

Development of a Physiologically Based Pharmacokinetic Model for Tegoprazan: Application for the Prediction of Drug–Drug Interactions with CYP3A4 Perpetrators

Abstract
:1. Introduction
2. Materials and Methods
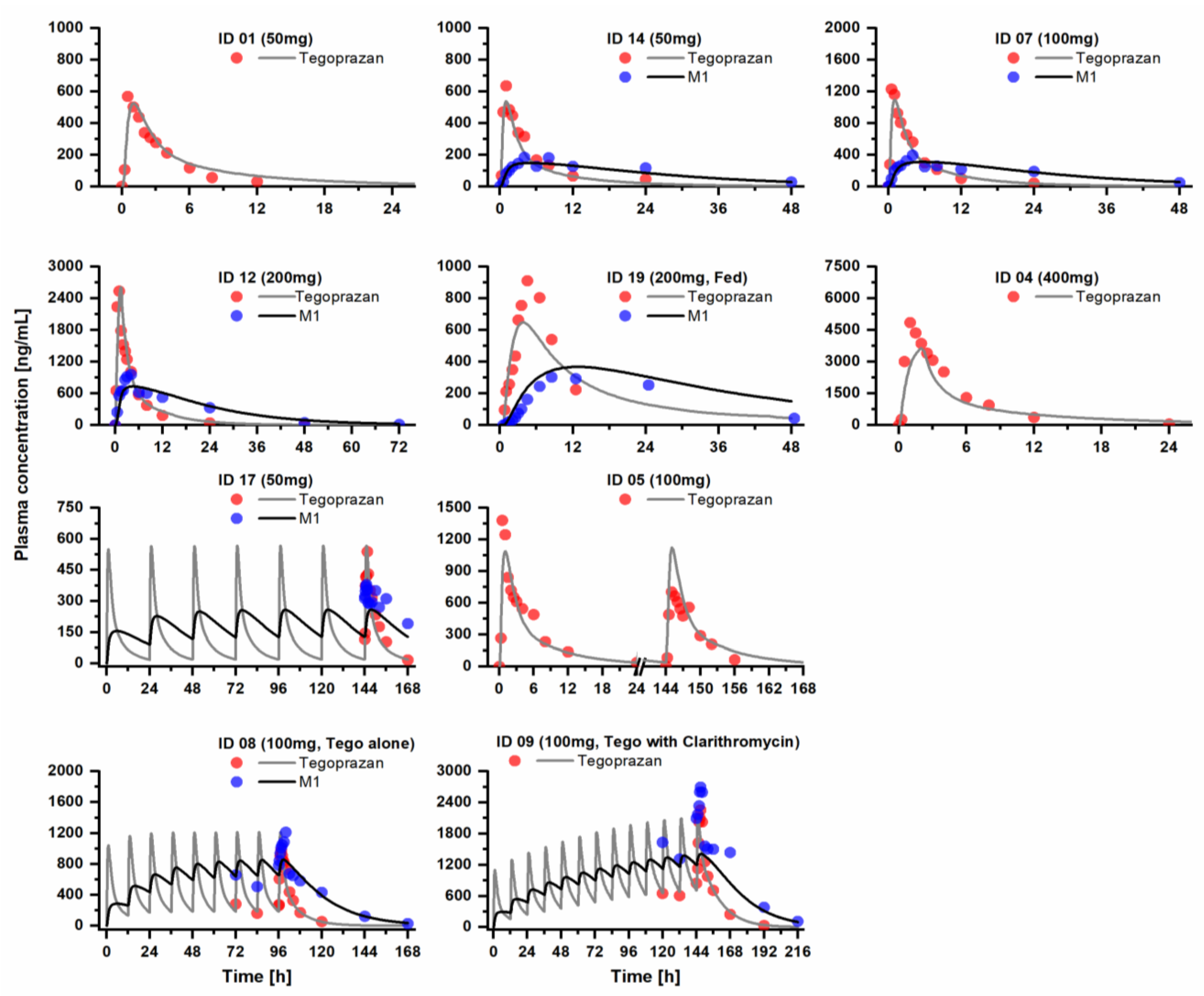
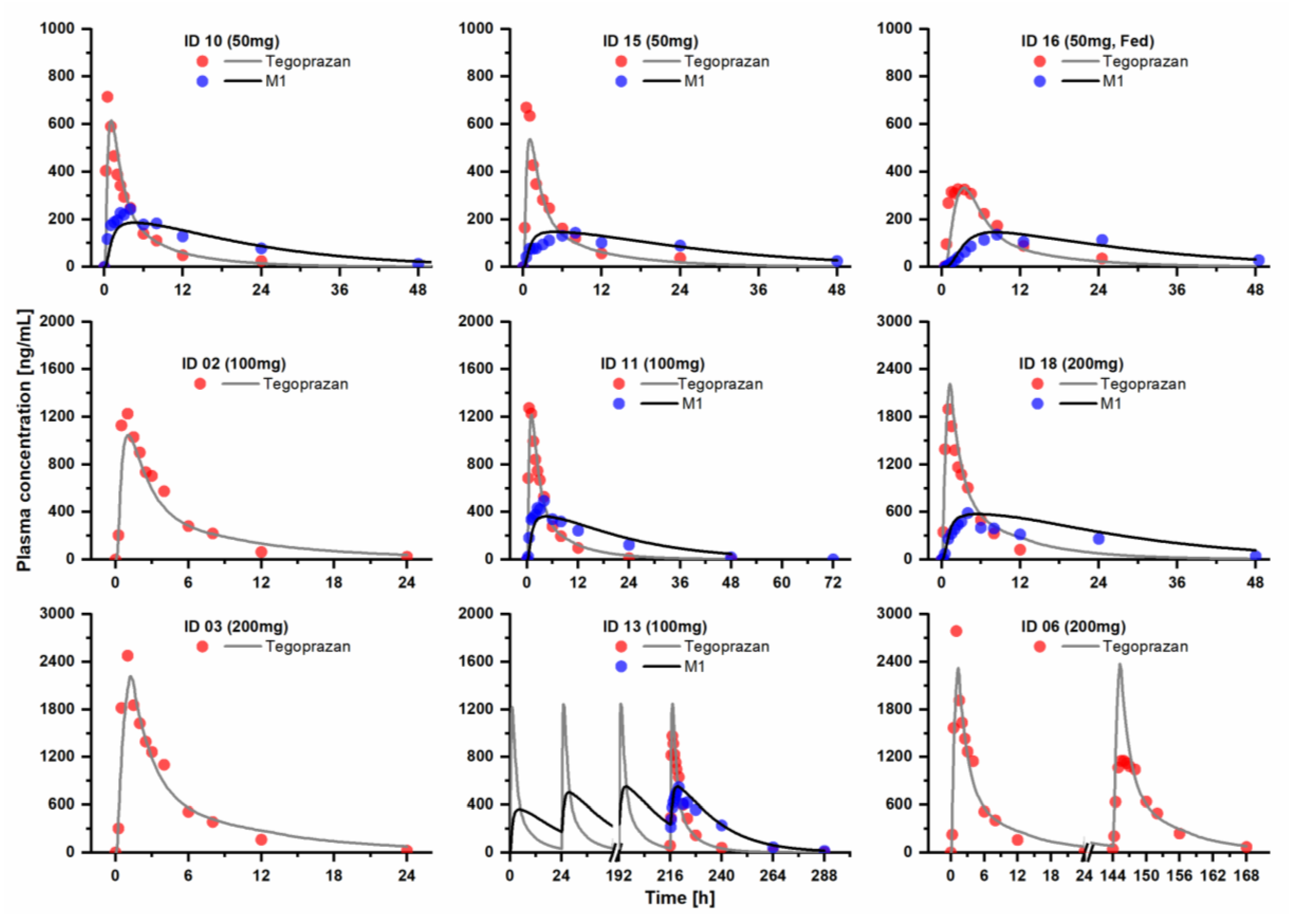
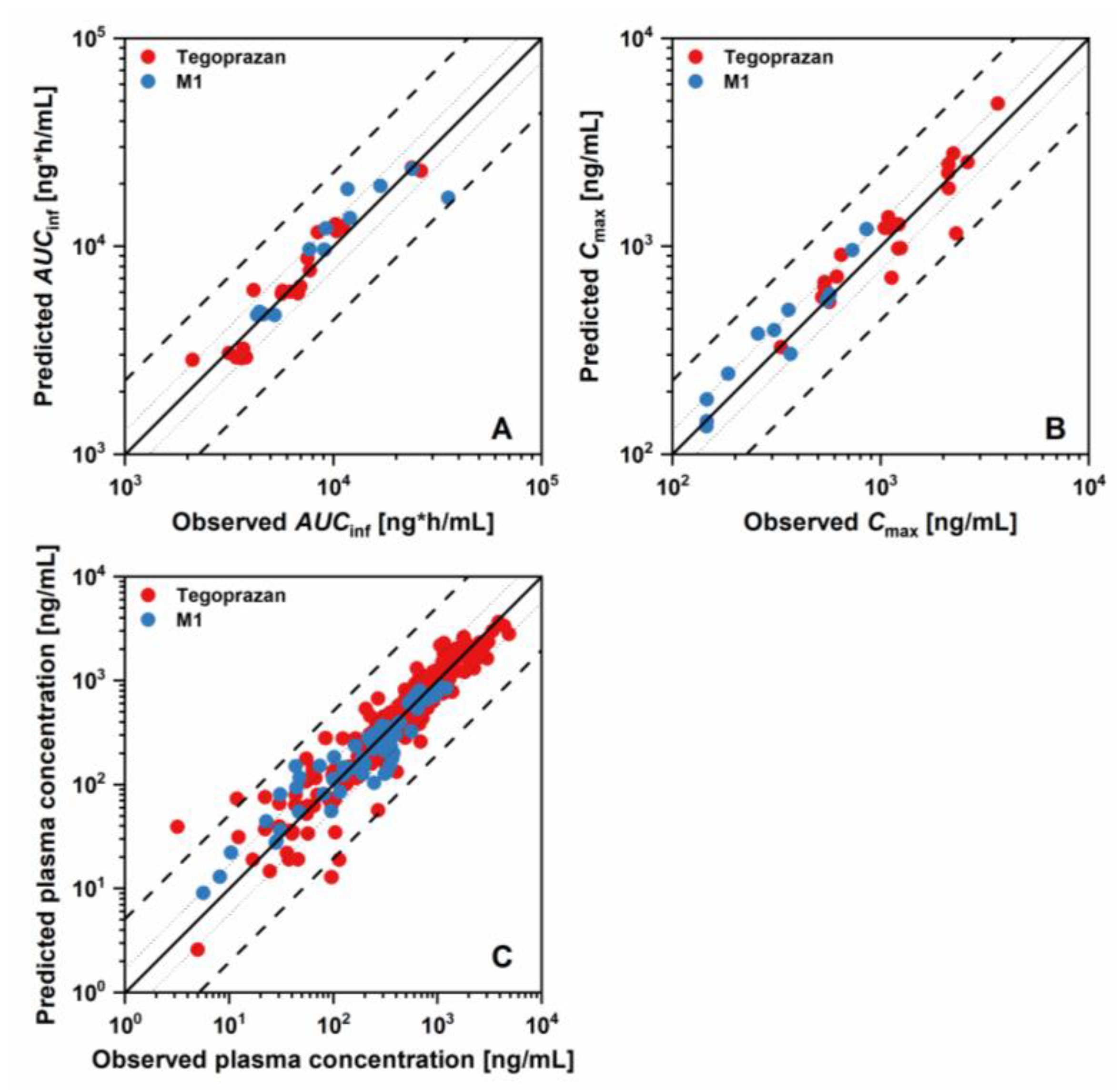
2.1. PBPK Model Development and Evaluation
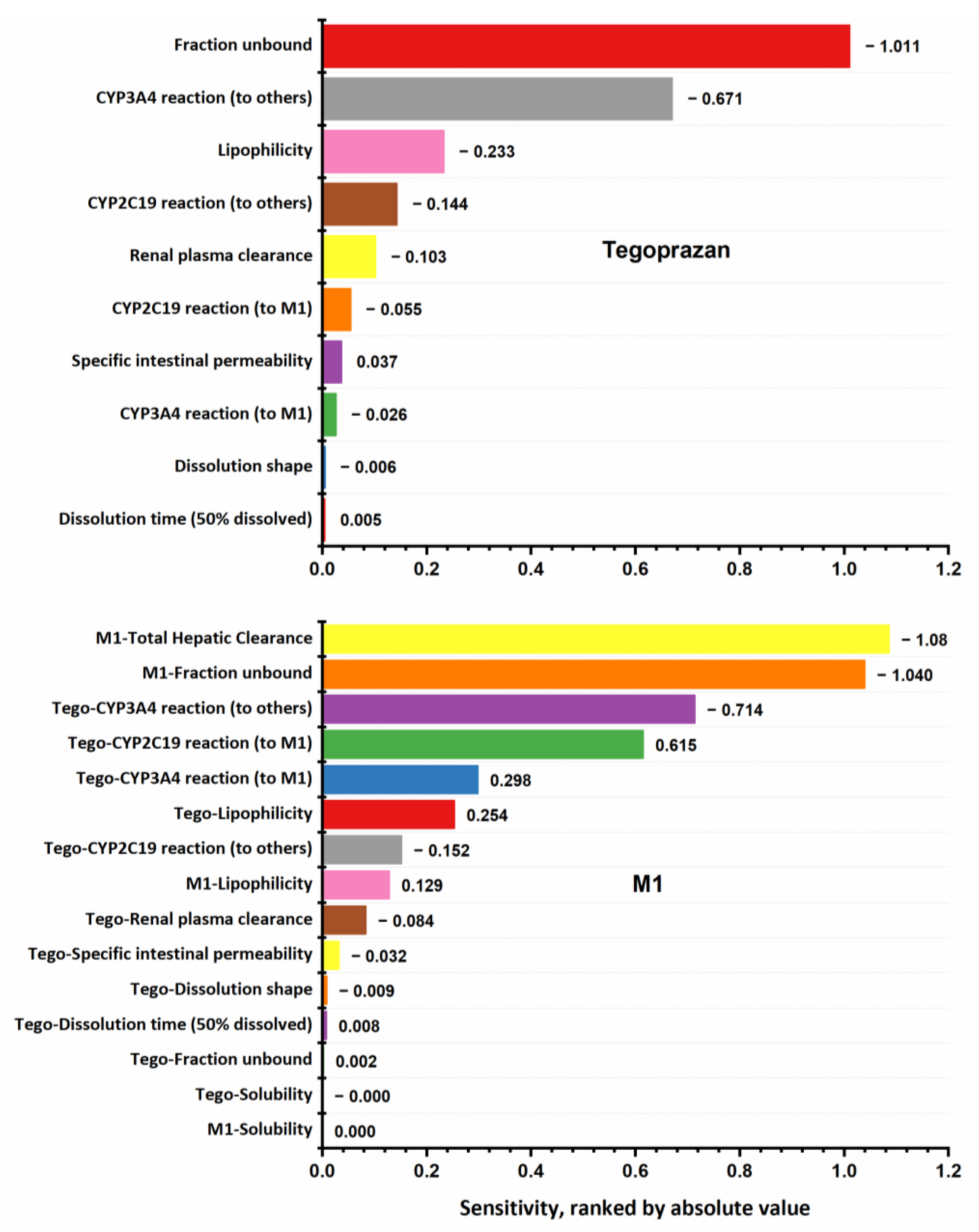
2.2. Sensitivity Analysis
2.3. Prediction DDI Profile of Tegoprazan
3. Results
3.1. PBPK Model Development and Evaluation
3.2. Sensitivity Analysis
3.3. Prediction DDI Profiles of Tegoprazan
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- He, J.; Cao, G.; Yu, J.; Wang, J.; Cheng, N.; Wu, J.; Zhang, J.; Wu, X.; Zhang, B.; Lu, J.; et al. Safety, Tolerability and Pharmacokinetics of Single Ascending and Multiple Oral Doses of Tegoprazan in Healthy Chinese Subjects. Clin. Drug. Investig. 2021, 41, 89–97. [Google Scholar] [CrossRef] [PubMed]
- K-CAB Tab. 50 mg. Available online: https://www.health.kr/searchDrug/result_drug.asp?drug_cd=2018070600004 (accessed on 15 February 2022).
- Tegoprazan (K-CAB). Available online: http://www.endotoday.com/endotoday02/temp.pdf (accessed on 18 March 2022).
- Jeong, H.-C.; Kim, M.-G.; Wei, Z.; Lee, K.-R.; Lee, J.; Song, I.-S.; Shin, K.-H. Integration of a Physiologically Based Pharmacokinetic and Pharmacodynamic Model for Tegoprazan and Its Metabolite: Application for Predicting Food Effect and Intragastric pH Alterations. Pharmaceutics 2022, 14, 1298. [Google Scholar] [CrossRef] [PubMed]
- Yoon, D.Y.; Lee, S.; Jang, I.-J.; Kim, M.; Lee, H.; Kim, S.; Kim, B.; Song, G.S.; Rhee, S. Prediction of Drug–Drug Interaction Potential of Tegoprazan Using Physiologically Based Pharmacokinetic Modeling and Simulation. Pharmaceutics 2021, 13, 1489. [Google Scholar] [CrossRef]
- Ghim, J.; Chin, M.C.; Jung, J.; Lee, J.; Kim, S.; Kim, B.; Song, G.S.; Choi, Y.; Shin, J. Pharmacokinetics and Pharmacodynamics of Tegoprazan Coadministered with Amoxicillin and Clarithromycin in Healthy Subjects. J. Clin. Pharmacol. 2021, 61, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.-Y.; Kim, S.-Y.; Moon, S.J.; Oh, K.; Lee, J.; Kim, B.; Song, G.S.; Kim, M.-G. Pharmacokinetic Interactions between Tegoprazan and Metronidazole/Tetracycline/Bismuth and Safety Assessment in Healthy Korean Male Subjects. Clin. Ther. 2021, 43, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Emoto, C.; McPhail, B.T.; Fukuda, T. Clinical Applications of Physiologically Based Pharmacokinetic Modeling: Perspectives on the Advantages and Challenges. Ther. Drug. Monit. 2020, 42, 157–158. [Google Scholar] [CrossRef] [PubMed]
- Einolf, H.J. Comparison of different approaches to predict metabolic drug-drug interactions. Xenobiotica 2007, 37, 1257–1294. [Google Scholar]
- Yeo, K.R.; Jamei, M.; Rostami-Hodjegan, A. Predicting drug–drug interactions: Application of physiologically based pharmacokinetic models under a systems biology approach. Expert. Rev. Clin. Pharmacol. 2013, 6, 143–157. [Google Scholar] [CrossRef]
- WebPlotDigitizer. Available online: https://automeris.io/WebPlotDigitizer/ (accessed on 15 February 2022).
- Chen, Y.; Liu, L.; Nguyen, K.; Fretland, A.J. Utility of Intersystem Extrapolation Factors in Early Reaction Phenotyping and the Quantitative Extrapolation of Human Liver Microsomal Intrinsic Clearance Using Recombinant Cytochromes P450. Drug. Metab. Dispos. 2011, 39, 373–382. [Google Scholar] [CrossRef] [Green Version]
- Proctor, N.J.; Tucker, G.T.; Rostami-Hodjegan, A. Predicting drug clearance from recombinantly expressed CYPs: Intersystem extrapolation factors. Xenobiotica 2004, 34, 151–178. [Google Scholar] [CrossRef] [PubMed]
- Open System Pharmacology PKsim—Formulations. Available online: https://docs.open-systems-pharmacology.org/working-with-pk-sim/pk-sim-documentation/pk-sim-formulations (accessed on 16 March 2022).
- Langenbucher, F. Letters to the Editor: Linearization of dissolution rate curves by the Weibull distribution. J. Pharm. Pharmacol. 2011, 24, 979–981. [Google Scholar] [CrossRef] [PubMed]
- National Library of Medicine—National Center for Biotechnology Information Tegoprazan. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Tegoprazan (accessed on 10 February 2022).
- DrugBank Tegoprazan. Available online: https://go.drugbank.com/drugs/DB16690 (accessed on 28 February 2022).
- Han, S.; Choi, H.Y.; Kim, Y.H.; Nam, J.Y.; Kim, B.; Song, G.S.; Lim, H.-S.; Bae, K.-S. Randomised clinical trial: Safety, tolerability, pharmacokinetics, and pharmacodynamics of single and multiple oral doses of tegoprazan (CJ-12420), a novel potassium-competitive acid blocker, in healthy male subjects. Aliment. Pharmacol. Ther. 2019, 50, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Moj, D.; Hanke, N.; Britz, H.; Frechen, S.; Kanacher, T.; Wendl, T.; Haefeli, W.E.; Lehr, T. Clarithromycin, Midazolam, and Digoxin: Application of PBPK Modeling to Gain New Insights into Drug–Drug Interactions and Co-medication Regimens. AAPS J. 2017, 19, 298–312. [Google Scholar] [CrossRef] [PubMed]
- Hanke, N.; Frechen, S.; Moj, D.; Britz, H.; Eissing, T.; Wendl, T.; Lehr, T. PBPK Models for CYP3A4 and P-gp DDI Prediction: A Modeling Network of Rifampicin, Itraconazole, Clarithromycin, Midazolam, Alfentanil, and Digoxin. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 647–659. [Google Scholar] [CrossRef] [Green Version]
- OSP PBPK Model Library. Clarithromycin. Available online: https://github.com/Open-Systems-Pharmacology/OSP-PBPK-Model-Library/tree/master/Clarithromycin (accessed on 14 April 2022).
- OSP PBPK Model Library. Rifampicin. Available online: https://github.com/Open-Systems-Pharmacology/OSP-PBPK-Model-Library/tree/master/Rifampicin (accessed on 14 April 2022).
- Türk, D.; Hanke, N.; Wolf, S.; Frechen, S.; Eissing, T.; Wendl, T.; Schwab, M.; Lehr, T. Physiologically Based Pharmacokinetic Models for Prediction of Complex CYP2C8 and OATP1B1 (SLCO1B1) Drug–Drug–Gene Interactions: A Modeling Network of Gemfibrozil, Repaglinide, Pioglitazone, Rifampicin, Clarithromycin and Itraconazole. Clin. Pharmacokinet. 2019, 58, 1595–1607. [Google Scholar] [CrossRef] [Green Version]
- Britz, H.; Hanke, N.; Volz, A.; Spigset, O.; Schwab, M.; Eissing, T.; Wendl, T.; Frechen, S.; Lehr, T. Physiologically-Based Pharmacokinetic Models for <scp>CYP</scp> 1A2 Drug–Drug Interaction Prediction: A Modeling Network of Fluvoxamine, Theophylline, Caffeine, Rifampicin, and Midazolam. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 296–307. [Google Scholar]
- Silverman, R.B. Mechanism-based enzyme inactivators. Methods Enzymol. 1995, 249, 240–283. [Google Scholar] [PubMed]
- Open System Pharmacology Definition of new Events in PK-Sim®. Available online: https://docs.open-systems-pharmacology.org/working-with-pk-sim/pk-sim-documentation/pk-sim-events#definition-of-new-events-in-pk-sim-r (accessed on 2 February 2022).
- Jin, K.H.; Jun, K.S.; Kyoung, L.M.; Ah, L.S.; Young, Y.M. Composition for Injection. U.S. Patent Application No. 16/629,082, 14 May 2020. [Google Scholar]
- Hwang, J.G.; Yoo, H.; Lee, J.W.; Song, G.S.; Lee, S.; Kim, M.-G. Comparison of pharmacokinetic characteristics of two Tegoprazan (CJ-12420) formulations in healthy male subjects. Transl. Clin. Pharmacol. 2019, 27, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, D.Y.; Sunwoo, J.; Shin, N.; Kim, A.R.; Kim, B.; Song, G.S.; Jang, I.; Lee, S. Effect of meal timing on pharmacokinetics and pharmacodynamics of tegoprazan in healthy male volunteers. Clin. Transl. Sci. 2021, 14, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Choi, H.Y.; Kim, Y.H.; Nam, J.Y.; Kim, B.; Song, G.S.; Lim, H.-S.; Bae, K.-S. Effect of Food on the Pharmacokinetics and Pharmacodynamics of a Single Oral Dose of Tegoprazan. Clin. Ther. 2021, 43, 1371–1380. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Final Value | Unit | Reference Value | Source of Reference |
|---|---|---|---|---|
| Tegoprazan | ||||
| physicochemical properties | ||||
| Molecular weight | 387.38 | g/mol | 387.38 | [16,17] |
| Lipophilicity (LogP) | 1.75 | - | 2.32 predicted by ChemAxon 2.91 predicted by ALOGPS | [17] |
| Fraction unbound in plasma | 8.7 | % | 8.7; 12.4 | [2] |
| Solubility | 45.3 | mg/L | 45.3 predicted by ALOGPS | [17] |
| Compound type/pKa Strongest basic Strongest acidic | 5.20 12.0 | 5.20/6.07 12.0/11.37 | [5,17] | |
| ADME parameters | ||||
| Absorption | ||||
| Specific intestinal permeability | cm/s | (Peff) (PAMPA Caco-2cell) | [5] | |
| Distribution | ||||
| Partition coefficients | PK-Sim® Standards | |||
| Cellular permeabilities | PK-Sim® Standards | |||
| Specific organ permeability | cm/s | PK-Sim® | ||
| Metabolism Intrinsic metabolic rate in the presence of CYPs | ||||
| CYP3A4 to M1 | μL/min/pmol recombinant enzyme | 0.134 | [4] | |
| CYP3A4 to other metabolites | 0.236 | |||
| CYP2C19 to M1 | 0.0921 | 0.154 | ||
| CYP2C19 to other metabolites | 0.242 | |||
| Transport and excretion | ||||
| Renal plasma clearance | 0.297 | mL/min/kg | 0.297 | [18] |
| Dissolution profile: Dissolution time and shape | Fasted: 0.942 h and 0.990 Fed: 4.23 h and 0.502 | |||
| M1 | ||||
| physicochemical properties | ||||
| Molecular weight | 373.40 | [4,7] | ||
| Lipophilicity (LogP) | 1.75 | 2.10 predicted by ChemAxon 2.67 predicted by ALOGPS | [4] | |
| Fraction unbound in plasma | 1 | % | 1 | [2] |
| Solubility | 1.00 | ng/mL | assumed | |
| Compound type/pKa (Strongest base) | 5.35 | [4] | ||
| ADME parameters | ||||
| Metabolism Total hepatic clearance | 0.140 | mL/min/kg |
| Parameters | Unit | Tegoprazan Alone | Tegoprazan with Clarithromycin 500 mg BID | Tegoprazan with Clarithromycin 500 mg TID | Tegoprazan with Rifampicin 600 mg QD | |||
|---|---|---|---|---|---|---|---|---|
| Value (Min–Max) | Value (Min–Max) | Fold Increase | Value (Min–Max) | Fold Increase | Value (Min–Max) | Fold Decrease | ||
| AUCfirst | ng × h/mL | 2895.5 | 7059.3 | 2.44 | 9092.3 | 3.14 | 578.2 | 5.01 |
| 1949.1–4155.1 | 3523.5–11,510.1 | 4708.8–14,461.4 | 398.5–845 | |||||
| AUCSS | ng × h/mL | 2994.4 | 8198.7 | 2.74 | 11312.0 | 3.78 | 568.8 | 5.26 |
| 2008.1–4536 | 3782.8–16,814.2 | 5213–21,020.6 | 392.8–804.6 | |||||
| Cmax_first | ng/mL | 576.2 | 826.0 | 1.43 | 893.0 | 1.55 | 183.7 | 3.14 |
| 426.1–740.3 | 570–1101.3 | 627.7–1161.1 | 127.7–254.8 | |||||
| Cmax_SS | ng/mL | 590.7 | 948.2 | 1.61 | 1066.3 | 1.81 | 180.1 | 3.28 |
| 428.8–763.3 | 616.4–1253.4 | 711.3–1434.6 | 125.2–248.4 | |||||
| Parameters | Unit | Tegoprazan Alone | Tegoprazan with Clarithromycin 500 mg BID | Tegoprazan with Clarithromycin 500 mg TID | Tegoprazan with Rifampicin 600 mg QD | |||
|---|---|---|---|---|---|---|---|---|
| Value (Min–Max) | Value (Min–Max) | Fold Increase | Value (Min–Max) | Fold Increase | Value (Min–Max) | Fold Increase | ||
| AUCfirst | ng × h/mL | 2866.9 | 6680.3 | 2.33 | 8900.1 | 3.10 | 575.6 | 4.98 |
| 1917.8–4157.4 | 3391.8–11,486.7 | 4638.9–15,811 | 397.6–845 | |||||
| AUCSS | ng × h/mL | 3452.5 | 10984.0 | 3.18 | 15673.1 | 4.54 | 605.1 | 5.71 |
| 2235.7–5593.3 | 4653.1–27,087.4 | 6398.6–34,092.7 | 425.6–804.6 | |||||
| Cmax_first | ng/mL | 576.2 | 826.0 | 1.43 | 893.0 | 1.55 | 183.7 | 3.14 |
| 426.1–740.3 | 570–1101.3 | 627.7–1161.1 | 127.7–254.8 | |||||
| Cmax_SS | ng/mL | 649.9 | 1163.9 | 1.79 | 1330.9 | 2.05 | 185.4 | 3.51 |
| 466.8–848.1 | 700.9–1625.3 | 851.9–1892.6 | 129–248.4 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ngo, L.T.; Lee, J.; Yun, H.-y.; Chae, J.-w. Development of a Physiologically Based Pharmacokinetic Model for Tegoprazan: Application for the Prediction of Drug–Drug Interactions with CYP3A4 Perpetrators. Pharmaceutics 2023, 15, 182. https://doi.org/10.3390/pharmaceutics15010182
Ngo LT, Lee J, Yun H-y, Chae J-w. Development of a Physiologically Based Pharmacokinetic Model for Tegoprazan: Application for the Prediction of Drug–Drug Interactions with CYP3A4 Perpetrators. Pharmaceutics. 2023; 15(1):182. https://doi.org/10.3390/pharmaceutics15010182
Chicago/Turabian StyleNgo, Lien Thi, Jaeyeon Lee, Hwi-yeol Yun, and Jung-woo Chae. 2023. "Development of a Physiologically Based Pharmacokinetic Model for Tegoprazan: Application for the Prediction of Drug–Drug Interactions with CYP3A4 Perpetrators" Pharmaceutics 15, no. 1: 182. https://doi.org/10.3390/pharmaceutics15010182