
Protein Modification Employing Non-Canonical Amino Acids to Prepare SUMOylation Detecting Bioconjugates

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. General
2.2. Mutagenesis
2.3. Protein Expression and Purification
2.4. Glaser–Hay Bioconjugation
2.5. Pull down Assay on Fluorescent Labeled UTAG
2.6. Plate Pull down Assay
2.7. Mammalian Cell Detection of SUMO with Bioconjugate
3. Results and Discussion
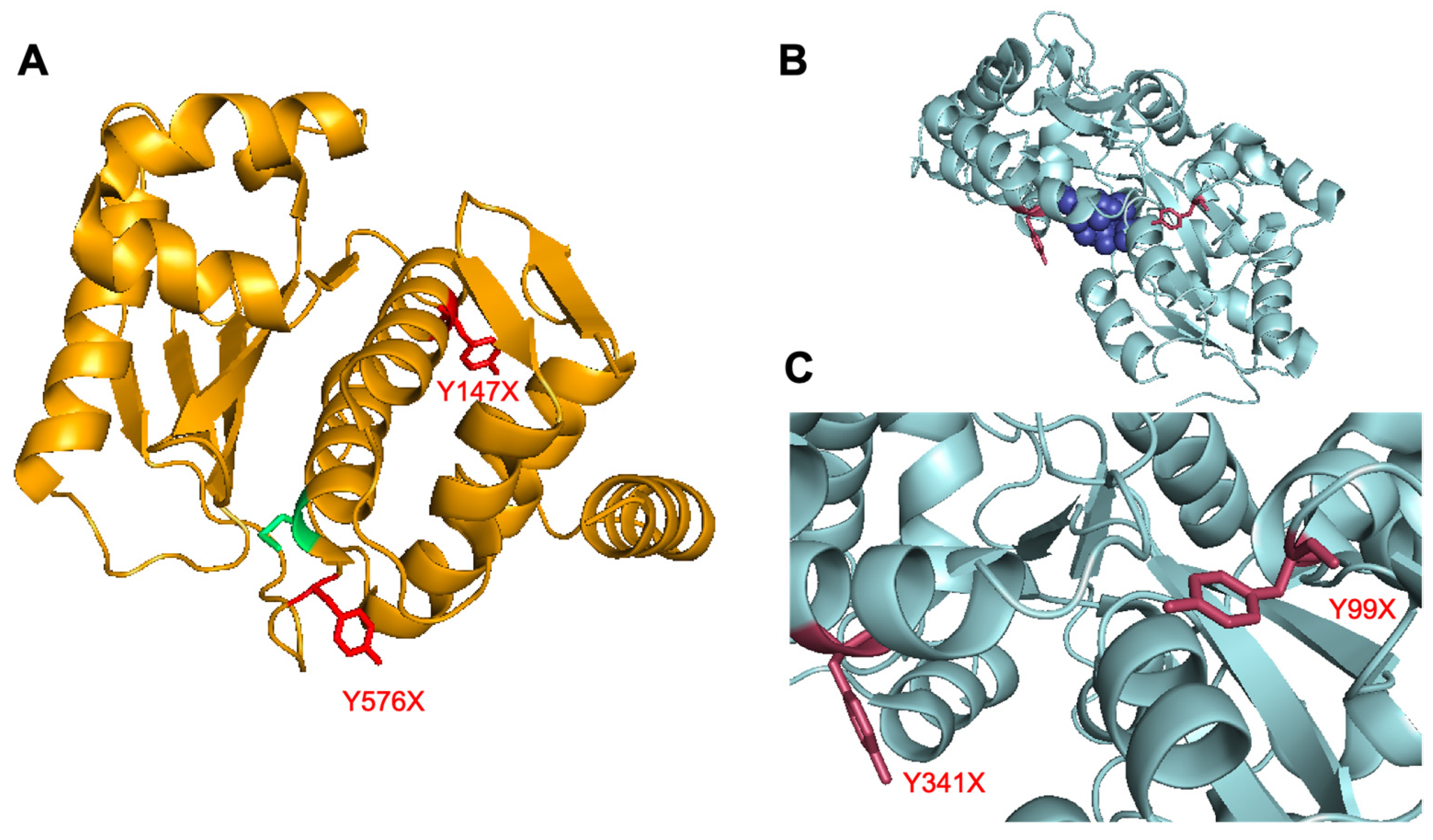
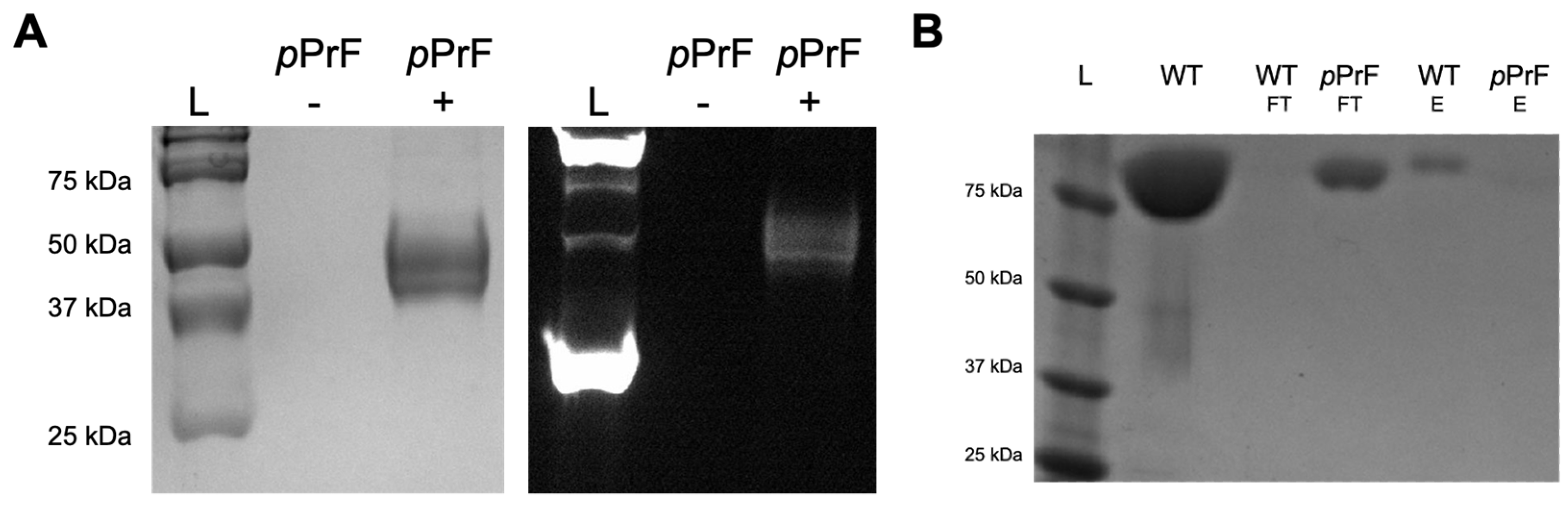
3.1. UTAG Mutagenesis and Assessment
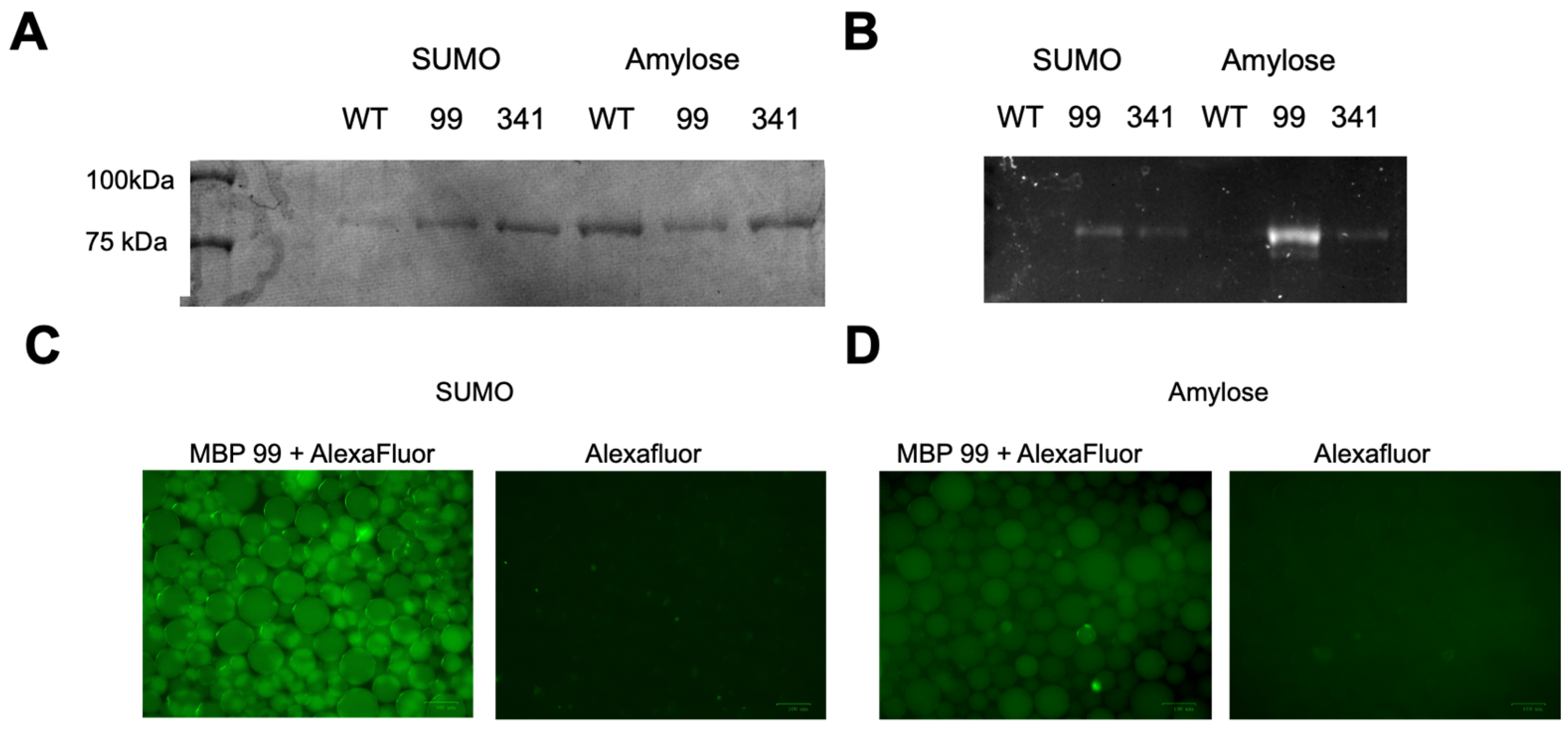
3.2. Maltose-Binding Protein Mutagenesis and Evaluation
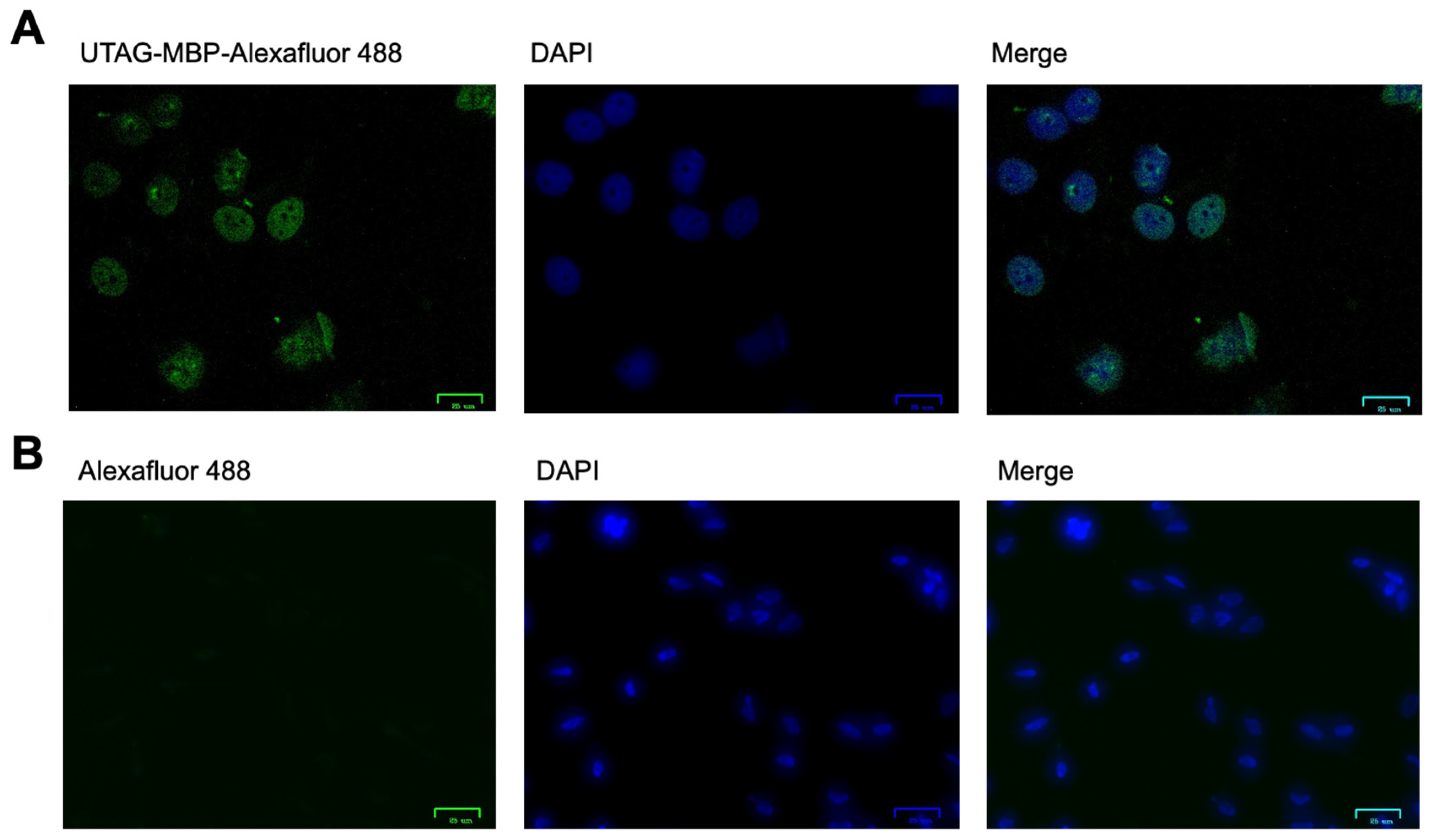
3.3. Assessment of SUMO Binding in Mammalian Cells
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cancer Stat Facts: Prostate Cancer. Available online: https://seer.cancer.gov/statfacts/html/prost.html (accessed on 22 October 2022).
- Schipper, M.; Wang, G.; Giles, N.; Ohrnberger, J. Novel prostate cancer biomarkers derived from autoantibody signatures. Transl. Oncol. 2015, 8, 106–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bander, N.; Nanus, D.; Milowsky, M.; Kostakoglu, L.; Vallabahajosula, S.; Goldsmith, S. Targeted systemic therapy of prostate cancer with a monoclonal antibody to prostate-specific membrane antigen. Semin. Oncol. 2003, 30, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Bander, N.; Nanus, D.; Tagawa, S.; Vallabhajosula, S.; Goldsmith, S.; Milowsky, M.; Morris, M. Phase 2a trial of 177lutetium radiolabeled antiprostate-specific membrane antigen (PSMA) monoclonal antibody J591 (177Lu-J591) in patients (PTS) with metastatic androgen-independent prostate cancer (AIPC). J. Urol. 2008, 179, 253. [Google Scholar] [CrossRef]
- Watts, F. Starting and stopping SUMOylation What regulates the regulator? Chromosoma 2013, 122, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dasso, M. SUMOylation and deSUMOylation at a glance. J. Cell Sci. 2009, 122, 4249–4252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, S.H. A novel link between SUMO modification and cancer metastasis. Cell Cycle 2006, 5, 1492–1495. [Google Scholar] [CrossRef]
- Kim, J.H.; Choi, H.J.; Kim, B.; Kim, M.H.; Lee, J.M.; Kim, I.S.; Lee, M.H.; Choi, S.J.; Kim, K.I.; Kim, S.I.; et al. Roles of sumoylation of a reptin chromatin-remodelling complex in cancer metastasis. Nat. Cell Biol. 2006, 8, 631–639. [Google Scholar] [CrossRef]
- Peek, J.; Harvey, C.; Gray, D.; Rosenberg, D.; Kolla, L.; Levy-Myers, R.; Yin, R.; McMurry, J.L.; Kerscher, O. SUMO targeting of a stress-tolerant Ulp1 SUMO protease. PLoS ONE 2018, 13, e0191391. [Google Scholar] [CrossRef] [Green Version]
- Elmore, Z.; Donaher, M.; Matson, B.; Murphy, H.; Westerbeck, J.; Kerscher, O. Sumo-dependent substrate targeting of the SUMO protease Ulp1. BMC Biol. 2011, 9, 74. [Google Scholar] [CrossRef] [Green Version]
- Young, D.D.; Schultz, P.G. Playing with the Molecules of Life. ACS Chem. Biol. 2018, 13, 854–870. [Google Scholar] [CrossRef]
- Young, T.S.; Schultz, P.G. Beyond the Canonical 20 Amino Acids: Expanding the Genetic Lexicon. J. Biol. Chem. 2010, 285, 11039–11044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.C.; Mack, A.V.; Tsao, M.L.; Mills, J.H.; Lee, H.S.; Choe, H.; Farzan, M.; Schultz, P.G.; Smider, V.V. Protein evolution with an expanded genetic code. Proc. Natl. Acad. Sci. USA 2008, 105, 17688–17693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deiters, A.; Cropp, T.A.; Summerer, D.; Mukherji, M.; Schultz, P.G. Site-specific PEGylation of proteins containing unnatural amino acids. Bioorg. Med. Chem. Lett. 2004, 14, 5743–5745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.H.; Axup, J.Y.; Lawson, B.R.; Yun, H.; Tardif, V.; Choi, S.H.; Zhou, Q.; Dubrovska, A.; Biroc, S.L.; Marsden, R.; et al. Bispecific small molecule-antibody conjugate targeting prostate cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 17796–17801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axup, J.Y.; Bajjuri, K.M.; Ritland, M.; Hutchins, B.M.; Kim, C.H.; Kazane, S.A.; Halder, R.; Forsyth, J.S.; Santidrian, A.F.; Stafin, K.; et al. Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proc. Natl. Acad. Sci. USA 2012, 109, 16101–16106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.H.; Axup, J.Y.; Dubrovska, A.; Kazane, S.A.; Hutchins, B.A.; Wold, E.D.; Smider, V.V.; Schultz, P.G. Synthesis of bispecific antibodies using genetically encoded unnatural amino acids. J. Am. Chem. Soc. 2012, 134, 9918–9921. [Google Scholar] [CrossRef] [Green Version]
- Glaser, C. Beiträge zur Kenntniss des Acetenylbenzols. Ber. Dtsch. Chem. Ges. 1896, 2, 422–424. [Google Scholar] [CrossRef] [Green Version]
- Lampkowski, J.S.; Villa, J.K.; Young, T.S.; Young, D.D. Development and Optimization of Glaser-Hay Bioconjugations. Angew. Chem. Int. Ed. Engl. 2015, 54, 9343–9346. [Google Scholar] [CrossRef]
- Maza, J.; Howard, C.; Vipani, M.; Travis, C.; Young, D. Utilization of alkyne bioconjugations to modulate protein function. Bioorg. Med. Chem. Lett. 2017, 27, 30–33. [Google Scholar] [CrossRef] [Green Version]
- Nimmo, Z.M.; Halonski, J.F.; Chatkewitz, L.E.; Young, D.D. Development of optimized conditions for Glaser-Hay bioconjugations. Bioorg. Chem. 2018, 76, 326–331. [Google Scholar] [CrossRef]
- Travis, C.R.; Mazur, L.E.; Peairs, E.M.; Gaunt, G.H.; Young, D.D. Mechanistic investigation and further optimization of the aqueous Glaser-Hay bioconjugation. Org. Biomol. Chem. 2019, 17, 3396–3402. [Google Scholar] [CrossRef] [PubMed]
- Yin, R.; Harvey, C.; Shakes, D.C.; Kerscher, O. Localization of SUMO-modified Proteins Using Fluorescent Sumo-trapping Proteins. J. Vis. Exp. 2019, 146, e59576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, D.; Young, T.; Jahnz, M.; Ahmad, I.; Spraggon, G.; Schultz, P. An Evolved Aminoacyl-tRNA Synthetase with Atypical Polysubstrate Specificity. Biochemistry 2011, 50, 1894–1900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, J.W.; Santoro, S.W.; Martin, A.B.; King, D.S.; Wang, L.; Schultz, P.G. Addition of p-azido-L-phenylalanine to the genetic code of Escherichia coli. J. Am. Chem. Soc. 2002, 124, 9026–9027. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, Z.; Brock, A.; Schultz, P.G. Addition of the keto functional group to the genetic code of Escherichia coli. Proc. Natl. Acad. Sci. USA 2003, 100, 56–61. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Lam, L.S.; Lam, L.H.; Chau, S.F.; Ng, T.B.; Au, S.W. Molecular basis of the redox regulation of SUMO proteases: A protective mechanism of intermolecular disulfide linkage against irreversible sulfhydryl oxidation. FASEB J. 2008, 22, 127–137. [Google Scholar] [CrossRef]
- Oldham, M.L.; Chen, J. Snapshots of the maltose transporter during ATP hydrolysis. Proc. Natl. Acad. Sci. USA 2011, 108, 15152–15156. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Williard, A.C.; Switzer, H.J.; Howard, C.A.; Yin, R.; Russell, B.L.; Sanyal, R.; Yu, S.; Myers, T.M.; Flood, B.M.; Kerscher, O.; et al. Protein Modification Employing Non-Canonical Amino Acids to Prepare SUMOylation Detecting Bioconjugates. Pharmaceutics 2022, 14, 2826. https://doi.org/10.3390/pharmaceutics14122826
Williard AC, Switzer HJ, Howard CA, Yin R, Russell BL, Sanyal R, Yu S, Myers TM, Flood BM, Kerscher O, et al. Protein Modification Employing Non-Canonical Amino Acids to Prepare SUMOylation Detecting Bioconjugates. Pharmaceutics. 2022; 14(12):2826. https://doi.org/10.3390/pharmaceutics14122826
Chicago/Turabian StyleWilliard, Alexander C., Hannah J. Switzer, Christina A. Howard, Rui Yin, Brent L. Russell, Ritwik Sanyal, Shaun Yu, Trinity M. Myers, Brian M. Flood, Oliver Kerscher, and et al. 2022. "Protein Modification Employing Non-Canonical Amino Acids to Prepare SUMOylation Detecting Bioconjugates" Pharmaceutics 14, no. 12: 2826. https://doi.org/10.3390/pharmaceutics14122826