
Transporter-Mediated Cellular Distribution of Tyrosine Kinase Inhibitors as a Potential Resistance Mechanism in Chronic Myeloid Leukemia
 , ,
, ,
Abstract
:1. Introduction
2. CML Stem Cells and Progenitor Cells
3. Resistance Mechanisms against TKIs in CML
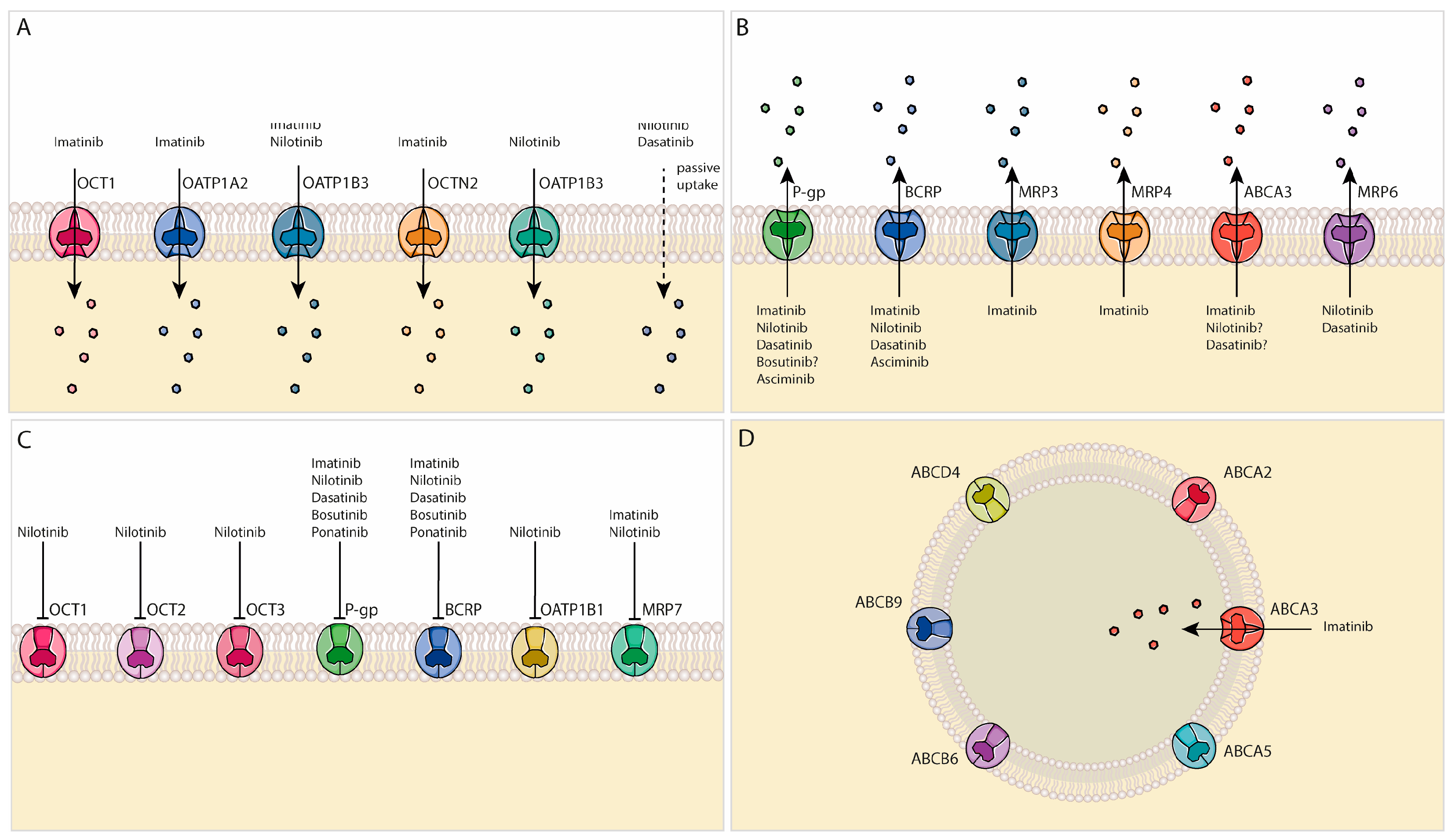
4. Drug Transporter Expression in HSCs and LSCs
5. TKIs and Cellular Transport Mechanisms
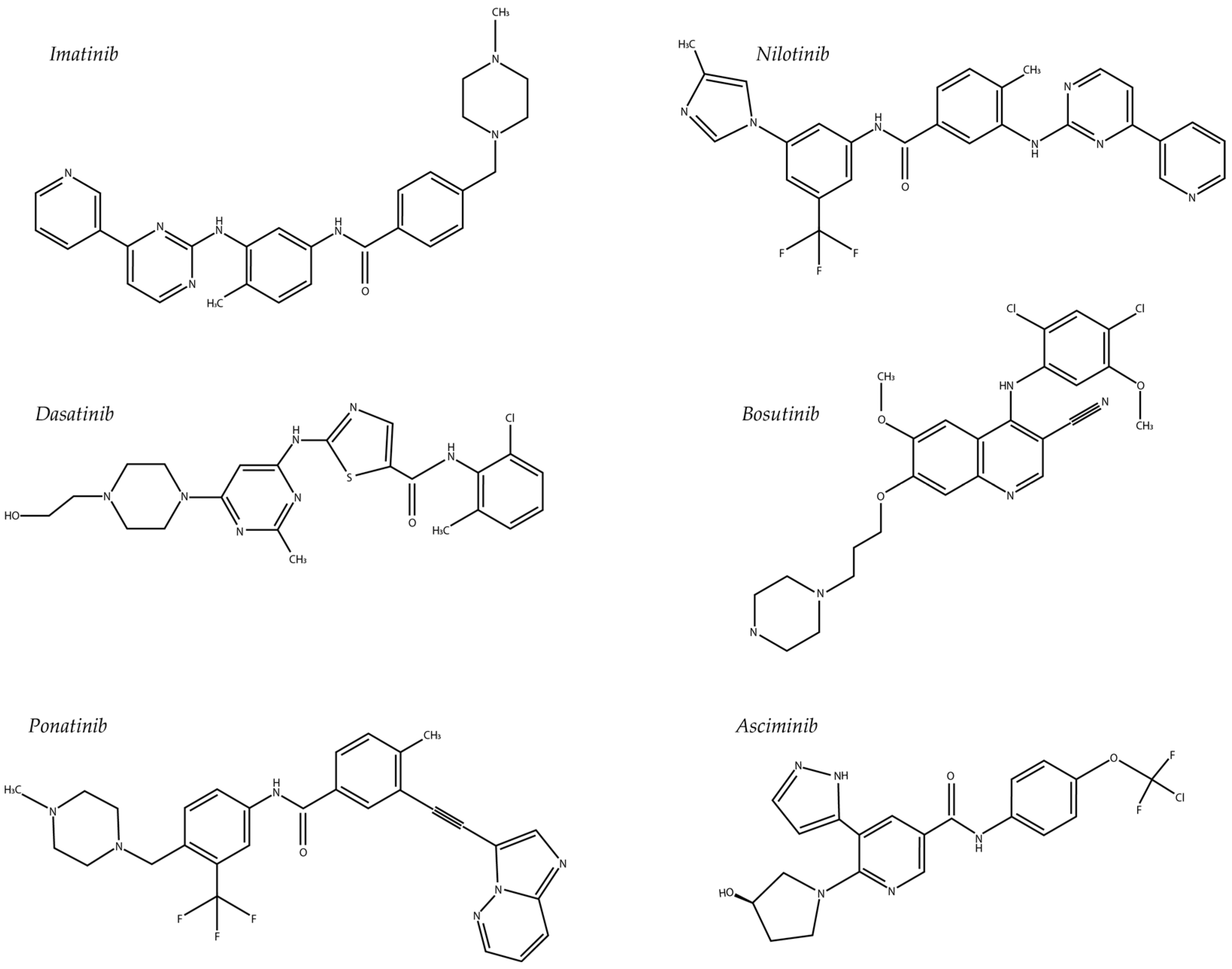
5.1. Imatinib
5.2. Nilotinib
5.3. Dasatinib
5.4. Ponatinib
5.5. Bosutinib
5.6. Asciminib
6. Lysosomal TKI Sequestration
7. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Heisterkamp, N.; Stephenson, J.R.; Groffen, J.; Hansen, P.F.; de Klein, A.; Bartram, C.R.; Grosveld, G. Localization of the c-ab1 oncogene adjacent to a translocation break point in chronic myelocytic leukaemia. Nature 1983, 306, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.J.; Liu, Y.F.; Xu, L.Z.; Long, Z.J.; Huang, D.; Yang, Y.; Liu, B.; Feng, J.X.; Pan, Y.J.; Yan, J.S.; et al. The Philadelphia chromosome in leukemogenesis. Chin. J. Cancer 2016, 35, 48. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, S.R.; Till, J.H. Protein tyrosine kinase structure and function. Annu. Rev. Biochem. 2000, 69, 373–398. [Google Scholar] [CrossRef]
- Baykal-Köse, S.; Acikgoz, E.; Yavuz, A.S.; Gönül Geyik, Ö.; Ateş, H.; Sezerman, O.U.; Özsan, G.H.; Yüce, Z. Adaptive phenotypic modulations lead to therapy resistance in chronic myeloid leukemia cells. PLoS ONE 2020, 15, e0229104. [Google Scholar] [CrossRef]
- Amarante-Mendes, G.P.; Rana, A.; Datoguia, T.S.; Hamerschlak, N.; Brumatti, G. BCR-ABL1 Tyrosine Kinase Complex Signaling Transduction: Challenges to Overcome Resistance in Chronic Myeloid Leukemia. Pharmaceutics 2022, 14, 215. [Google Scholar] [CrossRef]
- Jabbour, E.; Kantarjian, H. Chronic myeloid leukemia: 2020 update on diagnosis, therapy and monitoring. Am. J. Hematol. 2020, 95, 691–709. [Google Scholar] [CrossRef]
- Chereda, B.; Melo, J.V. Natural course and biology of CML. Ann. Hematol. 2015, 94 (Suppl. 2), S107–S121. [Google Scholar] [CrossRef]
- Tauchi, T.; Ohyashiki, K. Molecular mechanisms of resistance of leukemia to imatinib mesylate. Leuk. Res. 2004, 28, 39–45. [Google Scholar] [CrossRef]
- Hochhaus, A.; Larson, R.A.; Guilhot, F.; Radich, J.P.; Branford, S.; Hughes, T.P.; Baccarani, M.; Deininger, M.W.; Cervantes, F.; Fujihara, S.; et al. Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. N. Engl. J. Med. 2017, 376, 917–927. [Google Scholar] [CrossRef]
- García-Gutiérrez, V.; Hernández-Boluda, J.C. Tyrosine Kinase Inhibitors Available for Chronic Myeloid Leukemia: Efficacy and Safety. Front. Oncol. 2019, 9, 603. [Google Scholar] [CrossRef]
- Yeung, D.T.; Shanmuganathan, N.; Hughes, T.P. Asciminib: A new therapeutic option in chronic-phase CML with treatment failure. Blood 2022, 139, 3474–3479. [Google Scholar] [CrossRef] [PubMed]
- Ciftciler, R.; Haznedaroglu, I.C. Tailored tyrosine kinase inhibitor (TKI) treatment of chronic myeloid leukemia (CML) based on current evidence. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 7787–7798. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Lloyd, P.; Schran, H. Clinical pharmacokinetics of imatinib. Clin. Pharmacokinet. 2005, 44, 879–894. [Google Scholar] [CrossRef] [PubMed]
- Neul, C.; Schaeffeler, E.; Sparreboom, A.; Laufer, S.; Schwab, M.; Nies, A.T. Impact of Membrane Drug Transporters on Resistance to Small-Molecule Tyrosine Kinase Inhibitors. Trends Pharmacol. Sci. 2016, 37, 904–932. [Google Scholar] [CrossRef] [PubMed]
- DeGorter, M.K.; Xia, C.Q.; Yang, J.J.; Kim, R.B. Drug Transporters in Drug Efficacy and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 249–273. [Google Scholar] [CrossRef]
- Angelini, S.; Pantaleo, M.A.; Ravegnini, G.; Zenesini, C.; Cavrini, G.; Nannini, M.; Fumagalli, E.; Palassini, E.; Saponara, M.; Di Battista, M.; et al. Polymorphisms in OCTN1 and OCTN2 transporters genes are associated with prolonged time to progression in unresectable gastrointestinal stromal tumours treated with imatinib therapy. Pharmacol. Res. 2013, 68, 1–6. [Google Scholar] [CrossRef]
- Burger, H.; den Dekker, A.T.; Segeletz, S.; Boersma, A.W.; de Bruijn, P.; Debiec-Rychter, M.; Taguchi, T.; Sleijfer, S.; Sparreboom, A.; Mathijssen, R.H.; et al. Lysosomal Sequestration Determines Intracellular Imatinib Levels. Mol. Pharmacol. 2015, 88, 477–487. [Google Scholar] [CrossRef]
- Morrison, S.J.; Wandycz, A.M.; Hemmati, H.D.; Wright, D.E.; Weissman, I.L. Identification of a lineage of multipotent hematopoietic progenitors. Development 1997, 124, 1929–1939. [Google Scholar] [CrossRef]
- Bhatia, M.; Wang, J.C.; Kapp, U.; Bonnet, D.; Dick, J.E. Purification of primitive human hematopoietic cells capable of repopulating immune-deficient mice. Proc. Natl. Acad. Sci. USA 1997, 94, 5320–5325. [Google Scholar] [CrossRef]
- Holyoake, T.L.; Vetrie, D. The chronic myeloid leukemia stem cell: Stemming the tide of persistence. Blood 2017, 129, 1595–1606. [Google Scholar] [CrossRef]
- Graham, S.M.; Jørgensen, H.G.; Allan, E.; Pearson, C.; Alcorn, M.J.; Richmond, L.; Holyoake, T.L. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood 2002, 99, 319–325. [Google Scholar] [CrossRef]
- Atallah, E.; Schiffer, C.A.; Radich, J.P.; Weinfurt, K.P.; Zhang, M.J.; Pinilla-Ibarz, J.; Kota, V.; Larson, R.A.; Moore, J.O.; Mauro, M.J.; et al. Assessment of Outcomes After Stopping Tyrosine Kinase Inhibitors Among Patients With Chronic Myeloid Leukemia: A Nonrandomized Clinical Trial. JAMA Oncol. 2021, 7, 42–50. [Google Scholar] [CrossRef]
- Chu, S.; McDonald, T.; Lin, A.; Chakraborty, S.; Huang, Q.; Snyder, D.S.; Bhatia, R. Persistence of leukemia stem cells in chronic myelogenous leukemia patients in prolonged remission with imatinib treatment. Blood 2011, 118, 5565–5572. [Google Scholar] [CrossRef] [PubMed]
- Chomel, J.C.; Bonnet, M.L.; Sorel, N.; Bertrand, A.; Meunier, M.C.; Fichelson, S.; Melkus, M.; Bennaceur-Griscelli, A.; Guilhot, F.; Turhan, A.G. Leukemic stem cell persistence in chronic myeloid leukemia patients with sustained undetectable molecular residual disease. Blood 2011, 118, 3657–3660. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, R.; Holtz, M.; Niu, N.; Gray, R.; Snyder, D.S.; Sawyers, C.L.; Arber, D.A.; Slovak, M.L.; Forman, S.J. Persistence of malignant hematopoietic progenitors in chronic myelogenous leukemia patients in complete cytogenetic remission following imatinib mesylate treatment. Blood 2003, 101, 4701–4707. [Google Scholar] [CrossRef] [PubMed]
- Holyoake, T.; Jiang, X.; Eaves, C.; Eaves, A. Isolation of a highly quiescent subpopulation of primitive leukemic cells in chronic myeloid leukemia. Blood 1999, 94, 2056–2064. [Google Scholar] [CrossRef]
- Vetrie, D.; Helgason, G.V.; Copland, M. The leukaemia stem cell: Similarities, differences and clinical prospects in CML and AML. Nat. Rev. Cancer 2020, 20, 158–173. [Google Scholar] [CrossRef]
- Warfvinge, R.; Geironson, L.; Sommarin, M.N.E.; Lang, S.; Karlsson, C.; Roschupkina, T.; Stenke, L.; Stentoft, J.; Olsson-Strömberg, U.; Hjorth-Hansen, H.; et al. Single-cell molecular analysis defines therapy response and immunophenotype of stem cell subpopulations in CML. Blood 2017, 129, 2384–2394. [Google Scholar] [CrossRef]
- Giustacchini, A.; Thongjuea, S.; Barkas, N.; Woll, P.S.; Povinelli, B.J.; Booth, C.A.G.; Sopp, P.; Norfo, R.; Rodriguez-Meira, A.; Ashley, N.; et al. Single-cell transcriptomics uncovers distinct molecular signatures of stem cells in chronic myeloid leukemia. Nat. Med. 2017, 23, 692–702. [Google Scholar] [CrossRef]
- Herrmann, H.; Sadovnik, I.; Cerny-Reiterer, S.; Rülicke, T.; Stefanzl, G.; Willmann, M.; Hoermann, G.; Bilban, M.; Blatt, K.; Herndlhofer, S. Dipeptidylpeptidase IV (CD26) defines leukemic stem cells (LSC) in chronic myeloid leukemia. Blood J. Am. Soc. Hematol. 2014, 123, 3951–3962. [Google Scholar] [CrossRef]
- Bose, S.; Deininger, M.; Gora-Tybor, J.; Goldman, J.M.; Melo, J.V. The presence of typical and atypical BCR-ABL fusion genes in leukocytes of normal individuals: Biologic significance and implications for the assessment of minimal residual disease. Blood 1998, 92, 3362–3367. [Google Scholar] [CrossRef] [PubMed]
- Biernaux, C.; Loos, M.; Sels, A.; Huez, G.; Stryckmans, P. Detection of major bcr-abl gene expression at a very low level in blood cells of some healthy individuals. Blood 1995, 86, 3118–3122. [Google Scholar] [CrossRef] [PubMed]
- Quintás-Cardama, A.; Kantarjian, H.M.; Cortes, J.E. Mechanisms of primary and secondary resistance to imatinib in chronic myeloid leukemia. Cancer Control 2009, 16, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.B.; O’Hare, T.; Deininger, M.W. Mechanisms of Resistance to ABL Kinase Inhibition in Chronic Myeloid Leukemia and the Development of Next Generation ABL Kinase Inhibitors. Hematol. Oncol. Clin. N. Am. 2017, 31, 589–612. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, F.E.; Ibrahim, A.R.; Soverini, S.; Martinelli, G.; Müller, M.C.; Hochhaus, A.; Dufva, I.H.; Kim, D.-W.; Cortes, J.; Mauro, M.J. The BCR-ABLT315I mutation compromises survival in chronic phase chronic myelogenous leukemia patients resistant to tyrosine kinase inhibitors, in a matched pair analysis. Haematologica 2013, 98, 1510–1516. [Google Scholar] [CrossRef]
- Alves, R.; Gonçalves, A.C.; Rutella, S.; Almeida, A.M.; De Las Rivas, J.; Trougakos, I.P.; Sarmento Ribeiro, A.B. Resistance to Tyrosine Kinase Inhibitors in Chronic Myeloid Leukemia—From Molecular Mechanisms to Clinical Relevance. Cancers 2021, 13, 4820. [Google Scholar] [CrossRef]
- Qiu, S.; Sheth, V.; Yan, C.; Liu, J.; Chacko, B.K.; Li, H.; Crossman, D.K.; Fortmann, S.D.; Aryal, S.; Rennhack, A.; et al. Metabolic adaptation to tyrosine kinase inhibition in leukemia stem cells. Blood 2023, 142, 574–588. [Google Scholar] [CrossRef]
- Teo, Y.L.; Ho, H.K.; Chan, A. Metabolism-related pharmacokinetic drug-drug interactions with tyrosine kinase inhibitors: Current understanding, challenges and recommendations. Br. J. Clin. Pharmacol. 2015, 79, 241–253. [Google Scholar] [CrossRef]
- Soucek, P.; Anzenbacher, P.; Skoumalová, I.; Dvorák, M. Expression of cytochrome P450 genes in CD34+ hematopoietic stem and progenitor cells. Stem Cells 2005, 23, 1417–1422. [Google Scholar] [CrossRef]
- Rochat, B.; Zoete, V.; Grosdidier, A.; von Grünigen, S.; Marull, M.; Michielin, O. In vitro biotransformation of imatinib by the tumor expressed CYP1A1 and CYP1B1. Biopharm. Drug Dispos. 2008, 29, 103–118. [Google Scholar] [CrossRef]
- Kumar, V.; Singh, P.; Gupta, S.K.; Ali, V.; Verma, M. Transport and metabolism of tyrosine kinase inhibitors associated with chronic myeloid leukemia therapy: A review. Mol. Cell. Biochem. 2022, 477, 1261–1279. [Google Scholar] [CrossRef] [PubMed]
- White, D.L.; Saunders, V.A.; Dang, P.; Engler, J.; Zannettino, A.C.W.; Cambareri, A.C.; Quinn, S.R.; Manley, P.W.; Hughes, T.P. OCT-1–mediated influx is a key determinant of the intracellular uptake of imatinib but not nilotinib (AMN107): Reduced OCT-1 activity is the cause of low in vitro sensitivity to imatinib. Blood 2006, 108, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Forrest, D.; Nicolini, F.; Turhan, A.; Guilhot, J.; Yip, C.; Holyoake, T.; Jorgensen, H.; Lambie, K.; Saw, K.M.; et al. Properties of CD34+ CML stem/progenitor cells that correlate with different clinical responses to imatinib mesylate. Blood 2010, 116, 2112–2121. [Google Scholar] [CrossRef] [PubMed]
- Peeters, S.D.P.W.M.; van der Kolk, D.M.; de Haan, G.; Bystrykh, L.; Kuipers, F.; de Vries, E.G.E.; Vellenga, E. Selective expression of cholesterol metabolism genes in normal CD34+CD38− cells with a heterogeneous expression pattern in AML cells. Exp. Hematol. 2006, 34, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.M.; Roninson, I.B. Expression and activity of P-glycoprotein, a multidrug efflux pump, in human hematopoietic stem cells. Cell 1991, 66, 85–94. [Google Scholar] [CrossRef]
- Brendel, C.; Scharenberg, C.; Dohse, M.; Robey, R.W.; Bates, S.E.; Shukla, S.; Ambudkar, S.V.; Wang, Y.; Wennemuth, G.; Burchert, A.; et al. Imatinib mesylate and nilotinib (AMN107) exhibit high-affinity interaction with ABCG2 on primitive hematopoietic stem cells. Leukemia 2007, 21, 1267–1275. [Google Scholar] [CrossRef]
- Seelig, A. P-Glycoprotein: One Mechanism, Many Tasks and the Consequences for Pharmacotherapy of Cancers. Front. Oncol. 2020, 10, 576559. [Google Scholar] [CrossRef]
- Mao, Q.; Unadkat, J.D. Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport—An update. Aaps J. 2015, 17, 65–82. [Google Scholar] [CrossRef]
- Dohse, M.; Scharenberg, C.; Shukla, S.; Robey, R.W.; Volkmann, T.; Deeken, J.F.; Brendel, C.; Ambudkar, S.V.; Neubauer, A.; Bates, S.E. Comparison of ATP-binding cassette transporter interactions with the tyrosine kinase inhibitors imatinib, nilotinib, and dasatinib. Drug Metab. Dispos. 2010, 38, 1371–1380. [Google Scholar] [CrossRef]
- de Jonge-Peeters, S.D.P.W.M.; Kuipers, F.; de Vries, E.G.E.; Vellenga, E. ABC transporter expression in hematopoietic stem cells and the role in AML drug resistance. Crit. Rev. Oncol./Hematol. 2007, 62, 214–226. [Google Scholar] [CrossRef]
- Jiang, X.; Zhao, Y.; Smith, C.; Gasparetto, M.; Turhan, A.; Eaves, A.; Eaves, C. Chronic myeloid leukemia stem cells possess multiple unique features of resistance to BCR-ABL targeted therapies. Leukemia 2007, 21, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Eadie, L.N.; Dang, P.; Saunders, V.A.; Yeung, D.T.; Osborn, M.P.; Grigg, A.P.; Hughes, T.P.; White, D.L. The clinical significance of ABCB1 overexpression in predicting outcome of CML patients undergoing first-line imatinib treatment. Leukemia 2017, 31, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Raaijmakers, H.G.; Van Den Bosch, G.; Boezeman, J.; De Witte, T.; Raymakers, R.A. Single-cell image analysis to assess ABC-transporter-mediated efflux in highly purified hematopoietic progenitors. Cytometry 2002, 49, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Eadie, L.N.; Hughes, T.P.; White, D.L. ABCB1 Overexpression Is a Key Initiator of Resistance to Tyrosine Kinase Inhibitors in CML Cell Lines. PLoS ONE 2016, 11, e0161470. [Google Scholar] [CrossRef]
- Scharenberg, C.W.; Harkey, M.A.; Torok-Storb, B. The ABCG2 transporter is an efficient Hoechst 33342 efflux pump and is preferentially expressed by immature human hematopoietic progenitors. Blood 2002, 99, 507–512. [Google Scholar] [CrossRef]
- de Grouw, E.P.L.M.; Raaijmakers, M.H.G.P.; Boezeman, J.B.; van der Reijden, B.A.; van de Locht, L.T.F.; de Witte, T.J.M.; Jansen, J.H.; Raymakers, R.A.P. Preferential expression of a high number of ATP binding cassette transporters in both normal and leukemic CD34+CD38− cells. Leukemia 2006, 20, 750–754. [Google Scholar] [CrossRef]
- Oguro, H. The Roles of Cholesterol and Its Metabolites in Normal and Malignant Hematopoiesis. Front. Endocrinol. 2019, 10, 204. [Google Scholar] [CrossRef]
- Tabassum, A.; Samdani, M.N.; Dhali, T.C.; Alam, R.; Ahammad, F.; Samad, A.; Karpiński, T.M. Transporter associated with antigen processing 1 (TAP1) expression and prognostic analysis in breast, lung, liver, and ovarian cancer. J. Mol. Med. 2021, 99, 1293–1309. [Google Scholar] [CrossRef]
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef]
- Zhu, Z.; Hao, X.; Yan, M.; Yao, M.; Ge, C.; Gu, J.; Li, J. Cancer stem/progenitor cells are highly enriched in CD133+CD44+ population in hepatocellular carcinoma. Int. J. Cancer 2010, 126, 2067–2078. [Google Scholar] [CrossRef]
- Gromicho, M.; Dinis, J.; Magalhães, M.; Fernandes, A.R.; Tavares, P.; Laires, A.; Rueff, J.; Rodrigues, A.S. Development of imatinib and dasatinib resistance: Dynamics of expression of drug transporters ABCB1, ABCC1, ABCG2, MVP, and SLC22A1. Leuk. Lymphoma 2011, 52, 1980–1990. [Google Scholar] [CrossRef] [PubMed]
- Kitai, K.; Kawaguchi, K.; Tomohiro, T.; Morita, M.; So, T.; Imanaka, T. The lysosomal protein ABCD4 can transport vitamin B(12) across liposomal membranes in vitro. J. Biol. Chem. 2021, 296, 100654. [Google Scholar] [CrossRef] [PubMed]
- Ceraulo, A.; Lapillonne, H.; Cheok, M.H.; Preudhomme, C.; Dombret, H.; Terré, C.; Lambert, J.; Leverger, G.; Bertrand, Y.; Mortreux, F.; et al. Prognostic impact of ABCA3 expression in adult and pediatric acute myeloid leukemia: An ALFA-ELAM02 joint study. Blood Adv. 2022, 6, 2773–2777. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Bergevoet, S.M.; Gilissen, C.; de Witte, T.; Jansen, J.H.; van der Reijden, B.A.; Raymakers, R.A.P. Hematopoietic stem cells exhibit a specific ABC transporter gene expression profile clearly distinct from other stem cells. BMC Pharmacol. 2010, 10, 12. [Google Scholar] [CrossRef]
- Polillo, M.; Galimberti, S.; Baratè, C.; Petrini, M.; Danesi, R.; Di Paolo, A. Pharmacogenetics of BCR/ABL Inhibitors in Chronic Myeloid Leukemia. Int. J. Mol. Sci. 2015, 16, 22811–22829. [Google Scholar] [CrossRef]
- Liang, Y.; Li, S.; Chen, L. The physiological role of drug transporters. Protein Cell 2015, 6, 334–350. [Google Scholar] [CrossRef]
- Au, A.; Aziz Baba, A.; Goh, A.S.; Wahid Fadilah, S.A.; Teh, A.; Rosline, H.; Ankathil, R. Association of genotypes and haplotypes of multi-drug transporter genes ABCB1 and ABCG2 with clinical response to imatinib mesylate in chronic myeloid leukemia patients. Biomed. Pharmacother. 2014, 68, 343–349. [Google Scholar] [CrossRef]
- Lombardo, L.J.; Lee, F.Y.; Chen, P.; Norris, D.; Barrish, J.C.; Behnia, K.; Castaneda, S.; Cornelius, L.A.; Das, J.; Doweyko, A.M.; et al. Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J. Med. Chem. 2004, 47, 6658–6661. [Google Scholar] [CrossRef]
- Seeliger, M.A.; Ranjitkar, P.; Kasap, C.; Shan, Y.; Shaw, D.E.; Shah, N.P.; Kuriyan, J.; Maly, D.J. Equally potent inhibition of c-Src and Abl by compounds that recognize inactive kinase conformations. Cancer Res. 2009, 69, 2384–2392. [Google Scholar] [CrossRef]
- Sihto, H.; Sarlomo-Rikala, M.; Tynninen, O.; Tanner, M.; Andersson, L.C.; Franssila, K.; Nupponen, N.N.; Joensuu, H. KIT and Platelet-Derived Growth Factor Receptor Alpha Tyrosine Kinase Gene Mutations and KIT Amplifications in Human Solid Tumors. J. Clin. Oncol. 2005, 23, 49–57. [Google Scholar] [CrossRef]
- Okuda, K.; Weisberg, E.; Gilliland, D.G.; Griffin, J.D. ARG tyrosine kinase activity is inhibited by STI571. Blood 2001, 97, 2440–2448. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.J. Asciminib: The first-in-class allosteric inhibitor of BCR::ABL1 kinase. Blood Res. 2023, 58, S29–S36. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.P.; Mauro, M.J.; Cortes, J.E.; Minami, H.; Rea, D.; DeAngelo, D.J.; Breccia, M.; Goh, Y.T.; Talpaz, M.; Hochhaus, A.; et al. Asciminib in Chronic Myeloid Leukemia after ABL Kinase Inhibitor Failure. N. Engl. J. Med. 2019, 381, 2315–2326. [Google Scholar] [CrossRef]
- Branford, S. Molecular monitoring in chronic myeloid leukemia-how low can you go? Hematol. Am. Soc. Hematol. Educ. Program. 2016, 2016, 156–163. [Google Scholar] [CrossRef]
- Cross, N.C.; White, H.E.; Müller, M.C.; Saglio, G.; Hochhaus, A. Standardized definitions of molecular response in chronic myeloid leukemia. Leukemia 2012, 26, 2172–2175. [Google Scholar] [CrossRef]
- García-Ferrer, M.; Wojnicz, A.; Mejía, G.; Koller, D.; Zubiaur, P.; Abad-Santos, F. Utility of Therapeutic Drug Monitoring of Imatinib, Nilotinib, and Dasatinib in Chronic Myeloid Leukemia: A Systematic Review and Meta-analysis. Clin. Ther. 2019, 41, 2558–2570.e7. [Google Scholar] [CrossRef]
- Soverini, S.; Martinelli, G.; Iacobucci, I.; Baccarani, M. Imatinib mesylate for the treatment of chronic myeloid leukemia. Expert Rev. Anticancer. Ther. 2008, 8, 853–864. [Google Scholar] [CrossRef]
- Nardinelli, L.; Sanabani, S.S.; Didone, A.; de Barros Ferreira, P.; Serpa, M.; Novaes, M.M.; Marchiani, M.; Ruiz, A.L.; Lima, I.S.; de Alencar Fischer Chamone, D.; et al. Pretherapeutic expression of the hOCT1 gene predicts a complete molecular response to imatinib mesylate in chronic-phase chronic myeloid leukemia. Acta Haematol. 2012, 127, 228–234. [Google Scholar] [CrossRef]
- Deborah, L.W.; Jerald, R.; Simona, S.; Verity, A.S.; Amity, K.F.; Phuong, D.; Daniela, C.; Peter, L.; Lidia, M.; Richard, W.; et al. Chronic phase chronic myeloid leukemia patients with low OCT-1 activity randomized to high-dose imatinib achieve better responses and have lower failure rates than those randomized to standard-dose imatinib. Haematologica 2012, 97, 907–914. [Google Scholar] [CrossRef]
- White, D.L.; Saunders, V.A.; Dang, P.; Engler, J.; Venables, A.; Zrim, S.; Zannettino, A.; Lynch, K.; Manley, P.W.; Hughes, T. Most CML patients who have a suboptimal response to imatinib have low OCT-1 activity: Higher doses of imatinib may overcome the negative impact of low OCT-1 activity. Blood 2007, 110, 4064–4072. [Google Scholar] [CrossRef] [PubMed]
- Nies, A.T.; Schaeffeler, E.; van der Kuip, H.; Cascorbi, I.; Bruhn, O.; Kneba, M.; Pott, C.; Hofmann, U.; Volk, C.; Hu, S.; et al. Cellular uptake of imatinib into leukemic cells is independent of human organic cation transporter 1 (OCT1). Clin. Cancer Res. 2014, 20, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, Y.; Hamada, A.; Uchida, T.; Sato, D.; Yuki, M.; Hayashi, M.; Kawaguchi, T.; Saito, H. Distinct interaction of nilotinib and imatinib with P-Glycoprotein in intracellular accumulation and cytotoxicity in CML Cell Line K562 cells. Biol. Pharm. Bull. 2014, 37, 1330–1335. [Google Scholar] [CrossRef]
- Hu, S.; Franke, R.M.; Filipski, K.K.; Hu, C.; Orwick, S.J.; de Bruijn, E.A.; Burger, H.; Baker, S.D.; Sparreboom, A. Interaction of imatinib with human organic ion carriers. Clin. Cancer Res. 2008, 14, 3141–3148. [Google Scholar] [CrossRef] [PubMed]
- Hamada, A.; Miyano, H.; Watanabe, H.; Saito, H. Interaction of imatinib mesilate with human P-glycoprotein. J. Pharmacol. Exp. Ther. 2003, 307, 824–828. [Google Scholar] [CrossRef]
- Illmer, T.; Schaich, M.; Platzbecker, U.; Freiberg-Richter, J.; Oelschlägel, U.; von Bonin, M.; Pursche, S.; Bergemann, T.; Ehninger, G.; Schleyer, E. P-glycoprotein-mediated drug efflux is a resistance mechanism of chronic myelogenous leukemia cells to treatment with imatinib mesylate. Leukemia 2004, 18, 401–408. [Google Scholar] [CrossRef]
- Ammar, M.; Louati, N.; Frikha, I.; Medhaffar, M.; Ghozzi, H.; Elloumi, M.; Menif, H.; Zeghal, K.; Ben Mahmoud, L. Overexpression of P-glycoprotein and resistance to Imatinib in chronic myeloid leukemia patients. J. Clin. Lab. Anal. 2020, 34, e23374. [Google Scholar] [CrossRef]
- Hegedűs, T.; Őrfi, L.; Seprődi, A.; Váradi, A.; Sarkadi, B.; Kéri, G. Interaction of tyrosine kinase inhibitors with the human multidrug transporter proteins, MDR1 and MRP1. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2002, 1587, 318–325. [Google Scholar] [CrossRef]
- Eadie, L.N.; Hughes, T.P.; White, D.L. Interaction of the efflux transporters ABCB1 and ABCG2 with imatinib, nilotinib, and dasatinib. Clin. Pharmacol. Ther. 2014, 95, 294–306. [Google Scholar] [CrossRef]
- Hirayama, C.; Watanabe, H.; Nakashima, R.; Nanbu, T.; Hamada, A.; Kuniyasu, A.; Nakayama, H.; Kawaguchi, T.; Saito, H. Constitutive overexpression of P-glycoprotein, rather than breast cancer resistance protein or organic cation transporter 1, contributes to acquisition of imatinib-resistance in K562 cells. Pharm. Res. 2008, 25, 827–835. [Google Scholar] [CrossRef]
- Nakanishi, T.; Shiozawa, K.; Hassel, B.A.; Ross, D.D. Complex interaction of BCRP/ABCG2 and imatinib in BCR-ABL-expressing cells: BCRP-mediated resistance to imatinib is attenuated by imatinib-induced reduction of BCRP expression. Blood 2006, 108, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Houghton, P.J.; Germain, G.S.; Harwood, F.C.; Schuetz, J.D.; Stewart, C.F.; Buchdunger, E.; Traxler, P. Imatinib mesylate is a potent inhibitor of the ABCG2 (BCRP) transporter and reverses resistance to topotecan and SN-38 in vitro. Cancer Res. 2004, 64, 2333–2337. [Google Scholar] [CrossRef] [PubMed]
- Burger, H.; van Tol, H.; Boersma, A.W.; Brok, M.; Wiemer, E.A.; Stoter, G.; Nooter, K. Imatinib mesylate (STI571) is a substrate for the breast cancer resistance protein (BCRP)/ABCG2 drug pump. Blood 2004, 104, 2940–2942. [Google Scholar] [CrossRef] [PubMed]
- Burger, H.; van Tol, H.; Brok, M.; Wiemer, E.A.; de Bruijn, E.A.; Guetens, G.; de Boeck, G.; Sparreboom, A.; Verweij, J.; Nooter, K. Chronic imatinib mesylate exposure leads to reduced intracellular drug accumulation by induction of the ABCG2 (BCRP) and ABCB1 (MDR1) drug transport pumps. Cancer Biol. Ther. 2005, 4, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Breedveld, P.; Pluim, D.; Cipriani, G.; Wielinga, P.; van Tellingen, O.; Schinkel, A.H.; Schellens, J.H. The effect of Bcrp1 (Abcg2) on the in vivo pharmacokinetics and brain penetration of imatinib mesylate (Gleevec): Implications for the use of breast cancer resistance protein and P-glycoprotein inhibitors to enable the brain penetration of imatinib in patients. Cancer Res. 2005, 65, 2577–2582. [Google Scholar] [CrossRef]
- Liu, W.; Baer, M.R.; Bowman, M.J.; Pera, P.; Zheng, X.; Morgan, J.; Pandey, R.A.; Oseroff, A.R. The tyrosine kinase inhibitor imatinib mesylate enhances the efficacy of photodynamic therapy by inhibiting ABCG2. Clin. Cancer Res. 2007, 13, 2463–2470. [Google Scholar] [CrossRef]
- Jordanides, N.E.; Jorgensen, H.G.; Holyoake, T.L.; Mountford, J.C. Functional ABCG2 is overexpressed on primary CML CD34+ cells and is inhibited by imatinib mesylate. Blood 2006, 108, 1370–1373. [Google Scholar] [CrossRef]
- Orlando, B.J.; Liao, M. ABCG2 transports anticancer drugs via a closed-to-open switch. Nat. Commun. 2020, 11, 2264. [Google Scholar] [CrossRef]
- Mogi, M.; Yang, J.; Lambert, J.F.; Colvin, G.A.; Shiojima, I.; Skurk, C.; Summer, R.; Fine, A.; Quesenberry, P.J.; Walsh, K. Akt signaling regulates side population cell phenotype via Bcrp1 translocation. J. Biol. Chem. 2003, 278, 39068–39075. [Google Scholar] [CrossRef]
- Danisz, K.; Blasiak, J. Role of anti-apoptotic pathways activated by BCR/ABL in the resistance of chronic myeloid leukemia cells to tyrosine kinase inhibitors. Acta Biochim. Pol. 2013, 60, 503–514. [Google Scholar] [CrossRef]
- Giannoudis, A.; Davies, A.; Harris, R.J.; Lucas, C.M.; Pirmohamed, M.; Clark, R.E. The clinical significance of ABCC3 as an imatinib transporter in chronic myeloid leukaemia. Leukemia 2014, 28, 1360–1363. [Google Scholar] [CrossRef] [PubMed]
- Eadie, L.N.; Dang, P.; Goyne, J.M.; Hughes, T.P.; White, D.L. ABCC6 plays a significant role in the transport of nilotinib and dasatinib, and contributes to TKI resistance in vitro, in both cell lines and primary patient mononuclear cells. PLoS ONE 2018, 13, e0192180. [Google Scholar] [CrossRef] [PubMed]
- Beers, M.F.; Mulugeta, S. The biology of the ABCA3 lipid transporter in lung health and disease. Cell Tissue Res. 2017, 367, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Hupfeld, T.; Chapuy, B.; Schrader, V.; Beutler, M.; Veltkamp, C.; Koch, R.; Cameron, S.; Aung, T.; Haase, D.; Larosee, P.; et al. Tyrosinekinase inhibition facilitates cooperation of transcription factor SALL4 and ABC transporter A3 towards intrinsic CML cell drug resistance. Br. J. Haematol. 2013, 161, 204–213. [Google Scholar] [CrossRef]
- Manley, P.W.; Breitenstein, W.; Brüggen, J.; Cowan-Jacob, S.W.; Furet, P.; Mestan, J.; Meyer, T. Urea derivatives of STI571 as inhibitors of Bcr-Abl and PDGFR kinases. Bioorg. Med. Chem. Lett. 2004, 14, 5793–5797. [Google Scholar] [CrossRef]
- Tian, X.; Zhang, H.; Heimbach, T.; He, H.; Buchbinder, A.; Aghoghovbia, M.; Hourcade-Potelleret, F. Clinical Pharmacokinetic and Pharmacodynamic Overview of Nilotinib, a Selective Tyrosine Kinase Inhibitor. J. Clin. Pharmacol. 2018, 58, 1533–1540. [Google Scholar] [CrossRef]
- Tanaka, C.; Yin, O.Q.; Sethuraman, V.; Smith, T.; Wang, X.; Grouss, K.; Kantarjian, H.; Giles, F.; Ottmann, O.G.; Galitz, L.; et al. Clinical pharmacokinetics of the BCR-ABL tyrosine kinase inhibitor nilotinib. Clin. Pharmacol. Ther. 2010, 87, 197–203. [Google Scholar] [CrossRef]
- Weisberg, E.; Manley, P.W.; Breitenstein, W.; Brüggen, J.; Cowan-Jacob, S.W.; Ray, A.; Huntly, B.; Fabbro, D.; Fendrich, G.; Hall-Meyers, E.; et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell 2005, 7, 129–141. [Google Scholar] [CrossRef]
- Golemovic, M.; Verstovsek, S.; Giles, F.; Cortes, J.; Manshouri, T.; Manley, P.W.; Mestan, J.; Dugan, M.; Alland, L.; Griffin, J.D.; et al. AMN107, a novel aminopyrimidine inhibitor of Bcr-Abl, has in vitro activity against imatinib-resistant chronic myeloid leukemia. Clin. Cancer Res. 2005, 11, 4941–4947. [Google Scholar] [CrossRef]
- Saglio, G.; Kim, D.W.; Issaragrisil, S.; le Coutre, P.; Etienne, G.; Lobo, C.; Pasquini, R.; Clark, R.E.; Hochhaus, A.; Hughes, T.P.; et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N. Engl. J. Med. 2010, 362, 2251–2259. [Google Scholar] [CrossRef]
- Khurana, V.; Minocha, M.; Pal, D.; Mitra, A.K. Role of OATP-1B1 and/or OATP-1B3 in hepatic disposition of tyrosine kinase inhibitors. Drug Metab. Drug Interact. 2014, 29, 179–190. [Google Scholar] [CrossRef]
- Minematsu, T.; Giacomini, K.M. Interactions of tyrosine kinase inhibitors with organic cation transporters and multidrug and toxic compound extrusion proteins. Mol. Cancer Ther. 2011, 10, 531–539. [Google Scholar] [CrossRef]
- Hegedus, C.; Ozvegy-Laczka, C.; Apáti, A.; Magócsi, M.; Német, K.; Orfi, L.; Kéri, G.; Katona, M.; Takáts, Z.; Váradi, A.; et al. Interaction of nilotinib, dasatinib and bosutinib with ABCB1 and ABCG2: Implications for altered anti-cancer effects and pharmacological properties. Br. J. Pharmacol. 2009, 158, 1153–1164. [Google Scholar] [CrossRef]
- Davies, A.; Giannoudis, A.; Lucas, C.M.; Harris, R.J.; Manley, P.W.; Pirmohamed, M.; Clark, R.E. Characterisation of Nilotinib Transport in Chronic Myeloid Leukaemia Cells. Blood 2007, 110, 2364. [Google Scholar] [CrossRef]
- Mlejnek, P.; Kosztyu, P.; Dolezel, P.; Bates, S.E.; Ruzickova, E. Reversal of ABCB1 mediated efflux by imatinib and nilotinib in cells expressing various transporter levels. Chem. Biol. Interact. 2017, 273, 171–179. [Google Scholar] [CrossRef]
- Tiwari, A.K.; Sodani, K.; Wang, S.R.; Kuang, Y.H.; Ashby, C.R., Jr.; Chen, X.; Chen, Z.S. Nilotinib (AMN107, Tasigna) reverses multidrug resistance by inhibiting the activity of the ABCB1/Pgp and ABCG2/BCRP/MXR transporters. Biochem. Pharmacol. 2009, 78, 153–161. [Google Scholar] [CrossRef]
- Hiwase, D.K.; White, D.; Zrim, S.; Saunders, V.; Melo, J.V.; Hughes, T.P. Nilotinib-mediated inhibition of ABCB1 increases intracellular concentration of dasatinib in CML cells: Implications for combination TKI therapy. Leukemia 2010, 24, 658–660. [Google Scholar] [CrossRef]
- Blake, S.; Hughes, T.P.; Mayrhofer, G.; Lyons, A.B. The Src/ABL kinase inhibitor dasatinib (BMS-354825) inhibits function of normal human T-lymphocytes in vitro. Clin. Immunol. 2008, 127, 330–339. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Shah, N.P.; Cortes, J.E.; Baccarani, M.; Agarwal, M.B.; Undurraga, M.S.; Wang, J.; Ipiña, J.J.; Kim, D.W.; Ogura, M.; et al. Dasatinib or imatinib in newly diagnosed chronic-phase chronic myeloid leukemia: 2-year follow-up from a randomized phase 3 trial (DASISION). Blood 2012, 119, 1123–1129. [Google Scholar] [CrossRef]
- Cortes, J.E.; Saglio, G.; Kantarjian, H.M.; Baccarani, M.; Mayer, J.; Boqué, C.; Shah, N.P.; Chuah, C.; Casanova, L.; Bradley-Garelik, B.; et al. Final 5-Year Study Results of DASISION: The Dasatinib Versus Imatinib Study in Treatment-Naïve Chronic Myeloid Leukemia Patients Trial. J. Clin. Oncol. 2016, 34, 2333–2340. [Google Scholar] [CrossRef]
- Ishida, Y.; Murai, K.; Yamaguchi, K.; Miyagishima, T.; Shindo, M.; Ogawa, K.; Nagashima, T.; Sato, S.; Watanabe, R.; Yamamoto, S.; et al. Pharmacokinetics and pharmacodynamics of dasatinib in the chronic phase of newly diagnosed chronic myeloid leukemia. Eur. J. Clin. Pharmacol. 2016, 72, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.P.; Tran, C.; Lee, F.Y.; Chen, P.; Norris, D.; Sawyers, C.L. Overriding Imatinib Resistance with a Novel ABL Kinase Inhibitor. Science 2004, 305, 399–401. [Google Scholar] [CrossRef] [PubMed]
- O’Hare, T.; Walters, D.K.; Stoffregen, E.P.; Jia, T.; Manley, P.W.; Mestan, J.; Cowan-Jacob, S.W.; Lee, F.Y.; Heinrich, M.C.; Deininger, M.W.N.; et al. In vitro Activity of Bcr-Abl Inhibitors AMN107 and BMS-354825 against Clinically Relevant Imatinib-Resistant Abl Kinase Domain Mutants. Cancer Res. 2005, 65, 4500–4505. [Google Scholar] [CrossRef] [PubMed]
- Hiwase, D.K.; Saunders, V.; Hewett, D.; Frede, A.; Zrim, S.; Dang, P.; Eadie, L.; To, L.B.; Melo, J.; Kumar, S.; et al. Dasatinib cellular uptake and efflux in chronic myeloid leukemia cells: Therapeutic implications. Clin. Cancer Res. 2008, 14, 3881–3888. [Google Scholar] [CrossRef]
- Chapuy, B.; Koch, R.; Radunski, U.; Corsham, S.; Cheong, N.; Inagaki, N.; Ban, N.; Wenzel, D.; Reinhardt, D.; Zapf, A.; et al. Intracellular ABC transporter A3 confers multidrug resistance in leukemia cells by lysosomal drug sequestration. Leukemia 2008, 22, 1576–1586. [Google Scholar] [CrossRef]
- Pulte, E.D.; Chen, H.; Price, L.S.L.; Gudi, R.; Li, H.; Okusanya, O.O.; Ma, L.; Rodriguez, L.; Vallejo, J.; Norsworthy, K.J.; et al. FDA Approval Summary: Revised Indication and Dosing Regimen for Ponatinib Based on the Results of the OPTIC Trial. Oncologist 2022, 27, 149–157. [Google Scholar] [CrossRef]
- Musumeci, F.; Greco, C.; Grossi, G.; Molinari, A.; Schenone, S. Recent Studies on Ponatinib in Cancers Other Than Chronic Myeloid Leukemia. Cancers 2018, 10, 430. [Google Scholar] [CrossRef]
- Cortes, J.E.; Kim, D.W.; Pinilla-Ibarz, J.; le Coutre, P.; Paquette, R.; Chuah, C.; Nicolini, F.E.; Apperley, J.F.; Khoury, H.J.; Talpaz, M.; et al. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 2013, 369, 1783–1796. [Google Scholar] [CrossRef]
- Lu, L.; Saunders, V.A.; Leclercq, T.M.; Hughes, T.P.; White, D.L. Ponatinib is not transported by ABCB1, ABCG2 or OCT-1 in CML cells. Leukemia 2015, 29, 1792–1794. [Google Scholar] [CrossRef]
- Sen, R.; Natarajan, K.; Bhullar, J.; Shukla, S.; Fang, H.B.; Cai, L.; Chen, Z.S.; Ambudkar, S.V.; Baer, M.R. The novel BCR-ABL and FLT3 inhibitor ponatinib is a potent inhibitor of the MDR-associated ATP-binding cassette transporter ABCG2. Mol. Cancer Ther. 2012, 11, 2033–2044. [Google Scholar] [CrossRef]
- Sun, Y.L.; Kumar, P.; Sodani, K.; Patel, A.; Pan, Y.; Baer, M.R.; Chen, Z.S.; Jiang, W.Q. Ponatinib enhances anticancer drug sensitivity in MRP7-overexpressing cells. Oncol. Rep. 2014, 31, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Golas, J.M.; Arndt, K.; Etienne, C.; Lucas, J.; Nardin, D.; Gibbons, J.; Frost, P.; Ye, F.; Boschelli, D.H.; Boschelli, F. SKI-606, a 4-Anilino-3-quinolinecarbonitrile Dual Inhibitor of Src and Abl Kinases, Is a Potent Antiproliferative Agent against Chronic Myelogenous Leukemia Cells in Culture and Causes Regression of K562 Xenografts in Nude Mice. Cancer Res. 2003, 63, 375–381. [Google Scholar] [PubMed]
- Hsyu, P.H.; Mould, D.R.; Abbas, R.; Amantea, M. Population pharmacokinetic and pharmacodynamic analysis of bosutinib. Drug Metab. Pharmacokinet. 2014, 29, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Kantarjian, H.M.; Brümmendorf, T.H.; Kim, D.W.; Turkina, A.G.; Shen, Z.X.; Pasquini, R.; Khoury, H.J.; Arkin, S.; Volkert, A.; et al. Safety and efficacy of bosutinib (SKI-606) in chronic phase Philadelphia chromosome-positive chronic myeloid leukemia patients with resistance or intolerance to imatinib. Blood 2011, 118, 4567–4576. [Google Scholar] [CrossRef] [PubMed]
- Rossari, F.; Minutolo, F.; Orciuolo, E. Past, present, and future of Bcr-Abl inhibitors: From chemical development to clinical efficacy. J. Hematol. Oncol. 2018, 11, 84. [Google Scholar] [CrossRef]
- Brümmendorf, T.H.; Cortes, J.E.; Milojkovic, D.; Gambacorti-Passerini, C.; Clark, R.E.; le Coutre, P.; Garcia-Gutierrez, V.; Chuah, C.; Kota, V.; Lipton, J.H.; et al. Bosutinib versus imatinib for newly diagnosed chronic phase chronic myeloid leukemia: Final results from the BFORE trial. Leukemia 2022, 36, 1825–1833. [Google Scholar] [CrossRef]
- Abumiya, M.; Takahashi, N.; Takahashi, S.; Yoshioka, T.; Kameoka, Y.; Miura, M. Effects of SLC22A2 808G>T polymorphism and bosutinib concentrations on serum creatinine in patients with chronic myeloid leukemia receiving bosutinib therapy. Sci. Rep. 2021, 11, 6362. [Google Scholar] [CrossRef]
- Redaelli, S.; Perini, P.; Ceccon, M.; Piazza, R.; Rigolio, R.; Mauri, M.; Boschelli, F.; Giannoudis, A.; Gambacorti-Passerini, C. In vitro and in vivo identification of ABCB1 as an efflux transporter of bosutinib. J. Hematol. Oncol. 2015, 8, 81. [Google Scholar] [CrossRef]
- Hoch, M.; Zack, J.; Quinlan, M.; Huth, F.; Forte, S.; Dodd, S.; Aimone, P.; Hourcade-Potelleret, F. Pharmacokinetics of Asciminib When Taken With Imatinib or With Food. Clin. Pharmacol. Drug Dev. 2022, 11, 207–219. [Google Scholar] [CrossRef]
- Manley, P.W.; Barys, L.; Cowan-Jacob, S.W. The specificity of asciminib, a potential treatment for chronic myeloid leukemia, as a myristate-pocket binding ABL inhibitor and analysis of its interactions with mutant forms of BCR-ABL1 kinase. Leuk. Res. 2020, 98, 106458. [Google Scholar] [CrossRef]
- Wylie, A.A.; Schoepfer, J.; Jahnke, W.; Cowan-Jacob, S.W.; Loo, A.; Furet, P.; Marzinzik, A.L.; Pelle, X.; Donovan, J.; Zhu, W.; et al. The allosteric inhibitor ABL001 enables dual targeting of BCR–ABL1. Nature 2017, 543, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Schoepfer, J.; Jahnke, W.; Berellini, G.; Buonamici, S.; Cotesta, S.; Cowan-Jacob, S.W.; Dodd, S.; Drueckes, P.; Fabbro, D.; Gabriel, T.; et al. Discovery of Asciminib (ABL001), an Allosteric Inhibitor of the Tyrosine Kinase Activity of BCR-ABL1. J. Med. Chem. 2018, 61, 8120–8135. [Google Scholar] [CrossRef] [PubMed]
- Gleixner, K.V.; Filik, Y.; Berger, D.; Schewzik, C.; Stefanzl, G.; Sadovnik, I.; Degenfeld-Schonburg, L.; Eisenwort, G.; Schneeweiss-Gleixner, M.; Byrgazov, K.; et al. Asciminib and ponatinib exert synergistic anti-neoplastic effects on CML cells expressing BCR-ABL1 (T315I)-compound mutations. Am. J. Cancer Res. 2021, 11, 4470–4484. [Google Scholar] [PubMed]
- Eadie, L.N.; Saunders, V.A.; Branford, S.; White, D.L.; Hughes, T.P. The new allosteric inhibitor asciminib is susceptible to resistance mediated by ABCB1 and ABCG2 overexpression in vitro. Oncotarget 2018, 9, 13423–13437. [Google Scholar] [CrossRef] [PubMed]
- Breccia, M.; Colafigli, G.; Scalzulli, E.; Martelli, M. Asciminib: An investigational agent for the treatment of chronic myeloid leukemia. Expert Opin. Investig. Drugs 2021, 30, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Qiang, W.; Antelope, O.; Zabriskie, M.S.; Pomicter, A.D.; Vellore, N.A.; Szankasi, P.; Rea, D.; Cayuela, J.M.; Kelley, T.W.; Deininger, M.W.; et al. Mechanisms of resistance to the BCR-ABL1 allosteric inhibitor asciminib. Leukemia 2017, 31, 2844–2847. [Google Scholar] [CrossRef]
- Kazmi, F.; Hensley, T.; Pope, C.; Funk, R.S.; Loewen, G.J.; Buckley, D.B.; Parkinson, A. Lysosomal sequestration (trapping) of lipophilic amine (cationic amphiphilic) drugs in immortalized human hepatocytes (Fa2N-4 cells). Drug Metab. Dispos. 2013, 41, 897–905. [Google Scholar] [CrossRef]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomes as mediators of drug resistance in cancer. Drug Resist. Updat. 2016, 24, 23–33. [Google Scholar] [CrossRef]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomal accumulation of anticancer drugs triggers lysosomal exocytosis. Oncotarget 2017, 8, 45117–45132. [Google Scholar] [CrossRef]
- Szakacs, G.; Abele, R. An inventory of lysosomal ABC transporters. FEBS Lett. 2020, 594, 3965–3985. [Google Scholar] [CrossRef]
- Chapuy, B.; Panse, M.; Radunski, U.; Koch, R.; Wenzel, D.; Inagaki, N.; Haase, D.; Truemper, L.; Wulf, G.G. ABC transporter A3 facilitates lysosomal sequestration of imatinib and modulates susceptibility of chronic myeloid leukemia cell lines to this drug. Haematologica 2009, 94, 1528–1536. [Google Scholar] [CrossRef] [PubMed]
- Stefan, S.M.; Jansson, P.J.; Kalinowski, D.S.; Anjum, R.; Dharmasivam, M.; Richardson, D.R. The growing evidence for targeting P-glycoprotein in lysosomes to overcome resistance. Future Med. Chem. 2020, 12, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Bower, H.; Björkholm, M.; Dickman, P.W.; Höglund, M.; Lambert, P.C.; Andersson, T.M.L. Life expectancy of patients with chronic myeloid leukemia approaches the life expectancy of the general population. J. Clin. Oncol. 2016, 34, 2851–2857. [Google Scholar] [CrossRef]
- Zong, Y.; Zhou, S.; Sorrentino, B.P. Loss of P-glycoprotein expression in hematopoietic stem cells does not improve responses to imatinib in a murine model of chronic myelogenous leukemia. Leukemia 2005, 19, 1590–1596. [Google Scholar] [CrossRef] [PubMed]
- Loscocco, F.; Visani, G.; Ruzzo, A.; Bagaloni, I.; Fuligni, F.; Galimberti, S.; Di Paolo, A.; Stagno, F.; Pregno, P.; Annunziata, M.; et al. Clinical Relevance of ABCB1, ABCG2, and ABCC2 Gene Polymorphisms in Chronic Myeloid Leukemia Patients Treated With Nilotinib. Front. Oncol. 2021, 11, 672287. [Google Scholar] [CrossRef] [PubMed]
- Au, A.; Baba, A.A.; Azlan, H.; Norsa’adah, B.; Ankathil, R. Clinical impact of ABCC1 and ABCC2 genotypes and haplotypes in mediating imatinib resistance among chronic myeloid leukaemia patients. J. Clin. Pharm. Ther. 2014, 39, 685–690. [Google Scholar] [CrossRef]
- Prado-Carrillo, O.; Arenas-Ramírez, A.; Llaguno-Munive, M.; Jurado, R.; Pérez-Rojas, J.; Cervera-Ceballos, E.; Garcia-Lopez, P. Ketoconazole Reverses Imatinib Resistance in Human Chronic Myelogenous Leukemia K562 Cells. Int. J. Mol. Sci. 2022, 23, 7715. [Google Scholar] [CrossRef]
- Alves, R.; Gonçalves, A.C.; Jorge, J.; Almeida, A.M.; Sarmento-Ribeiro, A.B. Combination of Elacridar with Imatinib Modulates Resistance Associated with Drug Efflux Transporters in Chronic Myeloid Leukemia. Biomedicines 2022, 10, 1158. [Google Scholar] [CrossRef]
- Binkhathlan, Z.; Lavasanifar, A. P-glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer: Current status and future perspectives. Curr. Cancer Drug Targets 2013, 13, 326–346. [Google Scholar] [CrossRef]
- Mu, H.; Zhu, X.; Jia, H.; Zhou, L.; Liu, H. Combination Therapies in Chronic Myeloid Leukemia for Potential Treatment-Free Remission: Focus on Leukemia Stem Cells and Immune Modulation. Front. Oncol. 2021, 11, 643382. [Google Scholar] [CrossRef]
- Hochhaus, A.; Baccarani, M.; Silver, R.T.; Schiffer, C.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Deininger, M.W.; Guilhot, F.; et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia 2020, 34, 966–984. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Compound | Drug Transporting Protein | Direct/Indirect Evidence | Readout | Source |
|---|---|---|---|---|
| Imatinib | P-gp | Indirect | Increased intracellular uptake and retention in P-gp overexpressing LLC-PK1 cells upon addition of the specific P-gp inhibitor cyclosporin A. | [85] |
| BCRP | Direct | Decreased imatinib uptake in BCRP overexpressing cell lines and increased intracellular uptake and retention upon addition of the specific BCRP inhibitor Ko-143. | [93] | |
| ABCC3 | Direct | Increased efflux in ABCC3 overexpressing MDCKII cell monolayers that could be nullified by the addition of the ABCC3 inhibitor probenecid. | [101] | |
| ABCA3 | Indirect | Enhanced transporter expression in CD34+ BCR::ABL1+ leukemic cells after imatinib exposure. | [104] | |
| Nilotinib | P-gp | Indirect | No evidence of radiolabeled nilotinib efflux in P-gp overexpressing MDCKII cells, but significant upregulation of P-gp expression in nilotinib resistant K562 cells. | [54,114] |
| BCRP | Indirect | BCRP overexpression in K562 cells protects against nilotinib-mediated cell death. | [46,113] | |
| ABCA3 | Indirect | Enhanced transporter expression in CD34+ BCR::ABL1+ leukemic cells after nilotinib exposure. | [104] | |
| Dasatinib | P-gp | Direct | Reduced intracellular uptake and retention of dasatinib in P-gp overexpressing K562 cells, which could be reversed by a specific P-gp inhibitor. | [113,124] |
| BCRP | Direct | Reduced intracellular uptake and retention of dasatinib in BCRP overexpressing K562 cells, which could be reversed by a specific BCRP inhibitor. | [113,124] | |
| ABCA3 | Indirect | Enhanced transporter expression in CD34+ BCR::ABL1 leukemic cells after dasatinib exposure. | [104] | |
| Bosutinib | P-gp | Indirect | Lower intracellular uptake and retention of bosutinib in P-gp overexpressing K562 cells and reduced BCR::ABL1 phosphorylation upon co-treatment of bosutinib and a specific P-gp inhibitor. However, P-gp overexpression had no effect on bosutinib-mediated cellular toxicity. | [113,138] |
| BCRP | Indirect | Minor protective effect against bosutinib treatment in BCRP overexpressing K562 cells. | [113] | |
| Asciminib | P-gp | Indirect | Decreased asciminib-mediated cell death in P-gp overexpressing K562 cells, which was nullified upon inhibition of P-gp. | [144,145] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verhagen, N.E.; Koenderink, J.B.; Blijlevens, N.M.A.; Janssen, J.J.W.M.; Russel, F.G.M. Transporter-Mediated Cellular Distribution of Tyrosine Kinase Inhibitors as a Potential Resistance Mechanism in Chronic Myeloid Leukemia. Pharmaceutics 2023, 15, 2535. https://doi.org/10.3390/pharmaceutics15112535
Verhagen NE, Koenderink JB, Blijlevens NMA, Janssen JJWM, Russel FGM. Transporter-Mediated Cellular Distribution of Tyrosine Kinase Inhibitors as a Potential Resistance Mechanism in Chronic Myeloid Leukemia. Pharmaceutics. 2023; 15(11):2535. https://doi.org/10.3390/pharmaceutics15112535
Chicago/Turabian StyleVerhagen, Noor E., Jan B. Koenderink, Nicole M. A. Blijlevens, Jeroen J. W. M. Janssen, and Frans G. M. Russel. 2023. "Transporter-Mediated Cellular Distribution of Tyrosine Kinase Inhibitors as a Potential Resistance Mechanism in Chronic Myeloid Leukemia" Pharmaceutics 15, no. 11: 2535. https://doi.org/10.3390/pharmaceutics15112535