
Polymeric Nanomicelles Loaded with Anandamide and Their Renal Effects as a Therapeutic Alternative for Hypertension Treatment by Passive Targeting


Abstract
:1. Introduction
2. Materials and Methods
2.1. Micellar Preparation and Characterization
2.2. Radiolabeling of TPGS and PF127 Micellar Systems
2.3. Radiochemical Purity Control
2.4. Stability of Labeling
2.5. Biodistribution of Micellar Systems
2.5.1. Administration
2.5.2. Gamma Camera Scintigraphic Images
2.5.3. Ex Vivo Procedure
2.6. In Vivo Biological Activity Studies
2.7. Statistical Analysis
3. Results
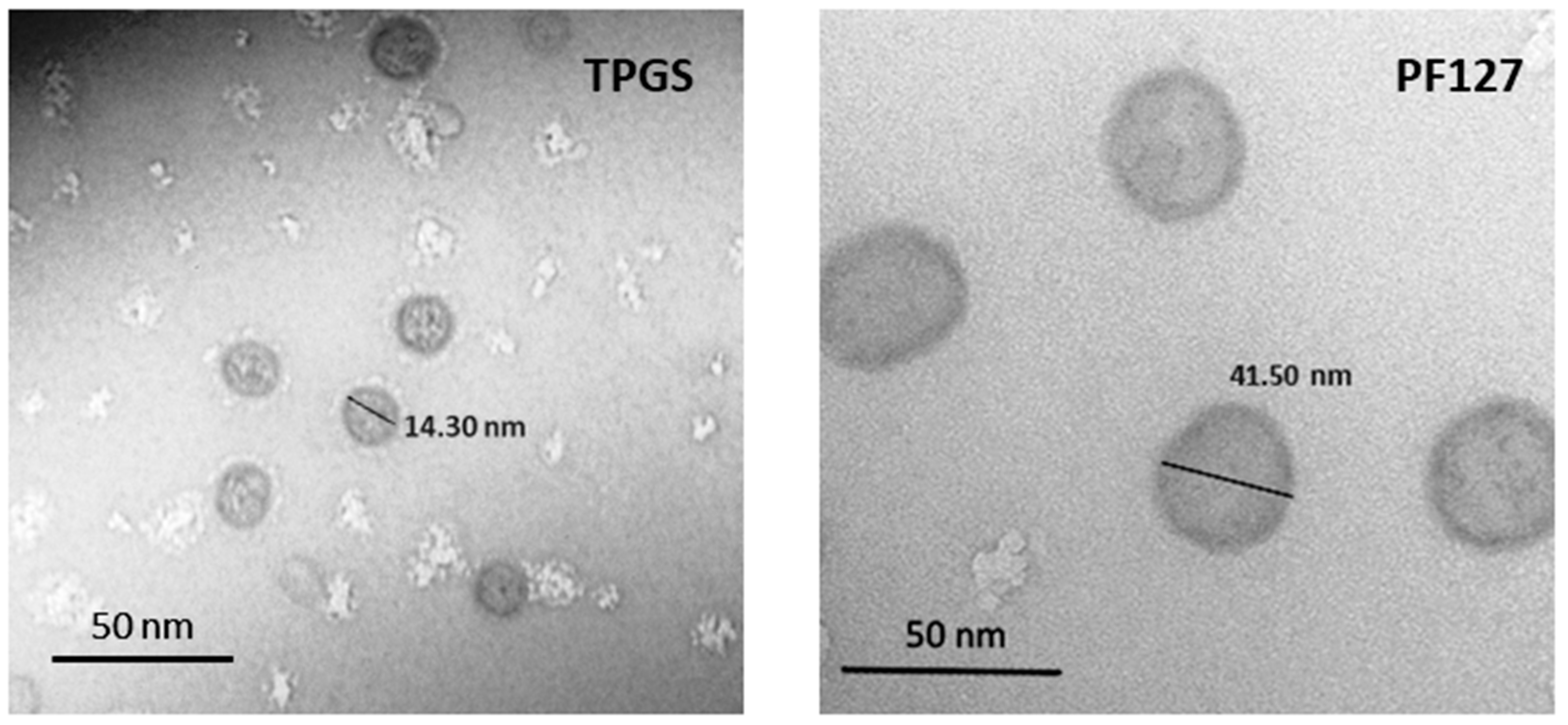
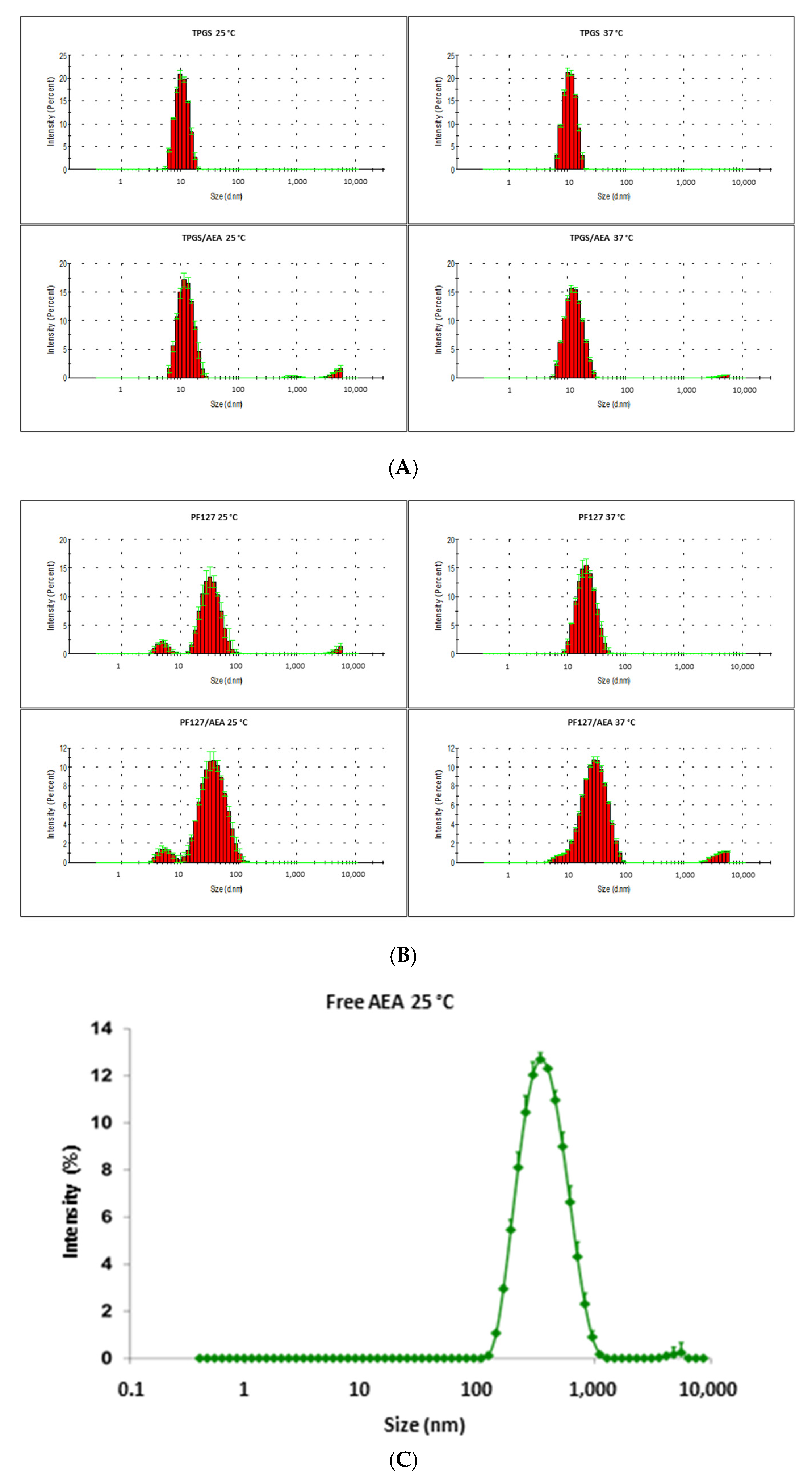
3.1. Characterization of the Micellar Systems
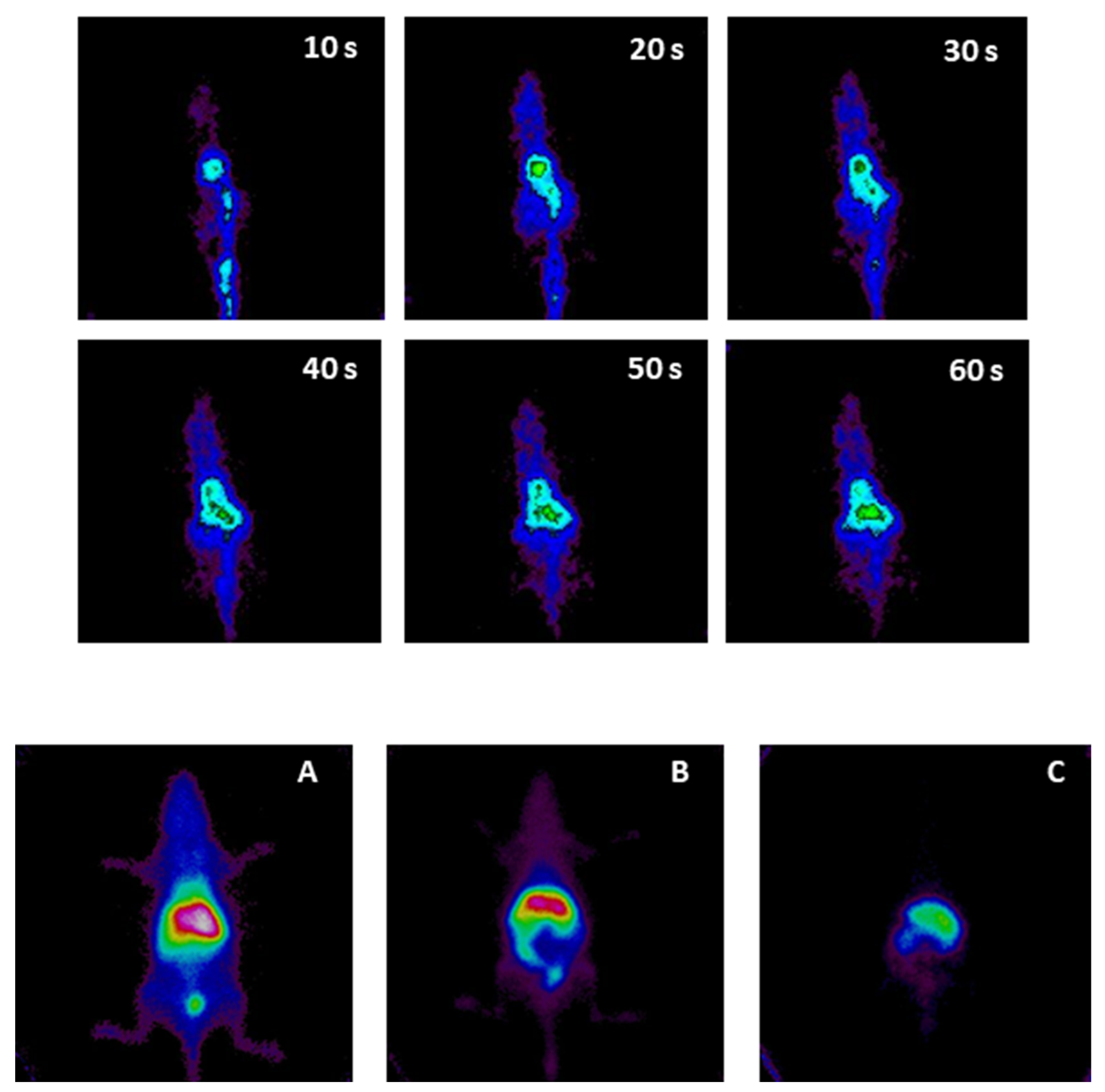
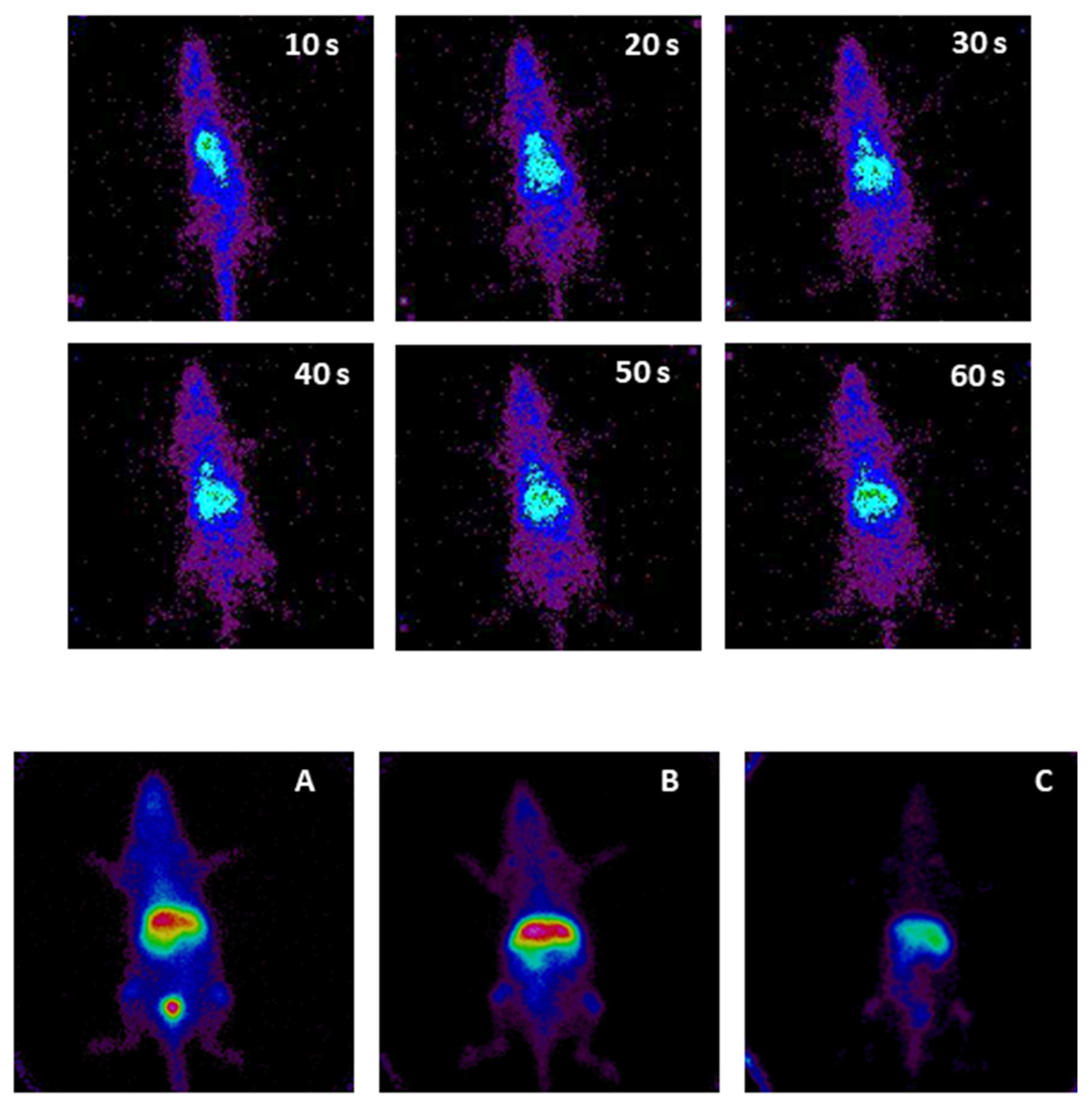
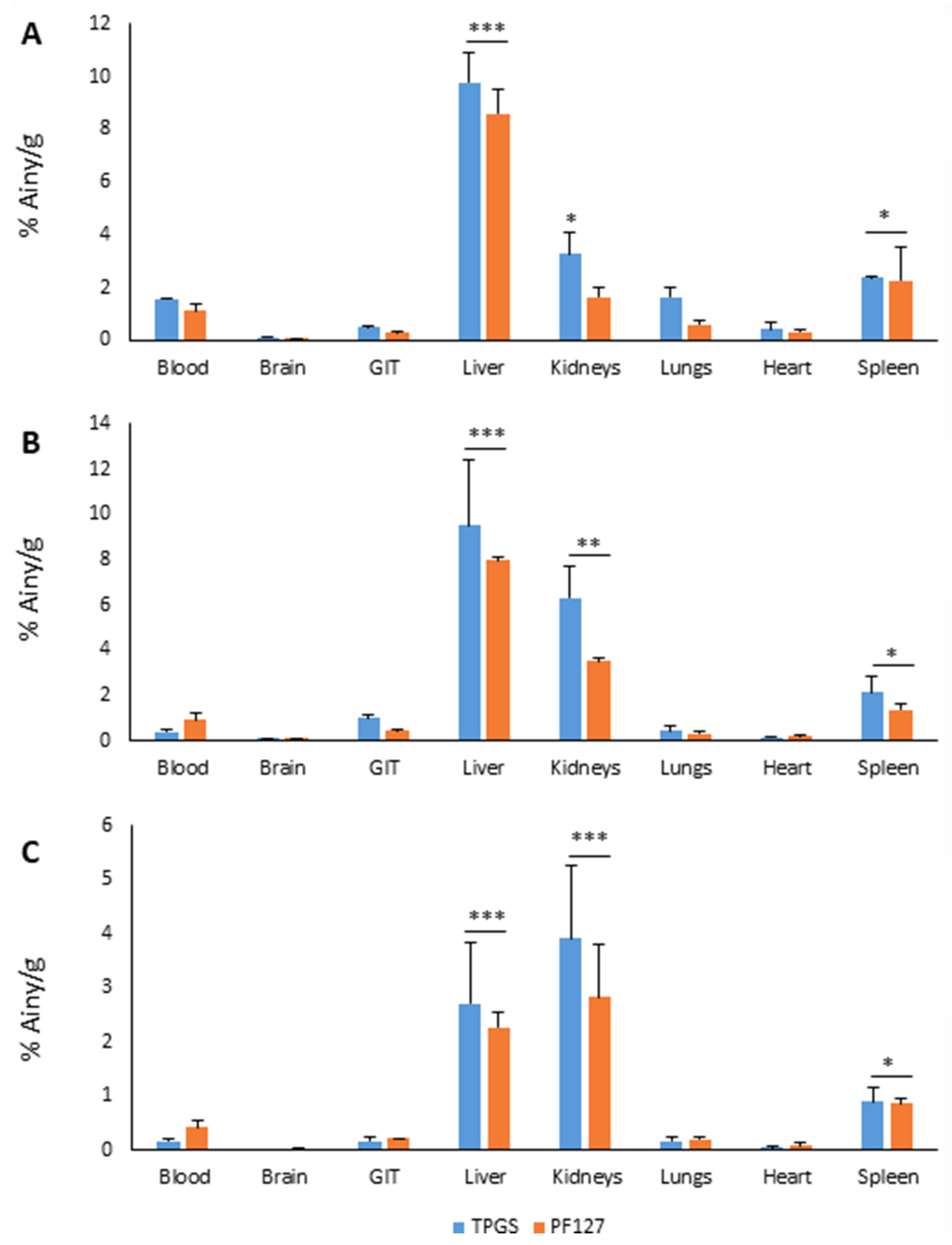
3.2. Micellar Biodistribution
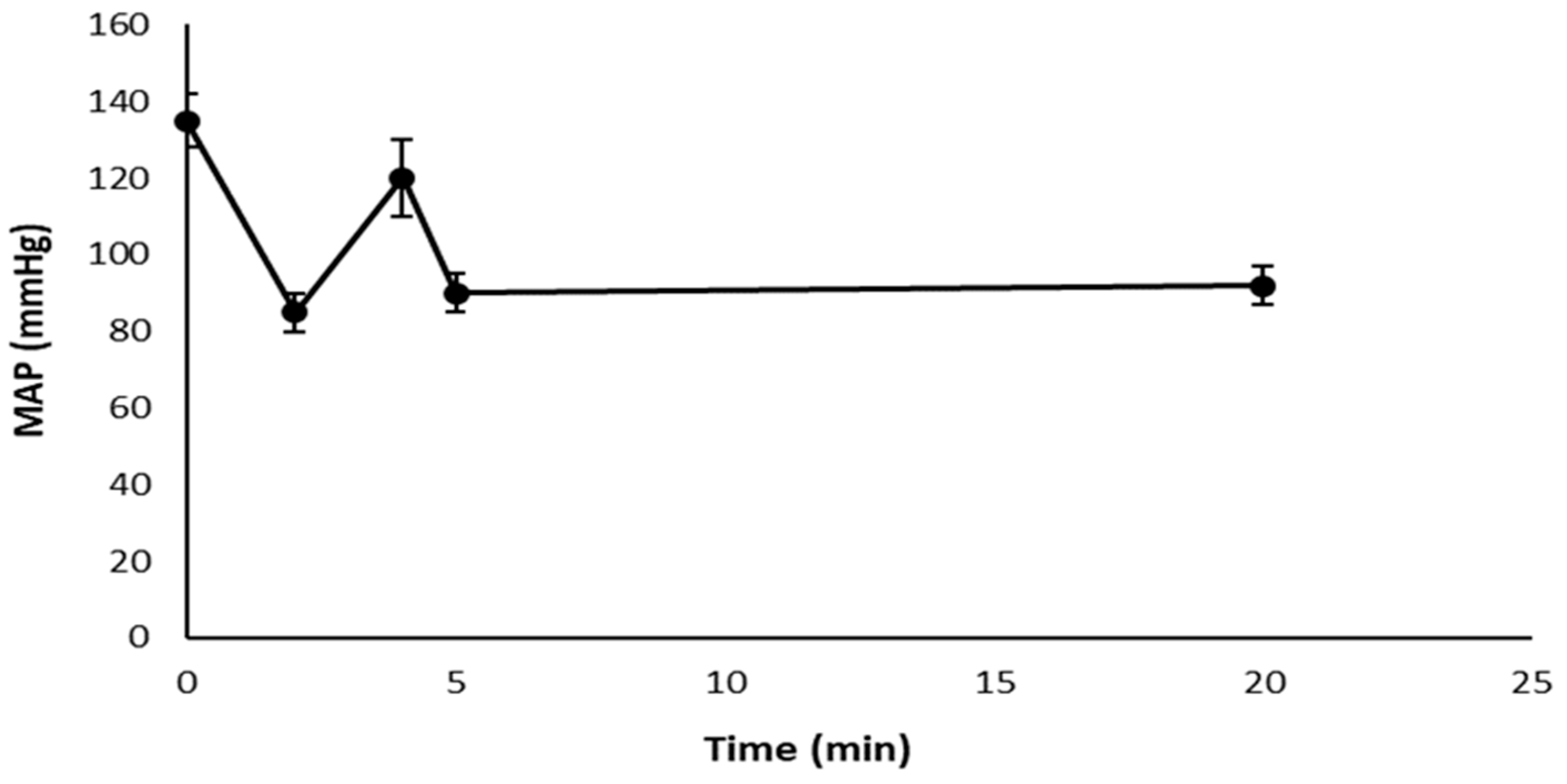
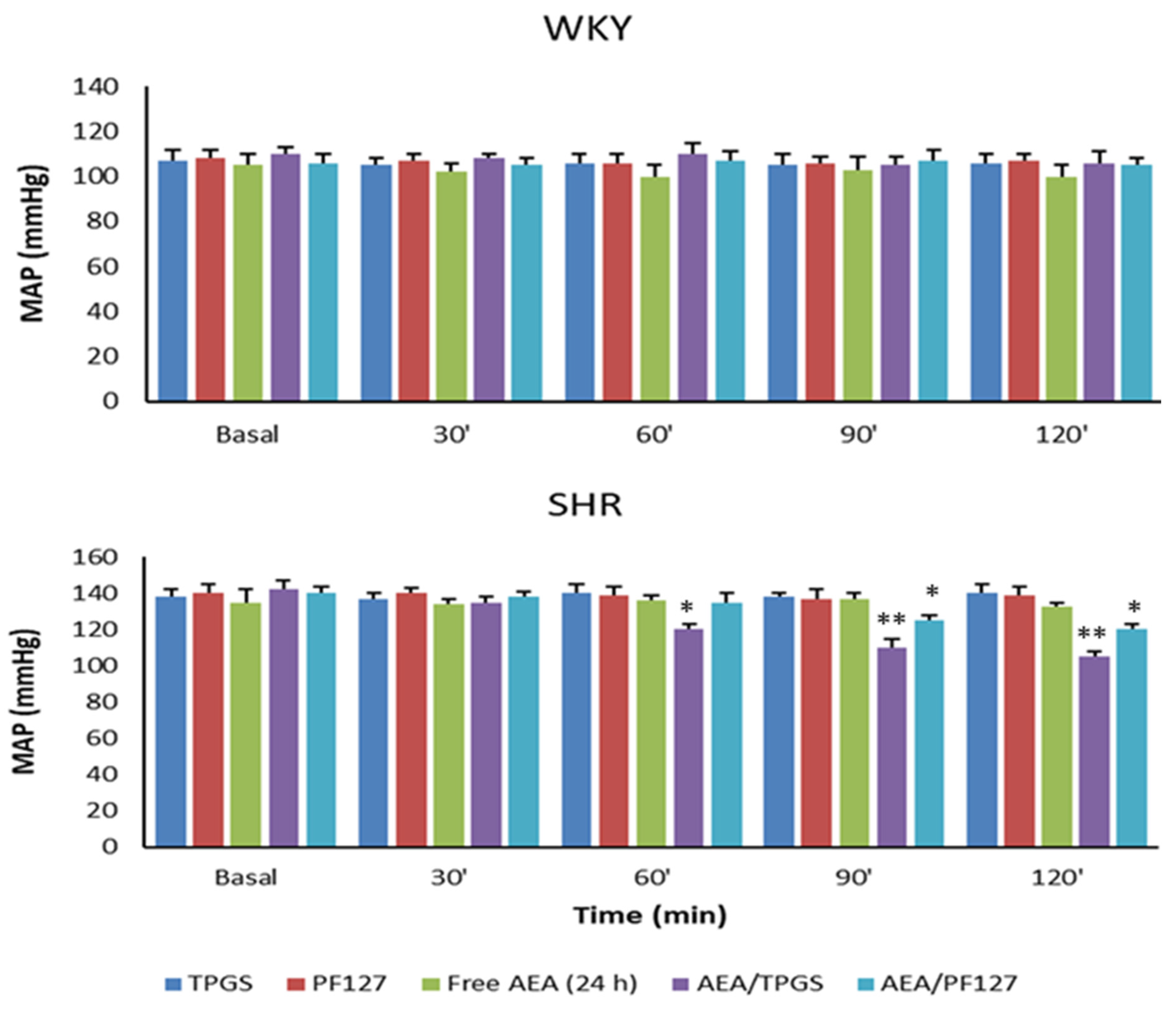
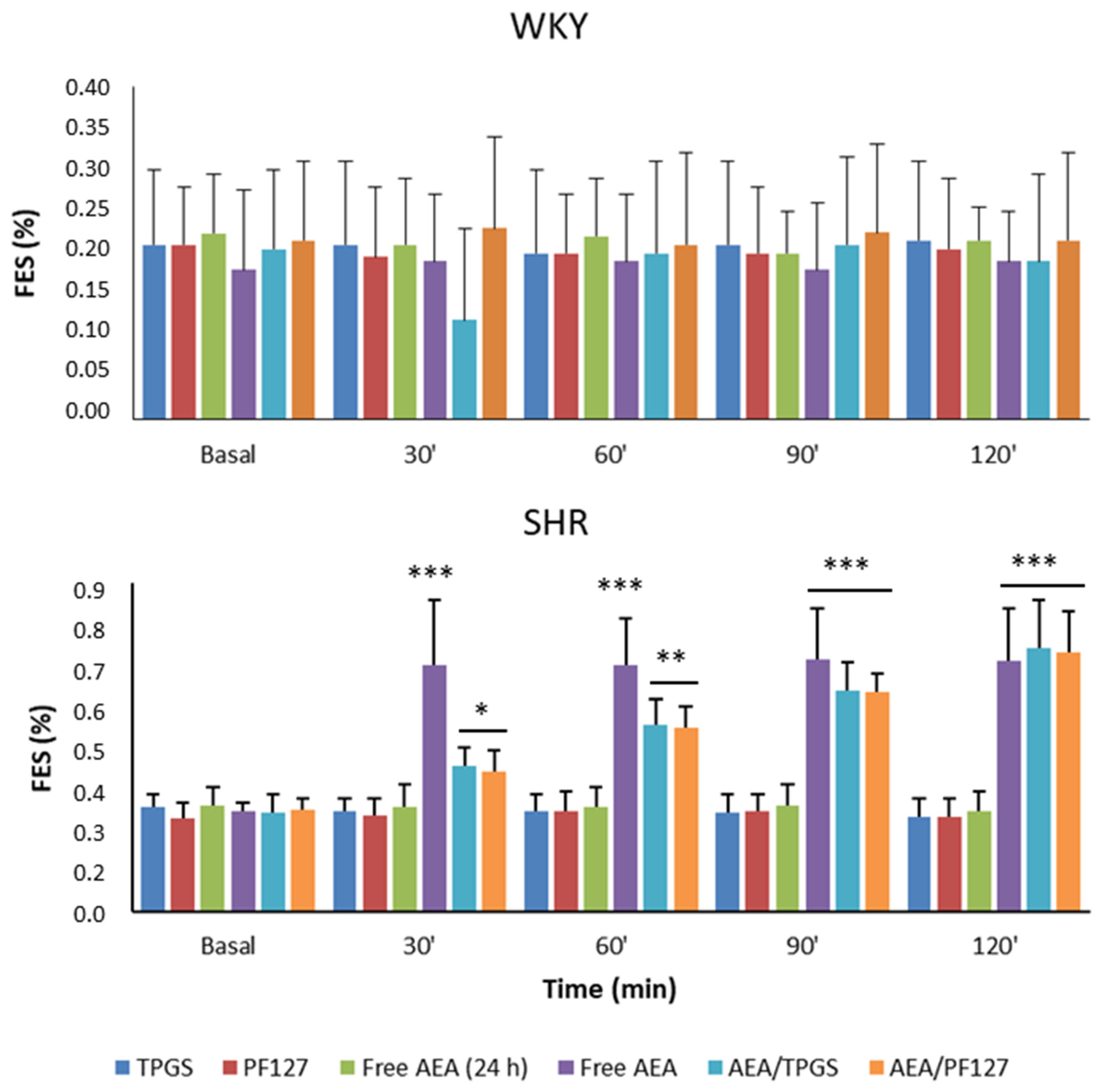
3.3. Mean Arterial Pressure Measurement, Plasmatic and Urinary Parameters Assessment, and Fractional Excretion of Sodium Determination
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shi, A.; Tao, Z.; Wei, P.; Zhao, J. Epidemiological aspects of heart diseases. Exp. Ther. Med. 2016, 12, 1645–1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Said, S.; Hernandez, G.T. The link between chronic kidney disease and cardiovascular disease. J. Nephropathol. 2014, 3, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Ivy, J.R.; Bailey, M.A. Pressure natriuresis and the renal control of arterial blood pressure. J. Physiol. 2014, 592, 3955–3967. [Google Scholar] [CrossRef] [PubMed]
- Giménez, V.M.M.; Kassuha, D.E.; Manucha, W. Nanomedicine applied to cardiovascular diseases: Latest developments. Ther. Adv. Cardiovasc. Dis. 2017, 11, 133–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giménez, V.M.M.; Fuentes, L.B.; Kassuha, D.E.; Manucha, W. Current Drug Nano-targeting Strategies for Improvement in the Diagnosis and Treatment of Prevalent Pathologies such as Cardiovascular and Renal Diseases. Curr. Drug Targets 2019, 20, 1496–1504. [Google Scholar] [CrossRef]
- Varga, K.; Lake, K.D.; Huangfu, D.; Guyenet, P.G.; Kunos, G. Mechanism of the Hypotensive Action of Anandamide in Anesthetized Rats. Hypertension 1996, 28, 682–686. [Google Scholar] [CrossRef]
- Giménez, V.M.M.; Díaz-Rodríguez, P.; Sanz, R.L.; Vivero-Lopez, M.; Concheiro, A.; Diez, E.; Prado, N.; Kassuha, D.E.; Alvarez-Lorenzo, C.; Manucha, W. Anandamide-nanoformulation obtained by electrospraying for cardiovascular therapy. Int. J. Pharm. 2019, 566, 1–10. [Google Scholar] [CrossRef]
- Aberturas, M.R.; De La Ossa, D.H.P.; Gil-Alegre, M.E.; Ligresti, L.; De Petrocellis, L.; Torres-Suárez, A.I.; Di Marzo, V.; Molpeceres, J. Anandamide-loaded nanoparticles: Preparation and characterization. J. Microencapsul. 2011, 28, 200–210. [Google Scholar] [CrossRef]
- Giménez, V.M.M.; Marón, F.J.M.; García, S.; Mazzei, L.; Guevara, M.; Yunes, R.; Manucha, W. Central nervous system, peripheral and hemodynamic effects of nanoformulated anandamide in hypertension. Adv. Med. Sci. 2020, 66, 72–80. [Google Scholar] [CrossRef]
- Giménez, V.M.M.; Noriega, S.E.; Kassuha, D.E.; Fuentes, L.B.; Manucha, W. Anandamide and endocannabinoid system: An attractive therapeutic approach for cardiovascular disease. Ther. Adv. Cardiovasc. Dis. 2018, 12, 177–190. [Google Scholar] [CrossRef]
- Giménez, V.M.M.; Russo, M.G.; Narda, G.E.; Fuentes, L.B.; Mazzei, L.; Gamarra-Luques, C.; Kassuha, D.E.; Manucha, W. Synthesis, physicochemical characterisation and biological activity of anandamide/ɛ-polycaprolactone nanoparticles obtained by electrospraying. IET Nanobiotechnology 2020, 14, 86–93. [Google Scholar] [CrossRef]
- Hoshyar, N.; Gray, S.; Han, H.; Bao, G. The effect of nanoparticle size on in vivo pharmacokinetics and cellular interaction. Nanomedicine 2016, 11, 673–692. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Masehi-Lano, J.J.; Chung, E.J. Peptide and antibody ligands for renal targeting: Nanomedicine strategies for kidney disease. Biomater. Sci. 2017, 5, 1450–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kataoka, K.; Harada, A.; Nagasaki, Y. Block copolymer micelles for drug delivery: Design, characterization and biological significance. Adv. Drug Deliv. Rev. 2001, 47, 113–131. [Google Scholar] [CrossRef]
- Owen, S.C.; Chan, D.P.; Shoichet, M.S. Polymeric micelle stability. Nano Today 2012, 7, 53–65. [Google Scholar] [CrossRef]
- Cagel, M.; Bernabeu, E.; Gonzalez, L.; Lagomarsino, E.; Zubillaga, M.; Moretton, M.A.; Chiappetta, D.A. Mixed micelles for encapsulation of doxorubicin with enhanced in vitro cytotoxicity on breast and ovarian cancer cell lines versus Doxil®. Biomed. Pharmacother. 2017, 95, 894–903. [Google Scholar] [CrossRef]
- Bernabeu, E.; Gonzalez, L.; Cagel, M.; Moretton, M.A.; Chiappetta, D.A. Deoxycholate-TPGS mixed nanomicelles for encapsulation of methotrexate with enhanced in vitro cytotoxicity on breast cancer cell lines. J. Drug Deliv. Sci. Technol. 2019, 50, 293–304. [Google Scholar] [CrossRef]
- Grotz, E.; Tateosian, N.L.; Salgueiro, J.; Bernabeu, E.; Gonzalez, L.; Manca, M.L.; Amiano, N.; Valenti, D.; Manconi, M.; García, V.; et al. Pulmonary delivery of rifampicin-loaded soluplus micelles against Mycobacterium tuberculosis. J. Drug Deliv. Sci. Technol. 2019, 53, 101170. [Google Scholar] [CrossRef]
- Hussein, Y.H.A.; Youssry, M. Polymeric Micelles of Biodegradable Diblock Copolymers: Enhanced Encapsulation of Hydrophobic Drugs. Materials 2018, 11, 688. [Google Scholar] [CrossRef] [Green Version]
- Chiappetta, D.A.; Sosnik, A. Poly(ethylene oxide)–poly(propylene oxide) block copolymer micelles as drug delivery agents: Improved hydrosolubility, stability and bioavailability of drugs. Eur. J. Pharm. Biopharm. 2007, 66, 303–317. [Google Scholar] [CrossRef]
- Bernabeu, E.; Chiappetta, D.A. Vitamin E TPGS Used as Emulsifier in the Preparation of Nanoparticulate Systems. J. Biomater. Tissue Eng. 2013, 3, 122–134. [Google Scholar] [CrossRef]
- Honda, S.; Yamamoto, T.; Tezuka, Y. Tuneable enhancement of the salt and thermal stability of polymeric micelles by cyclized amphiphiles. Nat. Commun. 2013, 4, 1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, S.; Sousa, J.; Pais, A.; Vitorino, C. Nanomedicine: Principles, Properties, and Regulatory Issues. Front. Chem. 2018, 6, 360. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.H.J.; Zuckerman, J.E.; Webster, P.; Davis, M.E. Targeting kidney mesangium by nanoparticles of defined size. Proc. Natl. Acad. Sci. USA 2011, 108, 6656–6661. [Google Scholar] [CrossRef] [Green Version]
- Jain, S.; Pandey, S.; Sola, P.; Pathan, H.; Patil, R.; Ray, D.; Aswal, V.K.; Bahadur, P.; Tiwari, S. Solubilization of Carbamazepine in TPGS Micelles: Effect of Temperature and Electrolyte Addition. AAPS PharmSciTech 2019, 20, 203. [Google Scholar] [CrossRef] [PubMed]
- Basak, R.; Bandyopadhyay, R. Encapsulation of Hydrophobic Drugs in Pluronic F127 Micelles: Effects of Drug Hydrophobicity, Solution Temperature, and pH. Langmuir 2013, 29, 4350–4356. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, T.A.; El-Say, K.M.; Ahmed, O.A.A.; Aljaeid, B.M. Superiority of TPGS-loaded micelles in the brain delivery of vinpocetine via administration of thermosensitive intranasal gel. Int. J. Nanomed. 2019, 14, 5555–5567. [Google Scholar] [CrossRef] [Green Version]
- Arranja, A.; Schroder, A.P.; Schmutz, M.; Waton, G.; Schosseler, F.; Mendes, E. Cytotoxicity and internalization of Pluronic micelles stabilized by core cross-linking. J. Control. Release 2014, 196, 87–95. [Google Scholar] [CrossRef]
- Kaus, N.H.M.; Shaarani, S.; Hamid, S.S. The Influence of pluronic F68 and F127 nanocarrier on physicochemical properties, in vitro release, and antiproliferative activity of thymoquinone drug. Pharmacogn. Res. 2017, 9, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Chiappetta, D.A.; Hocht, C.; Taira, C.; Sosnik, A. Efavirenz-loaded polymeric micelles for pediatric anti-HIV pharmacotherapy with significantly higher oral bioavailability. Nanomedicine 2010, 5, 11–23. [Google Scholar] [CrossRef]
- Hao, T.; Chen, D.; Liu, K.; Qi, Y.; Tian, Y.; Sun, P.; Liu, Y.; Li, Z. Micelles of d-α-Tocopheryl Polyethylene Glycol 2000 Succinate (TPGS 2K) for Doxorubicin Delivery with Reversal of Multidrug Resistance. ACS Appl. Mater. Interfaces 2015, 7, 18064–18075. [Google Scholar] [CrossRef]
- Longmire, M.; Choyke, P.L.; Kobayashi, H. Clearance properties of nano-sized particles and molecules as imaging agents: Considerations and caveats. Nanomedicine 2008, 3, 703–717. [Google Scholar] [CrossRef] [Green Version]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug Solubility: Importance and Enhancement Techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef] [Green Version]
- Pawar, A.; Thakkar, S.; Misra, M. A bird’s eye view of nanoparticles prepared by electrospraying: Advancements in drug delivery field. J. Control. Release 2018, 286, 179–200. [Google Scholar] [CrossRef]
- Muthu, M.S.; Kulkarni, S.A.; Liu, Y.; Feng, S.-S. Development of docetaxel-loaded vitamin E TPGS micelles: Formulation optimization, effects on brain cancer cells and biodistribution in rats. Nanomedicine 2012, 7, 353–364. [Google Scholar] [CrossRef]
- Martin Gimenez, V.M.; Mocayar Maron, F.J.; Kassuha, D.E.; Ferder, L.; Manucha, W. Key Aspects to Consider about Beneficial and Harmful Effects on the Central Nervous System by the Endocannabinoid Modulation Linked to New Cardiovascular Therapies. Ann. Pharmacol. Pharm. 2019, 4, 1168. [Google Scholar] [CrossRef]
- Tesan, F.C.; Portillo, M.G.; Moretton, M.A.; Bernabeu, E.; Chiappetta, D.A.; Salgueiro, M.J.; Zubillaga, M.B. Radiolabeling and biological characterization of TPGS-based nanomicelles by means of small animal imaging. Nucl. Med. Biol. 2017, 44, 62–68. [Google Scholar] [CrossRef]
- Tesan, F.; Cerqueira-Coutinho, C.; Salgueiro, J.; Albernaz, M.D.S.; Pinto, S.R.; Dos Reis, S.R.R.; Bernardes, E.S.; Chiapetta, D.; Zubillaga, M.; Santos-Oliveira, R. Characterization and biodistribution of bevacizumab TPGS-based nanomicelles: Preliminary studies. J. Drug Deliv. Sci. Technol. 2016, 36, 95–98. [Google Scholar] [CrossRef]
- Meng, X.; Liu, J.; Yu, X.; Li, J.; Lu, X.; Shen, T. Pluronic F127 and D-α-Tocopheryl Polyethylene Glycol Succinate (TPGS) Mixed Micelles for Targeting Drug Delivery across The Blood Brain Barrier. Sci. Rep. 2017, 7, 2964. [Google Scholar] [CrossRef]
- Pawar, A.; Singh, S.; Rajalakshmi, S.; Shaikh, K.; Bothiraja, C. Development of fisetin-loaded folate functionalized pluronic micelles for breast cancer targeting. Artif. Cells Nanomed. Biotechnol. 2018, 46, 347–361. [Google Scholar] [CrossRef]
- Malinowska, B.; Baranowska-Kuczko, M.; Schlicker, E. Triphasic blood pressure responses to cannabinoids: Do we understand the mechanism? Br. J. Pharmacol. 2012, 165, 2073–2088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gocen, T.; Bayari, S.H.; Guven, M.H. Conformational and vibrational studies of arachidonic acid, light and temperature effects on ATR-FTIR spectra. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 203, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Waterman, K.C.; Adami, R.C.; Alsante, K.M.; Antipas, A.S.; Arenson, D.R.; Carrier, R.; Hong, J.; Landis, M.S.; Lombardo, F.; Shah, J.C.; et al. Hydrolysis in Pharmaceutical Formulations. Pharm. Dev. Technol. 2002, 7, 113–146. [Google Scholar] [CrossRef] [PubMed]
- Silva, G.B.; Atchison, D.K.; Juncos, L.I.; García, N.H. Anandamide inhibits transport-related oxygen consumption in the loop of Henle by activating CB1 receptors. Am. J. Physiol. Physiol. 2013, 304, F376–F381. [Google Scholar] [CrossRef] [Green Version]
- Ghezzi, M.; Pescina, S.; Padula, C.; Santi, P.; Del Favero, E.; Cantù, L.; Nicoli, S. Polymeric micelles in drug delivery: An insight of the techniques for their characterization and assessment in biorelevant conditions. J. Control. Release 2021, 332, 312–336. [Google Scholar] [CrossRef]
- Pacher, P.; Bátkai, S.; Kunos, G. Blood pressure regulation by endocannabinoids and their receptors. Neuropharmacology 2005, 48, 1130–1138. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | AEA Presence | Temperature (°C) | Size | ZP (mV) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Peak 1 (nm) | Peak 2 (nm) | Peak 3 (nm) | PDI (±D.E.) | |||||||
| Dh (±D.E.) | % | Dh (±D.E.) | % | Dh (±D.E.) | % | |||||
| TPGS 3% | 25 | 10.9 (0.1) | 100 | - | - | - | - | 0.032 (0.013) | −1.455 (0.721) | |
| √ | 13.0 (0.4) | 100 | - | - | - | - | 0.234 (0.038) | −2.76 (1.27) | ||
| PF127 3% | 5.1 (0.5) | 8.7 | 35.4 (3.1) | 91.3 | - | - | 0.391 (0.041) | −1.87 (0.55) | ||
| √ | 5.8 (0.7) | 7.2 | 40.6 (1.5) | 92.8 | - | - | 0.299 (0.039) | −0.879 (0.414) | ||
| TPGS 3% | 37 | 11.2 (0.1) | 100 | - | - | - | - | 0.020 (0.008) | - | |
| √ | 13.4 (0.1) | 100 | - | - | - | - | 0.157 (0.008) | - | ||
| PF127 3% | 22.0 (1.2) | 100 | - | - | - | - | 0.154 (0.039) | - | ||
| √ | 31.2 (0.3) | 100 | - | - | - | - | 0.287 (0.007) | - | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martín Giménez, V.M.; Moretton, M.A.; Chiappetta, D.A.; Salgueiro, M.J.; Fornés, M.W.; Manucha, W. Polymeric Nanomicelles Loaded with Anandamide and Their Renal Effects as a Therapeutic Alternative for Hypertension Treatment by Passive Targeting. Pharmaceutics 2023, 15, 176. https://doi.org/10.3390/pharmaceutics15010176
Martín Giménez VM, Moretton MA, Chiappetta DA, Salgueiro MJ, Fornés MW, Manucha W. Polymeric Nanomicelles Loaded with Anandamide and Their Renal Effects as a Therapeutic Alternative for Hypertension Treatment by Passive Targeting. Pharmaceutics. 2023; 15(1):176. https://doi.org/10.3390/pharmaceutics15010176
Chicago/Turabian StyleMartín Giménez, Virna Margarita, Marcela Analía Moretton, Diego Andrés Chiappetta, María Jimena Salgueiro, Miguel Walter Fornés, and Walter Manucha. 2023. "Polymeric Nanomicelles Loaded with Anandamide and Their Renal Effects as a Therapeutic Alternative for Hypertension Treatment by Passive Targeting" Pharmaceutics 15, no. 1: 176. https://doi.org/10.3390/pharmaceutics15010176