
Novel Therapeutic Approaches Targeting Post-Translational Modifications in Lung Cancer

Abstract
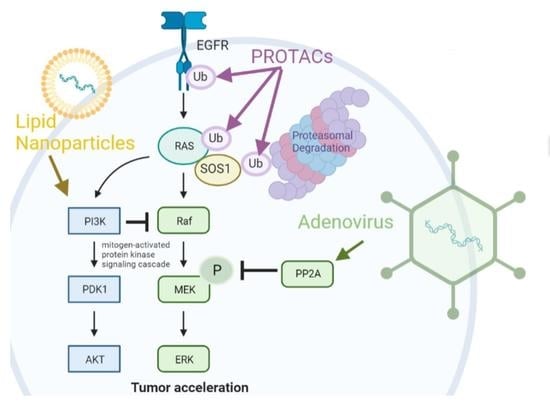
:
1. Introduction
2. Adenoviral Targeting of Oncogenic Protein Modifications
3. PROteolysis TArgeting Chimeras (PROTACs) Based Therapies
4. Lipid Nanoparticle-Based Therapies
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tursz, T.; Le Cesne, A.; Baldeyrou, P.; Gautier, E.; Opolon, P.; Schatz, C.; Pavirani, A.; Courtney, M.; Lamy, D.; Ragot, T.; et al. Phase I Study of a Recombinant Adenovirus-Mediated Gene Transfer in Lung Cancer Patients. Gynecol. Oncol. 1996, 88, 1857–1863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuler, M.; Rochlitz, C.; Horowitz, J.A.; Schlegel, J.; Perruchoud, A.P.; Kommoss, F.; Bolliger, C.T.; Kauczor, H.-U.; Dalquen, P.; Fritz, M.A.; et al. A Phase I Study of Adenovirus-Mediated Wild-Type p53 Gene Transfer in Patients with Advanced Non-Small Cell Lung Cancer. Hum. Gene Ther. 1998, 9, 2075–2082. [Google Scholar] [CrossRef] [PubMed]
- To Immunize Patients with Extensive Stage SCLC Combined with Chemo with or without All Trans Retinoic Acid. Available online: https://clinicaltrials.gov/show/NCT00617409 (accessed on 7 December 2022).
- Guan, Y.-S.; Liu, Y.; Zou, Q.; He, Q.; La, Z.; Yang, L.; Hu, Y. Adenovirus-mediated wild-type p53 gene transfer in combination with bronchial arterial infusion for treatment of advanced non-small-cell lung cancer, one year follow-up. J. Zhejiang Univ. B 2009, 10, 331–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kauczor, H.-U.; Schuler, M.; Heussel, C.P.; Von Weymarn, A.; Bongartz, G.; Rochlitz, C.; Huber, C.; Thelen, M. CT-guided intratumoral gene therapy in non-small-cell lung cancer. Eur. Radiol. 1999, 9, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://clinicaltrials.gov/show/NCT00004225 (accessed on 7 December 2022).
- Available online: https://clinicaltrials.gov/show/NCT00003649 (accessed on 7 December 2022).
- Available online: https://clinicaltrials.gov/show/NCT00049218 (accessed on 7 December 2022).
- Available online: https://clinicaltrials.gov/show/NCT03029871 (accessed on 7 December 2022).
- Available online: https://clinicaltrials.gov/show/NCT02140996 (accessed on 7 December 2022).
- Nemunaitis, J.; Meyers, T.; Senzer, N.; Cunningham, C.; West, H.; Vallieres, E.; Anthony, S.; Vukelja, S.; Berman, B.; Tully, H.; et al. Phase I Trial of Sequential Administration of Recombinant DNA and Adenovirus Expressing L523S Protein in Early Stage Non-Small-Cell Lung Cancer. Mol. Ther. 2006, 13, 1185–1191. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov/show/NCT00062907 (accessed on 7 December 2022).
- Available online: https://clinicaltrials.gov/show/NCT02879760 (accessed on 7 December 2022).
- Zhang, X.; Fang, B.; Mohan, R.; Chang, J.Y. Coxsackie-adenovirus receptor as a novel marker of stem cells in treatment-resistant non-small cell lung cancer. Radiother. Oncol. 2012, 105, 250–257. [Google Scholar] [CrossRef] [Green Version]
- Sakhawat, A.; Liu, Y.; Ma, L.; Muhammad, T.; Wang, S.; Zhang, L.; Cong, X.; Huang, Y. Upregulation of Coxsackie Adenovirus Receptor Sensitizes Cisplatin-Resistant Lung Cancer Cells to CRAd-Induced Inhibition. J. Cancer 2017, 8, 1425–1432. [Google Scholar] [CrossRef] [Green Version]
- Jin, C.; Lagoudas, G.K.; Zhao, C.; Bullman, S.; Bhutkar, A.; Hu, B.; Ameh, S.; Sandel, D.; Liang, X.S.; Mazzilli, S.; et al. Commensal microbiota promote lung cancer development via γδ T cells. Cell 2019, 176, 998–1013.e16. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Lu, Z.H.; Li, J.; Kashentseva, E.A.; Dmitriev, I.P.; Mendonca, S.A.; Curiel, D.T. Targeting Tumor Neoangiogenesis via Targeted Adenoviral Vector to Achieve Effective Cancer Gene Therapy for Disseminated Neoplastic Disease. Mol. Cancer Ther. 2020, 19, 966–971. [Google Scholar] [CrossRef] [Green Version]
- Harvey, B.-G.; McKinney, R.L.; Rosengart, T.; Lesser, M.L.; Crystal, R.G. Systemic Interleukin-6 Responses Following Administration of Adenovirus Gene Transfer Vectors to Humans by Different Routes. Mol. Ther. 2002, 6, 287–297. [Google Scholar] [CrossRef]
- Anwar, M.; Arendt, M.-L.; Ramachandran, M.; Carlsson, A.; Essand, M.; Akusjärvi, G.; Alusi, G.; Öberg, D. Ixovex-1, a novel oncolytic E1B-mutated adenovirus. Cancer Gene Ther. 2022, 29, 1628–1635. [Google Scholar] [CrossRef] [PubMed]
- Van Veen, J.E.; Scherzer, M.; Boshuizen, J.; Chu, M.; Liu, A.; Landman, A.; Green, S.; Trejo, C.; McMahon, M. Mutationally-activated PI3’-kinase-α promotes de-differentiation of lung tumors initiated by the BRAFV600E oncoprotein kinase. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Kauko, O.; O’Connor, C.M.; Kulesskiy, E.; Sangodkar, J.; Aakula, A.; Izadmehr, S.; Yetukuri, L.; Yadav, B.; Padzik, A.; Laajala, T.D.; et al. PP2A inhibition is a druggable MEK inhibitor resistance mechanism in KRAS-mutant lung cancer cells. Sci. Transl. Med. 2018, 10, eaaq1093. [Google Scholar] [CrossRef] [Green Version]
- Meeusen, B.; Cortesi, E.E.; Omella, J.D.; Sablina, A.; Ventura, J.-J.; Janssens, V. PPP2R4 dysfunction promotes KRAS-mutant lung adenocarcinoma development and mediates opposite responses to MEK and mTOR inhibition. Cancer Lett. 2021, 520, 57–67. [Google Scholar] [CrossRef]
- Li, D.; Zhang, Y.; Xie, Y.; Xiang, J.; Zhu, Y.; Yang, J. Enhanced tumor suppression by adenoviral PTEN gene therapy combined with cisplatin chemotherapy in small-cell lung cancer. Cancer Gene Ther. 2013, 20, 251–259. [Google Scholar] [CrossRef]
- Nieto-Jiménez, C.; Morafraile, E.C.; Alonso-Moreno, C.; Ocaña, A. Clinical considerations for the design of PROTACs in cancer. Mol. Cancer 2022, 21, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Xiong, Y.; Safaee, N.; Nowak, R.P.; Donovan, K.A.; Yuan, C.J.; Nabet, B.; Gero, T.W.; Feru, F.; Li, L.; et al. Exploring Targeted Degradation Strategy for Oncogenic KRASG12C. Cell Chem. Biol. 2020, 27, 19–31.e6. [Google Scholar] [CrossRef]
- Bond, M.J.; Chu, L.; Nalawansha, D.A.; Li, K.; Crews, C.M. Targeted Degradation of Oncogenic KRASG12C by VHL-Recruiting PROTACs. ACS Central Sci. 2020, 6, 1367–1375. [Google Scholar] [CrossRef]
- Zhou, C.; Fan, Z.; Zhou, Z.; Li, Y.; Cui, R.; Liu, C.; Zhou, G.; Diao, X.; Jiang, H.; Zheng, M.; et al. Discovery of the First-in-Class Agonist-Based SOS1 PROTACs Effective in Human Cancer Cells Harboring Various KRAS Mutations. J. Med. Chem. 2022, 65, 3923–3942. [Google Scholar] [CrossRef]
- Liu, J.; Xue, L.; Xu, X.; Luo, J.; Zhang, S. FAK-targeting PROTAC demonstrates enhanced antitumor activity against KRAS mutant non-small cell lung cancer. Exp. Cell Res. 2021, 408, 112868. [Google Scholar] [CrossRef]
- Qu, X.; Liu, H.; Song, X.; Sun, N.; Zhong, H.; Qiu, X.; Yang, X.; Jiang, B. Effective degradation of EGFRL858R+T790M mutant proteins by CRBN-based PROTACs through both proteosome and autophagy/lysosome degradation systems. Eur. J. Med. Chem. 2021, 218, 113328. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.-Y.; Wang, H.-P.; Mao, Y.-Z.; Zhang, H.; Xin, M.; Xi, X.-X.; Lei, H.; Mao, S.; Li, D.-H.; Zhang, S.-Q. Discovery of Potent PROTACs Targeting EGFR Mutants through the Optimization of Covalent EGFR Ligands. J. Med. Chem. 2022, 65, 4709–4726. [Google Scholar] [CrossRef]
- Hofmann, M.H.; Gmachl, M.; Ramharter, J.; Savarese, F.; Gerlach, D.; Marszalek, J.R.; Sanderson, M.P.; Kessler, D.; Trapani, F.; Arnhof, H.; et al. BI-3406, a Potent and Selective SOS1–KRAS Interaction Inhibitor, Is Effective in KRAS-Driven Cancers through Combined MEK Inhibition. Cancer Discov. 2021, 11, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://clinicaltrials.gov/ct2/show/NCT04111458 (accessed on 7 December 2022).
- Gerber, D.E.; Camidge, D.R.; Morgensztern, D.; Cetnar, J.; Kelly, R.J.; Ramalingam, S.S.; Spigel, D.R.; Jeong, W.; Scaglioni, P.P.; Zhang, S.; et al. Phase 2 study of the focal adhesion kinase inhibitor defactinib (VS-6063) in previously treated advanced KRAS mutant non-small cell lung cancer. Lung Cancer 2020, 139, 60–67. [Google Scholar] [CrossRef]
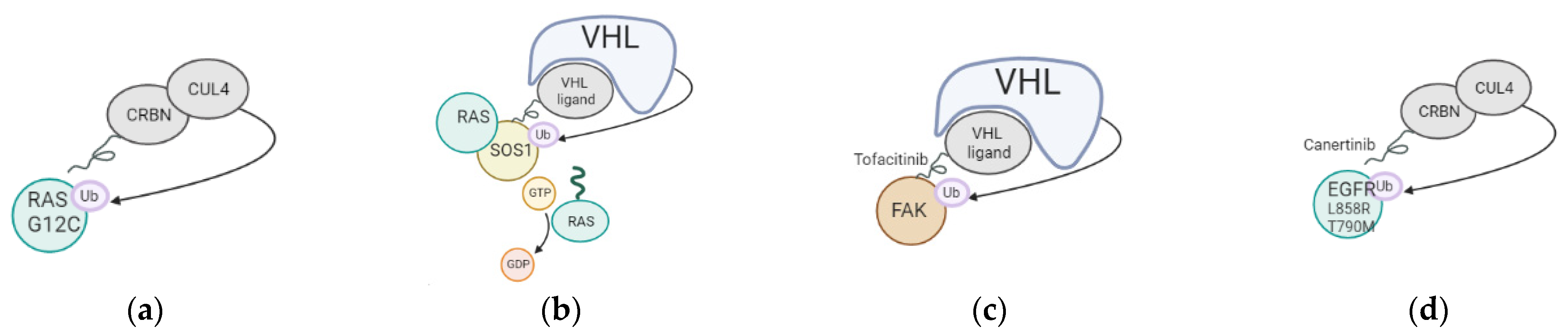
- Willson, J. DUBTACs for targeted protein stabilization. Nat. Rev. Drug Discov. 2022, 21, 258. [Google Scholar] [CrossRef]
- Henning, N.J.; Boike, L.; Spradlin, J.N.; Ward, C.C.; Liu, G.; Zhang, E.; Belcher, B.P.; Brittain, S.M.; Hesse, M.J.; Dovala, D.; et al. Deubiquitinase-targeting chimeras for targeted protein stabilization. Nat. Chem. Biol. 2022, 18, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Baietti, M.F.; Simicek, M.; Asbagh, L.A.; Radaelli, E.; Lievens, S.; Crowther, J.; Steklov, M.; Aushev, V.N.; García, D.M.; Tavernier, J.; et al. OTUB 1 triggers lung cancer development by inhibiting RAS monoubiquitination. EMBO Mol. Med. 2016, 8, 288–303. [Google Scholar] [CrossRef]
- Dunkle, L.M.; Kotloff, K.L.; Gay, C.L.; Áñez, G.; Adelglass, J.M.; Hernández, A.Q.B.; Harper, W.L.; Duncanson, D.M.; McArthur, M.A.; Florescu, D.F.; et al. Efficacy and Safety of NVX-CoV2373 in Adults in the United States and Mexico. N. Engl. J. Med. 2022, 386, 531–543. [Google Scholar] [CrossRef]
- Loquai, C.; Hassel, J.C.; Oehm, P.; Derhovanessian, E.; Jabulowsky, R.A.; Gold, M.; Schwarck-Kokarakis, D.; Attig, S.; Cuk, K.; Vogler, I.; et al. A shared tumor-antigen RNA-lipoplex vaccine with/without anti-PD1 in patients with checkpoint-inhibition experienced melanoma. J. Clin. Oncol. 2020, 38, 3136. [Google Scholar] [CrossRef]
- Xie, Y.J.; Dougan, M.; Jailkhani, N.; Ingram, J.; Fang, T.; Kummer, L.; Momin, N.; Pishesha, N.; Rickelt, S.; Hynes, R.O.; et al. Nanobody-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc. Natl. Acad. Sci. USA 2019, 116, 7624–7631. [Google Scholar] [CrossRef]
- Ribeiro, R.S.G.; Belderbos, S.; Danhier, P.; Gallo, J.; Manshian, B.B.; Gallez, B.; Bañobre, M.; de Cuyper, M.; Soenen, S.J.; Gsell, W.; et al. Targeting tumor cells and neovascularization using RGD-functionalized magnetoliposomes. Int. J. Nanomed. 2019, 14, 5911–5924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://clinicaltrials.gov/ct2/show/NCT02996214 (accessed on 7 December 2022).
- Goldinger, S.M.; Dummer, R.; Baumgaertner, P.; Mihic-Probst, D.; Schwarz, K.; Hammann-Haenni, A.; Willers, J.; Geldhof, C.; Prior, J.O.; Kündig, T.M.; et al. Nano-particle vaccination combined with TLR-7 and -9 ligands triggers memory and effector CD8+ T-cell responses in melanoma patients. Eur. J. Immunol. 2012, 42, 3049–3061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://clinicaltrials.gov/ct2/show/NCT04033354 (accessed on 7 December 2022).
- Available online: https://clinicaltrials.gov/ct2/show/NCT01792479 (accessed on 7 December 2022).
- Available online: https://clinicaltrials.gov/ct2/show/NCT02283320 (accessed on 7 December 2022).
- Graziani, S.R.; Vital, C.G.; Morikawa, A.T.; Van Eyll, B.M.; Junior, H.J.F.; Filho, R.K.; Maranhão, R.C. Phase II study of paclitaxel associated with lipid core nanoparticles (LDE) as third-line treatment of patients with epithelial ovarian carcinoma. Med Oncol. 2017, 34, 151. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://clinicaltrials.gov/ct2/show/NCT00729612 (accessed on 7 December 2022).
- Available online: https://clinicaltrials.gov/ct2/show/NCT00077246 (accessed on 7 December 2022).
- Available online: https://clinicaltrials.gov/ct2/show/NCT01380769 (accessed on 7 December 2022).
- Available online: https://clinicaltrials.gov/ct2/show/NCT02667743 (accessed on 7 December 2022).
- Available online: https://clinicaltrials.gov/ct2/show/NCT04486833 (accessed on 7 December 2022).
- Available online: https://clinicaltrials.gov/ct2/show/NCT04381910 (accessed on 7 December 2022).
- Available online: https://clinicaltrials.gov/ct2/show/NCT03088813 (accessed on 7 December 2022).
- Available online: https://clinicaltrials.gov/ct2/show/NCT02769962 (accessed on 7 December 2022).
- Available online: https://clinicaltrials.gov/ct2/show/NCT03670030 (accessed on 7 December 2022).
- Casaluce, F.; Sgambato, A.; Maione, P.; Ciardiello, F.; Gridelli, C. Emerging mitotic inhibitors for non-small cell carcinoma. Expert Opin. Emerg. Drugs 2013, 18, 97–107. [Google Scholar] [CrossRef]
- Hafezi, S.; Rahmani, M. Targeting BCL-2 in Cancer: Advances, Challenges, and Perspectives. Cancers 2021, 13, 1292. [Google Scholar] [CrossRef]
- Lin, Y.-X.; Wang, Y.; Ding, J.; Jiang, A.; Wang, J.; Yu, M.; Blake, S.; Liu, S.; Bieberich, C.J.; Farokhzad, O.C.; et al. Reactivation of the tumor suppressor PTEN by mRNA nanoparticles enhances antitumor immunity in preclinical models. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef]
- Tabernero, J.; Shapiro, G.I.; Lorusso, P.M.; Cervantes, A.; Schwartz, G.K.; Weiss, G.J.; Paz-Ares, L.; Cho, D.C.; Infante, J.R.; Alsina, M.; et al. First-in-Humans Trial of an RNA Interference Therapeutic Targeting VEGF and KSP in Cancer Patients with Liver Involvement. Cancer Discov. 2013, 3, 406–417. [Google Scholar] [CrossRef] [Green Version]
- Jansen, B.; Wacheck, V.; Heere-Ress, E.; Schlagbauer-Wadl, H.; Hoeller, C.; Lucas, T.; Hoermann, M.; Hollenstein, U.; Wolff, K.; Pehamberger, H. Chemosensitisation of malignant melanoma by BCL2 antisense therapy. Lancet 2000, 356, 1728–1733. [Google Scholar] [CrossRef]
- Hong, D.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; Zhou, T.; Schmidt, J.; Jo, M.; et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015, 7, 314ra185. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov/ct2/show/NCT05654623 (accessed on 7 December 2022).



{kind=link}
{kind=link}
{kind=link}
| Clinical Trial Name | Drug Tested | Status | Ref |
|---|---|---|---|
| Phase I study of recombinant adenovirus-mediated gene transfer in lung cancer patients. | replication-defective adenoviral expression vector encoding wild-type p53 | Completed | [1] |
| A phase I study of adenovirus-mediated wild-type p53 gene transfer in patients with advanced non-small cell lung cancer. | replication-defective adenoviral expression vector encoding wild-type p53 | Completed | [2] |
| Phase I trial of sequential administration of recombinant DNA and adenovirus expressing L523S protein in early-stage non-small-cell lung cancer. | L523S, an immunogenic lung cancer antigen delivered into an E1B-deleted adenovirus (Ad/L523S) | Completed | [11] |
| Phase II trial using dendritic cells transduced with an adenoviral vector containing the p53 gene to immunize patients with lung cancer | Efficacy of paclitaxel following the dendritic cell (DC)-based p53 vaccine (Ad.p53-DC vaccine), | Completed | [3] |
| Adenovirus-mediated wild-type p53 gene transfer in combination with bronchial arterial infusion for treatment of advanced non-small-cell lung cancer, one year follow-up | rAd-p53 and BAI | Completed | [4] |
| CT-guided intratumoral gene therapy in non-small-cell lung cancer. | replication-defective adenoviral expression vector encoding wild-type p53 | Completed | [5] |
| Gene therapy plus radiation therapy in treating patients with non-small cell lung cancer | adenovirus p53 gene therapy and radiotherapy | Completed | [6] |
| Phase I study of adenovirus p53 administered by bronchoalveolar lavage in patients with bronchioloalveolar cell lung carcinoma | replication-defective adenoviral expression vector encoding wild-type p53 | Completed | [7] |
| Chemotherapy followed by vaccine therapy in treating patients with extensive-stage small cell lung cancer | Dendritic cell (DC)-based p53 vaccine and trans-retinoic acid | Completed | [8] |
| Safety and efficacy of recombinant oncolytic sdenovirus L-IFN injection in relapsed/refractory solid tumors clinical study (YSCH-01) | Ad5-yCD/mutTKSR39rep-ADP adenovirus, 5-fluorocytosine (5-FC), valganciclovir (vGCV), 48Gy | Oncolytic effect, withdrawn | [9] |
| TNFα and IL-2 coding oncolytic adenovirus TILT-123 monotherapy (TUNIMO) | pVAX/L523S and two doses of Ad/L523S | T-cell activating, status unknown | [12] |
| Oncolytic MG1-MAGEA3 with Ad-MAGEA3 vaccine in combination with pembrolizumab for non-small cell lung cancer patients | Ad-MAGEA3 and MG1-MAGEA3 in combination with pembrolizumab | T-cell activating, status unknown | [13] |
| Safety study of human MUC-1 (Mucin-1) adenoviral vector vaccine for immunotherapy of epithelial cancers (MUC-1) | Ad-sig-hMUC-1/ecdCD40L vector encoding a fusion protein in which hMUC-1 epithelial antigen is attached to CD40L (CD40 ligand) | Dendritic cell activating, status unknown | [10] |
| Target of the Adenovirus | Oncogene Affected | Model Used | Delivery Route | Adenovirus Promoter | Refs. |
|---|---|---|---|---|---|
| Mutationally activated kinase PI3K (H1047R) | BRAF V600E signaling | BRAF V600E-driven lung adenocarcinoma tumors | Intratracheal injection | SFTPC Adenovirus | [20] |
| Phosphatase PP2A | KRAS G12D signaling | KRAS G12D induced lung adenocarcinoma tumors | Intratracheal injection | Constitutive Adenovirus | [21,22] |
| Phosphatase PTEN | KRAS signalling | Xenograft (NCI-H446 lung small cell cancer cell) | Intratumor injection | Constitutive adenovirus | [23] |
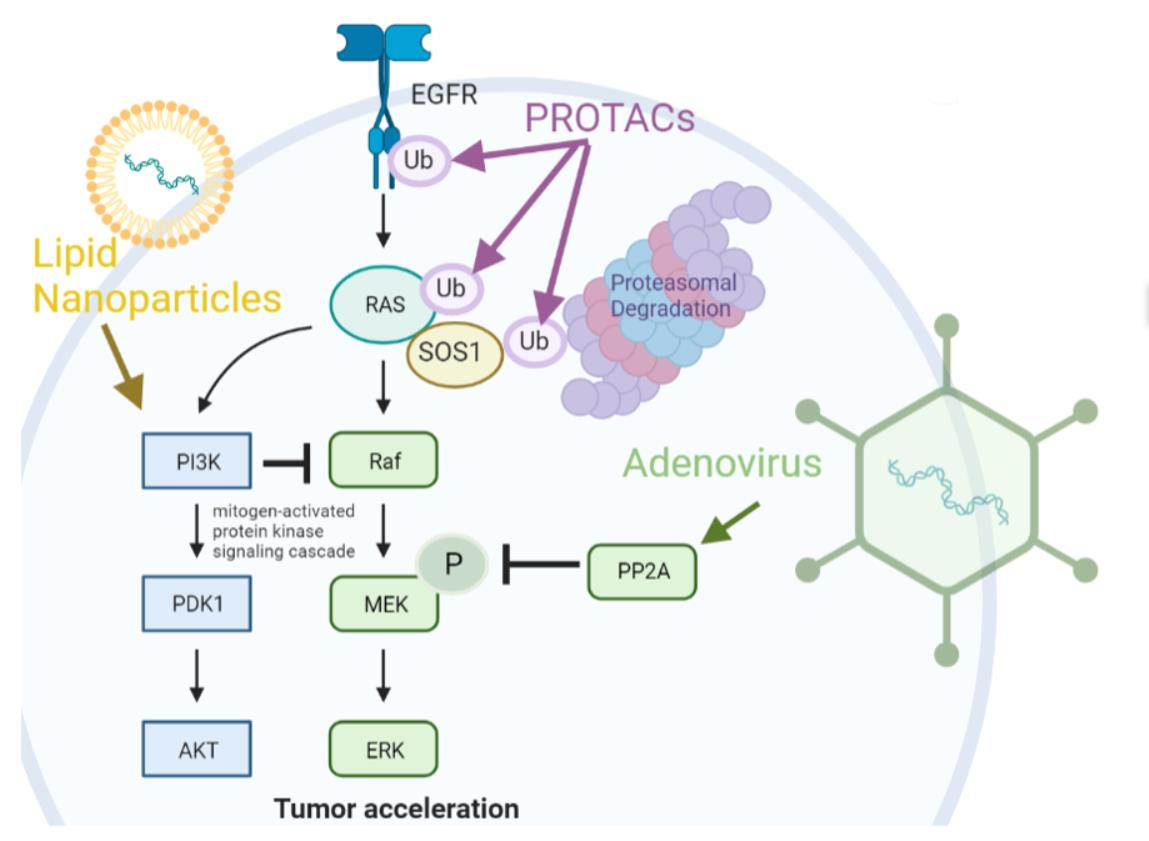
| Target | Degrader | E3 Ligase | References |
|---|---|---|---|
| KRAS G12C (a) | Cereblon CRBN | CUL4 | [25,26] |
| SOS1 (b) | SOS agonist | VHL | [27] |
| FAK (c) | Tofacitinib (FAK ligand) | VHL | [28] |
| EGFR L858R+T790M (d) | Cereblon CRBN | CUL4 | [29] |
| EGFR | novel covalent purine-containing EGFR ligand | [30] |
| Study Type | Target | Status | Reference |
|---|---|---|---|
| Phase II | BIND-014 (docetaxel nanoparticles) in lung cancer | Completed | [44] |
| Phase II | BIND-014 (docetaxel nanoparticles) in KRAS-mutated lung cancer | Completed | [45] |
| Phase II | Carboplatin and paclitaxel albumin-stabilized nanoparticle (ovarian) | Completed | [46] |
| Phase II | Paclitaxel albumin-stabilized nanoparticle in lung cancer | Completed | [47] |
| Phase I-II | ABI-007 (albumin-stabilised paclitaxel nanoparticle) in lung cancer | Completed | [48] |
| Phase II | CRLX101 (cyclodextrin-based polymer with camptothecin) in lung | Completed | [49] |
| Phase IV | Paclitaxel liposome in lung cancer | Completed | [41] |
| Phase III-IV | A-type CpG-oligonucleotides (CpG-ODN) coupled to peptide derived from Melan-A/MART-1 in melanoma | Completed | [42] |
| Phase III | Paclitaxel micelles in lung cancer | In progress | [50] |
| Phase I-II | GPX-001 (TUSC2 encapsulate in lipid nanoparticles) in lung cancer | In progress | [51] |
| Phase II | LY01610 (irinotecan hydrochloride liposome) in lung cancer | In progress | [52] |
| Phase II-III | ONIVYDE (irinotecan liposome) in lung cancer | In progress | [53] |
| Phase III | HLX10 (humanized—PD-1 receptor) liposome in lung cancer | In progress | [43] |
| Phase I-II | CRLX101 (cyclodextrin polymer with camptothecin) in lung cancer | In progress | [54] |
| Phase II | ABI-009 (albumin-bound mTOR inhibitor) in neuroendocrine tumor | Terminated | [55] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baietti, M.F.; Sewduth, R.N. Novel Therapeutic Approaches Targeting Post-Translational Modifications in Lung Cancer. Pharmaceutics 2023, 15, 206. https://doi.org/10.3390/pharmaceutics15010206
Baietti MF, Sewduth RN. Novel Therapeutic Approaches Targeting Post-Translational Modifications in Lung Cancer. Pharmaceutics. 2023; 15(1):206. https://doi.org/10.3390/pharmaceutics15010206
Chicago/Turabian StyleBaietti, Maria Francesca, and Raj Nayan Sewduth. 2023. "Novel Therapeutic Approaches Targeting Post-Translational Modifications in Lung Cancer" Pharmaceutics 15, no. 1: 206. https://doi.org/10.3390/pharmaceutics15010206