial biofilms with DNase I-loaded silver-doped mesoporous silica nanoparticles. Nanoscale 2020, 12, 2328–2332. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Yin, H. Alginate lyase: Review of major sources and classification, properties, structure-function analysis and applications. Bioengineered 2015, 6, 125–131. [Google Scholar] [CrossRef]
- Daboor, S.M.; Rohde, J.R.; Cheng, Z. Disruption of the extracellular polymeric network of Pseudomonas aeruginosa biofilms by alginate lyase enhances pathogen eradication by antibiotics. J. Cyst. Fibros. 2021, 20, 264–270. [Google Scholar] [CrossRef]
- Baker, P.; Hill, P.J.; Snarr, B.D.; Alnabelseya, N.; Pestrak, M.J.; Lee, M.J.; Jennings, L.K.; Tam, J.; Melnyk, R.A.; Parsek, M.R.; et al. Exopolysaccharide biosynthetic glycoside hydrolases can be utilized to disrupt and prevent Pseudomonas aeruginosa biofilms. Sci. Adv. 2016, 2, e1501632. [Google Scholar] [CrossRef]
- Thorn, C.R.; Raju, D.; Lacdao, I.; Gilbert, S.; Sivarajah, P.; Howell, P.L.; Prestidge, C.A.; Thomas, N. Protective Liquid Crystal Nanoparticles for Targeted Delivery of PslG: A Biofilm Dispersing Enzyme. ACS Infect. Dis. 2021, 7, 2102–2115. [Google Scholar] [CrossRef]
- Mukherjee, S.; Bassler, B.L. Bacterial quorum sensing in complex and dynamically changing environments. Nat. Rev. Microbiol. 2019, 17, 371–382. [Google Scholar] [CrossRef]
- Carradori, S.; Di Giacomo, N.; Lobefalo, M.; Luisi, G.; Campestre, C.; Sisto, F. Biofilm and Quorum Sensing inhibitors: The road so far. Expert Opin. Ther. Pat. 2020, 30, 917–930. [Google Scholar] [CrossRef]
- Duplantier, M.; Lohou, E.; Sonnet, P. Quorum Sensing Inhibitors to Quench P. aeruginosa Pathogenicity. Pharmaceuticals 2021, 14, 1262. [Google Scholar] [CrossRef]
- Zhao, X.; Yu, Z.; Ding, T. Quorum-Sensing Regulation of Antimicrobial Resistance in Bacteria. Microorganisms 2020, 8, 425. [Google Scholar] [CrossRef] [PubMed]
- Defoirdt, T. Quorum-Sensing Systems as Targets for Antivirulence Therapy. Trends Microbiol. 2018, 26, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Welsh, M.A.; Eibergen, N.R.; Moore, J.D.; Blackwell, H.E. Small molecule disruption of quorum sensing cross-regulation in Pseudomonas aeruginosa causes major and unexpected alterations to virulence phenotypes. J. Am. Chem. Soc. 2015, 137, 1510–1519. [Google Scholar] [CrossRef] [PubMed]
- Welsh, M.A.; Blackwell, H.E. Chemical probes of quorum sensing: From compound development to biological discovery. FEMS Microbiol. Rev. 2016, 40, 774–794. [Google Scholar] [CrossRef]
- Schutz, C.; Empting, M. Targeting the Pseudomonas quinolone signal quorum sensing system for the discovery of novel anti-infective pathoblockers. Beilstein J. Org. Chem. 2018, 14, 2627–2645. [Google Scholar] [CrossRef]
- Ho, D.K.; Murgia, X.; De Rossi, C.; Christmann, R.; Hufner de Mello Martins, A.G.; Koch, M.; Andreas, A.; Herrmann, J.; Muller, R.; Empting, M.; et al. Squalenyl Hydrogen Sulfate Nanoparticles for Simultaneous Delivery of Tobramycin and an Alkylquinolone Quorum Sensing Inhibitor Enable the Eradication of P. aeruginosa Biofilm Infections. Angew. Chem. Int. Ed. Engl. 2020, 59, 10292–10296. [Google Scholar] [CrossRef]
- Jenal, U.; Reinders, A.; Lori, C. Cyclic di-GMP: Second messenger extraordinaire. Nat. Rev. Microbiol. 2017, 15, 271–284. [Google Scholar] [CrossRef]
- Hengge, R. Principles of c-di-GMP signalling in bacteria. Nat. Rev. Microbiol. 2009, 7, 263–273. [Google Scholar] [CrossRef]
- Cai, Y.M.; Zhang, Y.D.; Yang, L. NO donors and NO delivery methods for controlling biofilms in chronic lung infections. Appl. Microbiol. Biotechnol. 2021, 105, 3931–3954. [Google Scholar] [CrossRef]
- Bartley, B.L.; Gardner, K.J.; Spina, S.; Hurley, B.P.; Campeau, D.; Berra, L.; Yonker, L.M.; Carroll, R.W. High-Dose Inhaled Nitric Oxide as Adjunct Therapy in Cystic Fibrosis Targeting Burkholderia multivorans. Case Rep. Pediat 2020, 2020, 1536714. [Google Scholar] [CrossRef]
- Miller, C.; Miller, M.; McMullin, B.; Regev, G.; Serghides, L.; Kain, K.; Road, J.; Av-Gay, Y. A phase I clinical study of inhaled nitric oxide in healthy adults. J. Cyst. Fibros. 2012, 11, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Deppisch, C.; Herrmann, G.; Graepler-Mainka, U.; Wirtz, H.; Heyder, S.; Engel, C.; Marschal, M.; Miller, C.C.; Riethmuller, J. Gaseous nitric oxide to treat antibiotic resistant bacterial and fungal lung infections in patients with cystic fibrosis: A phase I clinical study. Infection 2016, 44, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Poh, W.H.; Rice, S.A. Recent Developments in Nitric Oxide Donors and Delivery for Antimicrobial and Anti-Biofilm Applications. Molecules 2022, 27, 674. [Google Scholar] [CrossRef] [PubMed]
- Boce, M.; Tasse, M.; Mallet-Ladeira, S.; Pillet, F.; Da Silva, C.; Vicendo, P.; Lacroix, P.G.; Malfant, I.; Rols, M.P. Effect of trans(NO, OH)-[RuFT(Cl)(OH)NO](PF(6)) ruthenium nitrosyl complex on methicillin-resistant Staphylococcus epidermidis. Sci. Rep. 2019, 9, 4867. [Google Scholar] [CrossRef]
- Rineh, A.; Soren, O.; McEwan, T.; Ravikumar, V.; Poh, W.H.; Azamifar, F.; Naimi-Jamal, M.R.; Cheung, C.Y.; Elliott, A.G.; Zuegg, J.; et al. Discovery of Cephalosporin-3′-Diazeniumdiolates That Show Dual Antibacterial and Antibiofilm Effects against Pseudomonas aeruginosa Clinical Cystic Fibrosis Isolates and Efficacy in a Murine Respiratory Infection Model. ACS Infect. Dis. 2020, 6, 1460–1479. [Google Scholar] [CrossRef]
- Barraud, N.; Kardak, B.G.; Yepuri, N.R.; Howlin, R.P.; Webb, J.S.; Faust, S.N.; Kjelleberg, S.; Rice, S.A.; Kelso, M.J. Cephalosporin-3′-diazeniumdiolates: Targeted NO-donor prodrugs for dispersing bacterial biofilms. Angew. Chem. Int. Ed. Engl. 2012, 51, 9057–9060. [Google Scholar] [CrossRef]
- Adnan, N.N.M.; Sadrearhami, Z.; Bagheri, A.; Nguyen, T.K.; Wong, E.H.H.; Ho, K.K.K.; Lim, M.; Kumar, N.; Boyer, C. Exploiting the Versatility of Polydopamine-Coated Nanoparticles to Deliver Nitric Oxide and Combat Bacterial Biofilm. Macromol. Rapid Commun. 2018, 39, e1800159. [Google Scholar] [CrossRef]
- Nguyen, T.K.; Selvanayagam, R.; Ho, K.K.K.; Chen, R.; Kutty, S.K.; Rice, S.A.; Kumar, N.; Barraud, N.; Duong, H.T.T.; Boyer, C. Co-delivery of nitric oxide and antibiotic using polymeric nanoparticles. Chem. Sci. 2016, 7, 1016–1027. [Google Scholar] [CrossRef]
- Shen, Z.Q.; He, K.W.; Ding, Z.L.; Zhang, M.D.; Yu, Y.Q.; Hu, J.M. Visible-Light-Triggered Self-Reporting Release of Nitric Oxide (NO) for Bacterial Biofilm Dispersal. Macromolecules 2019, 52, 7668–7677. [Google Scholar] [CrossRef]
- Taccetti, G.; Francalanci, M.; Pizzamiglio, G.; Messore, B.; Carnovale, V.; Cimino, G.; Cipolli, M. Cystic Fibrosis: Recent Insights into Inhaled Antibiotic Treatment and Future Perspectives. Antibiotics 2021, 10, 338. [Google Scholar] [CrossRef]
- Suk, J.S.; Lai, S.K.; Boylan, N.J.; Dawson, M.R.; Boyle, M.P.; Hanes, J. Rapid transport of muco-inert nanoparticles in cystic fibrosis sputum treated with N-acetyl cysteine. Nanomedicine 2011, 6, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Ensign, L.M.; Schneider, C.; Suk, J.S.; Cone, R.; Hanes, J. Mucus penetrating nanoparticles: Biophysical tool and method of drug and gene delivery. Adv. Mater. 2012, 24, 3887–3894. [Google Scholar] [CrossRef] [PubMed]
- Huckaby, J.T.; Lai, S.K. PEGylation for enhancing nanoparticle diffusion in mucus. Adv. Drug Deliv. Rev. 2018, 124, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Suk, J.S.; Xu, Q.G.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef]
- Ernst, J.; Klinger-Strobel, M.; Arnold, K.; Thamm, J.; Hartung, A.; Pletz, M.W.; Makarewicz, O.; Fischer, D. Polyester-based particles to overcome the obstacles of mucus and biofilms in the lung for tobramycin application under static and dynamic fluidic conditions. Eur. J. Pharm. Biopharm. 2018, 131, 120–129. [Google Scholar] [CrossRef]
- Porsio, B.; Craparo, E.F.; Mauro, N.; Giammona, G.; Cavallaro, G. Mucus and Cell-Penetrating Nanoparticles Embedded in Nano-into-Micro Formulations for Pulmonary Delivery of Ivacaftor in Patients with Cystic Fibrosis. ACS Appl. Mater. Interfaces 2018, 10, 165–181. [Google Scholar] [CrossRef]
- Khutoryanskiy, V.V. Beyond PEGylation: Alternative surface-modification of nanoparticles with mucus-inert biomaterials. Adv. Drug Deliv. Rev. 2018, 124, 140–149. [Google Scholar] [CrossRef]
- Hu, S.S.; Yang, Z.X.; Wang, S.; Wang, L.P.; He, Q.Q.; Tang, H.; Ji, P.; Chen, T. Zwitterionic polydopamine modified nanoparticles as an efficient nanoplatform to overcome both the mucus and epithelial barriers. Chem. Eng. J. 2022, 428, 132107. [Google Scholar] [CrossRef]
- Islan, G.A.; Ruiz, M.E.; Morales, J.F.; Sbaraglini, M.L.; Enrique, A.V.; Burton, G.; Talevi, A.; Bruno-Blanch, L.E.; Castro, G.R. Hybrid inhalable microparticles for dual controlled release of levofloxacin and DNase: Physicochemical characterization and in vivo targeted delivery to the lungs. J. Mater. Chem. B 2017, 5, 3132–3144. [Google Scholar] [CrossRef]
- Suk, J.S.; Boylan, N.J.; Trehan, K.; Tang, B.C.; Schneider, C.S.; Lin, J.M.; Boyle, M.P.; Zeitlin, P.L.; Lai, S.K.; Cooper, M.J.; et al. N-acetylcysteine enhances cystic fibrosis sputum penetration and airway gene transfer by highly compacted DNA nanoparticles. Mol. Ther. 2011, 19, 1981–1989. [Google Scholar] [CrossRef]
- Deacon, J.; Abdelghany, S.M.; Quinn, D.J.; Schmid, D.; Megaw, J.; Donnelly, R.F.; Jones, D.S.; Kissenpfennig, A.; Elborn, J.S.; Gilmore, B.F.; et al. Antimicrobial efficacy of tobramycin polymeric nanoparticles for Pseudomonas aeruginosa infections in cystic fibrosis: Formulation, characterisation and functionalisation with dornase alfa (DNase). J. Control Release 2015, 198, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Lababidi, N.; Ofosu Kissi, E.; Elgaher, W.A.M.; Sigal, V.; Haupenthal, J.; Schwarz, B.C.; Hirsch, A.K.H.; Rades, T.; Schneider, M. Spray-drying of inhalable, multifunctional formulations for the treatment of biofilms formed in cystic fibrosis. J. Control Release 2019, 314, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Pinto, R.M.; Monteiro, C.; Costa Lima, S.A.; Casal, S.; Van Dijck, P.; Martins, M.C.L.; Nunes, C.; Reis, S. N-Acetyl-l-cysteine-Loaded Nanosystems as a Promising Therapeutic Approach Toward the Eradication of Pseudomonas aeruginosa Biofilms. ACS Appl. Mater. Interfaces 2021, 13, 42329–42343. [Google Scholar] [CrossRef] [PubMed]
- Morgan, L.E.; Jaramillo, A.M.; Shenoy, S.K.; Raclawska, D.; Emezienna, N.A.; Richardson, V.L.; Hara, N.; Harder, A.Q.; NeeDell, J.C.; Hennessy, C.E.; et al. Disulfide disruption reverses mucus dysfunction in allergic airway disease. Nat. Commun. 2021, 12, 249. [Google Scholar] [CrossRef] [PubMed]
- Ehre, C.; Rushton, Z.L.; Wang, B.; Hothem, L.N.; Morrison, C.B.; Fontana, N.C.; Markovetz, M.R.; Delion, M.F.; Kato, T.; Villalon, D.; et al. An Improved Inhaled Mucolytic to Treat Airway Muco-obstructive Diseases. Am. J. Respir. Crit. Care Med. 2019, 199, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Cazzola, M.; Calzetta, L.; Page, C.; Rogliani, P.; Matera, M.G. Thiol-Based Drugs in Pulmonary Medicine: Much More than Mucolytics. Trends Pharmacol. Sci. 2019, 40, 452–463. [Google Scholar] [CrossRef]
- Menzel, C.; Bernkop-Schnurch, A. Enzyme decorated drug carriers: Targeted swords to cleave and overcome the mucus barrier. Adv. Drug Deliv. Rev. 2018, 124, 164–174. [Google Scholar] [CrossRef]
- Hu, Y.L.; Ruan, X.H.; Lv, X.Y.; Xu, Y.; Wang, W.J.; Cai, Y.; Ding, M.; Dong, H.; Shao, J.J.; Yang, D.L.; et al. Biofilm microenvironment-responsive nanoparticles for the treatment of bacterial infection. Nano Today 2022, 46, 101602. [Google Scholar] [CrossRef]
- Chen, X.; Guo, R.; Wang, C.; Li, K.; Jiang, X.; He, H.; Hong, W. On-demand pH-sensitive surface charge-switchable polymeric micelles for targeting Pseudomonas aeruginosa biofilms development. J. Nanobiotechnol. 2021, 19, 99. [Google Scholar] [CrossRef]
- Chen, M.; Wei, J.; Xie, S.; Tao, X.; Zhang, Z.; Ran, P.; Li, X. Bacterial biofilm destruction by size/surface charge-adaptive micelles. Nanoscale 2019, 11, 1410–1422. [Google Scholar] [CrossRef]
- Fan, Q.; Wang, C.; Guo, R.; Jiang, X.; Li, W.; Chen, X.; Li, K.; Hong, W. Step-by-step dual stimuli-responsive nanoparticles for efficient bacterial biofilm eradication. Biomater. Sci. 2021, 9, 6889–6902. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Busscher, H.J.; Zhao, B.; Li, Y.; Zhang, Z.; van der Mei, H.C.; Ren, Y.; Shi, L. Surface-Adaptive, Antimicrobially Loaded, Micellar Nanocarriers with Enhanced Penetration and Killing Efficiency in Staphylococcal Biofilms. ACS Nano 2016, 10, 4779–4789. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.H.; Qiao, Z.Z.; Yan, D.P.; Yang, M.; Yang, L.J.; Wan, X.H.; Chen, H.L.; Luo, J.B.; Xiao, H.N. Ciprofloxacin conjugated gold nanorods with pH induced surface charge transformable activities to combat drug resistant bacteria and their biofilms. Mater. Sci. Eng. C-Mater. 2021, 128, 112292. [Google Scholar] [CrossRef]
- Li, B.; Mao, J.; Wu, J.; Mao, K.; Jia, Y.; Chen, F.; Liu, J. Nano—Bio Interactions: Biofilm—Targeted Antibacterial Nanomaterials. Small 2023, 2306135. [Google Scholar] [CrossRef] [PubMed]
- Wan, F.; Bohr, S.S.; Klodzinska, S.N.; Jumaa, H.; Huang, Z.; Nylander, T.; Thygesen, M.B.; Sorensen, K.K.; Jensen, K.J.; Sternberg, C.; et al. Ultrasmall TPGS-PLGA Hybrid Nanoparticles for Site-Specific Delivery of Antibiotics into Pseudomonas aeruginosa Biofilms in Lungs. ACS Appl. Mater. Interfaces 2020, 12, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yuan, Q.; Feng, W.; Pu, W.; Ding, J.; Zhang, H.; Li, X.; Yang, B.; Dai, Q.; Cheng, L.; et al. Targeted delivery of antibiotics to the infected pulmonary tissues using ROS-responsive nanoparticles. J. Nanobiotechnol. 2019, 17, 103. [Google Scholar] [CrossRef]
- Ye, M.; Zhao, Y.; Wang, Y.; Zhao, M.; Yodsanit, N.; Xie, R.; Andes, D.; Gong, S. A Dual-Responsive Antibiotic-Loaded Nanoparticle Specifically Binds Pathogens and Overcomes Antimicrobial-Resistant Infections. Adv. Mater. 2021, 33, e2006772. [Google Scholar] [CrossRef]
- Macia, M.D.; Rojo-Molinero, E.; Oliver, A. Antimicrobial susceptibility testing in biofilm-growing bacteria. Clin. Microbiol. Infect. 2014, 20, 981–990. [Google Scholar] [CrossRef]
- Hasan, S.; Albayaty, Y.N.S.; Thierry, B.; Prestidge, C.A.; Thomas, N. Mechanistic studies of the antibiofilm activity and synergy with antibiotics of isosorbide mononitrate. Eur. J. Pharm. Sci. 2018, 115, 50–56. [Google Scholar] [CrossRef]
- Di Bonaventura, G.; Pompilio, A. In Vitro Antimicrobial Susceptibility Testing of Biofilm-Growing Bacteria: Current and Emerging Methods. Adv. Exp. Med. Biol. 2022, 1369, 33–51. [Google Scholar] [CrossRef] [PubMed]
- Haagensen, J.A.; Regenberg, B.; Sternberg, C. Advanced microscopy of microbial cells. Adv. Biochem. Eng. Biotechnol. 2011, 124, 21–54. [Google Scholar] [CrossRef] [PubMed]
- Diaz Iglesias, Y.; Wilms, T.; Vanbever, R.; Van Bambeke, F. Activity of Antibiotics against Staphylococcus aureus in an In Vitro Model of Biofilms in the Context of Cystic Fibrosis: Influence of the Culture Medium. Antimicrob. Agents Chemother. 2019, 63, 10–128. [Google Scholar] [CrossRef]
- Crabbe, A.; Liu, Y.; Matthijs, N.; Rigole, P.; De La Fuente-Nunez, C.; Davis, R.; Ledesma, M.A.; Sarker, S.; Van Houdt, R.; Hancock, R.E.; et al. Antimicrobial efficacy against Pseudomonas aeruginosa biofilm formation in a three-dimensional lung epithelial model and the influence of fetal bovine serum. Sci. Rep. 2017, 7, 43321. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.; Muruli, A.; Higgins, S.; Diggle, S.P. Development of an ex vivo porcine lung model for studying growth, virulence, and signaling of Pseudomonas aeruginosa. Infect. Immun. 2014, 82, 3312–3323. [Google Scholar] [CrossRef] [PubMed]
- Doryab, A.; Groll, J. Biomimetic In Vitro Lung Models: Current Challenges and Future Perspective. Adv. Mater. 2023, 35, e2210519. [Google Scholar] [CrossRef]
- Ning, E.; Turnbull, G.; Clarke, J.; Picard, F.; Riches, P.; Vendrell, M.; Graham, D.; Wark, A.W.; Faulds, K.; Shu, W. 3D bioprinting of mature bacterial biofilms for antimicrobial resistance drug testing. Biofabrication 2019, 11, 045018. [Google Scholar] [CrossRef]
- Huh, D.; Kim, H.J.; Fraser, J.P.; Shea, D.E.; Khan, M.; Bahinski, A.; Hamilton, G.A.; Ingber, D.E. Microfabrication of human organs-on-chips. Nat. Protoc. 2013, 8, 2135–2157. [Google Scholar] [CrossRef]
- Kukavica-Ibrulj, I.; Bragonzi, A.; Paroni, M.; Winstanley, C.; Sanschagrin, F.o.; O’Toole, G.A.; Levesque, R.C. In Vivo Growth of Pseudomonas aeruginosa Strains PAO1 and PA14 and the Hypervirulent Strain LESB58 in a Rat Model of Chronic Lung Infection. J. Bacteriol. 2008, 190, 2804–2813. [Google Scholar] [CrossRef]
- Hu, D.; Deng, Y.; Jia, F.; Jin, Q.; Ji, J. Surface Charge Switchable Supramolecular Nanocarriers for Nitric Oxide Synergistic Photodynamic Eradication of Biofilms. ACS Nano 2020, 14, 347–359. [Google Scholar] [CrossRef]
- Chen, S.; Huang, X.; Xue, Y.; Álvarez-Benedicto, E.; Shi, Y.; Chen, W.; Koo, S.; Siegwart, D.J.; Dong, Y.; Tao, W. Nanotechnology-based mRNA vaccines. Nat. Rev. Methods Primers 2023, 3, 63. [Google Scholar] [CrossRef]
- Lotz, O.; McKenzie, D.R.; Bilek, M.M.; Akhavan, B. Biofunctionalized 3D printed structures for biomedical applications: A critical review of recent advances and future prospects. Prog. Mater. Sci. 2023, 137, 101124. [Google Scholar] [CrossRef]
- Tabatabaei Rezaei, N.; Kumar, H.; Liu, H.; Lee, S.S.; Park, S.S.; Kim, K. Recent Advances in Organ-on-Chips Integrated with Bioprinting Technologies for Drug Screening. Adv. Healthc. Mater. 2023, 12, e2203172. [Google Scholar] [CrossRef]
- D’Angelo, F.; Baldelli, V.; Halliday, N.; Pantalone, P.; Polticelli, F.; Fiscarelli, E.; Williams, P.; Visca, P.; Leoni, L.; Rampioni, G. Identification of FDA-Approved Drugs as Antivirulence Agents Targeting the pqs Quorum-Sensing System of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2018, 62, 10–128. [Google Scholar] [CrossRef] [PubMed]
- Collalto, D.; Giallonardi, G.; Fortuna, A.; Meneghini, C.; Fiscarelli, E.; Visca, P.; Imperi, F.; Rampioni, G.; Leoni, L. In vitro Activity of Antivirulence Drugs Targeting the las or pqs Quorum Sensing Against Cystic Fibrosis Pseudomonas aeruginosa Isolates. Front. Microbiol. 2022, 13, 845231. [Google Scholar] [CrossRef] [PubMed]
- Miro-Canturri, A.; Ayerbe-Algaba, R.; Smani, Y. Drug Repurposing for the Treatment of Bacterial and Fungal Infections. Front. Microbiol. 2019, 10, 41. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Microorganism | Associated Disease | References |
|---|---|---|---|
| Bacteria G+ | Staphylococcus aureus | Ventilator-associated pneumonia (VAP), CF | [35,36] |
| Streptococcus pneumoniae | Invasive pneumococcal disease | [37] | |
| Nocardia | Pulmonary nocardiosis | [38] | |
| Bacteria G− | Pseudomonas aeruginosa | CF, COPD | [39,40] |
| Escherichia coli | VAP | [41] | |
| Salmonella | Typhoid fever | [42] | |
| Klebsiella pneumoniae | COPD | [43] | |
| Haemophilus influenza | COPD | [44] | |
| Fungi | Aspergillus fumigatus | Aspergillosis | [45,46] |
| Blastomyces dermatitidis | Blastomycosis | [47] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Mao, Z.; Ran, F.; Sun, J.; Zhang, J.; Chai, G.; Wang, J. Nanotechnology-Based Drug Delivery Systems to Control Bacterial-Biofilm-Associated Lung Infections. Pharmaceutics 2023, 15, 2582. https://doi.org/10.3390/pharmaceutics15112582
Guo Y, Mao Z, Ran F, Sun J, Zhang J, Chai G, Wang J. Nanotechnology-Based Drug Delivery Systems to Control Bacterial-Biofilm-Associated Lung Infections. Pharmaceutics. 2023; 15(11):2582. https://doi.org/10.3390/pharmaceutics15112582
Chicago/Turabian StyleGuo, Yutong, Zeyuan Mao, Fang Ran, Jihong Sun, Jingfeng Zhang, Guihong Chai, and Jian Wang. 2023. "Nanotechnology-Based Drug Delivery Systems to Control Bacterial-Biofilm-Associated Lung Infections" Pharmaceutics 15, no. 11: 2582. https://doi.org/10.3390/pharmaceutics15112582