
Novel αO-conotoxin GeXIVA[1,2] Nonaddictive Analgesic with Pharmacokinetic Modelling-Based Mechanistic Assessment

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Experimental Animals
2.3. PK Studies in Animals
2.3.1. PK Studies in Rats
2.3.2. PK Studies in Beagle Dogs
2.4. Plasma Protein Binding Assay
2.5. Bioanalytical Assays
2.6. Mechanism-Based PK Modelling
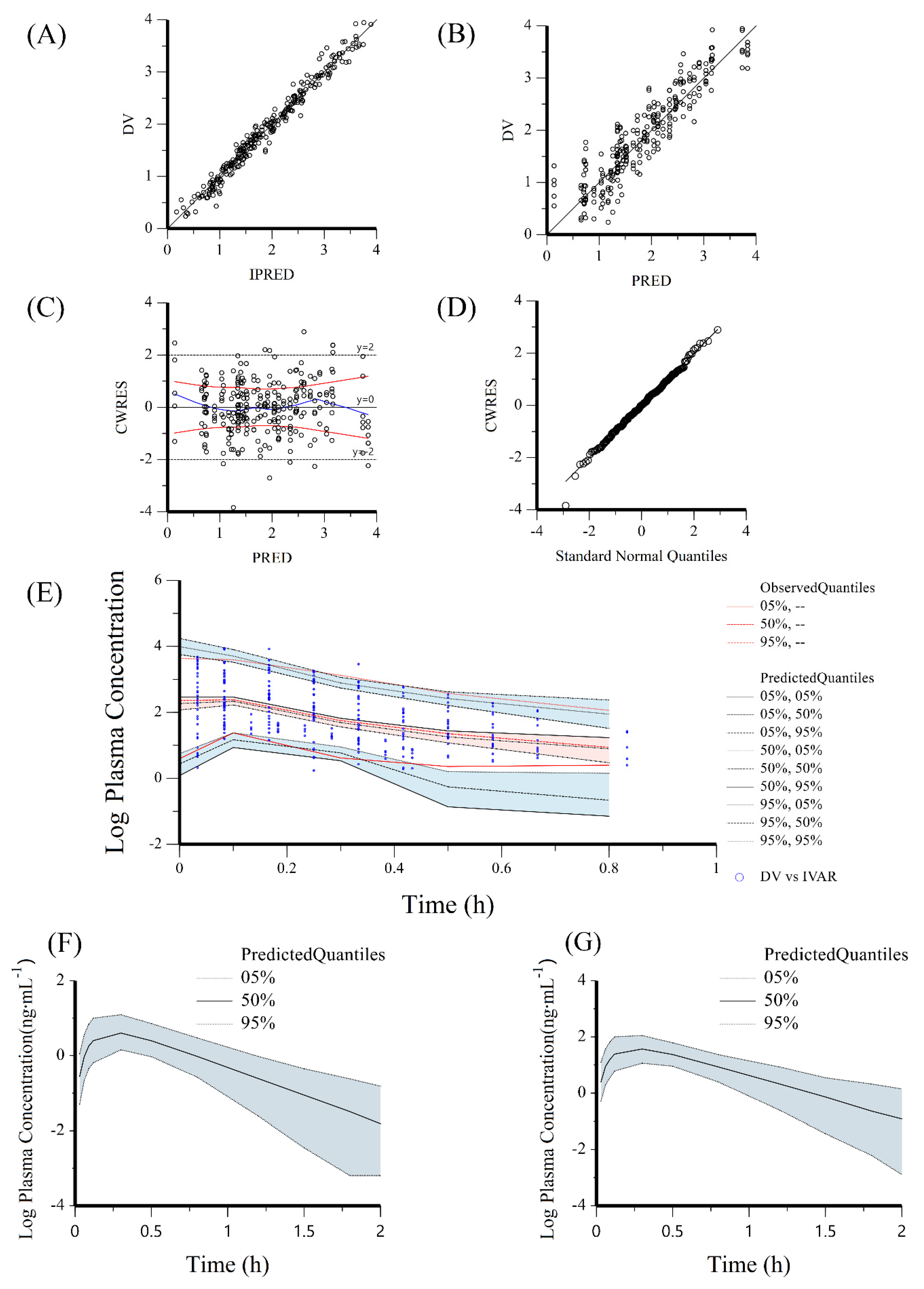
2.6.1. Population PK Modelling and Human PK Prediction
Structural Model
Statistical Model
Covariate Model
Model Evaluation and Validation
Model-Based Simulations
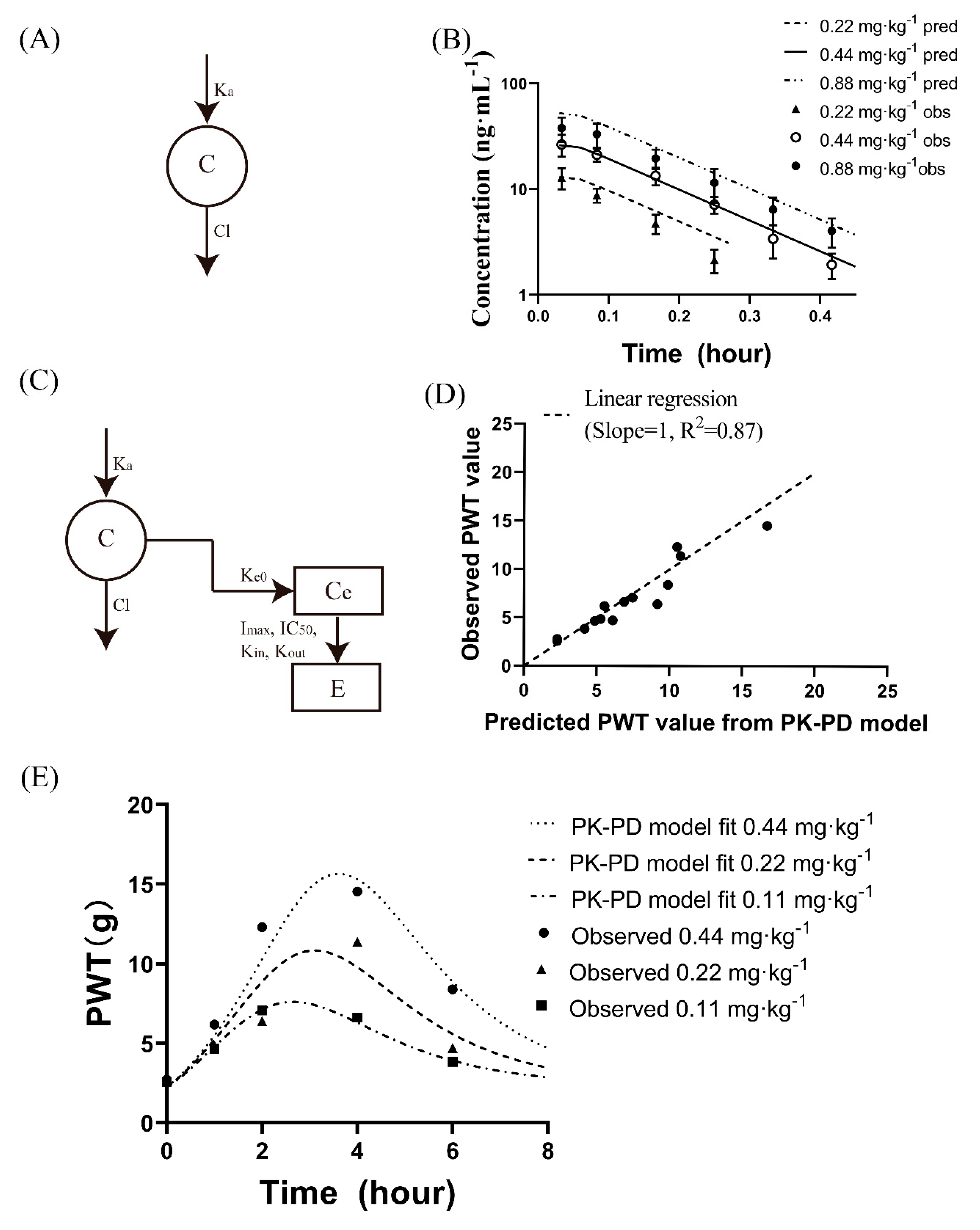
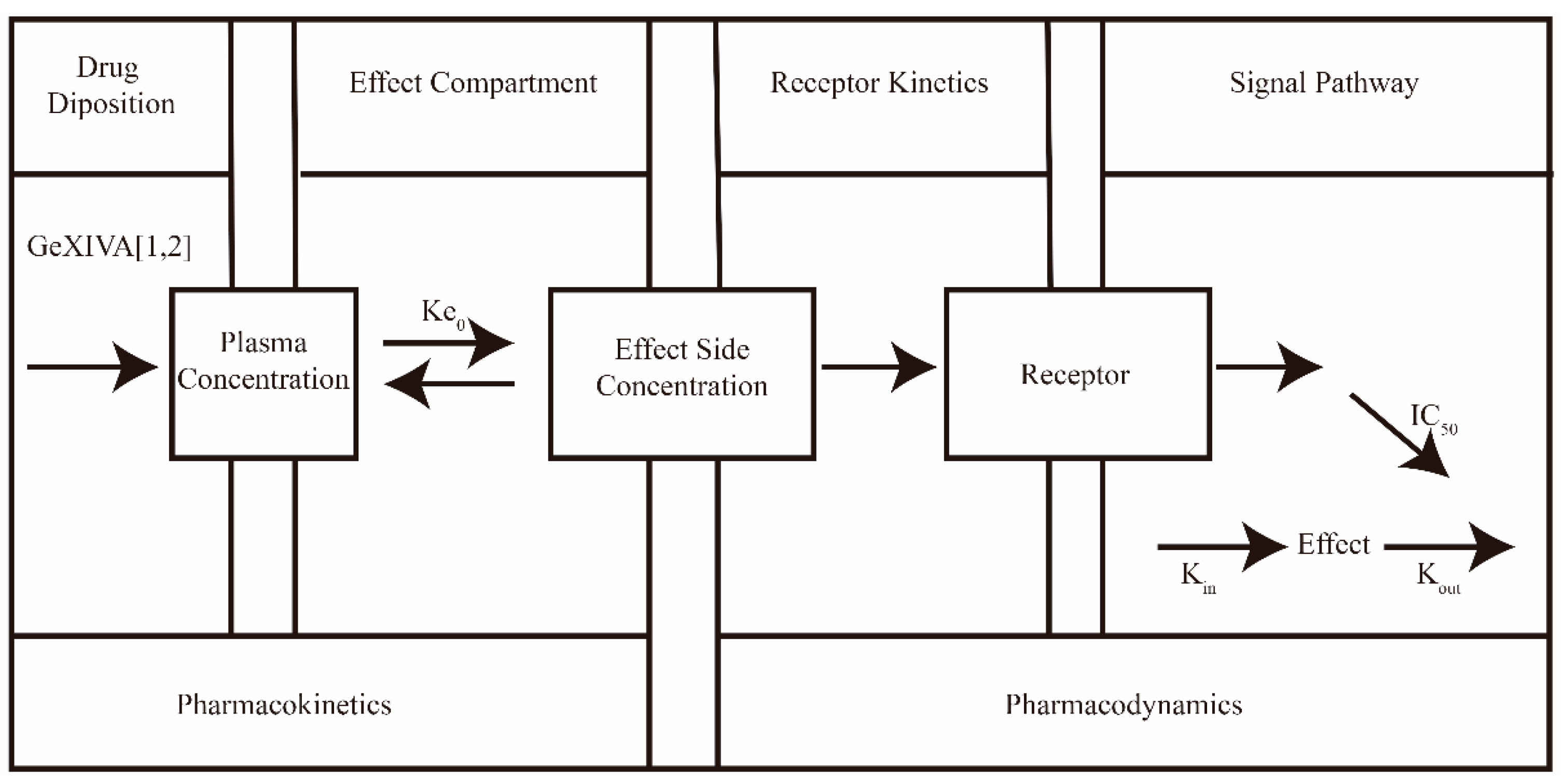
2.6.2. PK-PD Modelling
2.7. Data and Statistical Analysis
3. Results
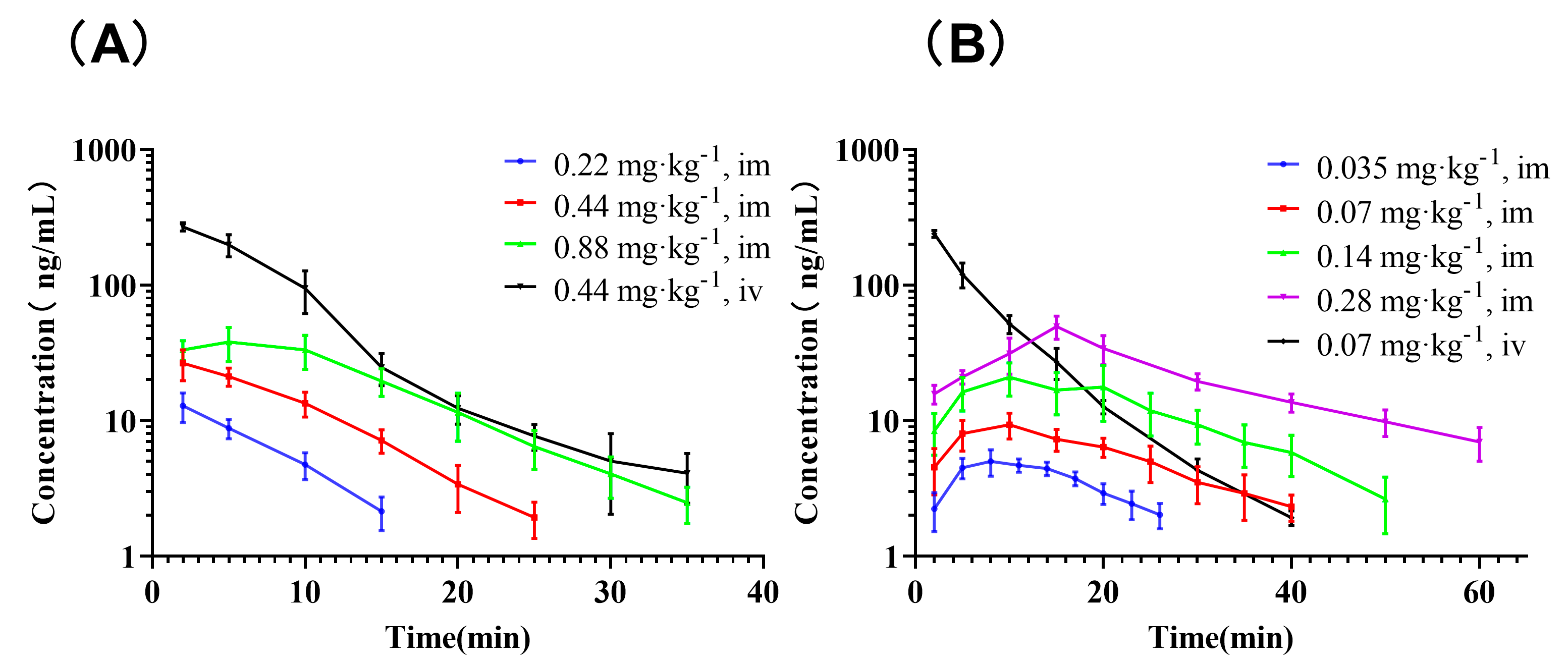
3.1. PK Studies
3.2. Plasma Protein Binding Rate
3.3. Multiscale PK Modelling
3.3.1. Population PK Modelling for Human Dose and PK Prediction
3.3.2. Mechanistic PK-PD Modelling
3.4. Potential Mechanism of GeXIVA[1,2] Based on Multiscale PK Modelling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| nAChRs | Nicotinic acetylcholine receptors |
| PK | pharmacokinetics |
| PopPK | population pharmacokinetics |
| PD | pharmacodynamics |
| CI | confidence interval |
| AUC | area under the curve |
| CL | clearance |
| Vz | the volume of distribution during the terminal elimination phase |
| Tmax | the peak concentration time |
| Cmax | peak concentration |
| t1/2 | terminal half-life |
| WT | body weight |
| NLME | nonlinear mixed-effects model |
| OFV | objective function value |
| AIC | Akaike Information Criterion |
| BIC | Bayesian Information Criterion |
| VPC | visual predictive check |
| RED | Rapid Equilibrium Dialysis |
| PI | prediction intervals |
| PRED | population predicted concentrations |
| IPRED | individual predicted concentrations |
| NCA | noncompartmental analysis |
References
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic pain. Nat. Rev. Dis. Primers 2017, 3, 17002. [Google Scholar] [CrossRef] [PubMed]
- Gilron, I.; Baron, R.; Jensen, T. Neuropathic pain: Principles of diagnosis and treatment. Mayo Clin. Proc. 2015, 90, 532–545. [Google Scholar] [CrossRef] [PubMed]
- Gierthmuhlen, J.; Baron, R. Neuropathic pain. Semin. Neurol. 2016, 36, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Tae, H.S.; Chu, Y.; Jiang, T.; Adams, D.J.; Yu, R. Medicinal chemistry, pharmacology, and therapeutic potential of α-conotoxins antagonizing the α9α10 nicotinic acetylcholine receptor. Pharmacol. Ther. 2020, 222, 107792. [Google Scholar] [CrossRef]
- Xu, P.; Kaas, Q.; Wu, Y.; Zhu, X.; Li, X.; Harvey, P.J.; Zhangsun, D.; Craik, D.J.; Luo, S. Structure and activity studies of disulfide-deficient analogues of alphaO-conotoxin GeXIVA. J. Med. Chem. 2020, 63, 1564–1575. [Google Scholar] [CrossRef]
- Christensen, S.B.; Hone, A.J.; Roux, I.; Kniazeff, J.; Pin, J.P.; Upert, G.; Servent, D.; Glowatzki, E.; McIntosh, J.M. RgIA4 potently blocks mouse α9α10 nAChRs and provides long lasting protection against oxaliplatin-induced cold allodynia. Front. Cell. Neurosci. 2017, 11, 219. [Google Scholar] [CrossRef]
- Pacini, A.; Micheli, L.; Maresca, M.; Branca, J.J.; McIntosh, J.M.; Ghelardini, C.; Di Cesare Mannelli, L. The α9α10 nicotinic receptor antagonist α-conotoxin RgIA prevents neuropathic pain induced by oxaliplatin treatment. Exp. Neurol. 2016, 282, 37–48. [Google Scholar] [CrossRef]
- McIntosh, J.M.; Absalom, N.; Chebib, M.; Elgoyhen, A.B.; Vincler, M. Alpha9 nicotinic acetylcholine receptors and the treatment of pain. Biochem. Pharmacol. 2009, 78, 693–702. [Google Scholar] [CrossRef]
- Fu, Y.; Li, C.; Dong, S.; Wu, Y.; Zhangsun, D.; Luo, S. Discovery Methodology of Novel Conotoxins from Conus Species. Mar. Drugs 2018, 16, 417. [Google Scholar] [CrossRef]
- Deer, T.R.; Pope, J.E.; Hayek, S.M.; Bux, A.; Buchser, E.; Eldabe, S.; De Andres, J.A.; Erdek, M.; Patin, D.; Grider, J.S.; et al. The Polyanalgesic Consensus Conference (PACC): Recommendations on Intrathecal Drug Infusion Systems Best Practices and Guidelines. Neuromodulation 2017, 20, 96–132. [Google Scholar] [CrossRef]
- Li, X.; Hu, Y.; Wu, Y.; Huang, Y.; Yu, S.; Ding, Q.; Zhangsun, D.; Luo, S. Anti-hypersensitive effect of intramuscular administration of alphaO-conotoxin GeXIVA[1,2] and GeXIVA[1,4] in rats of neuropathic pain. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 66, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Zhangsun, D.; Harvey, P.J.; Kaas, Q.; Wu, Y.; Zhu, X.; Hu, Y.; Li, X.; Tsetlin, V.I.; Christensen, S.; et al. Cloning, synthesis, and characterization of alphaO-conotoxin GeXIVA, a potent alpha9alpha10 nicotinic acetylcholine receptor antagonist. Proc. Natl. Acad. Sci. USA 2015, 112, E4026–E4035. [Google Scholar] [CrossRef] [PubMed]
- Zhangsun, D.; Zhu, X.; Kaas, Q.; Wu, Y.; Craik, D.J.; McIntosh, J.M.; Luo, S. αO-conotoxin GeXIVA disulfide bond isomers exhibit differential sensitivity for various nicotinic acetylcholine receptors but retain potency and selectivity for the human α9α10 subtype. Neuropharmacology 2017, 127, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Han, X.; Hong, X.; Li, X.; Gao, J.; Zhang, H.; Zheng, A. Lyophilization serves as an effective strategy for drug development of the α9α10 nicotinic acetylcholine receptor antagonist α-conotoxin GeXIVA[1,2]. Mar. Drugs 2021, 19, 121. [Google Scholar] [CrossRef]
- Wang, H.; Li, X.; Zhangsun, D.; Yu, G.; Su, R.; Luo, S. The α9α10 nicotinic acetylcholine receptor antagonist αO-conotoxin GeXIVA[1,2] alleviates and reverses chemotherapy-induced neuropathic pain. Mar. Drugs 2019, 17, 265. [Google Scholar] [CrossRef]
- Zhangsun, D.; Luo, S. Preclinical research of analgesic αO-conotoxin GeXIVA without addiction side effect. Toxicon 2020, 177 (Suppl. 1), S3. [Google Scholar] [CrossRef]
- du Sert, N.P.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biol. 2020, 18, e3000410. [Google Scholar] [CrossRef]
- Nair, A.; Morsy, M.A.; Jacob, S. Dose translation between laboratory animals and human in preclinical and clinical phases of drug development. Drug Dev. Res. 2018, 79, 373–382. [Google Scholar] [CrossRef]
- Yuan, Z.-Q.-Y.; Qiao, C.; Yang, Z.-C.; Yu, L.; Sun, L.-N.; Qian, Y.; Zhang, X.-H.; Meng, L.; Zhang, X.-Y.; Wang, Y.-Q. The impact of plasma protein binding characteristics and unbound concentration of voriconazole on its adverse drug reactions. Front. Pharmacol. 2020, 11, 505. [Google Scholar] [CrossRef]
- Zhu, X.Y.; Yuan, M.; Luo, S.L.; Che, J.J. Development and validation of antibody sandwich enzyme-linked immunosorbent assay method for quantitation of GeXIV A[1,2] in plasma of rats and beagle dogs. Acta Pharm. Sin. 2021, 56, 2378–2382. [Google Scholar] [CrossRef]
- Matsuura, K.; Sakai, A.; Watanabe, Y.; Mikahara, Y.; Sakamoto, A.; Suzuki, H. Endothelin receptor type A is involved in the development of oxaliplatin-induced mechanical allodynia and cold allodynia acting through spinal and peripheral mechanisms in rats. Mol. Pain 2021, 17. [Google Scholar] [CrossRef] [PubMed]
- Blivis, D.; Haspel, G.; Mannes, P.Z.; O’Donovan, M.J.; Iadarola, M.J. Identification of a novel spinal nociceptive-motor gate control for adelta pain stimuli in rats. Elife 2017, 6, e23584. [Google Scholar] [CrossRef] [PubMed]
- Wallas, T.R.; Winterson, B.J.; Ransil, B.J.; Bove, G.M. Paw withdrawal thresholds and persistent hindlimb flexion in experimental mononeuropathies. J. Pain 2003, 4, 222–230. [Google Scholar] [CrossRef]
- Deuis, J.R.; Dvorakova, L.S.; Vetter, I. Methods used to evaluate pain behaviors in rodents. Front. Mol. Neurosci. 2017, 10, 284. [Google Scholar] [CrossRef] [PubMed]
- Anonymous. Editorial: Bioavailability after intramuscular injection. Lancet 1975, 1, 261. [Google Scholar]
- Meibohm, B. Pharmacokinetics and Pharmacodynamics of Therapeutic Peptides and Proteins. In Pharmaceutical Biotechnology: Fundamentals and Applications; Crommelin, D.J.A., Sindelar, R.D., Meibohm, B., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 105–137. [Google Scholar]
- Davies, B.; Morris, T. Physiological parameters in laboratory animals and humans. Pharm. Res. 1993, 10, 1093–1095. [Google Scholar] [CrossRef]
- Yu, S.; Wu, Y.; Xu, P.; Wang, S.; Zhangsun, D.; Luo, S. Effects of serum, enzyme, thiol, and forced degradation on the stabilities of αO-conotoxin GeXIVA[1,2] and GeXIVA [1,4]. Chem. Biol. Drug Des. 2018, 91, 1030–1041. [Google Scholar] [CrossRef]
- Li, X.; Li, L.; Wang, X.; Ren, Y.; Zhou, T.; Lu, W. Application of model-based methods to characterize exenatide-loaded double-walled microspheres: In vivo release, pharmacokinetic/pharmacodynamic model, and in vitro and in vivo correlation. J. Pharm. Sci. 2012, 101, 3946–3961. [Google Scholar] [CrossRef]
- Li, X.G.; Zhou, T.Y.; Zhao, Z.G.; Lu, W. Introduction of mechanism-based pharmacokinetic/pharmacodynamic models. Chin. J. New Drugs 2013, 22, 1179-1185-1201. [Google Scholar]
- Dayneka, N.L.; Garg, V.; Jusko, W.J. Comparison of four basic models of indirect pharmacodynamic responses. J. Pharmacokinet. Biopharm. 1993, 21, 457–478. [Google Scholar] [CrossRef]
- Jin, A.H.; Muttenthaler, M.; Dutertre, S.; Himaya, S.W.A.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Conotoxins: Chemistry and Biology. Chem. Rev. 2019, 119, 11510–11549. [Google Scholar] [CrossRef] [PubMed]
- Segura-Campos, M.; Chel-Guerrero, L.; Betancur-Ancona, D.; Hernandez-Escalante, V.M. Bioavailability of Bioactive Peptides. Food Rev. Int. 2011, 27, 213–226. [Google Scholar] [CrossRef]
- Li, X.; Wang, S.; Zhu, X.; Zhangsun, D.; Wu, Y.; Luo, S. Effects of Cyclization on Activity and Stability of α-Conotoxin TxIB. Mar. Drugs 2020, 18, 180. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Xiong, Y.; Zhangsun, D.; Luo, S. DSPE-PEG Modification of α-Conotoxin TxID. Mar. Drugs 2019, 17, 342. [Google Scholar] [CrossRef] [PubMed]
- Jolling, K.; Perez Ruixo, J.J.; Hemeryck, A.; Vermeulen, A.; Greway, T. Mixed-effects modelling of the interspecies pharmacokinetic scaling of pegylated human erythropoietin. Eur. J. Pharm. Sci. 2005, 24, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Martin-Jimenez, T.; Riviere, J.E. Mixed-effects modeling of the interspecies pharmacokinetic scaling of oxytetracycline. J. Pharm. Sci. 2002, 91, 331–341. [Google Scholar] [CrossRef]
- Jin, T.; Wu, D.; Liu, X.-M.; Xu, J.-T.; Ma, B.-J.; Ji, Y.; Jin, Y.-Y.; Wu, S.-Y.; Wu, T.; Ma, K. Intra-articular delivery of celastrol by hollow mesoporous silica nanoparticles for pH-sensitive anti-inflammatory therapy against knee osteoarthritis. J. Nanobiotechnol. 2020, 18, 94. [Google Scholar] [CrossRef] [PubMed]
- Hone, A.J.; Servent, D.; McIntosh, J.M. alpha9-containing nicotinic acetylcholine receptors and the modulation of pain. Br. J. Pharmacol. 2018, 175, 1915–1927. [Google Scholar] [CrossRef]
- Sisignano, M.; Lotsch, J.; Parnham, M.J.; Geisslinger, G. Potential biomarkers for persistent and neuropathic pain therapy. Pharmacol. Ther. 2019, 199, 16–29. [Google Scholar] [CrossRef]
- Flores-Murrieta, F.J.; Ko, H.C.; Flores-Acevedo, D.M.; Lopez-Munoz, F.J.; Jusko, W.J.; Sale, M.E.; Castaneda-Hernandez, G. Pharmacokinetic-pharmacodynamic modeling of tolmetin antinociceptive effect in the rat using an indirect response model: A population approach. J. Pharmacokinet. Biopharm. 1998, 26, 547–557. [Google Scholar] [CrossRef]
- Yassen, A.; Passier, P.; Furuichi, Y.; Dahan, A. Translational PK-PD modeling in pain. J. Pharmacokinet. Pharmacodyn. 2013, 40, 401–418. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spices | Dose (mg·kg−1) | Weight (kg) | Number and Sex | PK Sampling Scheme (min) |
|---|---|---|---|---|
| Rat | IM, 0.22 | 0.25 (0.24–0.26) | 3 M + 3 F | 2, 5, 10, 15 |
| IM, 0.44 | 3 M + 3 F | 2, 5, 10, 15, 20, 25 | ||
| IM, 0.88 | 3 M + 3 F | 2, 5, 10, 15, 20, 25, 30, 35 | ||
| IV, 0.44 | 3 M + 3 F | 2, 5, 10, 15, 20, 25, 30, 35 | ||
| Dog | IM, 0.035 | 10 (8–12) | 3 M + 3 F | 2, 5, 8, 11, 14, 17, 20, 22, 26 |
| IM, 0.07 | 3 M + 3 F | 2, 5, 10, 15, 20, 25, 30, 35 | ||
| IM, 0.14 | 3 M + 3F | 2, 5, 10, 15, 20, 25, 30, 35, 40, 50 | ||
| IM, 0.28 | 3 M + 2 F | 2, 5, 10, 15, 20, 25, 30, 40, 50, 60 | ||
| IV, 0.07 | 3 M + 3 F | 2, 5, 10, 15, 20, 30, 40 |
| Species | Route | Dose (mg·kg−1) | AUC(0-inf) (ng·min·mL−1) | CL 1 (mL·min−1·kg−1) | Vz 2 (mL·kg−1) | t1/2 (min) | Cmax (ng·mL−1) | Tmax (min) |
|---|---|---|---|---|---|---|---|---|
| Rat | IV | 0.44 | 2472.84 ± 272.29 | 179.81 ± 20.50 | 1396.15 ± 328.79 | 5.37 ± 0.99 | 269.17 ± 18.68 | 2.00 ± 0.00 |
| IM | 0.22 | 109.84 ± 17.72 | 2047.91 ± 337.14 | 14,572.0 ± 2910.81 | 4.98 ± 0.91 | 12.86 ± 3.17 | 2.00 ± 0.00 | |
| IM | 0.44 | 283.52 ± 39.76 | 1578.12 ± 225.36 | 11,686.1 ± 1221.54 | 5.20 ± 0.78 | 27.17 ± 5.79 | 3.00 ± 1.55 | |
| IM | 0.88 | 637.17 ± 101.91 | 1415.13 ± 258.46 | 17,933.3 ± 8602.70 | 8.67 ± 3.70 | 41.65 ± 7.63 | 5.67 ± 3.61 | |
| Dog | IV | 0.07 | 1943.66 ± 132.18 | 36.15 ± 2.48 | 353.53 ± 72.14 | 6.76 ± 1.15 | 238.57 ± 13.90 | 2.00 ± 0.00 |
| IM | 0.035 | 121.23 ± 19.58 | 295.01 ± 47.07 | 4261.72 ± 538.69 | 10.24 ± 2.32 | 5.32 ± 0.88 | 9.50 ± 3.15 | |
| IM | 0.07 | 259.78 ± 59.90 | 281.62 ± 63.48 | 4968.49 ± 1021.32 | 12.50 ± 2.38 | 9.40 ± 2.00 | 9.17 ± 2.04 | |
| IM | 0.14 | 600.42 ± 172.99 | 251.61 ± 78.50 | 4170.37 ± 1411.30 | 11.50 ± 1.31 | 21.82 ± 7.09 | 11.67 ± 4.08 | |
| IM | 0.28 | 1440.13 ± 128.75 | 195.67 ± 17.27 | 5565.50 ± 1948.35 | 19.65 ± 6.21 | 49.36 ± 9.50 | 15.00 ± 0.00 |
| Model | Description | nParm | −2LL | AIC | Δ−2LL | ΔAIC |
|---|---|---|---|---|---|---|
| Absorption Model | ||||||
| 01 | Tlag | 9 | 2195.57 | 2213.57 | ||
| 02 a | No Tlag | 7 | 1535.18 | 1549.18 | −660.39 | −664.39 |
| Residual Model | ||||||
| 02-01 | Additive | 7 | 1535.18 | 1549.18 | ||
| 02-02 a | Log additive | 7 | 230.50 | 244.50 | −1304.68 | −1304.68 |
| 02-03 | Muliticative | 7 | 1259.60 | 1273.60 | 1029.10 | 1029.10 |
| IIV Model | ||||||
| 02-02-01 a | Do not remove | 230.50 | 244.50 | |||
| 02-02-02 | Remove Ka | 6 | 213.50 | 225.50 | −17.00 | −19.00 |
| 02-02-03 | Remove V | 6 | 419.64 | 431.64 | 206.14 | 206.14 |
| 02-02-04 | Remove Cl | 5 | 501.89 | 513.89 | 82.25 | 82.25 |
| 02-02-05 | Remove Ka and V | 5 | 531.09 | 541.09 | 29.20 | 27.20 |
| 02-02-06 | Remove Ka and CL | 5 | 729.47 | 739.47 | 198.39 | 198.39 |
| 02-02-07 | Remove V and CL | 5 | 789.12 | 799.12 | 59.65 | 59.65 |
| Covariates Model | ||||||
| 02-02-01-01 | WT on CL | 8 | 115.95 | 131.95 | −114.56 | −112.56 |
| 02-02-01-02 | WT on V | 8 | 75.52 | 91.52 | −154.98 | −152.98 |
| 02-02-01-03 b | WT on CL and V | 9 | −33.82 | −15.82 | −264.33 | −260.33 |
| Parameter | Final Model | Bootstrap | ||||
|---|---|---|---|---|---|---|
| Estimate | SE | RSE (%) | Shrinkage | Median | 95% CI | |
| Katv | 7.55 | 0.48 | 6.35 | 7.54 | 6.54~8.50 | |
| Vtv | 15.87 | 1.30 | 8.22 | 15.80 | 12.91~18.53 | |
| CLtv | 113.71 | 5.01 | 4.40 | 113.48 | 104.13~124.14 | |
| dCLdwt | 0.52 | 0.02 | 3.75 | 0.52 | 0.48~0.56 | |
| dVdwt | 1.10 | 0.05 | 4.50 | 1.10 | 1.01~1.21 | |
| ω2Ka | 0.10 | 0.05 | 47.46 | 0.25 | 0.16 | |
| ω2V | 0.04 | 0.02 | 36.00 | 0.25 | ||
| ω2Cl | 0.04 | 0.01 | 18.26 | 0.04 | ||
| σ | 0.16 | 0.01 | 8.23 | 0.16 | 0.13~0.18 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, X.; Yuan, M.; Wang, H.; Zhangsun, D.; Yu, G.; Che, J.; Luo, S. Novel αO-conotoxin GeXIVA[1,2] Nonaddictive Analgesic with Pharmacokinetic Modelling-Based Mechanistic Assessment. Pharmaceutics 2022, 14, 1789. https://doi.org/10.3390/pharmaceutics14091789
Zhu X, Yuan M, Wang H, Zhangsun D, Yu G, Che J, Luo S. Novel αO-conotoxin GeXIVA[1,2] Nonaddictive Analgesic with Pharmacokinetic Modelling-Based Mechanistic Assessment. Pharmaceutics. 2022; 14(9):1789. https://doi.org/10.3390/pharmaceutics14091789
Chicago/Turabian StyleZhu, Xiaoyu, Mei Yuan, Huanbai Wang, Dongting Zhangsun, Gang Yu, Jinjing Che, and Sulan Luo. 2022. "Novel αO-conotoxin GeXIVA[1,2] Nonaddictive Analgesic with Pharmacokinetic Modelling-Based Mechanistic Assessment" Pharmaceutics 14, no. 9: 1789. https://doi.org/10.3390/pharmaceutics14091789