
Dimeric Lectin Chimeras as Novel Candidates for Gb3-Mediated Transcytotic Drug Delivery through Cellular Barriers
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
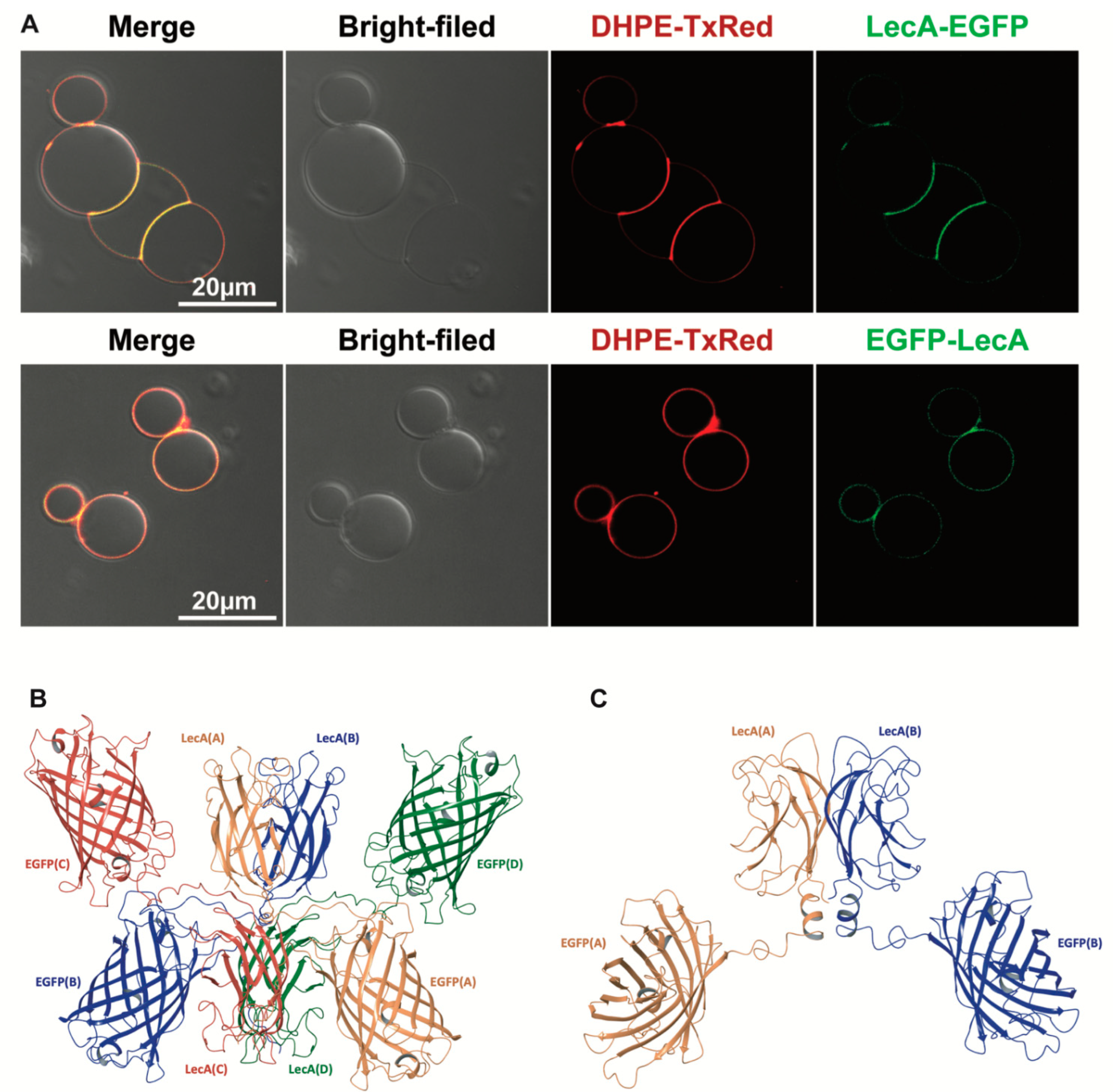{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Lectin Labeling
2.3. Immunofluorescence Microscopy
2.4. Pulse-Chase Assay
2.5. Release Efficiency Measurements (Medium-to-Medium Transcytosis)
2.6. Production, Purification, and Validation of Fusion Proteins
2.7. Molecular Dynamics Simulations
2.8. Statistical Analysis
3. Results
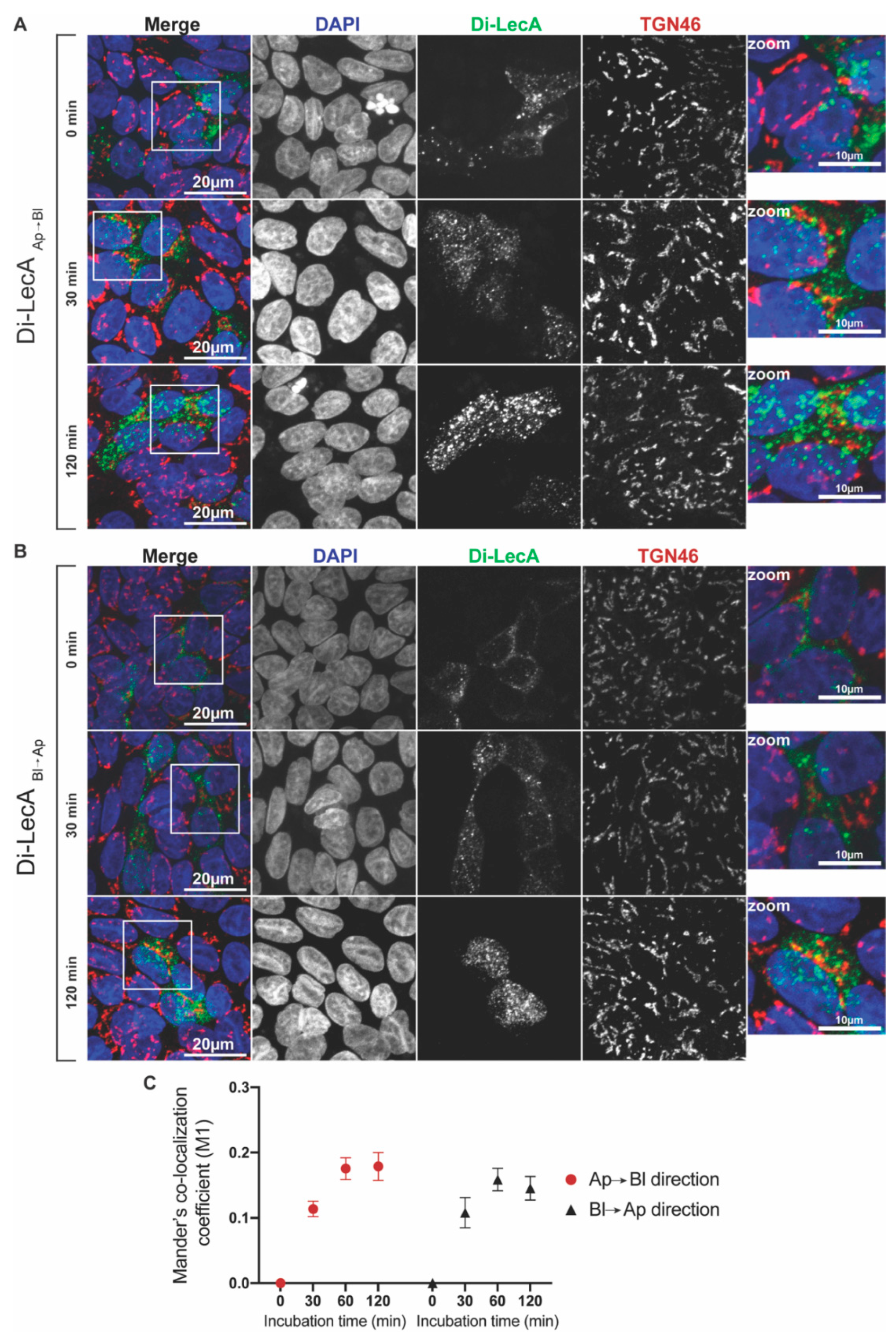
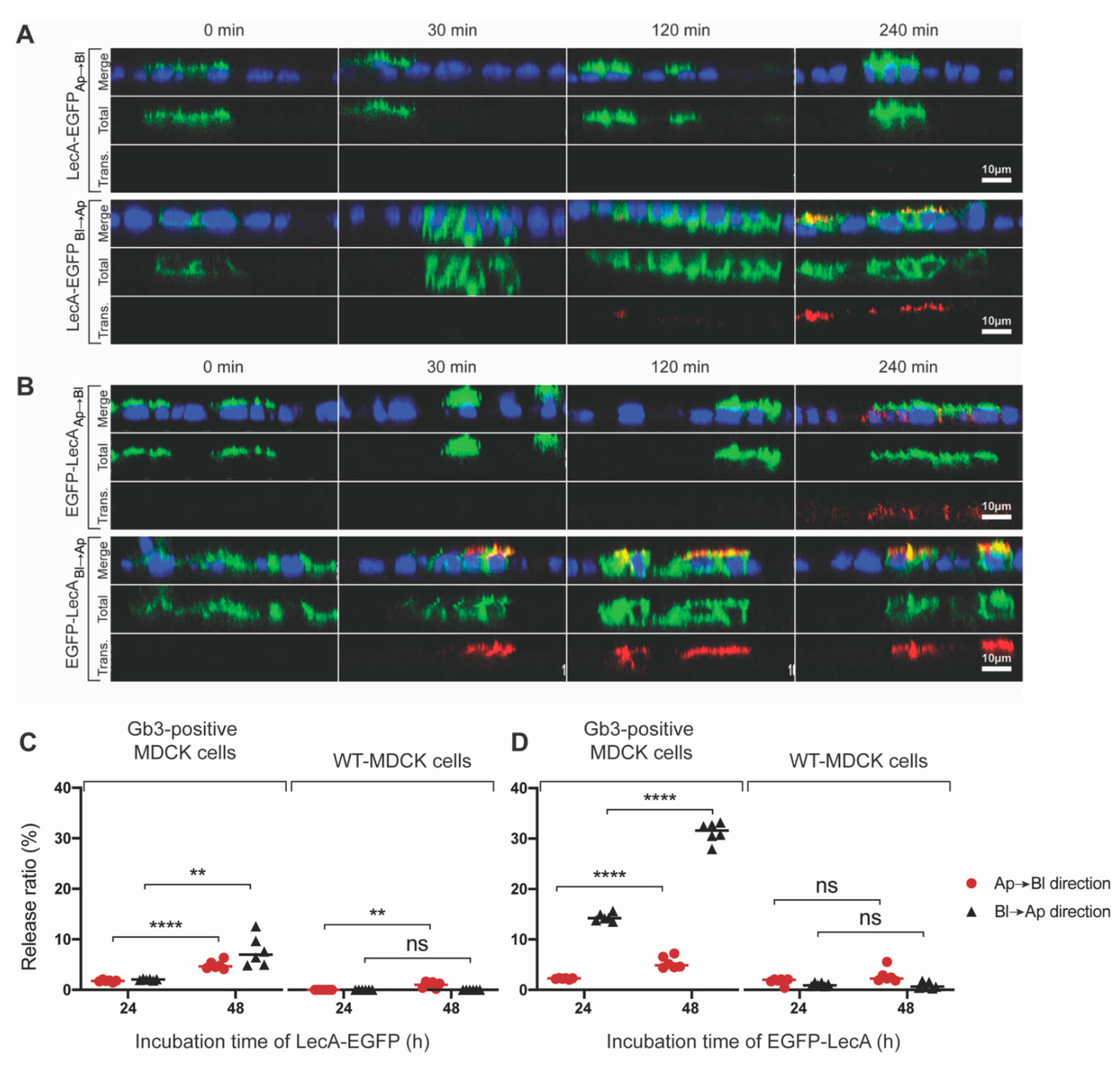
3.1. Di-LecA Undergoes Faster Transcytosis Than Tetra-LecA
3.2. Di-LecA Mainly Bypasses the Trans-Golgi Network
3.3. Small Quantities of Di-LecA Arrive in Lysosomes
3.4. Di-LecA Is Slightly Better Released from Its Receptor Than Tetra-LecA
3.5. Transcytosis of Model LecA Fusion Proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Janssen, K.-P.; Vignjevic, D.; Boisgard, R.; Falguières, T.; Bousquet, G.; Decaudin, D.; Dollé, F.; Louvard, D.; Tavitian, B.; Robine, S.; et al. In Vivo Tumor Targeting Using a Novel Intestinal Pathogen-Based Delivery Approach. Cancer Res. 2006, 66, 7230–7236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez Porras, M.A.; Durfee, P.N.; Gregory, A.M.; Sieck, G.C.; Brinker, C.J.; Mantilla, C.B. A Novel Approach for Targeted Delivery to Motoneurons Using Cholera Toxin-B Modified Protocells. J. Neurosci. Methods 2016, 273, 160–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, K.; Jia, X.; Zhou, M.; Wang, K.; Conde, J.; He, J.; Tian, J.; Yan, X. Ferritin Nanocarrier Traverses the Blood Brain Barrier and Kills Glioma. ACS Nano 2018, 12, 4105–4115. [Google Scholar] [CrossRef]
- Oliveira, S.; Heukers, R.; Sornkom, J.; Kok, R.J.; Van Bergen En Henegouwen, P.M.P. Targeting Tumors with Nanobodies for Cancer Imaging and Therapy. J. Control. Release 2013, 172, 607–617. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, R.; Han, L.; Ke, W.; Shao, K.; Ye, L.; Lou, J.; Jiang, C. Brain-Targeting Gene Delivery and Cellular Internalization Mechanisms for Modified Rabies Virus Glycoprotein RVG29 Nanoparticles. Biomaterials 2009, 30, 4195–4202. [Google Scholar] [CrossRef]
- Haqqani, A.S.; Delaney, C.E.; Brunette, E.; Baumann, E.; Farrington, G.K.; Sisk, W.; Eldredge, J.; Ding, W.; Tremblay, T.-L.; Stanimirovic, D.B. Endosomal Trafficking Regulates Receptor-Mediated Transcytosis of Antibodies across the Blood Brain Barrier. J. Cereb. Blood Flow Metab. 2018, 38, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Abdul Razzak, R.; Florence, G.J.; Gunn-Moore, F.J. Approaches to CNS Drug Delivery with a Focus on Transporter-Mediated Transcytosis. Int. J. Mol. Sci. 2019, 20, 3108. [Google Scholar] [CrossRef] [Green Version]
- Pulgar, V.M. Transcytosis to Cross the Blood Brain Barrier, New Advancements and Challenges. Front. Neurosci. 2018, 12, 1019. [Google Scholar] [CrossRef]
- Lajoie, J.M.; Shusta, E.V. Targeting Receptor-Mediated Transport for Delivery of Biologics across the Blood-Brain Barrier. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 613–631. [Google Scholar] [CrossRef] [Green Version]
- Thuenauer, R.; Müller, S.K.; Römer, W. Pathways of Protein and Lipid Receptor-Mediated Transcytosis in Drug Delivery. Expert Opin. Drug Deliv. 2017, 14, 341–351. [Google Scholar] [CrossRef]
- Tuma, P.L.; Hubbard, A.L. Transcytosis: Crossing Cellular Barriers. Physiol. Rev. 2003, 83, 871–932. [Google Scholar] [CrossRef] [PubMed]
- Griffitts, J.S.; Haslam, S.M.; Yang, T.; Garczynski, S.F.; Mulloy, B.; Morris, H.; Cremer, P.S.; Dell, A.; Adang, M.J.; Aroian, R.V. Glycolipids as Receptors for Bacillus Thuringiensis Crystal Toxin. Science 2005, 307, 922–925. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Vilanova, A.; Chan, J.; Torrelles, J.B. Underestimated Manipulative Roles of Mycobacterium Tuberculosis Cell Envelope Glycolipids During Infection. Front. Immunol. 2019, 10, 2909. [Google Scholar] [CrossRef] [PubMed]
- Uraki, R.; Kawaoka, Y. Host Glycolipids in SARS-CoV-2 Entry. Nat. Chem. Biol. 2022, 18, 6–7. [Google Scholar] [CrossRef]
- Aigal, S.; Claudinon, J.; Römer, W. Plasma Membrane Reorganization: A Glycolipid Gateway for Microbes. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 858–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siukstaite, L.; Imberty, A.; Römer, W. Structural Diversities of Lectins Binding to the Glycosphingolipid Gb3. Front. Mol. Biosci. 2021, 8, 704685. [Google Scholar] [CrossRef]
- Eierhoff, T.; Bastian, B.; Thuenauer, R.; Madl, J.; Audfray, A.; Aigal, S.; Juillot, S.; Rydell, G.E.; Müller, S.; de Bentzmann, S.; et al. A Lipid Zipper Triggers Bacterial Invasion. Proc. Natl. Acad. Sci. USA 2014, 111, 12895–12900. [Google Scholar] [CrossRef] [Green Version]
- Müller, S.K.; Wilhelm, I.; Schubert, T.; Zittlau, K.; Imberty, A.; Madl, J.; Eierhoff, T.; Thuenauer, R.; Römer, W. Gb3-Binding Lectins as Potential Carriers for Transcellular Drug Delivery. Expert Opin. Drug Deliv. 2017, 14, 141–153. [Google Scholar] [CrossRef]
- Arnaud, J.; Claudinon, J.; Tröndle, K.; Trovaslet, M.; Larson, G.; Thomas, A.; Varrot, A.; Römer, W.; Imberty, A.; Audfray, A. Reduction of Lectin Valency Drastically Changes Glycolipid Dynamics in Membranes but Not Surface Avidity. ACS Chem. Biol. 2013, 8, 1918–1924. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, J.; Tröndle, K.; Claudinon, J.; Audfray, A.; Varrot, A.; Römer, W.; Imberty, A. Membrane Deformation by Neolectins with Engineered Glycolipid Binding Sites. Angew. Chem. Int. Ed. 2014, 53, 9267–9270. [Google Scholar] [CrossRef]
- Keogh, D.; Thompson, R.; Larragy, R.; McMahon, K.; O’Connell, M.; O’Connor, B.; Clarke, P. Generating Novel Recombinant Prokaryotic Lectins with Altered Carbohydrate Binding Properties through Mutagenesis of the PA-IL Protein from Pseudomonas Aeruginosa. Biochim. Biophys. Acta 2014, 1840, 2091–2104. [Google Scholar] [CrossRef]
- Sali, A.; Tom, L.B. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Shaw, D.E.; Maragakis, P.; Lindorff-Larsen, K.; Piana, S.; Dror, R.O.; Eastwood, M.P.; Bank, J.A.; Jumper, J.M.; Salmon, J.K.; Shan, Y.; et al. Atomic-Level Characterization of the Structural Dynamics of Proteins. Science 2010, 330, 341–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Koukos, P.I.; Glykos, N.M. Grcarma: A Fully Automated Task-Oriented Interface for the Analysis of Molecular Dynamics Trajectories. J. Comput. Chem. 2013, 34, 2310–2312. [Google Scholar] [CrossRef] [PubMed]
- Amessou, M.; Popoff, V.; Yelamos, B.; Saint-Pol, A.; Johannes, L. Measuring Retrograde Transport to the Trans-Golgi Network. Curr. Protoc. Cell Biol. 2006, 32, 15.10.1–15.10.21. [Google Scholar] [CrossRef] [PubMed]
- Mallard, F.; Antony, C.; Tenza, D.; Salamero, J.; Goud, B.; Johannes, L. Direct Pathway from Early/Recycling Endosomes to the Golgi Apparatus Revealed through the Study of Shiga Toxin B-Fragment Transport. J. Cell Biol. 1998, 143, 973–990. [Google Scholar] [CrossRef]
- Sandvig, K.; Garred, Ø.; Prydz, K.; Kozlov, J.V.; Hansen, S.H.; van Deurs, B. Retrograde Transport of Endocytosed Shiga Toxin to the Endoplasmic Reticulum. Nature 1992, 358, 510–512. [Google Scholar] [CrossRef] [PubMed]
- Schubert, T.; Sych, T.; Madl, J.; Xu, M.; Omidvar, R.; Patalag, L.J.; Ries, A.; Kettelhoit, K.; Brandel, A.; Mely, Y.; et al. Differential Recognition of Lipid Domains by Two Gb3-Binding Lectins. Sci. Rep. 2020, 10, 9752. [Google Scholar] [CrossRef]
- Vellodi, A. Lysosomal Storage Disorders. Br. J. Haematol. 2005, 128, 413–431. [Google Scholar] [CrossRef]
- Schulze, H.; Kolter, T.; Sandhoff, K. Principles of Lysosomal Membrane Degradation: Cellular Topology and Biochemistry of Lysosomal Lipid Degradation. Biochim. Biophys. Acta 2009, 1793, 674–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.-T.; Xie, Y.-X.; Zhou, B.; Huang, N.; Farfel-Becker, T.; Sheng, Z.-H. Revisiting LAMP1 as a Marker for Degradative Autophagy-Lysosomal Organelles in the Nervous System. Autophagy 2018, 14, 1472–1474. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.J.; Zhang, Y.; Kenrick, M.; Hoyte, K.; Luk, W.; Lu, Y.; Atwal, J.; Elliott, J.M.; Prabhu, S.; Watts, R.J.; et al. Boosting Brain Uptake of a Therapeutic Antibody by Reducing Its Affinity for a Transcytosis Target. Sci. Transl. Med. 2011, 3, 84ra44. [Google Scholar] [CrossRef] [PubMed]
- Villringer, S.; Madl, J.; Sych, T.; Manner, C.; Imberty, A.; Römer, W. Lectin-Mediated Protocell Crosslinking to Mimic Cell-Cell Junctions and Adhesion. Sci. Rep. 2018, 8, 1932. [Google Scholar] [CrossRef] [Green Version]
- Miao, Y.; Feixas, F.; Eun, C.; McCammon, J.A. Accelerated Molecular Dynamics Simulations of Protein Folding. J. Comput. Chem. 2015, 36, 1536–1549. [Google Scholar] [CrossRef] [Green Version]
- Makshakova, O.N.; Bogdanova, L.R.; Faizullin, D.A.; Ermakova, E.A.; Zuev, Y.F.; Sedov, I.A. Interaction-Induced Structural Transformation of Lysozyme and Kappa-Carrageenan in Binary Complexes. Carbohydr. Polym. 2021, 252, 117181. [Google Scholar] [CrossRef]
- Ermakova, E.; Makshakova, O.; Zuev, Y.; Sedov, I. Beta-Rich Intermediates in Denaturation of Lysozyme: Accelerated Molecular Dynamics Simulations. J. Biomol. Struct. Dyn. 2021, 40, 13953–13964. [Google Scholar] [CrossRef]
- Ermakova, E.A.; Makshakova, O.N.; Zuev, Y.F.; Sedov, I.A. Fibril Fragments from the Amyloid Core of Lysozyme: An Accelerated Molecular Dynamics Study. J. Mol. Graph. Model. 2021, 106, 107917. [Google Scholar] [CrossRef]
- Chen, Y.; Fan, H.; Xu, C.; Hu, W.; Yu, B. Efficient Cholera Toxin B Subunit-Based Nanoparticles with MRI Capability for Drug Delivery to the Brain Following Intranasal Administration. Macromol. Biosci. 2019, 19, e18003402019. [Google Scholar] [CrossRef]
- Danielewicz, N.; Rosato, F.; Dai, W.; Römer, W.; Turnbull, W.B.; Mairhofer, J. Microbial Carbohydrate-Binding Toxins—From Etiology to Biotechnological Application. Biotechnol. Adv. 2022, 59, 107951. [Google Scholar] [CrossRef]
- Lencer, W.I.; Moe, S.; Rufo, P.A.; Madara, J.L. Transcytosis of Cholera Toxin Subunits across Model Human Intestinal Epithelia. Proc. Natl. Acad. Sci. USA 1995, 92, 10094–10098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svennerholm, L. Interaction of Cholera Toxin and Ganglioside G(M1). Adv. Exp. Med. Biol. 1976, 71, 191–204. [Google Scholar] [CrossRef]
- Pina, D.G.; Johannes, L. Cholera and Shiga Toxin B-Subunits: Thermodynamic and Structural Considerations for Function and Biomedical Applications. Toxicon 2005, 45, 389–393. [Google Scholar] [CrossRef]
- Singh, P.; Prabakaran, D.; Jain, S.; Mishra, V.; Jaganathan, K.S.; Vyas, S.P. Cholera Toxin B Subunit Conjugated Bile Salt Stabilized Vesicles (Bilosomes) for Oral Immunization. Int. J. Pharm. 2004, 278, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Baldauf, K.J.; Royal, J.M.; Hamorsky, K.T.; Matoba, N. Cholera Toxin B: One Subunit with Many Pharmaceutical Applications. Toxins 2015, 7, 974–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, J.; Zhang, Z.; Hu, X.; Yang, Y.; Chai, Z.; Liu, X.; Liu, J.; Gao, B.; Lu, W.; Qian, J.; et al. Cholera Toxin Subunit B Enabled Multifunctional Glioma-Targeted Drug Delivery. Adv. Health Mater. 2017, 6, 1700709. [Google Scholar] [CrossRef]
- Saslowsky, D.E.; te Welscher, Y.M.; Chinnapen, D.J.F.; Wagner, J.S.; Wan, J.; Kern, E.; Lencer, W.I. Ganglioside GM1-Mediated Transcytosis of Cholera Toxin Bypasses the Retrograde Pathway and Depends on the Structure of the Ceramide Domain. J. Biol. Chem. 2013, 288, 25804–25809. [Google Scholar] [CrossRef] [Green Version]
- Batisse, C.; Dransart, E.; Ait Sarkouh, R.; Brulle, L.; Bai, S.K.; Godefroy, S.; Johannes, L.; Schmidt, F. A New Delivery System for Auristatin in STxB-Drug Conjugate Therapy. Eur. J. Med. Chem. 2015, 95, 483–491. [Google Scholar] [CrossRef]
- Stimmer, L.; Dehay, S.; Nemati, F.; Massonnet, G.; Richon, S.; Decaudin, D.; Klijanienko, J.; Johannes, L. Human Breast Cancer and Lymph Node Metastases Express Gb3 and Can Be Targeted by STxB-Vectorized Chemotherapeutic Compounds. BMC Cancer 2014, 14, 1–11. [Google Scholar] [CrossRef]
- Meléndez, A.V.; Velasco Cárdenas, R.M.H.; Lagies, S.; Strietz, J.; Siukstaite, L.; Thomas, O.S.; Tomisch, J.; Weber, W.; Kammerer, B.; Römer, W.; et al. Novel Lectin-Based Chimeric Antigen Receptors Target Gb3-Positive Tumour Cells. Cell. Mol. Life Sci. 2022, 79, 513. [Google Scholar] [CrossRef]
- Rosato, F.; Pasupuleti, R.; Tomisch, J.; Meléndez, A.V.; Kolanovic, D.; Makshakova, O.N.; Wiltschi, B.; Römer, W. A Bispecific, Crosslinking Lectibody Activates Cytotoxic T Cells and Induces Cancer Cell Death. J. Transl. Med. 2022, 20, 1–26. [Google Scholar] [CrossRef]
- Brandel, A.; Aigal, S.; Lagies, S.; Schlimpert, M.; Meléndez, A.V.; Xu, M.; Lehmann, A.; Hummel, D.; Fisch, D.; Madl, J.; et al. The Gb3-Enriched CD59/Flotillin Plasma Membrane Domain Regulates Host Cell Invasion by Pseudomonas Aeruginosa. Cell. Mol. Life Sci. 2021, 78, 3637–3656. [Google Scholar] [CrossRef]
- Tartour, E.; Johannes, L. STxB as an Antigen Delivery Tool for Mucosal Vaccination. Toxins 2022, 14, 202. [Google Scholar] [CrossRef] [PubMed]
- Sych, T.; Omidvar, R.; Ostmann, R.; Schubert, T.; Brandel, A.; Richert, L.; Mely, Y.; Madl, J.; Römer, W. The Bacterial Lectin LecA from P. Aeruginosa Alters Membrane Organization by Dispersing Ordered Domains. bioRxiv 2022. 2022.04.17.488572. [Google Scholar] [CrossRef]
- Falguières, T.; Maak, M.; Von Weyhern, C.; Sarr, M.; Sastre, X.; Poupon, M.F.; Robine, S.; Johannes, L.; Janssen, K.P. Human Colorectal Tumors and Metastases Express Gb3 and Can Be Targeted by an Intestinal Pathogen-Based Delivery Tool. Mol. Cancer Ther. 2008, 7, 2498–2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; de Waard, A.A.; Wuhrer, M.; Spaapen, R.M. The Role of Glycosphingolipids in Immune Cell Functions. Front. Immunol. 2019, 10, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanley, P. Golgi Glycosylation. Cold Spring Harb. Perspect. Biol. 2011, 3, a005199. [Google Scholar] [CrossRef]
- Pier, G.B.; Grout, M.; Zaidi, T.S. Cystic Fibrosis Transmembrane Conductance Regulator Is an Epithelial Cell Receptor for Clearance of Pseudomonas Aeruginosa from the Lung. Proc. Natl. Acad. Sci. USA 1997, 94, 12088–12093. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Huang, X.; Tang, Y.; Lin, H.; Zhou, N. Effects of Panax Notoginseng Saponins on the Osteogenic Differentiation of Rabbit Bone Mesenchymal Stem Cells through TGF-Β1 Signaling Pathway. BMC Complement. Altern. Med. 2016, 16, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Jayagopal, A.; Sussman, E.M.; Shastri, V.P. Functionalized Solid Lipid Nanoparticles for Transendothelial Delivery. IEEE Trans. Nanobiosci. 2008, 7, 28–34. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber Biomolecular Simulation Programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jean-Paul Ryckaert, G.C.A.H.J.C.B. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Cioci, G.; Mitchell, E.P.; Gautier, C.; Wimmerová, M.; Sudakevitz, D.; Pérez, S.; Gilboa-Garber, N.; Imberty, A. Structural Basis of Calcium and Galactose Recognition by the Lectin PA-IL of Pseudomonas Aeruginosa. FEBS Lett. 2003, 555, 297–301. [Google Scholar] [CrossRef] [Green Version]
- Duan, L.; Guo, X.; Cong, Y.; Feng, G.; Li, Y.; Zhang, J.Z.H. Accelerated Molecular Dynamics Simulation for Helical Proteins Folding in Explicit Water. Front. Chem. 2019, 7. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-W.; Ying, T.-L.; Liao, L.-F. Molecular Modeling and Dynamics Simulation of a Histidine-Tagged Cytochrome B5. J. Mol. Model 2011, 17, 971–978. [Google Scholar] [CrossRef]
- Booth, W.T.; Schlachter, C.R.; Pote, S.; Ussin, N.; Mank, N.J.; Klapper, V.; Offermann, L.R.; Tang, C.; Hurlburt, B.K.; Chruszcz, M. Impact of an N-terminal Polyhistidine Tag on Protein Thermal Stability. ACS Omega 2018, 3, 760–768. [Google Scholar] [CrossRef]
- Carson, M.; Johnson, D.H.; McDonald, H.; Brouillette, C.; DeLucas, L.J. His-Tag Impact on Structure. Acta Crystallogr. D Biol. Crystallogr. 2007, 63, 295–301. [Google Scholar] [CrossRef]
- Thielges, M.C.; Chung, J.K.; Axup, J.Y.; Fayer, M.D. Influence of Histidine Tag Attachment on Picosecond Protein Dynamics. Biochemistry 2011, 50, 5799–5805. [Google Scholar] [CrossRef] [Green Version]
- Amor-Mahjoub, M.; Suppini, J.-P.; Gomez-Vrielyunck, N.; Ladjimi, M. The Effect of the Hexahistidine-Tag in the Oligomerization of HSC70 Constructs. J. Chromatogr. B 2006, 844, 328–334. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, M.; Antonova, M.; Salavei, P.; Illek, K.; Meléndez, A.V.; Omidvar, R.; Thuenauer, R.; Makshakova, O.; Römer, W. Dimeric Lectin Chimeras as Novel Candidates for Gb3-Mediated Transcytotic Drug Delivery through Cellular Barriers. Pharmaceutics 2023, 15, 225. https://doi.org/10.3390/pharmaceutics15010225
Xu M, Antonova M, Salavei P, Illek K, Meléndez AV, Omidvar R, Thuenauer R, Makshakova O, Römer W. Dimeric Lectin Chimeras as Novel Candidates for Gb3-Mediated Transcytotic Drug Delivery through Cellular Barriers. Pharmaceutics. 2023; 15(1):225. https://doi.org/10.3390/pharmaceutics15010225
Chicago/Turabian StyleXu, Maokai, Maria Antonova, Pavel Salavei, Katharina Illek, Ana Valeria Meléndez, Ramin Omidvar, Roland Thuenauer, Olga Makshakova, and Winfried Römer. 2023. "Dimeric Lectin Chimeras as Novel Candidates for Gb3-Mediated Transcytotic Drug Delivery through Cellular Barriers" Pharmaceutics 15, no. 1: 225. https://doi.org/10.3390/pharmaceutics15010225