
Targeting Histone Deacetylases 6 in Dual-Target Therapy of Cancer
 , , , ,
, , , ,  ,
,  and
and
Abstract
:1. Introduction
- (1)
- Increasing the selectivity toward one HDAC isoform (HDAC6) among others with the goal of reducing adverse effects;
- (2)
- Developing dual-target HDAC inhibitors in order to increase the efficacy and decrease the dose of HDAC6 inhibitors due to synergistic and additive effects.
- (1)
- (2)
- (3)
- (4)
2. Multi-Target Therapy as an Approach in Cancer Treatment
3. Dual HDAC6 Inhibitors
3.1. Dual HDAC6/PI3K Inhibitors
3.2. Dual HDAC6/mTOR Inhibitors
3.3. Dual HDAC6/BRD4 Inhibitors
3.4. Dual HDAC6/AR Inhibitors
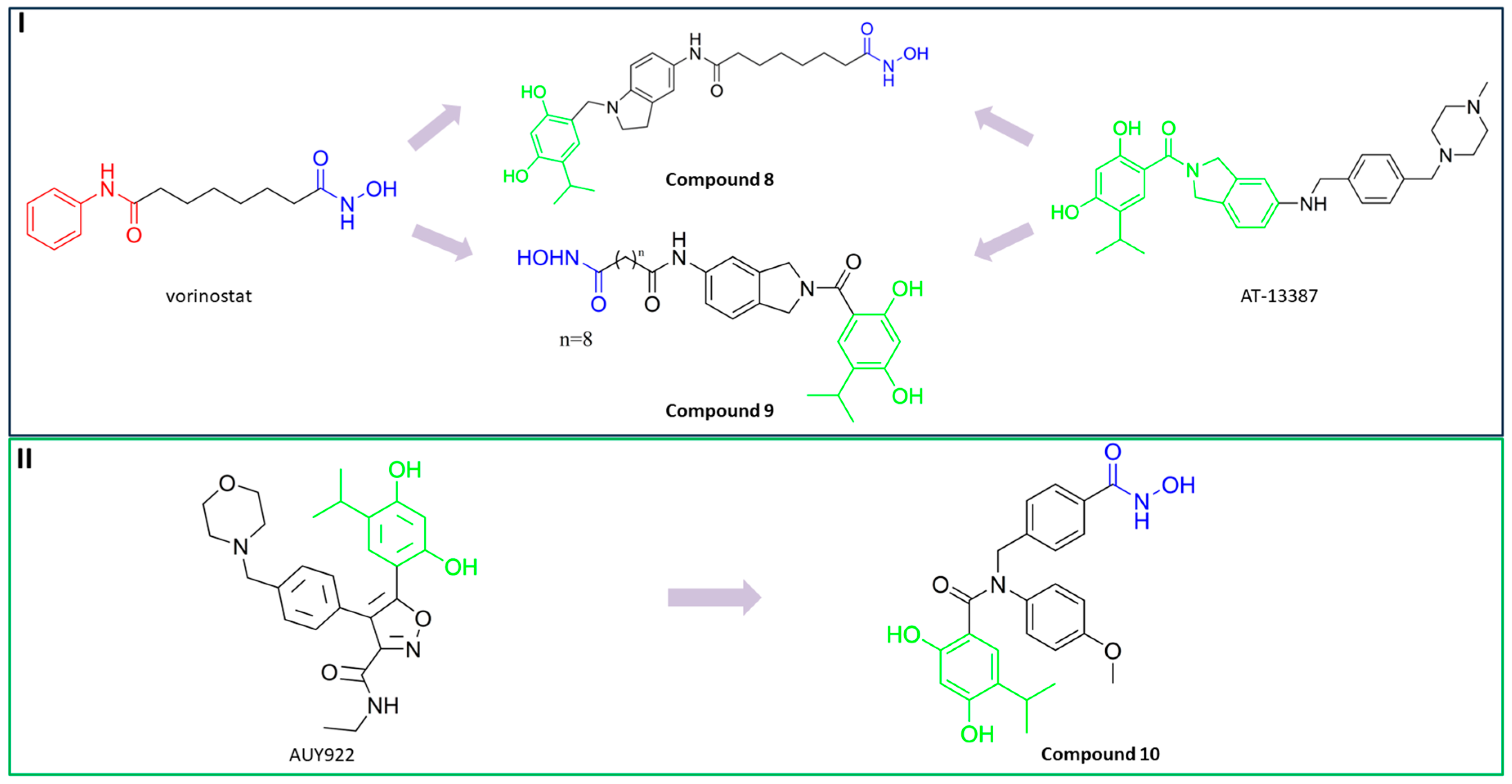
3.5. Dual HDAC6/HSP90 Inhibitors
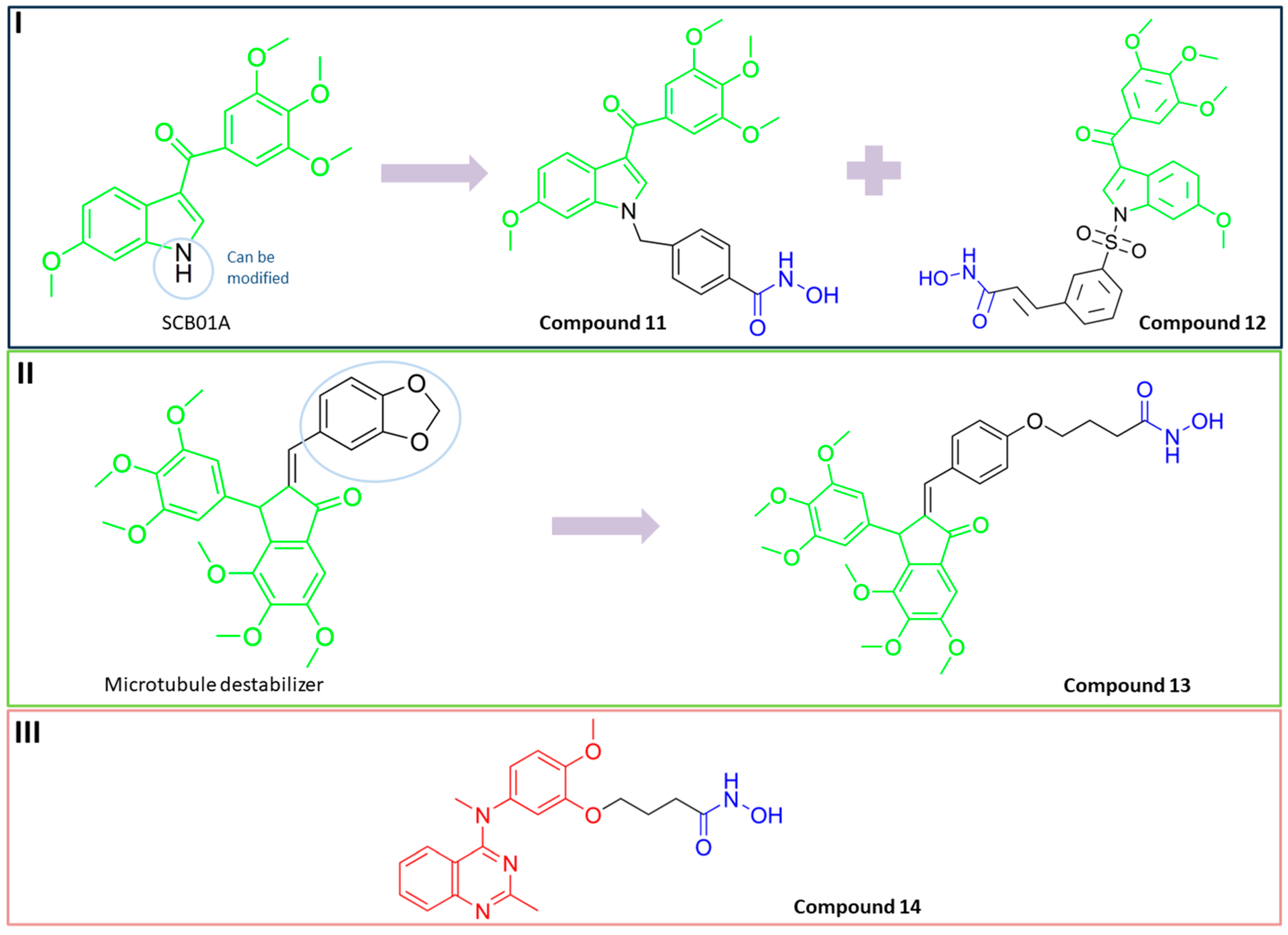
3.6. Dual HDAC6/Tubulin Inhibitors
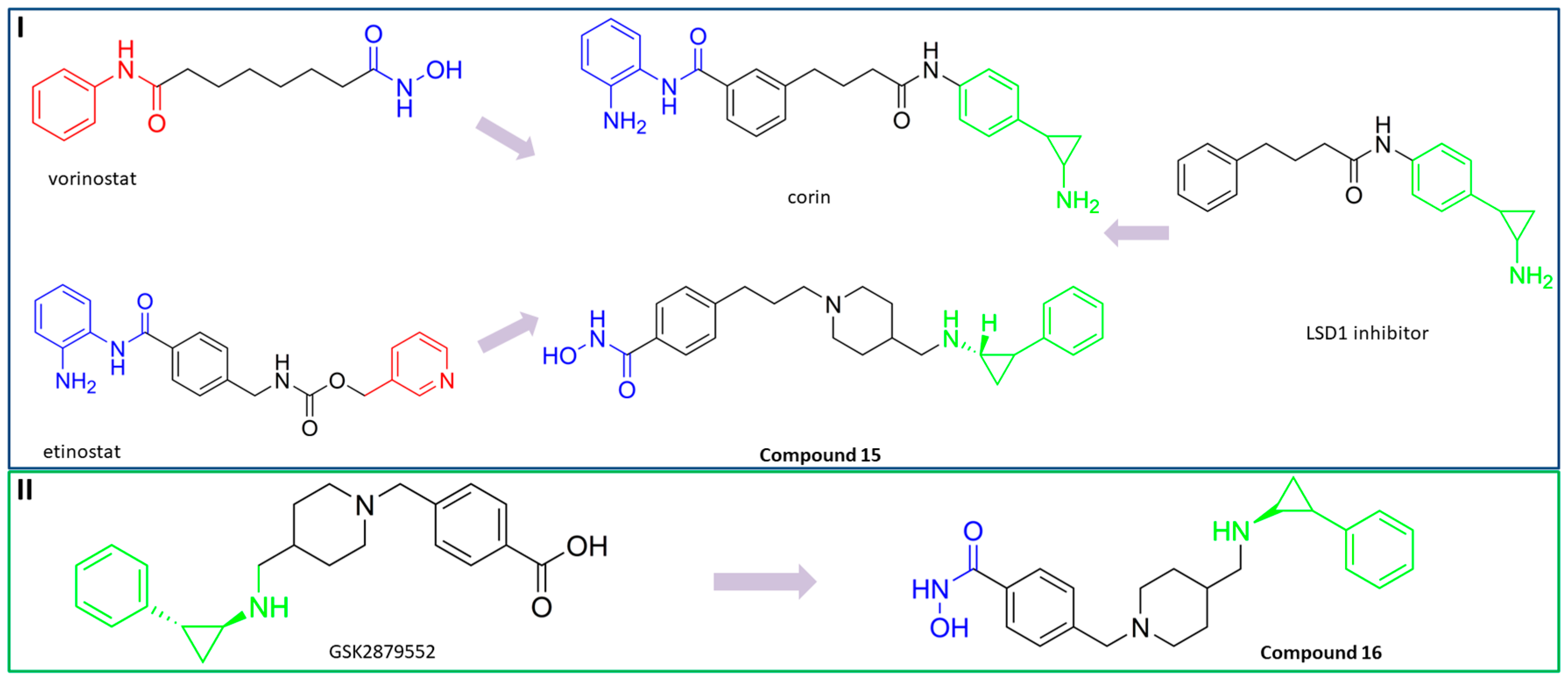
3.7. Dual HDAC6/LSD1 Inhibitors
3.8. Dual HDAC6/1, HDAC6/3, HDAC6/8
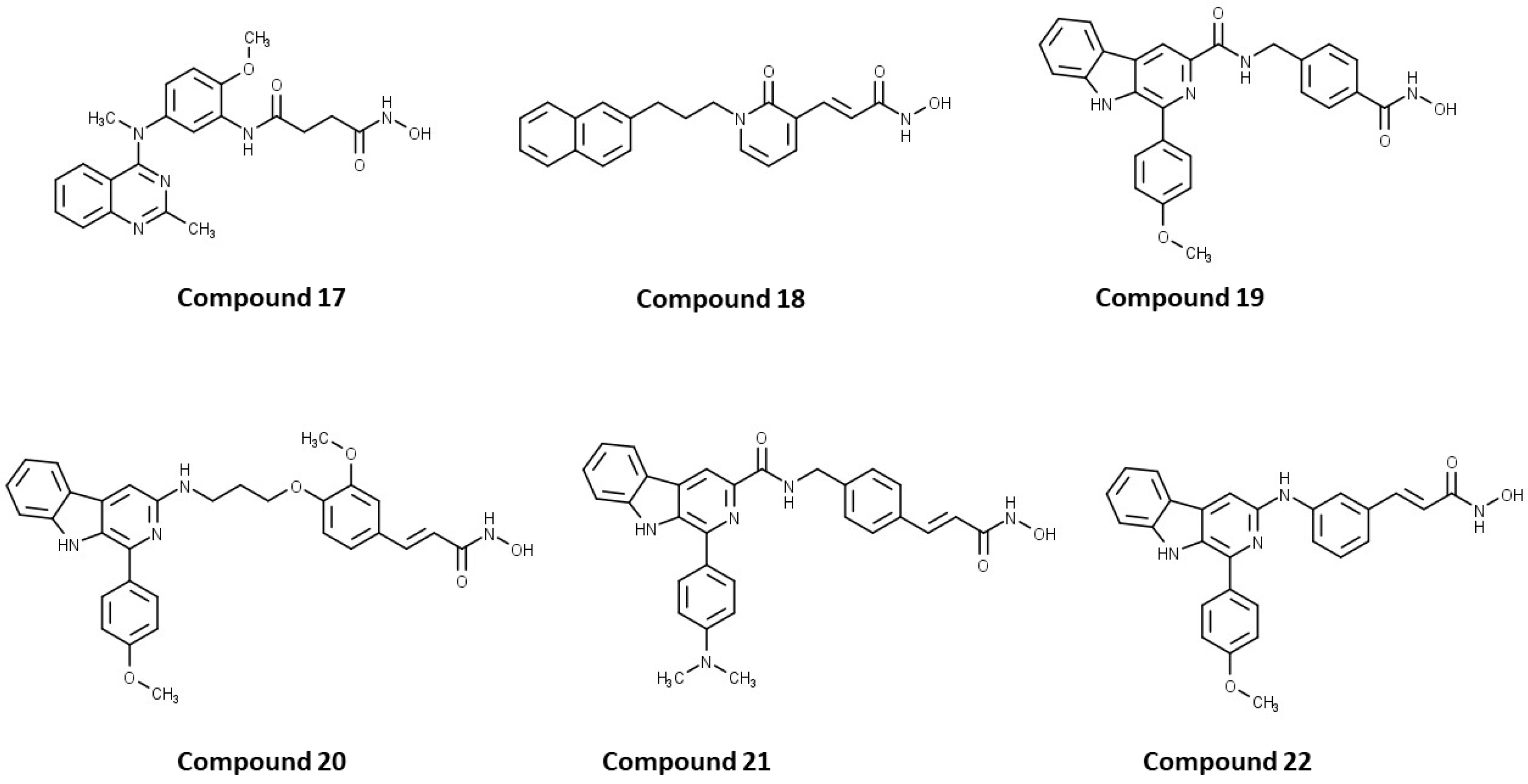
3.8.1. Dual HDAC6/1 Inhibitors
3.8.2. Dual HDAC6/3 Inhibitors
3.8.3. Dual HDAC6/8 Inhibitors
3.9. Dual HDAC6/PAK1 Inhibitors
3.10. Dual HDAC6/FAK Inhibitors
4. Present and Future Perspectives in Development of Dual/Multi-Target HDA6 Inhibitors
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Park, J.W.; Han, J.-W. Targeting Epigenetics for Cancer Therapy. Arch. Pharm. Res. 2019, 42, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.; Belwal, T.; Efferth, T.; Farooqi, A.A.; Sanches-Silva, A.; Vacca, R.A.; Nabavi, S.F.; Khan, F.; Prasad Devkota, H.; Barreca, D.; et al. Targeting Epigenetics in Cancer: Therapeutic Potential of Flavonoids. Crit. Rev. Food Sci. Nutr. 2021, 61, 1616–1639. [Google Scholar] [CrossRef] [PubMed]
- Rousseaux, S.; Khochbin, S. Histone Acylation beyond Acetylation: Terra Incognita in Chromatin Biology. Cell J. 2015, 17, 1–6. [Google Scholar]
- Wolffe, A.P.; Hayes, J.J. Chromatin Disruption and Modification. Nucleic Acids Res. 1999, 27, 711–720. [Google Scholar] [CrossRef]
- Turner, B.M. Histone Acetylation and an Epigenetic Code. BioEssays 2000, 22, 836–845. [Google Scholar] [CrossRef]
- Ruzic, D.; Ellinger, B.; Djokovic, N.; Santibanez, J.F.; Gul, S.; Beljkas, M.; Djuric, A.; Ganesan, A.; Pavic, A.; Srdic-Rajic, T.; et al. Discovery of 1-Benzhydryl-Piperazine-Based HDAC Inhibitors with Anti-Breast Cancer Activity: Synthesis, Molecular Modeling, In Vitro and In Vivo Biological Evaluation. Pharmaceutics 2022, 14, 2600. [Google Scholar] [CrossRef]
- Bertrand, P. Inside HDAC with HDAC Inhibitors. Eur. J. Med. Chem. 2010, 45, 2095–2116. [Google Scholar] [CrossRef]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.-F.; Yao, T.-P. HDAC6 Is a Microtubule-Associated Deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef]
- Patel, A.S.; Schechter, G.L.; Wasilenko, W.J.; Somers, K.D. Overexpression of EMS1/Cortactin in NIH3T3 Fibroblasts Causes Increased Cell Motility and Invasion in Vitro. Oncogene 1998, 16, 3227–3232. [Google Scholar] [CrossRef]
- Kovacs, J.J.; Murphy, P.J.M.; Gaillard, S.; Zhao, X.; Wu, J.-T.; Nicchitta, C.V.; Yoshida, M.; Toft, D.O.; Pratt, W.B.; Yao, T.-P. HDAC6 Regulates Hsp90 Acetylation and Chaperone-Dependent Activation of Glucocorticoid Receptor. Mol. Cell 2005, 18, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, C.; Jarzembowski, J.A.; Opipari, A.W.; Castle, V.P.; Kwok, R.P.S. HDAC6 Deacetylates Ku70 and Regulates Ku70-Bax Binding in Neuroblastoma. Neoplasia 2011, 13, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Riolo, M.T.; Cooper, Z.A.; Holloway, M.P.; Cheng, Y.; Bianchi, C.; Yakirevich, E.; Ma, L.; Chin, Y.E.; Altura, R.A. Histone Deacetylase 6 (HDAC6) Deacetylates Survivin for Its Nuclear Export in Breast Cancer. J. Biol. Chem. 2012, 287, 10885–10893. [Google Scholar] [CrossRef] [PubMed]
- Seigneurin-Berny, D.; Verdel, A.; Curtet, S.; Lemercier, C.; Garin, J.; Rousseaux, S.; Khochbin, S. Identification of Components of the Murine Histone Deacetylase 6 Complex: Link between Acetylation and Ubiquitination Signaling Pathways. Mol. Cell. Biol. 2001, 21, 8035–8044. [Google Scholar] [CrossRef] [PubMed]
- Ding, G.; Liu, H.-D.; Huang, Q.; Liang, H.-X.; Ding, Z.-H.; Liao, Z.-J.; Huang, G. HDAC6 Promotes Hepatocellular Carcinoma Progression by Inhibiting P53 Transcriptional Activity. FEBS Lett. 2013, 587, 880–886. [Google Scholar] [CrossRef]
- Lissanu Deribe, Y.; Wild, P.; Chandrashaker, A.; Curak, J.; Schmidt, M.H.H.; Kalaidzidis, Y.; Milutinovic, N.; Kratchmarova, I.; Buerkle, L.; Fetchko, M.J.; et al. Regulation of Epidermal Growth Factor Receptor Trafficking by Lysine Deacetylase HDAC6. Sci. Signal. 2009, 2, ra84. [Google Scholar] [CrossRef]
- Zhang, Z.; Yamashita, H.; Toyama, T.; Sugiura, H.; Omoto, Y.; Ando, Y.; Mita, K.; Hamaguchi, M.; Hayashi, S.; Iwase, H. HDAC6 Expression Is Correlated with Better Survival in Breast Cancer. Clin. Cancer Res. 2004, 10, 6962–6968. [Google Scholar] [CrossRef]
- Dai, H.-Y.; Chang, L.-S.; Yang, S.-F.; Wang, S.-N.; Su, S.-J.; Yeh, Y.-T. HDAC6 Promotes Aggressive Development of Liver Cancer by Improving Egfr mRNA Stability. Neoplasia 2022, 35, 100845. [Google Scholar] [CrossRef]
- Deng, Y.; Gao, J.; Xu, G.; Yao, Y.; Sun, Y.; Shi, Y.; Hao, X.; Niu, L.; Li, H. HDAC6-Dependent Deacetylation of AKAP12 Dictates Its Ubiquitination and Promotes Colon Cancer Metastasis. Cancer Lett. 2022, 549, 215911. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, N.; Xie, S.; He, X.; Null, T.; Zhou, J.; Liu, M.; Li, D. HDAC6 Regulates Neuroblastoma Cell Migration and May Play a Role in the Invasion Process. Cancer Biol. Ther. 2014, 15, 1561–1570. [Google Scholar] [CrossRef]
- Zuo, Q.; Wu, W.; Li, X.; Zhao, L.; Chen, W. HDAC6 and SIRT2 Promote Bladder Cancer Cell Migration and Invasion by Targeting Cortactin. Oncol. Rep. 2012, 27, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Keusch, J.J.; Wang, L.; Saito, M.; Hess, D.; Wang, X.; Melancon, B.J.; Helquist, P.; Gut, H.; Matthias, P. Structural Insights into HDAC6 Tubulin Deacetylation and Its Selective Inhibition. Nat. Chem. Biol. 2016, 12, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Hai, Y.; Christianson, D.W. Histone Deacetylase 6 Structure and Molecular Basis of Catalysis and Inhibition. Nat. Chem. Biol. 2016, 12, 741–747. [Google Scholar] [CrossRef]
- Melesina, J.; Simoben, C.V.; Praetorius, L.; Bülbül, E.F.; Robaa, D.; Sippl, W. Strategies to Design Selective Histone Deacetylase Inhibitors. ChemMedChem 2021, 16, 1336–1359. [Google Scholar] [CrossRef]
- Amengual, J.E.; Lue, J.K.; Ma, H.; Lichtenstein, R.; Shah, B.; Cremers, S.; Jones, S.; Sawas, A. First-in-Class Selective HDAC6 Inhibitor (ACY-1215) Has a Highly Favorable Safety Profile in Patients with Relapsed and Refractory Lymphoma. Oncologist 2021, 26, e184–e366. [Google Scholar] [CrossRef] [PubMed]
- Columbia University Multi-Center Phase IB Trial of ACY-1215 (Ricolinostat) Combined with Nab-Paclitaxel in Unresectable or Metastatic Breast Cancer. 2021. Available online: https://clinicaltrials.gov/study/NCT02632071 (accessed on 20 September 2023).
- Celgene A Phase 1b Study of the Selective HDAC6 Inhibitor ACY-241 in Combination with Ipilimumab and Nivolumab in Patients with Unresectable Stage III or Stage IV Melanoma. 2017. Available online: https://clinicaltrials.gov/study/NCT02935790 (accessed on 20 September 2023).
- Celgene A Phase 1b Study of the Selective HDAC6 Inhibitor ACY-241 in Combination with Nivolumab in Patients with Unresectable Non-Small Cell Lung Cancer. 2023. Available online: https://clinicaltrials.gov/study/NCT02635061 (accessed on 20 September 2023).
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA Approval Summary: Vorinostat for Treatment of Advanced Primary Cutaneous T-Cell Lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-Z.; Kwitkowski, V.E.; Del Valle, P.L.; Ricci, M.S.; Saber, H.; Habtemariam, B.A.; Bullock, J.; Bloomquist, E.; Li Shen, Y.; Chen, X.-H.; et al. FDA Approval: Belinostat for the Treatment of Patients with Relapsed or Refractory Peripheral T-Cell Lymphoma. Clin. Cancer Res. 2015, 21, 2666–2670. [Google Scholar] [CrossRef]
- Raedler, L.A. Farydak (Panobinostat): First HDAC Inhibitor Approved for Patients with Relapsed Multiple Myeloma. Am. Health Drug Benefits 2016, 9, 84–87. [Google Scholar]
- Lu, X.; Ning, Z.; Li, Z.; Cao, H.; Wang, X. Development of Chidamide for Peripheral T-Cell Lymphoma, the First Orphan Drug Approved in China. Intractable Rare Dis. Res. 2016, 5, 185–191. [Google Scholar] [CrossRef]
- Grant, C.; Rahman, F.; Piekarz, R.; Peer, C.; Frye, R.; Robey, R.W.; Gardner, E.R.; Figg, W.D.; Bates, S.E. Romidepsin: A New Therapy for Cutaneous T-Cell Lymphoma and a Potential Therapy for Solid Tumors. Expert Rev. Anticancer Ther. 2010, 10, 997–1008. [Google Scholar] [CrossRef]
- Depetter, Y.; Geurs, S.; De Vreese, R.; Goethals, S.; Vandoorn, E.; Laevens, A.; Steenbrugge, J.; Meyer, E.; de Tullio, P.; Bracke, M.; et al. Selective Pharmacological Inhibitors of HDAC6 Reveal Biochemical Activity but Functional Tolerance in Cancer Models. Int. J. Cancer 2019, 145, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, M.B.; Villuendas, R.; Bischoff, J.R.; Aparicio, C.B.; Martínez Leal, J.F.; de La Cueva, P.; Rodriguez, M.E.; Herreros, B.; Martin-Perez, D.; Longo, M.I.; et al. Vorinostat Interferes with the Signaling Transduction Pathway of T-Cell Receptor and Synergizes with Phosphoinositide-3 Kinase Inhibitors in Cutaneous T-Cell Lymphoma. Haematologica 2010, 95, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Dawson, J.C.; Serrels, B.; Byron, A.; Muir, M.T.; Makda, A.; García-Muñoz, A.; von Kriegsheim, A.; Lietha, D.; Carragher, N.O.; Frame, M.C. A Synergistic Anticancer FAK and HDAC Inhibitor Combination Discovered by a Novel Chemical–Genetic High-Content Phenotypic Screen. Mol. Cancer Ther. 2020, 19, 637–649. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Jeon, Y.H.; Lee, D.H.; Kim, G.W.; Lee, S.W.; Kim, S.Y.; Park, J.; Kwon, S.H. HDAC6-Selective Inhibitors Enhance Anticancer Effects of Paclitaxel in Ovarian Cancer Cells. Oncol. Lett. 2021, 21, 1. [Google Scholar] [CrossRef]
- Makarević, J.; Tawanaie, N.; Juengel, E.; Reiter, M.; Mani, J.; Tsaur, I.; Bartsch, G.; Haferkamp, A.; Blaheta, R.A. Cross-Communication between Histone H3 and H4 Acetylation and Akt-mTOR Signalling in Prostate Cancer Cells. J. Cell. Mol. Med. 2014, 18, 1460–1466. [Google Scholar] [CrossRef]
- Carew, J.S.; Espitia, C.M.; Zhao, W.; Visconte, V.; Anwer, F.; Kelly, K.R.; Nawrocki, S.T. Rational Cotargeting of HDAC6 and BET Proteins Yields Synergistic Antimyeloma Activity. Blood Adv. 2019, 3, 1318–1329. [Google Scholar] [CrossRef]
- Gibbs, A.; Schwartzman, J.; Deng, V.; Alumkal, J. Sulforaphane Destabilizes the Androgen Receptor in Prostate Cancer Cells by Inactivating Histone Deacetylase 6. Proc. Natl. Acad. Sci. USA 2009, 106, 16663–16668. [Google Scholar] [CrossRef]
- Liu, P.; Xiao, J.; Wang, Y.; Song, X.; Huang, L.; Ren, Z.; Kitazato, K.; Wang, Y. Posttranslational Modification and beyond: Interplay between Histone Deacetylase 6 and Heat-Shock Protein 90. Mol. Med. 2021, 27, 110. [Google Scholar] [CrossRef]
- Luo, J.; Su, F.; Chen, D.; Shiloh, A.; Gu, W. Deacetylation of P53 Modulates Its Effect on Cell Growth and Apoptosis. Nature 2000, 408, 377–381. [Google Scholar] [CrossRef]
- Mikami, D.; Kobayashi, M.; Uwada, J.; Yazawa, T.; Kamiyama, K.; Nishimori, K.; Nishikawa, Y.; Nishikawa, S.; Yokoi, S.; Taniguchi, T.; et al. β-Hydroxybutyrate Enhances the Cytotoxic Effect of Cisplatin via the Inhibition of HDAC/Survivin Axis in Human Hepatocellular Carcinoma Cells. J. Pharmacol. Sci. 2020, 142, 1–8. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, H.; Jeoung, D. Tubulin Beta3 Serves as a Target of HDAC3 and Mediates Resistance to Microtubule-Targeting Drugs. Mol. Cells 2015, 38, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Vanaja, G.R.; Ramulu, H.G.; Kalle, A.M. Overexpressed HDAC8 in Cervical Cancer Cells Shows Functional Redundancy of Tubulin Deacetylation with HDAC6. Cell Commun. Signal. 2018, 16, 20. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, S.; Cleary, R.A.; Wang, R.; Gannon, O.J.; Seto, E.; Tang, D.D. Histone Deacetylase 8 Regulates Cortactin Deacetylation and Contraction in Smooth Muscle Tissues. Am. J. Physiol. Cell Physiol. 2014, 307, C288–C295. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.-C.; Ma, Y.-C.; Qin, W.-P.; Ding, L.-N.; Zheng, Y.-C.; Zhu, Y.-L.; Zhai, X.-Y.; Yang, J.; Ma, C.-Y.; Guan, Y.-Y. Design and Synthesis of Tranylcypromine Derivatives as Novel LSD1/HDACs Dual Inhibitors for Cancer Treatment. Eur. J. Med. Chem. 2017, 140, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Strebhardt, K.; Ullrich, A. Paul Ehrlich’s Magic Bullet Concept: 100 Years of Progress. Nat. Rev. Cancer 2008, 8, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Han-Chung, W.; Chang, D.-K.; Chia-Ting, H. Targeted Therapy for Cancer. J. Cancer Mol. 2006, 2, 57–66. [Google Scholar]
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular Targeted Therapy: Treating Cancer with Specificity. Eur. J. Pharmacol. 2018, 834, 188–196. [Google Scholar] [CrossRef]
- Jin, H.; Wang, L.; Bernards, R. Rational Combinations of Targeted Cancer Therapies: Background, Advances and Challenges. Nat. Rev. Drug Discov. 2023, 22, 213–234. [Google Scholar] [CrossRef]
- Gerber, D.E. Targeted Therapies: A New Generation of Cancer Treatments. Target. Ther. 2008, 77, 311–319. [Google Scholar]
- Benson, J.D.; Chen, Y.-N.P.; Cornell-Kennon, S.A.; Dorsch, M.; Kim, S.; Leszczyniecka, M.; Sellers, W.R.; Lengauer, C. Validating Cancer Drug Targets. Nature 2006, 441, 451–456. [Google Scholar] [CrossRef]
- Mobashir, M.; Turunen, S.P.; Izhari, M.A.; Ashankyty, I.M.; Helleday, T.; Lehti, K. An Approach for Systems-Level Understanding of Prostate Cancer from High-Throughput Data Integration to Pathway Modeling and Simulation. Cells 2022, 11, 4121. [Google Scholar] [CrossRef] [PubMed]
- Giordano, S.; Petrelli, A. From Single- to Multi-Target Drugs in Cancer Therapy: When Aspecificity Becomes an Advantage. Curr. Med. Chem. 2008, 15, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Raghavendra, N.M.; Pingili, D.; Kadasi, S.; Mettu, A.; Prasad, S.V.U.M. Dual or Multi-Targeting Inhibitors: The next Generation Anticancer Agents. Eur. J. Med. Chem. 2018, 143, 1277–1300. [Google Scholar] [CrossRef] [PubMed]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and Opportunities in Drug Discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Jiang, X.; He, S.; Jiang, H.; Feng, F.; Liu, W.; Qu, W.; Sun, H. Rational Design of Multitarget-Directed Ligands: Strategies and Emerging Paradigms. J. Med. Chem. 2019, 62, 8881–8914. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Li, M.; Li, H.; Chen, L. Entrectinib, a New Multi-Target Inhibitor for Cancer Therapy. Biomed. Pharmacother. 2022, 150, 112974. [Google Scholar] [CrossRef]
- Morphy, R.; Rankovic, Z. Designing Multiple Ligands—Medicinal Chemistry Strategies and Challenges. Curr. Pharm. Des. 2009, 15, 587–600. [Google Scholar] [CrossRef]
- Kaliszczak, M.; Trousil, S.; Ali, T.; Aboagye, E.O. AKT Activation Controls Cell Survival in Response to HDAC6 Inhibition. Cell Death Dis. 2016, 7, e2286. [Google Scholar] [CrossRef]
- Rascio, F.; Spadaccino, F.; Rocchetti, M.T.; Castellano, G.; Stallone, G.; Netti, G.S.; Ranieri, E. The Pathogenic Role of PI3K/AKT Pathway in Cancer Onset and Drug Resistance: An Updated Review. Cancers 2021, 13, 3949. [Google Scholar] [CrossRef]
- Yang, Q.; Modi, P.; Newcomb, T.; Quéva, C.; Gandhi, V. Idelalisib: First-in-Class PI3K Delta Inhibitor for the Treatment of Chronic Lymphocytic Leukemia, Small Lymphocytic Leukemia, and Follicular Lymphoma. Clin. Cancer Res. 2015, 21, 1537–1542. [Google Scholar] [CrossRef]
- Liu, N.; Rowley, B.R.; Bull, C.O.; Schneider, C.; Haegebarth, A.; Schatz, C.A.; Fracasso, P.R.; Wilkie, D.P.; Hentemann, M.; Wilhelm, S.M.; et al. BAY 80-6946 Is a Highly Selective Intravenous PI3K Inhibitor with Potent P110α and P110δ Activities in Tumor Cell Lines and Xenograft Models. Mol. Cancer Ther. 2013, 12, 2319–2330. [Google Scholar] [CrossRef] [PubMed]
- Flinn, I.W.; O’Brien, S.; Kahl, B.; Patel, M.; Oki, Y.; Foss, F.F.; Porcu, P.; Jones, J.; Burger, J.A.; Jain, N.; et al. Duvelisib, a Novel Oral Dual Inhibitor of PI3K-δ,γ, Is Clinically Active in Advanced Hematologic Malignancies. Blood 2018, 131, 877–887. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Alpelisib: First Global Approval. Drugs 2019, 79, 1249–1253. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; Liu, S.; Anees, M.; Mehrotra, N.; Thakur, A.; Tawa, G.J.; Grewal, G.; Stone, R.; Kharbanda, S.; Singh, H. QuatramerTM Encapsulation of Dual-Targeted PI3-Kδ/HDAC6 Inhibitor, HSB-510, Suppresses Growth of Breast Cancer. Bioeng. Transl. Med. 2023, 8, e10541. [Google Scholar] [CrossRef]
- Kotian, S.; Zhang, L.; Boufraqech, M.; Gaskins, K.; Gara, S.K.; Quezado, M.; Nilubol, N.; Kebebew, E. Dual Inhibition of HDAC and Tyrosine Kinase Signaling Pathways with CUDC-907 Inhibits Thyroid Cancer Growth and Metastases. Clin. Cancer Res. 2017, 23, 5044–5054. [Google Scholar] [CrossRef]
- Qian, C.; Lai, C.-J.; Bao, R.; Wang, D.-G.; Wang, J.; Xu, G.-X.; Atoyan, R.; Qu, H.; Yin, L.; Samson, M.; et al. Cancer Network Disruption by a Single Molecule Inhibitor Targeting Both Histone Deacetylase Activity and Phosphatidylinositol 3-Kinase Signaling. Clin. Cancer Res. 2012, 18, 4104–4113. [Google Scholar] [CrossRef]
- Li, Z.; Zhao, C.; He, G.; Wang, Y.; Wang, Y.; Ma, X. Identification of PI3K/HDAC Dual-Targeted Inhibitors with Subtype Selectivity as Potential Therapeutic Agents against Solid Tumors: Building HDAC6 Potency in a Quinazolinone-Based PI3Kδ-Selective Template. Bioorg. Med. Chem. 2022, 73, 117028. [Google Scholar] [CrossRef]
- Rodrigues, D.A.; Guerra, F.S.; Sagrillo, F.S.; Pinheiro, P.d.S.M.; Alves, M.A.; Thota, S.; Chaves, L.S.; Sant’Anna, C.M.R.; Fernandes, P.D.; Fraga, C.A.M. Design, Synthesis, and Pharmacological Evaluation of First-in-Class Multitarget N-Acylhydrazone Derivatives as Selective HDAC6/8 and PI3Kα Inhibitors. ChemMedChem 2020, 15, 539–551. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, J.; Wu, J.; Liu, S.; Zhang, X.; Lv, X. Discovery of (S)-N1-(Thiazol-2-Yl) Pyrrolidine-1,2-Dicarboxamide Derivatives Targeting PI3Ka/HDAC6 for the Treatment of Cancer. Bioorg. Med. Chem. Lett. 2023, 94, 129462. [Google Scholar] [CrossRef]
- Yao, D.; Jiang, J.; Zhang, H.; Huang, Y.; Huang, J.; Wang, J. Design, Synthesis and Biological Evaluation of Dual mTOR/HDAC6 Inhibitors in MDA-MB-231 Cells. Bioorg. Med. Chem. Lett. 2021, 47, 128204. [Google Scholar] [CrossRef]
- Bissler, J.J.; McCormack, F.X.; Young, L.R.; Elwing, J.M.; Chuck, G.; Leonard, J.M.; Schmithorst, V.J.; Laor, T.; Brody, A.S.; Bean, J.; et al. Sirolimus for Angiomyolipoma in Tuberous Sclerosis Complex or Lymphangioleiomyomatosis. N. Engl. J. Med. 2008, 358, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.C.; Pavel, M.; Lombard-Bohas, C.; van Cutsem, E.; Lam, D.; Kunz, T.; Brandt, U.; Capdevila, J.; De Vries, E.G.E.; Hobday, T.; et al. 1132O—Everolimus (Eve) for the Treatment of Advanced Pancreatic Neuroendocrine Tumors (Pnet): Final Overall Survival (Os) Results of a Randomized, Double-Blind, Placebo (Pbo)-Controlled, Multicenter Phase III Trial (Radiant-3). Ann. Oncol. 2014, 25, iv394. [Google Scholar] [CrossRef]
- Hudes, G.; Carducci, M.; Tomczak, P.; Dutcher, J.; Figlin, R.; Kapoor, A.; Staroslawska, E.; Sosman, J.; McDermott, D.; Bodrogi, I.; et al. Temsirolimus, Interferon Alfa, or Both for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2007, 356, 2271–2281. [Google Scholar] [CrossRef] [PubMed]
- Panwar, V.; Singh, A.; Bhatt, M.; Tonk, R.K.; Azizov, S.; Raza, A.S.; Sengupta, S.; Kumar, D.; Garg, M. Multifaceted Role of mTOR (Mammalian Target of Rapamycin) Signaling Pathway in Human Health and Disease. Signal Transduct. Target. Ther. 2023, 8, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.; Li, C.; Jiang, J.; Huang, J.; Wang, J.; He, Z.; Zhang, J. Design, Synthesis and Biological Evaluation of Novel HDAC Inhibitors with Improved Pharmacokinetic Profile in Breast Cancer. Eur. J. Med. Chem. 2020, 205, 112648. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective Inhibition of BET Bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef]
- Faivre, E.J.; McDaniel, K.F.; Albert, D.H.; Mantena, S.R.; Plotnik, J.P.; Wilcox, D.; Zhang, L.; Bui, M.H.; Sheppard, G.S.; Wang, L.; et al. Selective Inhibition of the BD2 Bromodomain of BET Proteins in Prostate Cancer. Nature 2020, 578, 306–310. [Google Scholar] [CrossRef]
- Albrecht, B.K.; Gehling, V.S.; Hewitt, M.C.; Vaswani, R.G.; Côté, A.; Leblanc, Y.; Nasveschuk, C.G.; Bellon, S.; Bergeron, L.; Campbell, R.; et al. Identification of a Benzoisoxazoloazepine Inhibitor (CPI-0610) of the Bromodomain and Extra-Terminal (BET) Family as a Candidate for Human Clinical Trials. J. Med. Chem. 2016, 59, 1330–1339. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Zhang, J.; Zhang, M.; Wei, A.; Liu, H.; Xie, Z.; Ren, W.; Duan, W.; Zhang, Z.; et al. Discovery of Selective HDAC/BRD4 Dual Inhibitors as Epigenetic Probes. Eur. J. Med. Chem. 2021, 209, 112868. [Google Scholar] [CrossRef]
- Fujita, K.; Nonomura, N. Role of Androgen Receptor in Prostate Cancer: A Review. World J. Men’s Health 2019, 37, 288–295. [Google Scholar] [CrossRef]
- Ai, J.; Wang, Y.; Dar, J.A.; Liu, J.; Liu, L.; Nelson, J.B.; Wang, Z. HDAC6 Regulates Androgen Receptor Hypersensitivity and Nuclear Localization via Modulating Hsp90 Acetylation in Castration-Resistant Prostate Cancer. Mol. Endocrinol. 2009, 23, 1963–1972. [Google Scholar] [CrossRef]
- Chen, L.; Meng, S.; Wang, H.; Bali, P.; Bai, W.; Li, B.; Atadja, P.; Bhalla, K.N.; Wu, J. Chemical Ablation of Androgen Receptor in Prostate Cancer Cells by the Histone Deacetylase Inhibitor LAQ824. Mol. Cancer Ther. 2005, 4, 1311–1319. [Google Scholar] [CrossRef]
- Zhou, M.; Zheng, H.; Li, Y.; Huang, H.; Min, X.; Dai, S.; Zhou, W.; Chen, Z.; Xu, G.; Chen, Y. Discovery of a Novel AR/HDAC6 Dual Inhibitor for Prostate Cancer Treatment. Aging 2021, 13, 6982–6998. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-N.; Luo, Y. HSP90 Inhibitors and Cancer: Prospects for Use in Targeted Therapies (Review). Oncol. Rep. 2023, 49, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, H.; Liu, Y.; Zhao, K.; Wei, S.; Sugarman, E.T.; Liu, L.; Zhang, G. Targeting HSP90 as a Novel Therapy for Cancer: Mechanistic Insights and Translational Relevance. Cells 2022, 11, 2778. [Google Scholar] [CrossRef] [PubMed]
- Ojha, R.; Nepali, K.; Chen, C.-H.; Chuang, K.-H.; Wu, T.-Y.; Lin, T.E.; Hsu, K.-C.; Chao, M.-W.; Lai, M.-J.; Lin, M.-H.; et al. Isoindoline Scaffold-Based Dual Inhibitors of HDAC6 and HSP90 Suppressing the Growth of Lung Cancer in Vitro and in Vivo. Eur. J. Med. Chem. 2020, 190, 112086. [Google Scholar] [CrossRef]
- de Zoeten, E.F.; Wang, L.; Butler, K.; Beier, U.H.; Akimova, T.; Sai, H.; Bradner, J.E.; Mazitschek, R.; Kozikowski, A.P.; Matthias, P.; et al. Histone Deacetylase 6 and Heat Shock Protein 90 Control the Functions of Foxp3+ T-Regulatory Cells. Mol. Cell. Biol. 2011, 31, 2066–2078. [Google Scholar] [CrossRef]
- Ojha, R.; Huang, H.-L.; HuangFu, W.-C.; Wu, Y.-W.; Nepali, K.; Lai, M.-J.; Su, C.-J.; Sung, T.-Y.; Chen, Y.-L.; Pan, S.-L.; et al. 1-Aroylindoline-Hydroxamic Acids as Anticancer Agents, Inhibitors of HSP90 and HDAC. Eur. J. Med. Chem. 2018, 150, 667–677. [Google Scholar] [CrossRef]
- Wu, T.-Y.; Chen, M.; Chen, I.-C.; Chen, Y.-J.; Chen, C.-Y.; Wang, C.-H.; Cheng, J.-J.; Nepali, K.; Chuang, K.-H.; Liou, J.-P. Rational Design of Synthetically Tractable HDAC6/HSP90 Dual Inhibitors to Destroy Immune-Suppressive Tumor Microenvironment. J. Adv. Res. 2023, 46, 159–171. [Google Scholar] [CrossRef]
- Maliekal, T.T.; Dharmapal, D.; Sengupta, S. Tubulin Isotypes: Emerging Roles in Defining Cancer Stem Cell Niche. Front. Immunol. 2022, 13, 876278. [Google Scholar] [CrossRef]
- Ebenezer, O.; Shapi, M.; Tuszynski, J.A. A Review of the Recent Developments of Molecular Hybrids Targeting Tubulin Polymerization. Int. J. Mol. Sci. 2022, 23, 4001. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-Y.; Lee, J.-F.; Kumar, S.; Wu, Y.-W.; HuangFu, W.-C.; Lai, M.-J.; Li, Y.-H.; Huang, H.-L.; Kuo, F.-C.; Hsiao, C.-J.; et al. 3-Aroylindoles Display Antitumor Activity in Vitro and in Vivo: Effects of N1-Substituents on Biological Activity. Eur. J. Med. Chem. 2017, 125, 1268–1278. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-W.; Hsu, K.-C.; Lee, H.-Y.; Huang, T.-C.; Lin, T.E.; Chen, Y.-L.; Sung, T.-Y.; Liou, J.-P.; Hwang-Verslues, W.W.; Pan, S.-L.; et al. A Novel Dual HDAC6 and Tubulin Inhibitor, MPT0B451, Displays Anti-Tumor Ability in Human Cancer Cells in Vitro and in Vivo. Front. Pharmacol. 2018, 9, 205. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Zhou, Z.; Jiang, Z.; Zhu, W.; Qiao, D. Indole-Based Tubulin Inhibitors: Binding Modes and SARs Investigations. Molecules 2022, 27, 1587. [Google Scholar] [CrossRef]
- Kumar, K.; Das, R.; Thapa, B.; Rakhecha, B.; Srivastava, S.; Savita, K.; Israr, M.; Chanda, D.; Banerjee, D.; Shanker, K.; et al. Dual Targeted 2-Benzylideneindanone Pendant Hydroxamic Acid Group Exhibits Selective HDAC6 Inhibition along with Tubulin Stabilization Effect. Bioorg. Med. Chem. 2023, 86, 117300. [Google Scholar] [CrossRef]
- Wang, F.; Zheng, L.; Yi, Y.; Yang, Z.; Qiu, Q.; Wang, X.; Yan, W.; Bai, P.; Yang, J.; Li, D.; et al. SKLB-23bb, A HDAC6-Selective Inhibitor, Exhibits Superior and Broad-Spectrum Antitumor Activity via Additionally Targeting Microtubules. Mol. Cancer Ther. 2018, 17, 763–775. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, T.; Wang, F.; Niu, T.; Liu, Z.; Chen, X.; Long, C.; Tang, M.; Cao, D.; Wang, X.; et al. Discovery of Selective Histone Deacetylase 6 Inhibitors Using the Quinazoline as the Cap for the Treatment of Cancer. J. Med. Chem. 2016, 59, 1455–1470. [Google Scholar] [CrossRef]
- Ustinova, K.; Novakova, Z.; Saito, M.; Meleshin, M.; Mikesova, J.; Kutil, Z.; Baranova, P.; Havlinova, B.; Schutkowski, M.; Matthias, P.; et al. The Disordered N-Terminus of HDAC6 Is a Microtubule-Binding Domain Critical for Efficient Tubulin Deacetylation. J. Biol. Chem. 2020, 295, 2614–2628. [Google Scholar] [CrossRef]
- Binda, C.; Valente, S.; Romanenghi, M.; Pilotto, S.; Cirilli, R.; Karytinos, A.; Ciossani, G.; Botrugno, O.A.; Forneris, F.; Tardugno, M.; et al. Biochemical, Structural, and Biological Evaluation of Tranylcypromine Derivatives as Inhibitors of Histone Demethylases LSD1 and LSD2. J. Am. Chem. Soc. 2010, 132, 6827–6833. [Google Scholar] [CrossRef]
- Malagraba, G.; Yarmohammadi, M.; Javed, A.; Barceló, C.; Rubio-Tomás, T. The Role of LSD1 and LSD2 in Cancers of the Gastrointestinal System: An Update. Biomolecules 2022, 12, 462. [Google Scholar] [CrossRef]
- Dai, X.-J.; Liu, Y.; Xiong, X.-P.; Xue, L.-P.; Zheng, Y.-C.; Liu, H.-M. Tranylcypromine Based Lysine-Specific Demethylase 1 Inhibitor: Summary and Perspective. J. Med. Chem. 2020, 63, 14197–14215. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Zhang, P.; Yu, B. Advances toward LSD1 Inhibitors for Cancer Therapy. Future Med. Chem. 2017, 9, 1227–1242. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Vasilatos, S.N.; Boric, L.; Shaw, P.G.; Davidson, N.E. Inhibitors of Histone Demethylation and Histone Deacetylation Cooperate in Regulating Gene Expression and Inhibiting Growth in Human Breast Cancer Cells. Breast Cancer Res. Treat. 2012, 131, 777–789. [Google Scholar] [CrossRef]
- Fang, Y.; Yang, C.; Yu, Z.; Li, X.; Mu, Q.; Liao, G.; Yu, B. Natural Products as LSD1 Inhibitors for Cancer Therapy. Acta Pharm. Sin. B 2021, 11, 621–631. [Google Scholar] [CrossRef]
- Song, Y.; Zhang, H.; Yang, X.; Shi, Y.; Yu, B. Annual Review of Lysine-Specific Demethylase 1 (LSD1/KDM1A) Inhibitors in 2021. Eur. J. Med. Chem. 2022, 228, 114042. [Google Scholar] [CrossRef] [PubMed]
- Kalin, J.H.; Wu, M.; Gomez, A.V.; Song, Y.; Das, J.; Hayward, D.; Adejola, N.; Wu, M.; Panova, I.; Chung, H.J.; et al. Targeting the CoREST Complex with Dual Histone Deacetylase and Demethylase Inhibitors. Nat. Commun. 2018, 9, 53. [Google Scholar] [CrossRef]
- Bennesch, M.A.; Segala, G.; Wider, D.; Picard, D. LSD1 Engages a Corepressor Complex for the Activation of the Estrogen Receptor α by Estrogen and cAMP. Nucleic Acids Res. 2016, 44, 8655–8670. [Google Scholar] [CrossRef]
- Gajendran, C.; Tantry, S.J.; Sadhu, M.N.; Mohammed, Z.; Dewang, P.; Hallur, M.; Nair, S.; Vaithilingam, K.; Nagayya, B.; Rajagopal, S.; et al. Novel Dual LSD1/HDAC6 Inhibitor for the Treatment of Cancer. PLoS ONE 2023, 18, e0279063. [Google Scholar] [CrossRef]
- Naveen Sadhu, M.; Sivanandhan, D.; Gajendran, C.; Tantry, S.; Dewang, P.; Murugan, K.; Chickamunivenkatappa, S.; Zainuddin, M.; Nair, S.; Vaithilingam, K.; et al. Novel Dual LSD1/HDAC6 Inhibitors for the Treatment of Multiple Myeloma. Bioorg. Med. Chem. Lett. 2021, 34, 127763. [Google Scholar] [CrossRef]
- Bulut, I.; Lee, A.; Cevatemre, B.; Ruzic, D.; Belle, R.; Kawamura, A.; Gul, S.; Nikolic, K.; Ganesan, A.; Acilan, C. Dual LSD1 and HDAC6 Inhibition Induces Doxorubicin Sensitivity in Acute Myeloid Leukemia Cells. Cancers 2022, 14, 6014. [Google Scholar] [CrossRef]
- Nalawansha, D.A.; Pflum, M.K.H. LSD1 Substrate Binding and Gene Expression Are Affected by HDAC1-Mediated Deacetylation. ACS Chem. Biol. 2017, 12, 254–264. [Google Scholar] [CrossRef]
- Martínez-Balbás, M.A.; Bauer, U.-M.; Nielsen, S.J.; Brehm, A.; Kouzarides, T. Regulation of E2F1 Activity by Acetylation. EMBO J. 2000, 19, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Nalawansha, D.A.; Gomes, I.D.; Wambua, M.K.; Pflum, M.K.H. HDAC Inhibitor-Induced Mitotic Arrest Is Mediated by Eg5/KIF11 Acetylation. Cell Chem. Biol. 2017, 24, 481–492.e5. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-H.; Kwon, H.J.; Yoon, B.-I.; Kim, J.-H.; Han, S.U.; Joo, H.J.; Kim, D.-Y. Expression Profile of Histone Deacetylase 1 in Gastric Cancer Tissues. Jpn. J. Cancer Res. 2001, 92, 1300–1304. [Google Scholar] [CrossRef] [PubMed]
- Halkidou, K.; Gaughan, L.; Cook, S.; Leung, H.Y.; Neal, D.E.; Robson, C.N. Upregulation and Nuclear Recruitment of HDAC1 in Hormone Refractory Prostate Cancer. Prostate 2004, 59, 177–189. [Google Scholar] [CrossRef]
- Rivas, M.; Johnston, M.E.; Gulati, R.; Kumbaji, M.; Margues Aguiar, T.F.; Timchenko, L.; Krepischi, A.; Shin, S.; Bondoc, A.; Tiao, G.; et al. HDAC1-Dependent Repression of Markers of Hepatocytes and P21 Is Involved in Development of Pediatric Liver Cancer. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 1669–1682. [Google Scholar] [CrossRef]
- Kawai, H.; Li, H.; Avraham, S.; Jiang, S.; Avraham, H.K. Overexpression of Histone Deacetylase HDAC1 Modulates Breast Cancer Progression by Negative Regulation of Estrogen Receptor α. Int. J. Cancer 2003, 107, 353–358. [Google Scholar] [CrossRef]
- Park, S.Y.; Jun, J.A.; Jeong, K.J.; Heo, H.J.; Sohn, J.S.; Lee, H.Y.; Park, C.G.; Kang, J. Histone Deacetylases 1, 6 and 8 Are Critical for Invasion in Breast Cancer. Oncol. Rep. 2011, 25, 1677–1681. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wei, M.; Wang, C.; Sun, D.; Liu, P.; Zhong, X.; He, Q.; Yu, W. The Histone Deacetylase HDAC1 Activates HIF1α/VEGFA Signal Pathway in Colorectal Cancer. Gene 2020, 754, 144851. [Google Scholar] [CrossRef]
- Ramakrishnan, S.; Ku, S.; Ciamporcero, E.; Miles, K.M.; Attwood, K.; Chintala, S.; Shen, L.; Ellis, L.; Sotomayor, P.; Swetzig, W.; et al. HDAC 1 and 6 Modulate Cell Invasion and Migration in Clear Cell Renal Cell Carcinoma. BMC Cancer 2016, 16, 617. [Google Scholar] [CrossRef]
- Cao, L.-L.; Song, X.; Pei, L.; Liu, L.; Wang, H.; Jia, M. Histone Deacetylase HDAC1 Expression Correlates with the Progression and Prognosis of Lung Cancer. Medicine 2017, 96, e7663. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yu, Y.; Zhang, J.; Lu, C.; Wang, L.; Liu, P.; Song, H. HDAC1 Silencing in Ovarian Cancer Enhances the Chemotherapy Response. Cell. Physiol. Biochem. 2018, 48, 1505–1518. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Xie, C.; Edwards, H.; Zhou, H.; Buck, S.A.; Ge, Y. Inhibition of Histone Deacetylases 1 and 6 Enhances Cytarabine-Induced Apoptosis in Pediatric Acute Myeloid Leukemia Cells. PLoS ONE 2011, 6, e17138. [Google Scholar] [CrossRef]
- Chen, J.; Sang, Z.; Jiang, Y.; Yang, C.; He, L. Design, Synthesis, and Biological Evaluation of Quinazoline Derivatives as Dual HDAC1 and HDAC6 Inhibitors for the Treatment of Cancer. Chem. Biol. Drug Des. 2019, 93, 232–241. [Google Scholar] [CrossRef]
- Cho, M.; Choi, E.; Yang, J.S.; Lee, C.; Seo, J.J.; Kim, B.S.; Oh, S.J.; Kim, H.M.; Lee, K.; Park, S.-K.; et al. Discovery of Pyridone-Based Histone Deacetylase Inhibitors: Approaches for Metabolic Stability. ChemMedChem 2013, 8, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.-F.; Peng, Y.-F.; Chen, S.; Gao, W.-J.; Yang, Q.-X.; Zhu, P.; Guo, J.; Tao, J.; Luo, L.; Zhang, Y.; et al. A Novel Harmine Derivative, N-(4-(Hydroxycarbamoyl)Benzyl)-1-(4- Methoxyphenyl)-9H-Pyrido [3,4-b]Indole-3-Carboxamide (HBC), as Histone Deacetylase Inhibitor: In Vitro Antiproliferation, Apoptosis Induction, Cell Cycle Arrest, and Antimetastatic Effects. Eur. J. Pharmacol. 2018, 824, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.; Li, Y.; Zhu, R.; Qian, J.; Liu, J.; Gao, W.; Meng, C.; Miao, J.; Xiong, B.; Qiu, X.; et al. Hydroxamic Acid Derivatives of β-Carboline/Hydroxycinnamic Acid Hybrids Inducing Apoptosis and Autophagy through the PI3K/Akt/mTOR Pathways. J. Nat. Prod. 2019, 82, 1442–1450. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Meng, C.; Wu, H.; Shan, W.; Wang, H.; Ling, C.; Zhang, J.; Yang, T. Novel Hybrid CHC from β-Carboline and N-Hydroxyacrylamide Overcomes Drug-Resistant Hepatocellular Carcinoma by Promoting Apoptosis, DNA Damage, and Cell Cycle Arrest. Front. Pharmacol. 2021, 11, 626065. [Google Scholar] [CrossRef]
- Ling, Y.; Gao, W.-J.; Ling, C.; Liu, J.; Meng, C.; Qian, J.; Liu, S.; Gan, H.; Wu, H.; Tao, J.; et al. β-Carboline and N-Hydroxycinnamamide Hybrids as Anticancer Agents for Drug-Resistant Hepatocellular Carcinoma. Eur. J. Med. Chem. 2019, 168, 515–526. [Google Scholar] [CrossRef]
- Chen, L.; Fischle, W.; Verdin, E.; Greene, W.C. Duration of Nuclear NF-κB Action Regulated by Reversible Acetylation. Science 2001, 293, 1653–1657. [Google Scholar] [CrossRef]
- Zeng, L.; Xiao, Q.; Margariti, A.; Zhang, Z.; Zampetaki, A.; Patel, S.; Capogrossi, M.C.; Hu, Y.; Xu, Q. HDAC3 Is Crucial in Shear- and VEGF-Induced Stem Cell Differentiation toward Endothelial Cells. J. Cell Biol. 2006, 174, 1059–1069. [Google Scholar] [CrossRef]
- Grégoire, S.; Xiao, L.; Nie, J.; Zhang, X.; Xu, M.; Li, J.; Wong, J.; Seto, E.; Yang, X.-J. Histone Deacetylase 3 Interacts with and Deacetylates Myocyte Enhancer Factor 2. Mol. Cell. Biol. 2007, 27, 1280–1295. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Yoon, H.-G.; Qin, J.; Wong, J. Regulation of P-TEFb Elongation Complex Activity by CDK9 Acetylation. Mol. Cell. Biol. 2007, 27, 4641–4651. [Google Scholar] [CrossRef] [PubMed]
- Cha, T.-L.; Chuang, M.-J.; Wu, S.-T.; Sun, G.-H.; Chang, S.-Y.; Yu, D.-S.; Huang, S.-M.; Huan, S.K.-H.; Cheng, T.-C.; Chen, T.-T.; et al. Dual Degradation of Aurora A and B Kinases by the Histone Deacetylase Inhibitor LBH589 Induces G2-M Arrest and Apoptosis of Renal Cancer Cells. Clin. Cancer Res. 2009, 15, 840–850. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.J.; Byun, D.-S.; Popova, N.; Murray, L.B.; L’Italien, K.; Sowa, Y.; Arango, D.; Velcich, A.; Augenlicht, L.H.; Mariadason, J.M. Histone Deacetylase 3 (HDAC3) and Other Class I HDACs Regulate Colon Cell Maturation and P21 Expression and Are Deregulated in Human Colon Cancer. J. Biol. Chem. 2006, 281, 13548–13558. [Google Scholar] [CrossRef]
- Beyer, M.; Romanski, A.; Mustafa, A.-H.M.; Pons, M.; Büchler, I.; Vogel, A.; Pautz, A.; Sellmer, A.; Schneider, G.; Bug, G.; et al. HDAC3 Activity Is Essential for Human Leukemic Cell Growth and the Expression of β-Catenin, MYC, and WT1. Cancers 2019, 11, 1436. [Google Scholar] [CrossRef]
- Zhang, L.; Cai, M.; Gong, Z.; Zhang, B.; Li, Y.; Guan, L.; Hou, X.; Li, Q.; Liu, G.; Xue, Z.; et al. Geminin Facilitates FoxO3 Deacetylation to Promote Breast Cancer Cell Metastasis. J. Clin. Investig. 2017, 127, 2159–2175. [Google Scholar] [CrossRef]
- Routholla, G.; Pulya, S.; Patel, T.; Abdul Amin, S.; Adhikari, N.; Biswas, S.; Jha, T.; Ghosh, B. Synthesis, Biological Evaluation, and Molecular Docking Analysis of Novel Linker-Less Benzamide Based Potent and Selective HDAC3 Inhibitors. Bioorganic Chem. 2021, 114, 105050. [Google Scholar] [CrossRef]
- Huang, M.; Xie, X.; Gong, P.; Wei, Y.; Du, H.; Xu, Y.; Xu, Q.; Jing, Y.; Zhao, L. A 18β-Glycyrrhetinic Acid Conjugate with Vorinostat Degrades HDAC3 and HDAC6 with Improved Antitumor Effects. Eur. J. Med. Chem. 2020, 188, 111991. [Google Scholar] [CrossRef]
- Soumyanarayanan, U.; Ramanujulu, P.M.; Mustafa, N.; Haider, S.; Fang Nee, A.H.; Tong, J.X.; Tan, K.S.W.; Chng, W.J.; Dymock, B.W. Discovery of a Potent Histone Deacetylase (HDAC) 3/6 Selective Dual Inhibitor. Eur. J. Med. Chem. 2019, 184, 111755. [Google Scholar] [CrossRef]
- Yan, W.; Liu, S.; Xu, E.; Zhang, J.; Zhang, Y.; Chen, X. Histone Deacetylase Inhibitors Suppress Mutant P53 Transcription via Histone Deacetylase 8. Oncogene 2013, 32, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, T.; Antony, J.; Braithwaite, A.W.; Horsfield, J.A. HDAC8 Inhibition Blocks SMC3 Deacetylation and Delays Cell Cycle Progression without Affecting Cohesin-Dependent Transcription in MCF7 Cancer Cells. J. Biol. Chem. 2016, 291, 12761–12770. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.J.; Tremblay, A.M.; Deblois, G.; Sylvain-Drolet, G.; Giguère, V. An Acetylation Switch Modulates the Transcriptional Activity of Estrogen-Related Receptor α. Mol. Endocrinol. 2010, 24, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Spreafico, M.; Gruszka, A.M.; Valli, D.; Mazzola, M.; Deflorian, G.; Quintè, A.; Totaro, M.G.; Battaglia, C.; Alcalay, M.; Marozzi, A.; et al. HDAC8: A Promising Therapeutic Target for Acute Myeloid Leukemia. Front. Cell Dev. Biol. 2020, 8, 844. [Google Scholar] [CrossRef]
- Kang, Y.; Nian, H.; Rajendran, P.; Kim, E.; Dashwood, W.M.; Pinto, J.T.; Boardman, L.A.; Thibodeau, S.N.; Limburg, P.J.; Löhr, C.V.; et al. HDAC8 and STAT3 Repress BMF Gene Activity in Colon Cancer Cells. Cell Death Dis. 2014, 5, e1476. [Google Scholar] [CrossRef]
- An, P.; Chen, F.; Li, Z.; Ling, Y.; Peng, Y.; Zhang, H.; Li, J.; Chen, Z.; Wang, H. HDAC8 Promotes the Dissemination of Breast Cancer Cells via AKT/GSK-3β/Snail Signals. Oncogene 2020, 39, 4956–4969. [Google Scholar] [CrossRef]
- Wu, J.; Du, C.; Lv, Z.; Ding, C.; Cheng, J.; Xie, H.; Zhou, L.; Zheng, S. The Up-Regulation of Histone Deacetylase 8 Promotes Proliferation and Inhibits Apoptosis in Hepatocellular Carcinoma. Dig. Dis. Sci. 2013, 58, 3545–3553. [Google Scholar] [CrossRef]
- Oehme, I.; Deubzer, H.E.; Wegener, D.; Pickert, D.; Linke, J.-P.; Hero, B.; Kopp-Schneider, A.; Westermann, F.; Ulrich, S.M.; von Deimling, A.; et al. Histone Deacetylase 8 in Neuroblastoma Tumorigenesis. Clin. Cancer Res. 2008, 15, 91–99. [Google Scholar] [CrossRef]
- Chotitumnavee, J.; Yamashita, Y.; Takahashi, Y.; Takada, Y.; Iida, T.; Oba, M.; Itoh, Y.; Suzuki, T. Selective Degradation of Histone Deacetylase 8 Mediated by a Proteolysis Targeting Chimera (PROTAC). Chem. Commun. 2022, 58, 4635–4638. [Google Scholar] [CrossRef]
- Watters, J.M.; Wright, G.; Smith, M.A.; Shah, B.; Wright, K.L. Histone Deacetylase 8 Inhibition Suppresses Mantle Cell Lymphoma Viability While Preserving Natural Killer Cell Function. Biochem. Biophys. Res. Commun. 2021, 534, 773–779. [Google Scholar] [CrossRef]
- Rettig, I.; Koeneke, E.; Trippel, F.; Mueller, W.C.; Burhenne, J.; Kopp-Schneider, A.; Fabian, J.; Schober, A.; Fernekorn, U.; von Deimling, A.; et al. Selective Inhibition of HDAC8 Decreases Neuroblastoma Growth in Vitro and in Vivo and Enhances Retinoic Acid-Mediated Differentiation. Cell Death Dis. 2015, 6, e1657. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Han, S.Y.; Yoo, J.; Kim, G.W.; Jeon, Y.H.; Lee, S.W.; Park, J.; Kwon, S.H. HDAC8-Selective Inhibition by PCI-34051 Enhances the Anticancer Effects of ACY-241 in Ovarian Cancer Cells. Int. J. Mol. Sci. 2022, 23, 8645. [Google Scholar] [CrossRef] [PubMed]
- Olson, D.E.; Wagner, F.F.; Kaya, T.; Gale, J.P.; Aidoud, N.; Davoine, E.L.; Lazzaro, F.; Weïwer, M.; Zhang, Y.-L.; Holson, E.B. Discovery of the First Histone Deacetylase 6/8 Dual Inhibitors. J. Med. Chem. 2013, 56, 4816–4820. [Google Scholar] [CrossRef]
- Rodrigues, D.A.; Ferreira-Silva, G.À.; Ferreira, A.C.S.; Fernandes, R.A.; Kwee, J.K.; Sant’Anna, C.M.R.; Ionta, M.; Fraga, C.A.M. Design, Synthesis, and Pharmacological Evaluation of Novel N-Acylhydrazone Derivatives as Potent Histone Deacetylase 6/8 Dual Inhibitors. J. Med. Chem. 2016, 59, 655–670. [Google Scholar] [CrossRef]
- Tang, G.; Wong, J.C.; Zhang, W.; Wang, Z.; Zhang, N.; Peng, Z.; Zhang, Z.; Rong, Y.; Li, S.; Zhang, M.; et al. Identification of a Novel Aminotetralin Class of HDAC6 and HDAC8 Selective Inhibitors. J. Med. Chem. 2014, 57, 8026–8034. [Google Scholar] [CrossRef] [PubMed]
- Negmeldin, A.T.; Knoff, J.R.; Pflum, M.K.H. The Structural Requirements of Histone Deacetylase Inhibitors: C4-Modified SAHA Analogs Display Dual HDAC6/HDAC8 Selectivity. Eur. J. Med. Chem. 2018, 143, 1790–1806. [Google Scholar] [CrossRef] [PubMed]
- Negmeldin, A.T.; Pflum, M.K.H. The Structural Requirements of Histone Deacetylase Inhibitors: SAHA Analogs Modified at the C5 Position Display Dual HDAC6/8 Selectivity. Bioorg. Med. Chem. Lett. 2017, 27, 3254–3258. [Google Scholar] [CrossRef]
- Negmeldin, A.T.; Padige, G.; Bieliauskas, A.V.; Pflum, M.K.H. Structural Requirements of HDAC Inhibitors: SAHA Analogues Modified at the C2 Position Display HDAC6/8 Selectivity. ACS Med. Chem. Lett. 2017, 8, 281–286. [Google Scholar] [CrossRef]
- Federico, S.; Khan, T.; Fontana, A.; Brogi, S.; Benedetti, R.; Sarno, F.; Carullo, G.; Pezzotta, A.; Saraswati, A.P.; Passaro, E.; et al. Azetidin-2-One-Based Small Molecules as Dual hHDAC6/HDAC8 Inhibitors: Investigation of Their Mechanism of Action and Impact of Dual Inhibition Profile on Cell Viability. Eur. J. Med. Chem. 2022, 238, 114409. [Google Scholar] [CrossRef]
- Yao, D.; Li, C.; Rajoka, M.S.R.; He, Z.; Huang, J.; Wang, J.; Zhang, J. P21-Activated Kinase 1: Emerging Biological Functions and Potential Therapeutic Targets in Cancer. Theranostics 2020, 10, 9741–9766. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, X.; Chen, G.; Wang, H.; Jia, L.; Hao, Y.; Yao, D. Identification of a Novel PAK1/HDAC6 Dual Inhibitor ZMF-23 That Triggers Tubulin-Stathmin Regulated Cell Death in Triple Negative Breast Cancer. Int. J. Biol. Macromol. 2023, 251, 126348. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.Y.; Timpson, P.; Horvath, L.G.; Daly, R.J. FAK Signaling in Human Cancer as a Target for Therapeutics. Pharmacol. Ther. 2015, 146, 132–149. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Liu, X.; Zhang, Y.-F.; Tian, X.-Y.; Deng, M.-Y.; Huang, C.-Z.; Zhang, S.-Y. The Dual FAK-HDAC Inhibitor MY-1259 Displays Potent Activities in Gastric Cancers in Vitro and in Vivo. Bioorganic Chem. 2023, 131, 106328. [Google Scholar] [CrossRef] [PubMed]
- Curis, Inc. A Phase I Open-Label, Multiple Dose, Sequential Dose Escalation Study to Investigate the Safety and Pharmacokinetics of Intravenous CUDC-101 in Subjects with Advanced and Refractory Solid Tumors. 2018. Available online: https://clinicaltrials.gov/study/NCT00728793 (accessed on 27 September 2023).
- Cai, X.; Zhai, H.-X.; Wang, J.; Forrester, J.; Qu, H.; Yin, L.; Lai, C.-J.; Bao, R.; Qian, C. Discovery of 7-(4-(3-Ethynylphenylamino)-7-Methoxyquinazolin-6-Yloxy)-N-Hydroxyheptanamide (CUDC-101) as a Potent Multi-Acting HDAC, EGFR, and HER2 Inhibitor for the Treatment of Cancer. J. Med. Chem. 2010, 53, 2000–2009. [Google Scholar] [CrossRef] [PubMed]
- Mundipharma Research Limited A Phase 1/2 Study to Investigate the Safety, Pharmacokinetics and Efficacy of EDO-S101, a First-in-Class Alkylating Histone Deacetylase Inhibition (HDACi) Fusion Molecule, in Patients with Advanced Solid Tumors. 2023. Available online: https://clinicaltrials.gov/study/NCT03345485 (accessed on 28 September 2023).
- Mundipharma Research Limited A Phase 1 Trial to Investigate the Safety, Pharmacokinetic Profiles and the Efficacy of Tinostamustine, a First-in-Class Alkylating Histone Deacetylase Inhibition (HDACi) Fusion Molecule, in Relapsed/Refractory Hematologic Malignancies. 2023. Available online: https://clinicaltrials.gov/study/NCT02576496 (accessed on 28 September 2023).
- Mehrling, T.; Chen, Y. The Alkylating-HDAC Inhibition Fusion Principle: Taking Chemotherapy to the Next Level with the First in Class Molecule EDO-S101. Anti-Cancer Agents Med. Chem. 2016, 16, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Shulman, D.S. Phase 1 Trial of CUDC-907 in Children and Young Adults with Relapsed or Refractory Solid Tumors, CNS Tumors, or Lymphoma. 2023. Available online: https://clinicaltrials.gov/study/NCT02909777 (accessed on 28 September 2023).
- Curis, Inc. Phase I Open Label, Multi-Center Study to Assess the Safety, Tolerability and Pharmacokinetics of Orally Administered CUDC-907, an HDAC and PI3K Inhibitor, in Subjects with Advanced/Relapsed Solid Tumors. 2019. Available online: https://clinicaltrials.gov/study/NCT02307240 (accessed on 28 September 2023).
- Machado de Oliveira, R.; Sarkander, J.; Kazantsev, A.; Outeiro, T. SIRT2 as a Therapeutic Target for Age-Related Disorders. Front. Pharmacol. 2012, 3, 82. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, J.; Hong, T.; Chen, X.; Cui, L. SIRT2: Controversy and Multiple Roles in Disease and Physiology. Ageing Res. Rev. 2019, 55, 100961. [Google Scholar] [CrossRef]
- Yang, M.H.; Laurent, G.; Bause, A.S.; Spang, R.; German, N.; Haigis, M.C.; Haigis, K.M. HDAC6 and SIRT2 Regulate the Acetylation State and Oncogenic Activity of Mutant K-RAS. Mol. Cancer Res. 2013, 11, 1072–1077. [Google Scholar] [CrossRef]
- North, B.J.; Marshall, B.L.; Borra, M.T.; Denu, J.M.; Verdin, E. The Human Sir2 Ortholog, SIRT2, Is an NAD+-Dependent Tubulin Deacetylase. Mol. Cell 2003, 11, 437–444. [Google Scholar] [CrossRef]
- Sinatra, L.; Vogelmann, A.; Friedrich, F.; Tararina, M.; Neuwirt, E.; Colcerasa, A.; König, P.; Toy, L.; Yesiloglu, T.; Zhang, L.; et al. Development of First-in-Class Dual Sirt2/HDAC6 Inhibitors as Molecular Tools for Dual Inhibition of Tubulin Deacetylation. 2023. Available online: https://chemrxiv.org/engage/chemrxiv/article-details/6516a83b00659409121c0bc2 (accessed on 28 September 2023).
- Chen, Y.; Tan, L.; Gao, J.; Lin, C.; Wu, F.; Li, Y.; Zhang, J. Targeting Glutaminase 1 (GLS1) by Small Molecules for Anticancer Therapeutics. Eur. J. Med. Chem. 2023, 252, 115306. [Google Scholar] [CrossRef]
- Masisi, B.K.; El Ansari, R.; Alfarsi, L.; Rakha, E.A.; Green, A.R.; Craze, M.L. The Role of Glutaminase in Cancer. Histopathology 2020, 76, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Q.; Yang, L.; Feng, Y.; Zhu, Z.; Li, N.; Zheng, L.; Sun, Y.; Pan, C.; Qiu, H.; Cui, X.; et al. HDAC I/IIb Selective Inhibitor Purinostat Mesylate Combined with GLS1 Inhibition Effectively Eliminates CML Stem Cells. Bioact. Mater. 2023, 21, 483–498. [Google Scholar] [CrossRef] [PubMed]

















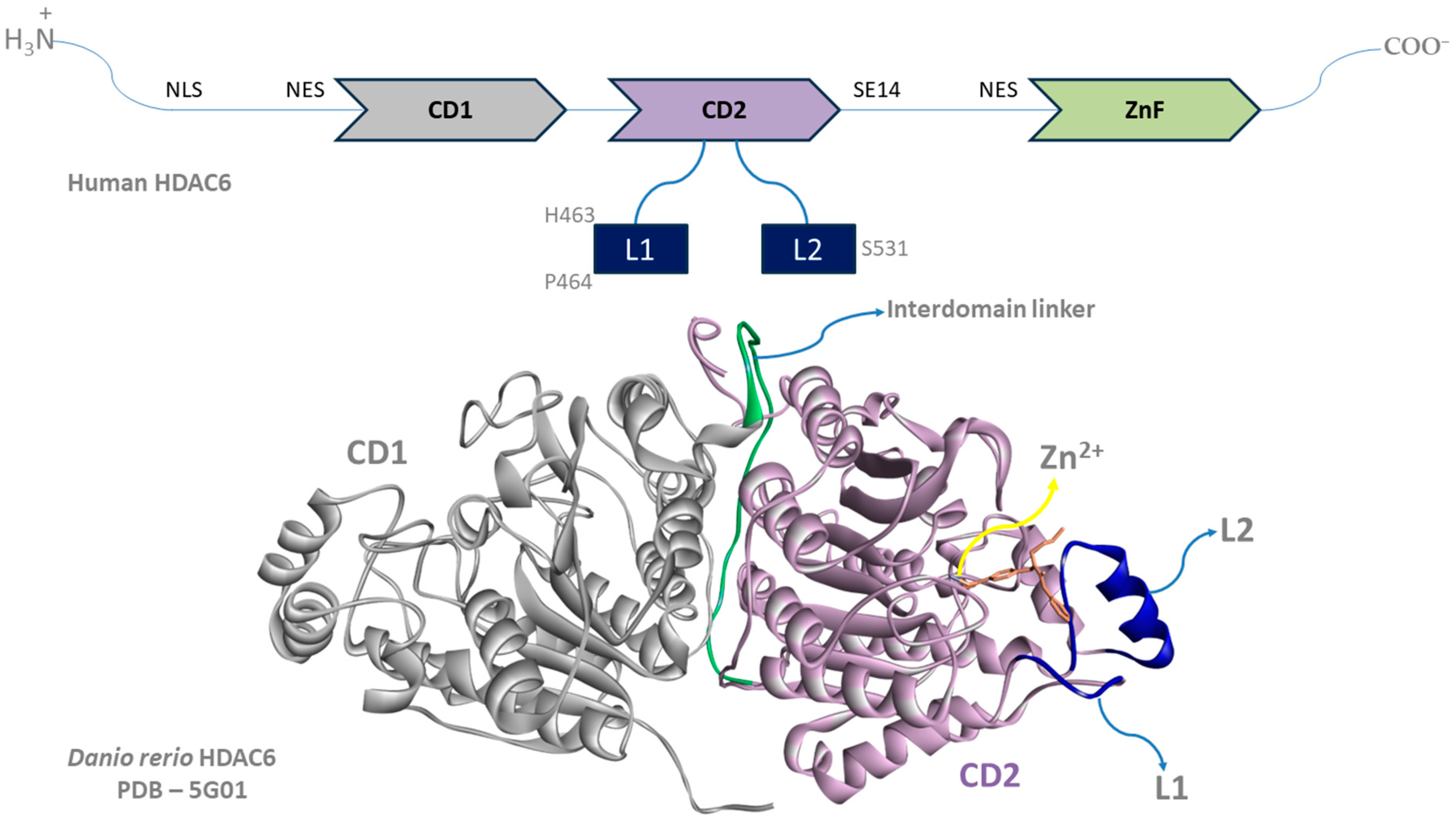{kind=link}
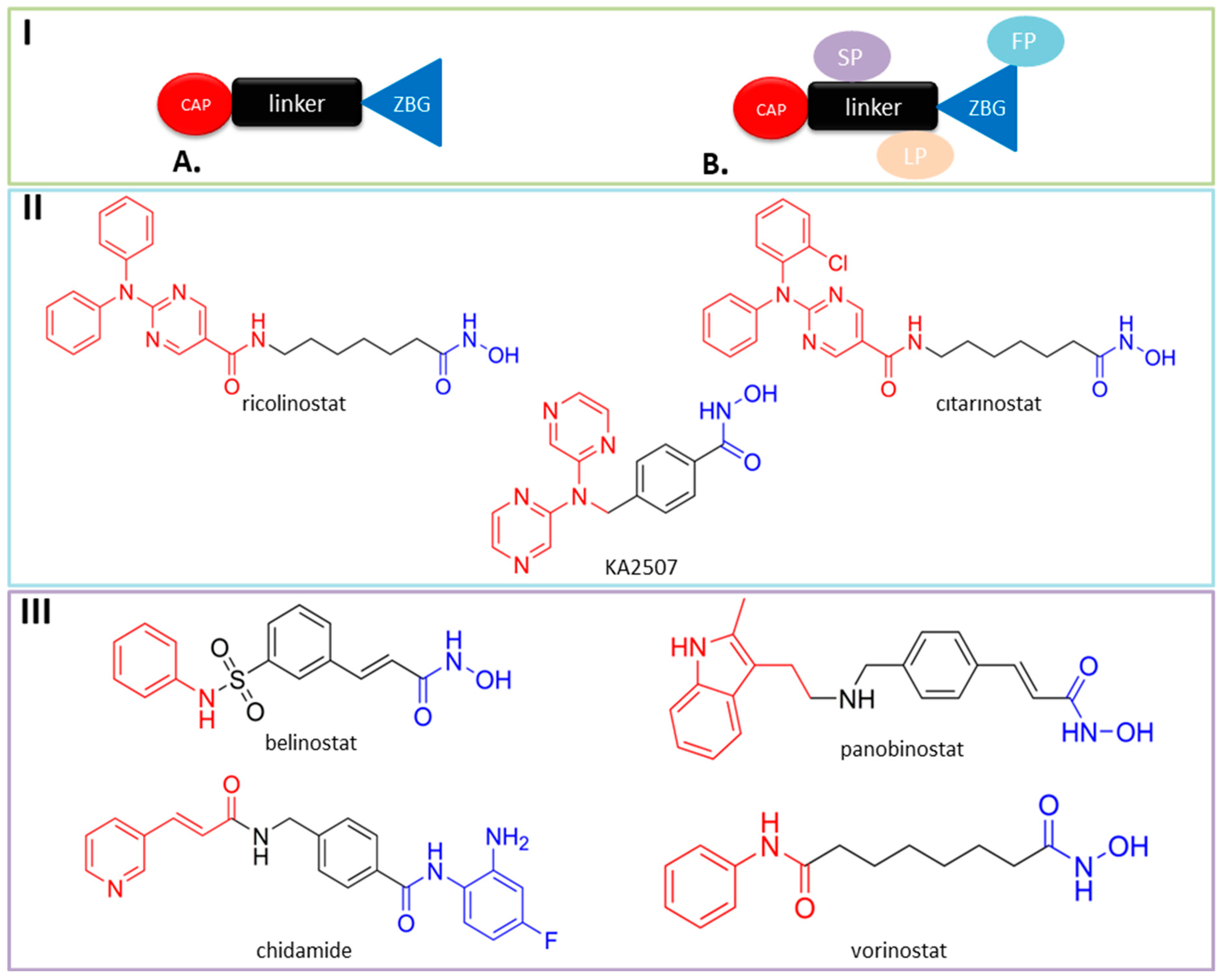{kind=link}
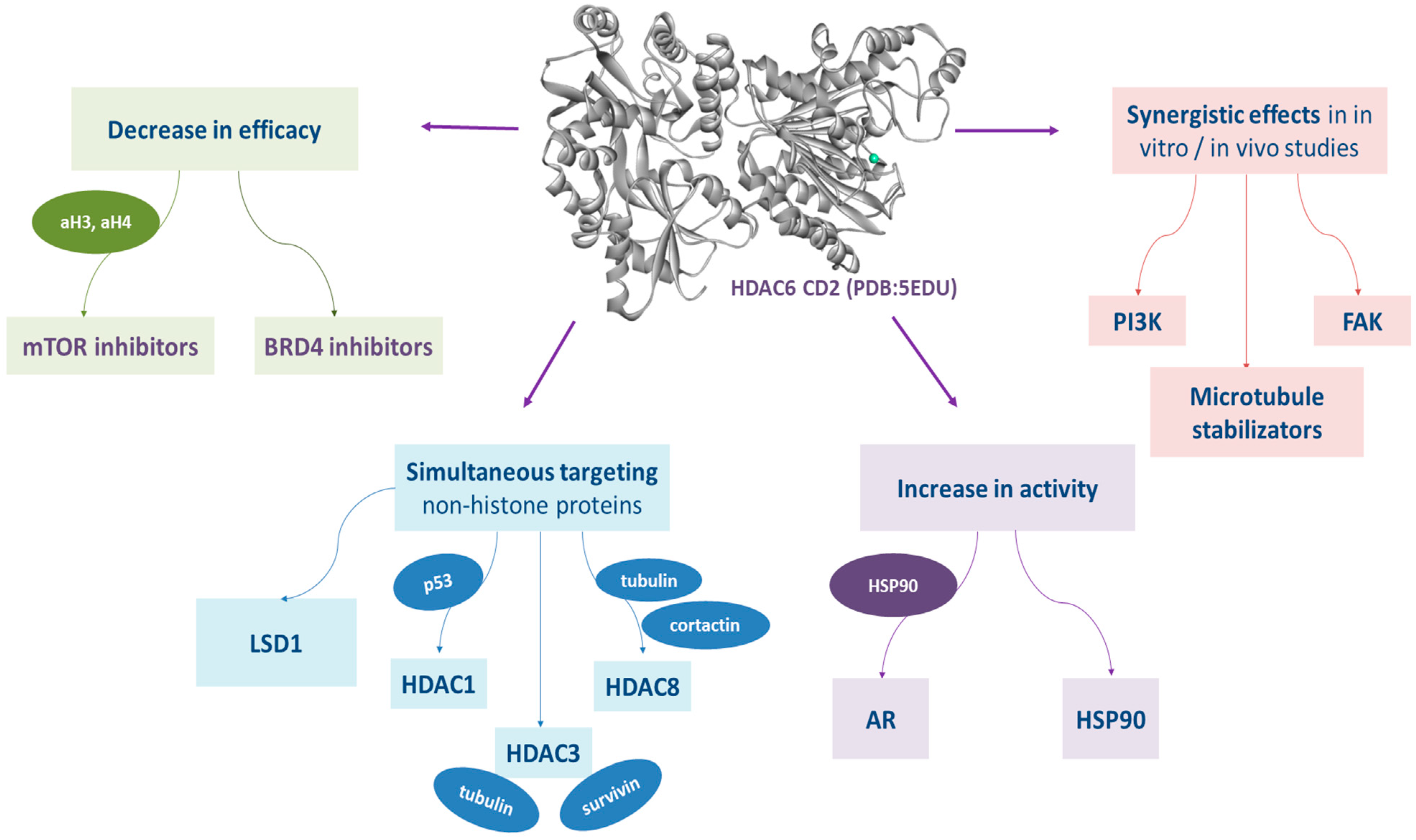{kind=link}
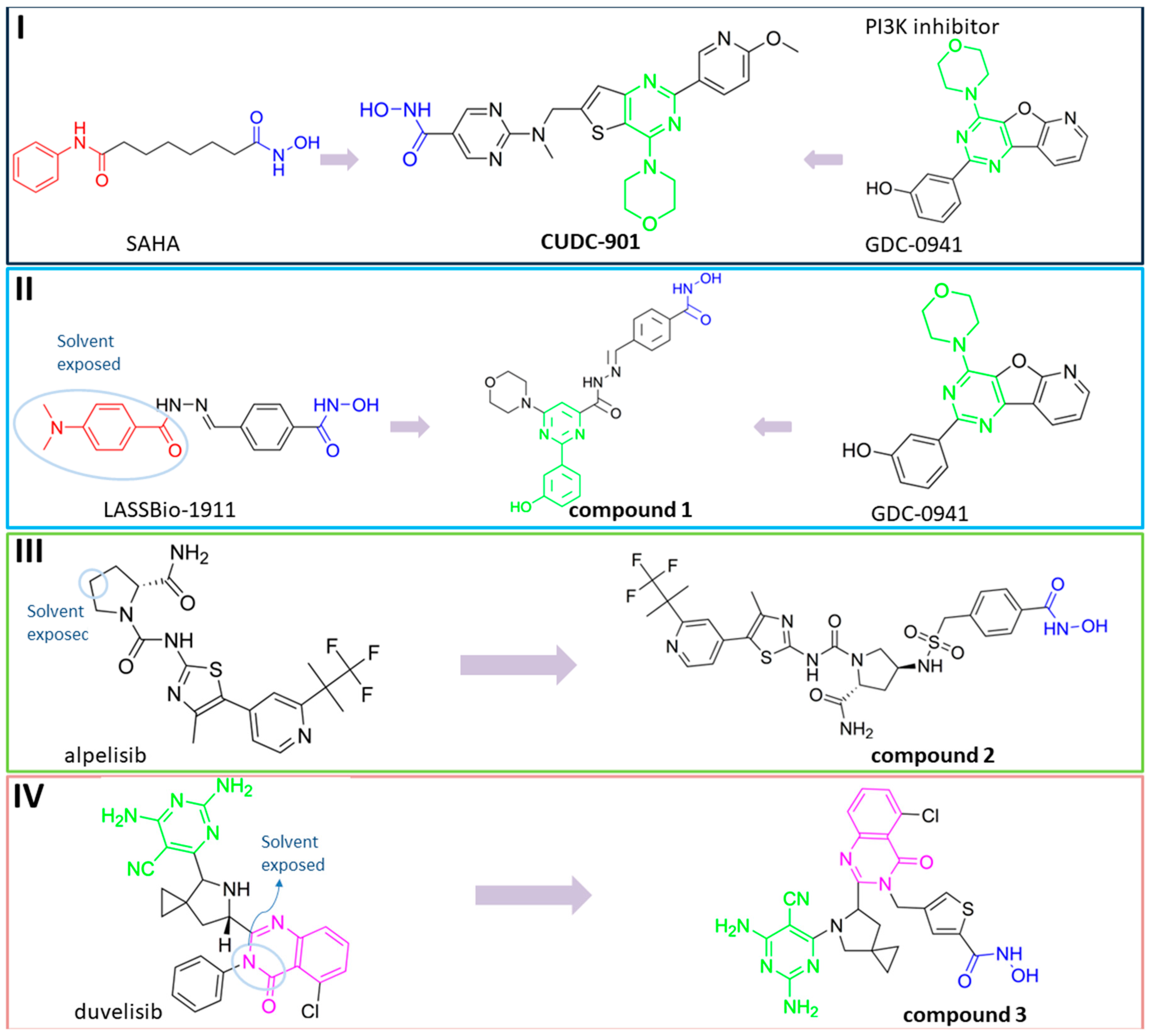{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure/PubChem ID | Targets | IC50 Values | In Vivo | Ref. |
|---|---|---|---|---|
 PubChem CID: 168289467 (1) | HDAC6/PI3Kα | 15.3 nM (HDAC6) 46.3 nM (PI3Kα) | No data | [71] |
 2 | HDAC6/PI3Kα | 26 nM (HDAC6) 2.9 nM (PI3Kα) | No data | [72] |
 PubChem CID: 168298545 (3) | HDAC6/PI3Kδ | 2.3 nM (PI3Kδ) 13 nM (HDAC6) | No data | [70] |
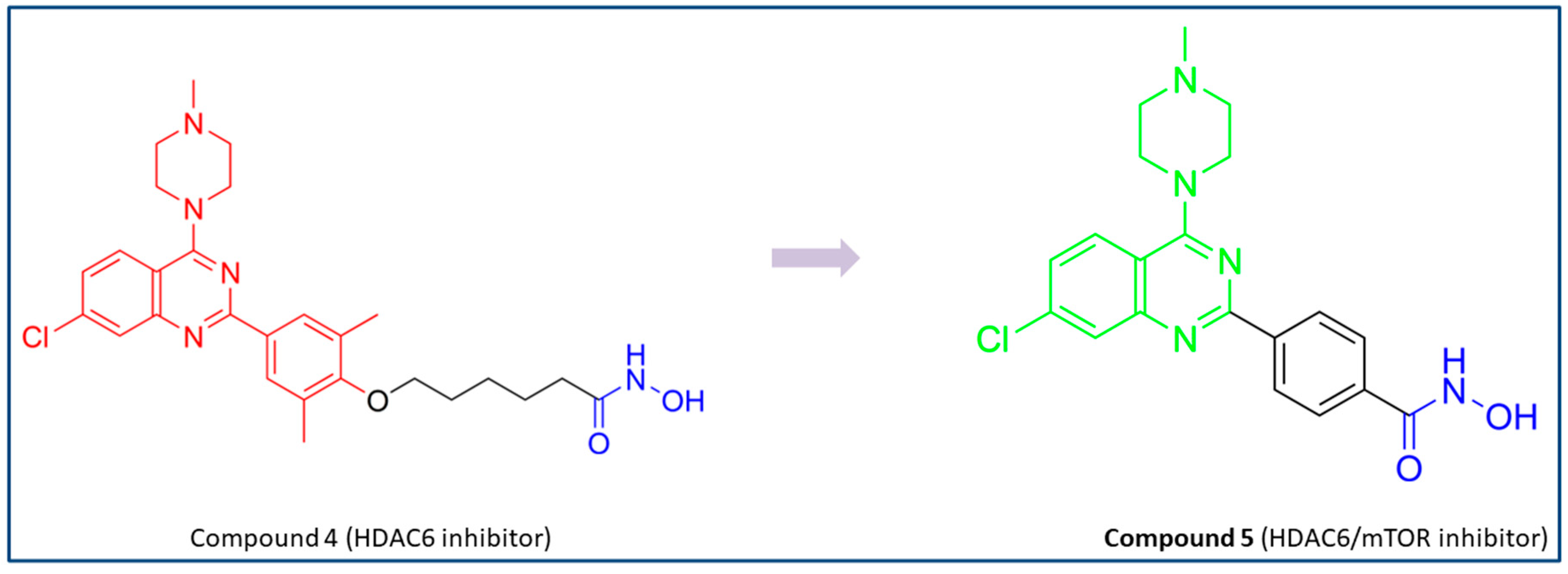
 PubChem CID: 163322242 (5) | HDAC6/mTOR | 56 nM (HDAC6) 133.7 nM (mTOR) | No data | [73] |
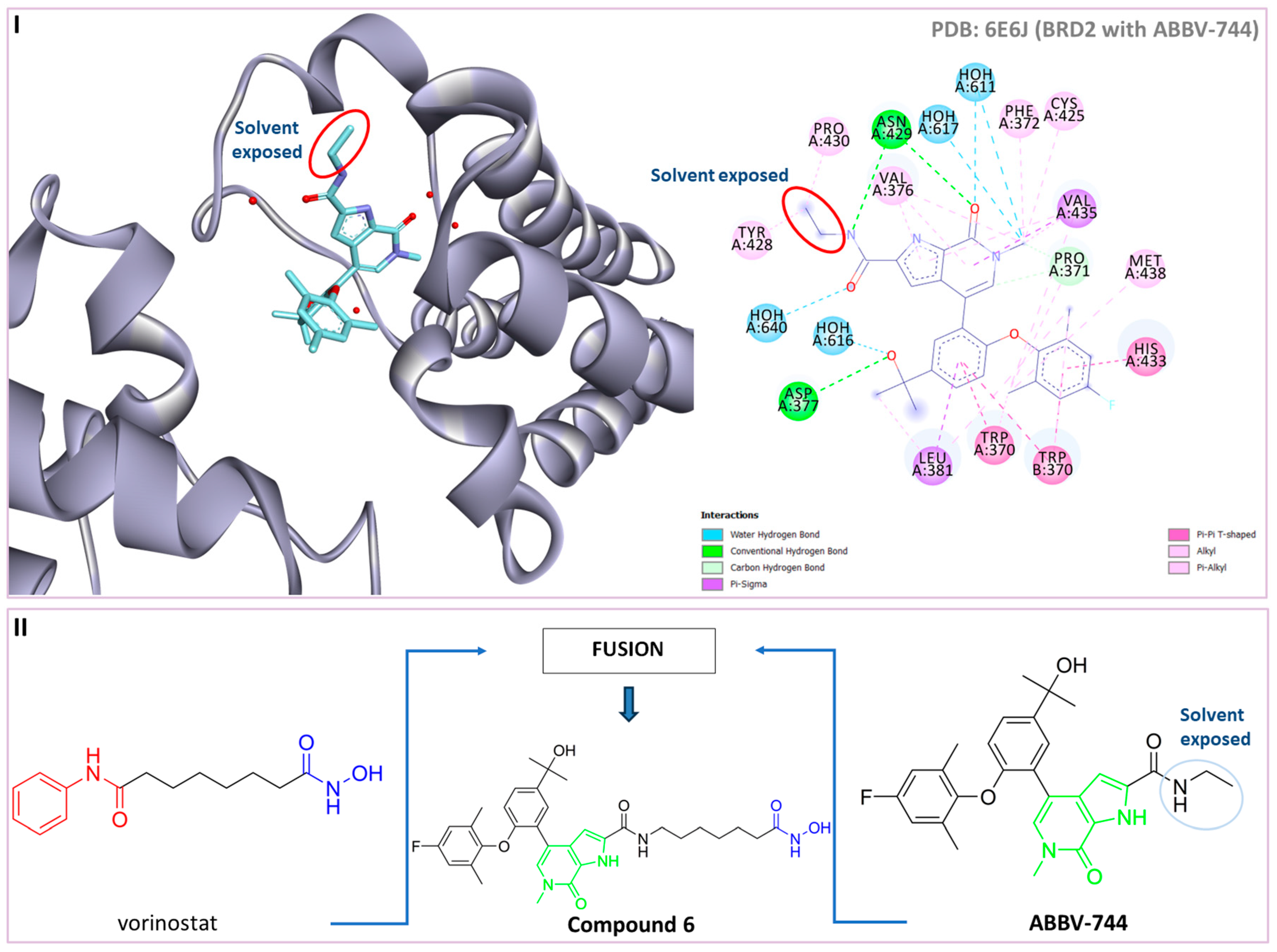
 PubChem CID: 155431228 (6) | HDAC6/ BRD4 | 17.2 nM (HDAC6) 1.2 µM (BRD4) | No data | [82] |
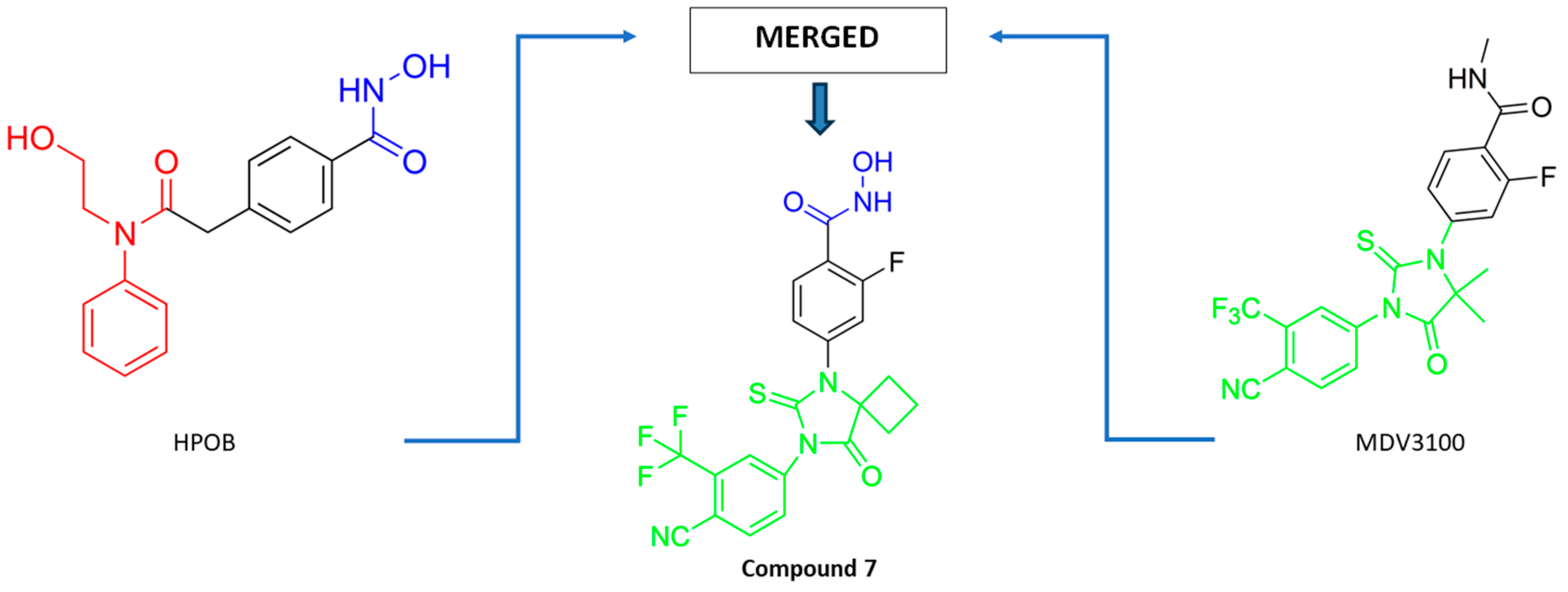
 PubChem CID: 146434737 (7) | HDAC6/AR | HDAC6 (0.98 µM) AR (0.63 µM) | Stronger antitumor activity compared to positive control (MDV3100) in CRPC xenograft model | [86] |
 PubChem CID: 145985243 (8) | HDAC6/HSP90 | 1.15 nM (HDAC6) 46.3 nM (HSP90) | No data | [91] |
 PubChem CID: 145984665 (9) | HDAC6/HSP90 | 4.32 nM (HDAC6) 46.8 nM (HSP90) | TGI = 44.8% (50 mg/kg of 9); TGI = 69.7% (100 mg/kg of 9); TGI = 72.3% (50 mg/kg of 9) in combination with afatinib (20 mg/kg); human NSCLC H1975 xenograft model | [89] |
 PubChem CID: 167505967 (10) | HDAC6/HSP90 | 4.56 nM (HDAC6) 52 nM (HSP90) | TGI = 83.9% (50 mg/kg of 10) in combination with anti-PD-1 (programmed cell death protein 1) antibodies (200 µg); BALB/c mice-bearing CT26 tumor (colorectal cancer) | [92] |
 PubChem CID: 137654946 (11) | HDAC6/ tubulin | 64.5 nM (HDAC6) | TGI = 24.8% (100 mg/kg) and TGI = 68.5% (200 mg/kg)—human prostate xenograft nude mouse model (PC3); TGI = 35.5% (50 mg/kg) and TGI = 58.2% (100 mg/kg)—multiple myeloma xenograft model (RPMI-8226) | [95] |
 PubChem CID: 137645287 (12) | HDAC6/ tubulin | 275.35 nM (HDAC6) | TGI = 40.9% human leukemia mouse xenograft model (HL-60) TGI = 31.1%—PC3 grafted mice model | [96] |
 13 | HDAC6/ tubulin | residual HDAC6 activity at 20 µM was 3% | No data | [98] |
 PubChem CID: 122550211 (14) | HDAC6/ tubulin | 17 nM (HDAC6) | TGI = 66.05% (50 mg/kg of 14)—HCT116 model; TGI = 77.39% (25 mg/kg of 14)—A2780s model; TGI = 65.65% (50 mg/kg of 14)—MCF-7 model | [99] |
 PubChem CID: 132138171 (15) | HDAC6/LSD1 | 48 nM (HDAC6) 6 nM (LSD1) | TGI = 67% (25 mg/kg of 15)—MM 1.S xenograft model | [112] |
 PubChem CID: 132118589 (16) | HDAC6/LSD1 | 0.11 µM (HDAC6) 0.54 µM (LSD1) | No data | [113] |
 17 | HDAC6/HDAC1 | 16.15 nM (HDAC6) 31.1 nM (HDAC1) | No data | [127] |
 PubChem CID: 49847150 (18) | HDAC6/HDAC1 | 2.46 nM (HDAC6) 19.4 nM (HDAC1) | No data | [128] |
 19 | HDAC6/HDAC1 | 2.6 nM (HDAC6) 4.1 nM (HDAC1) | Treatment with 19 inhibited growth of hepatoma tumor that was comparable to positive control SAHA at the same dose | [129] |
 PubChem CID: 155515836 (20) | HDAC6/HDAC1 | 13 nM (HDAC6) 27 nM (HDAC1) | No data | [130] |
 21 | HDAC6/HDAC1 | 7.6 nM (HDAC6) 29 nM (HDAC1) | TGI = 65% (70 µmol/kg of 21)—Bel7402/5-FU xenograft tumor model | [131] |
 22 | HDAC6/HDAC1 | 3.1 nM (HDAC6) 1.3 nM (HDAC1) | No data | [132] |
 PubChem CID: 155556138 (23) | HDAC6/HDAC3 | 34 nM (HDAC6) 2.6 nM (HDAC3) | No data | [143] |
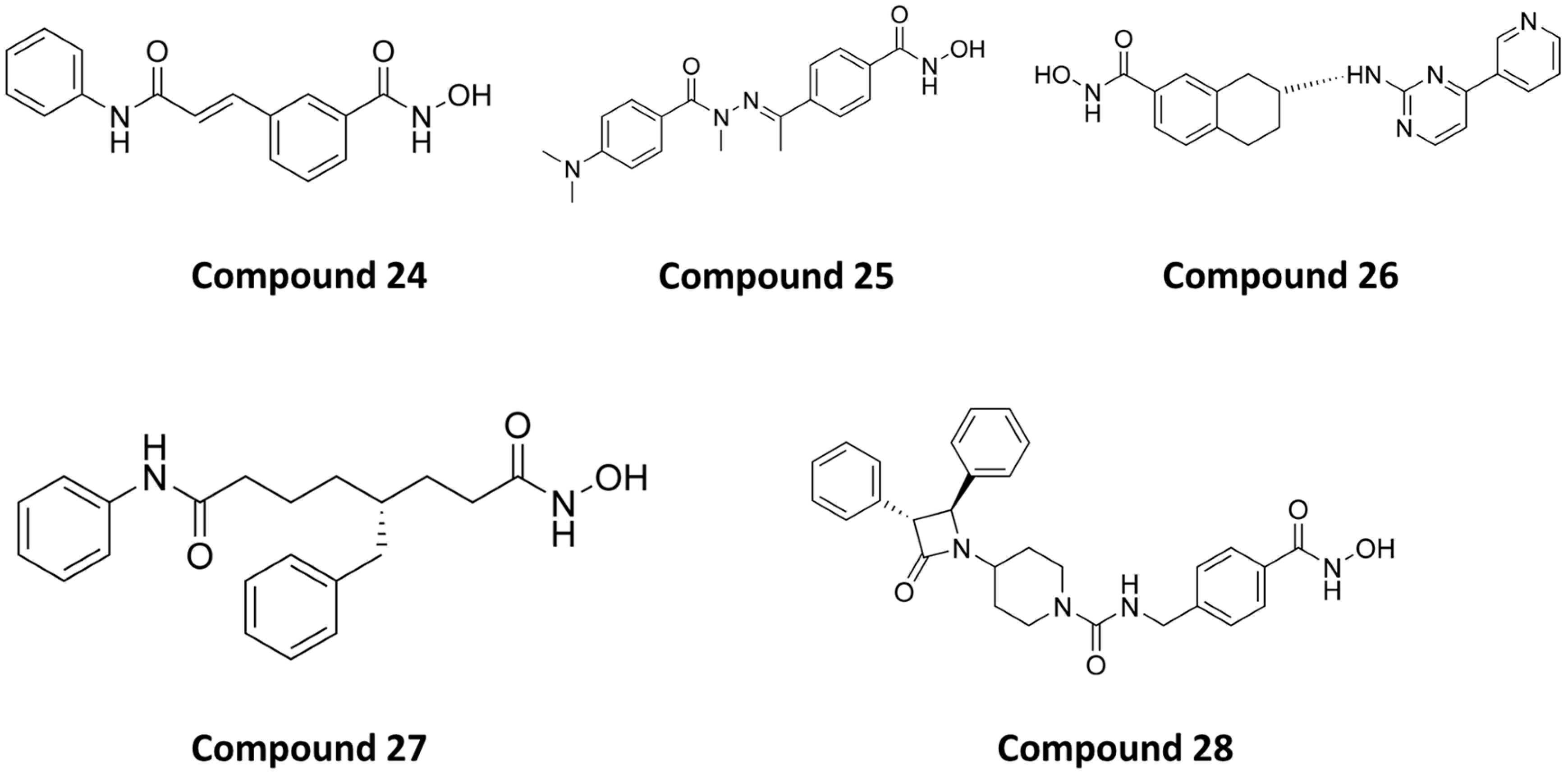
 PubChem CID: 71681069 (24) | HDAC6/HDAC8 | 21 nM (HDAC6) 37 nM (HDAC8) | No data | [156] |
 PubChem CID: 154487896 (25) | HDAC6/HDAC8 | 97 nM (HDAC6) 54 nM (HDAC8) | No data | [157] |
 PubChem CID: 92045024 (26) | HDAC6/HDAC8 | 50 nM (HDAC6) 80 nM (HDAC8) | No data | [158] |
 PubChem CID: 134283800 (27) | HDAC6/HDAC8 | 48 nM (HDAC6) 27 nM (HDAC8) | No data | [159] |
 PubChem CID: 168282542 (28) | HDAC6/HDAC8 | 21 nM (HDAC6) 42 nM (HDAC8) | Chemical toxicity was evaluated by zebrafish model. 28 was well tolerated up to 75 µM, which confirmed its safety profile | [162] |
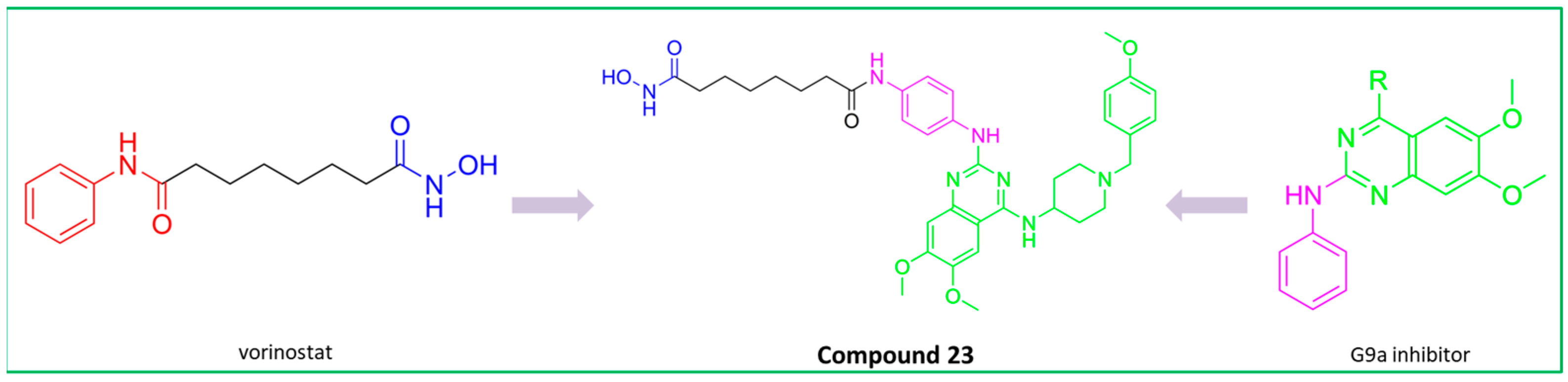
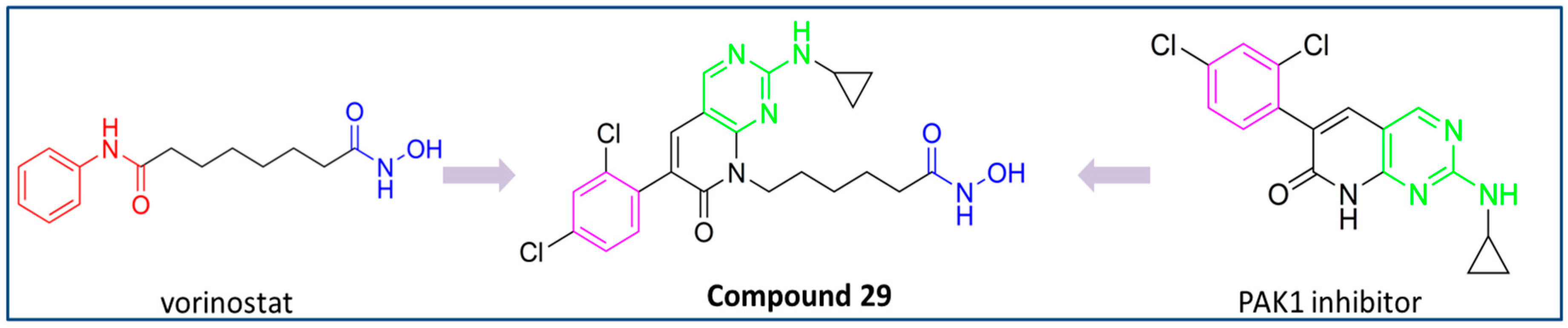
 PubChem CID: 168510343 (29) | HDAC6/PAK1 | 38.23 nM (HDAC6) 13.62 nM (PAK1) | Great potential of 29 was confirmed by in vivo studies—in the TNBC xenograft zebrafish and mouse model | [164] |
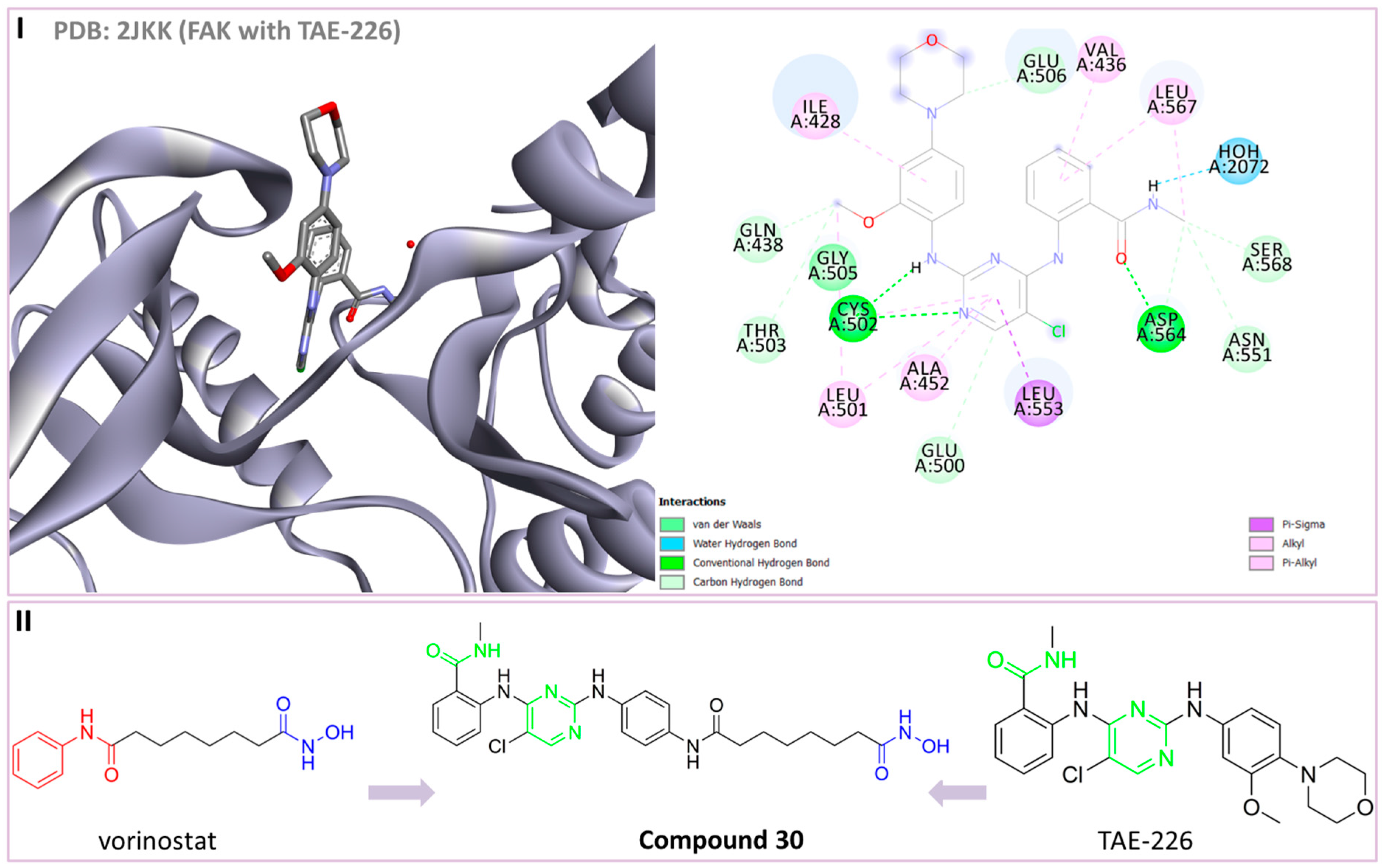
 30 | HDAC /FAK | 16 nM (HDAC6) 132 nM (FAK) | TGI = 53.5% (20 mg/kg of 30)—gastric cancer cells MGC-803 xenograft model | [166] |
| Cd | Interactions with HDAC6 | Interactions with Another Target | Ref. |
|---|---|---|---|
| 1 | / | PI3Kα: hydrogen bonds with Val851, tyrosine 836, aspartate 810. | [71] |
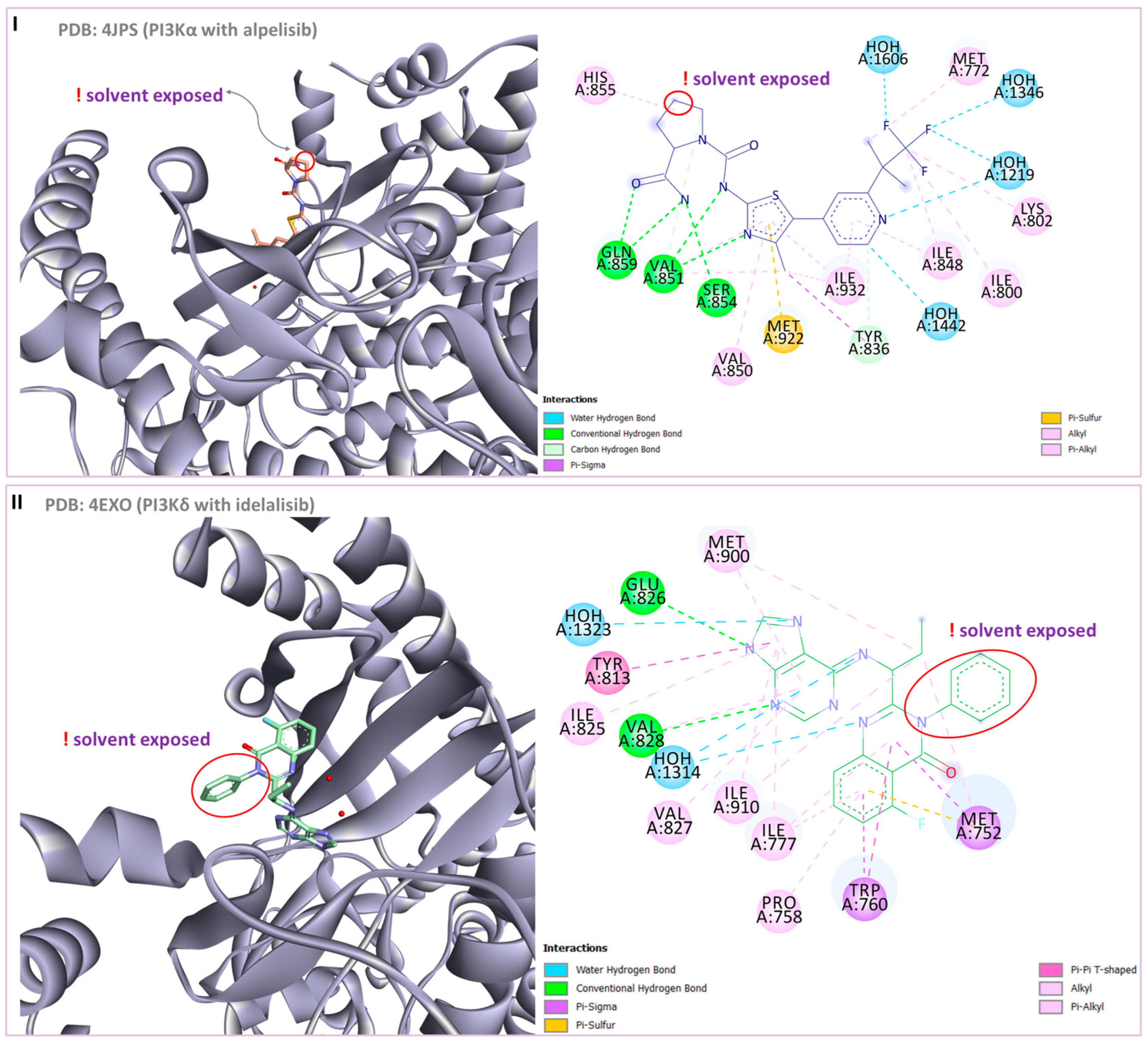
| 2 | PDB:5EDU—hydroxamic acid coordinates the zinc ion; hydrogen bonds with His610, His611 and Tyr782; π-π interactions are established with Phe620 and Phe680. | PI3Kα (PDB:4JPS): key hydrogen bonds with Val851, Ser854 and Gln859; nitrogen atom of pyridine forms hydrogen bonds with Lys802 and Asp810; hydrogen bond is established between fluorine atoms and Lys802 | [72] |
| 3 | Hydroxamic acid coordinates zinc; this complex is additionally stabilized by hydrogen bond with Tyr745; π-π interactions are established between the linker and Phe583, His614, Phe643 residues; π-π interactions are established between CAP group and His463 and Pro464. | PI3Kδ: hydrogen bonds with hinge region (Glu826 and Val828); π-π interactions are established with Met752 and Trp760. | [70] |
| 5 | Hydroxamic acid chelates the zinc ion, and this complex is additionally stabilized by hydrogen bond with the His573; π-π interactions are formed with Phe583 and Phe643. | mTOR: salt bridge interactions are observed with Asp2195, Asp2357 and Glu2190 residues and hydrogen bonds with Val2240 and Trp2239 residues. | [73] |
| 7 | Compound 7 interacts with HDAC6 in a manner similar to that of already known HDAC6 inhibitor—HOBP. | AR: compound 7 inserts in AR in a manner similar to that of bicalutamide. | [86] |
| 9 | Hydrogen bonds are described between CAP group and Ser546, Phe566 and Ile569; hydrophobic interactions are established with Phe620; hydroxamic acid coordinates zinc; and this complex is stabilized by hydrogen bonds with His610 and Gly619. | HSP90: hydrogen bonds are established between Asn51, Lys58 Asp93, Gly108, Thr184 and 2,4-dihydroxy-5-isopropybenzoyl moiety; hydrophobic interactions are established with Ala55, Met98, Thr109. | [89] |
| 12 | PDB:5EDU—N-hydroxyformamide moiety forms a complex with the zinc ion; hydrogen bonds are established with residues His610 and His611; hydrophobic interactions are established with residues Ser568, Gly619, Phe620, His651 and Phe680. | Colchicine binding site of tubulin (PDB:4O2B): hydrophobic interactions are formed with Met259, Ala316, Ile318 and Ile378; hydrogen bonds are established with Ser178 and Asp329 residues. | [96] |
| 17 | Π-cation interaction is observed between the quinazoline group and the phenyl group of Tyr1022; hydrophobic interactions are established with Asp1044 and Tyr1055 residues; hydroxamic acid forms a complex with the zinc ion. | HDAC1: hydrophobic interactions are formed with Lys331, Arg270 and Arg306 residues; hydroxamic acid forms a complex with the zinc ion. | [127] |
| 25 | Docking procedure is performed with a homology model built from HDAC7 (PDB ID: 1C0Z). | HDAC8 (PDB ID:1VKG)—a complex with the zinc ion is established as well as hydrogen bonds with His142, His143 and Tyr306; hydrophobic interactions are formed with Phe152 and Phe208 residues. | [157] |
| 26 | Hydrophobic interactions are formed with Phe620 and Phe680; hydroxamic acid coordinates the zinc ion. | HDAC8 (PDB ID:1VKG): hydrophobic interactions are established with Pro35, Phe152 and Tyr306 of HDAC8 | [158] |
| 28 | Hydroxamic acid coordinates the zinc ion and forms hydrogen bonds with Gly619 and Tyr782; hydrogen bonds are established with Phe680; and π-π interactions are established between benzyl linker and His651, Phe620 and Phe680. | HDAC8: in addition to coordinating the zinc ion, hydroxamic acid forms hydrogen bonds with Gly151 and Tyr306; π-π interactions are formed between benzyl linker and Phe152 and Phe208. Π-cation interaction is established between phenyl substituent of β-lactam moiety and Lys202 | [162] |
| 29 | Hydroxamic acid group forms two hydrogen bonds with the His573 residue and generates a chelate product with the zinc ion; π-alkyl interactions are established with Pro464, Pro711 and Leu712 residues. | PAK1: two conserved hydrogen interactions are observed with the key Leu3347 residue from the kinase hinge; hydrophobic interactions are formed with Val284, Met344, Val342 and Lys299 residues; π-sulfur interaction is established between phenyl-moiety and Met344, and a π-cation interaction is formed within Lys299; hydrogen bonds are described between hydroxamic acid and Asp393 and Asn394 residues. | [164] |
| Compound | Structure | Phase of Clinical Trials | HDAC (IC50 in nM) | Other Targets (IC50 in nM) | Clinical Use |
|---|---|---|---|---|---|
| CUDC-101 |  | Phase 1 [167] | HDAC1 (4.5); HDAC2 (12.6); HDAC3 (9.1); HDAC8 (79.8); HDAC4 (13.2); HDAC5 (11.4); HDAC6 (5.1); HDAC7 (373); HDAC9 (67.2); HDAC10 (26.1) [168] | EGFR (2.4); HER2 (15.7) [168] | Advanced solid tumors [167] |
| Tinostamustine (EDO-S101, NL-101) |  | Phase ½ [169,170] | HDAC1 (9); HDAC2 (9); HDAC3 (25); HDAC8 (107); HDAC6 (6); HDAC10 (72) [171] | DNA alkylating agent [171] | Malignant hematological diseases and advanced solid tumors [169,170] |
| Fimepinostat (CUDC-907) |  | Phase ½ (completed) Phase 1—active [172,173] | HDAC1 (1.7); HDAC2 (5.0); HDAC3 (1.8); HDAC8 (191); HDAC4 (409); HDAC5 (674); HDAC6 (27); HDAC7 (426); HDAC9 (554); HDAC10 (2.8); HDAC1 (5.4) [69] | PI3Kα (19); PI3Kβ (54); PI3Kδ (39); PI3Kγ (311) [69] | Advanced, relapsed and refractory solid tumors, CNS tumors, lymphoma [172,173] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beljkas, M.; Ilic, A.; Cebzan, A.; Radovic, B.; Djokovic, N.; Ruzic, D.; Nikolic, K.; Oljacic, S. Targeting Histone Deacetylases 6 in Dual-Target Therapy of Cancer. Pharmaceutics 2023, 15, 2581. https://doi.org/10.3390/pharmaceutics15112581
Beljkas M, Ilic A, Cebzan A, Radovic B, Djokovic N, Ruzic D, Nikolic K, Oljacic S. Targeting Histone Deacetylases 6 in Dual-Target Therapy of Cancer. Pharmaceutics. 2023; 15(11):2581. https://doi.org/10.3390/pharmaceutics15112581
Chicago/Turabian StyleBeljkas, Milan, Aleksandra Ilic, Alen Cebzan, Branko Radovic, Nemanja Djokovic, Dusan Ruzic, Katarina Nikolic, and Slavica Oljacic. 2023. "Targeting Histone Deacetylases 6 in Dual-Target Therapy of Cancer" Pharmaceutics 15, no. 11: 2581. https://doi.org/10.3390/pharmaceutics15112581