
Galactosed and Reduction-Responsive Nanoparticles Assembled from Trimethylchitosan–Camptothecin Conjugates for Enhanced Hepatocellular Carcinoma Therapy


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials, Cell Lines, and Animals
2.2. Synthesis and Characterization of GT-ss-CPT Conjugate
2.3. Preparation and Characterization of GT-ss-CPT NPs
2.4. In Vitro Drug Release
2.5. In Vitro Cytotoxicity
2.6. Cell Apoptosis
2.7. Hemolysis
2.8. In Vivo Antitumor Efficacy
2.9. Statistical Analysis
3. Results and Discussion
3.1. Synthesis and Characterization of GT-ss-CPT Conjugate
3.2. Preparation and Characterization of GT-ss-CPT NPs
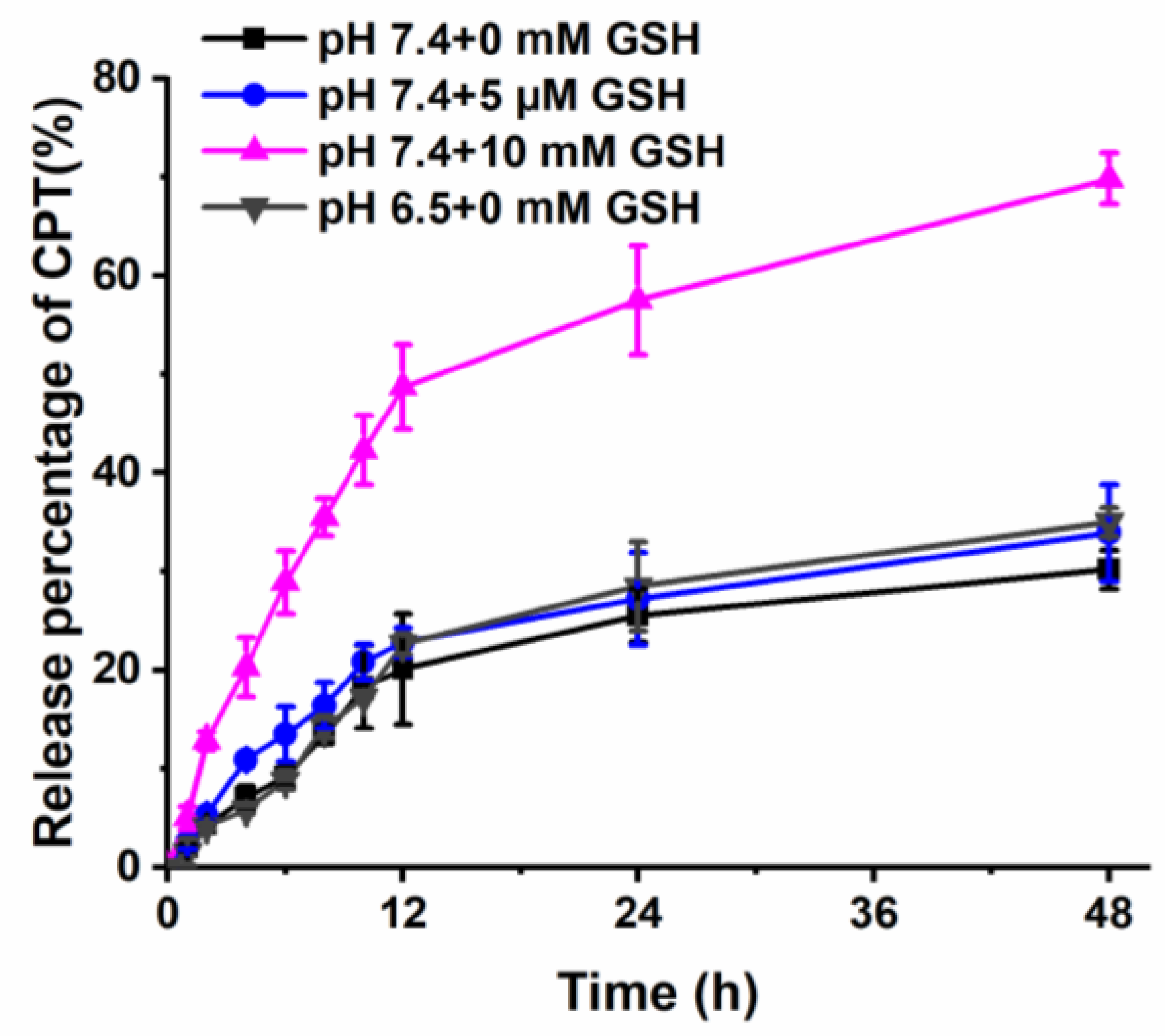
3.3. In Vitro Drug Release
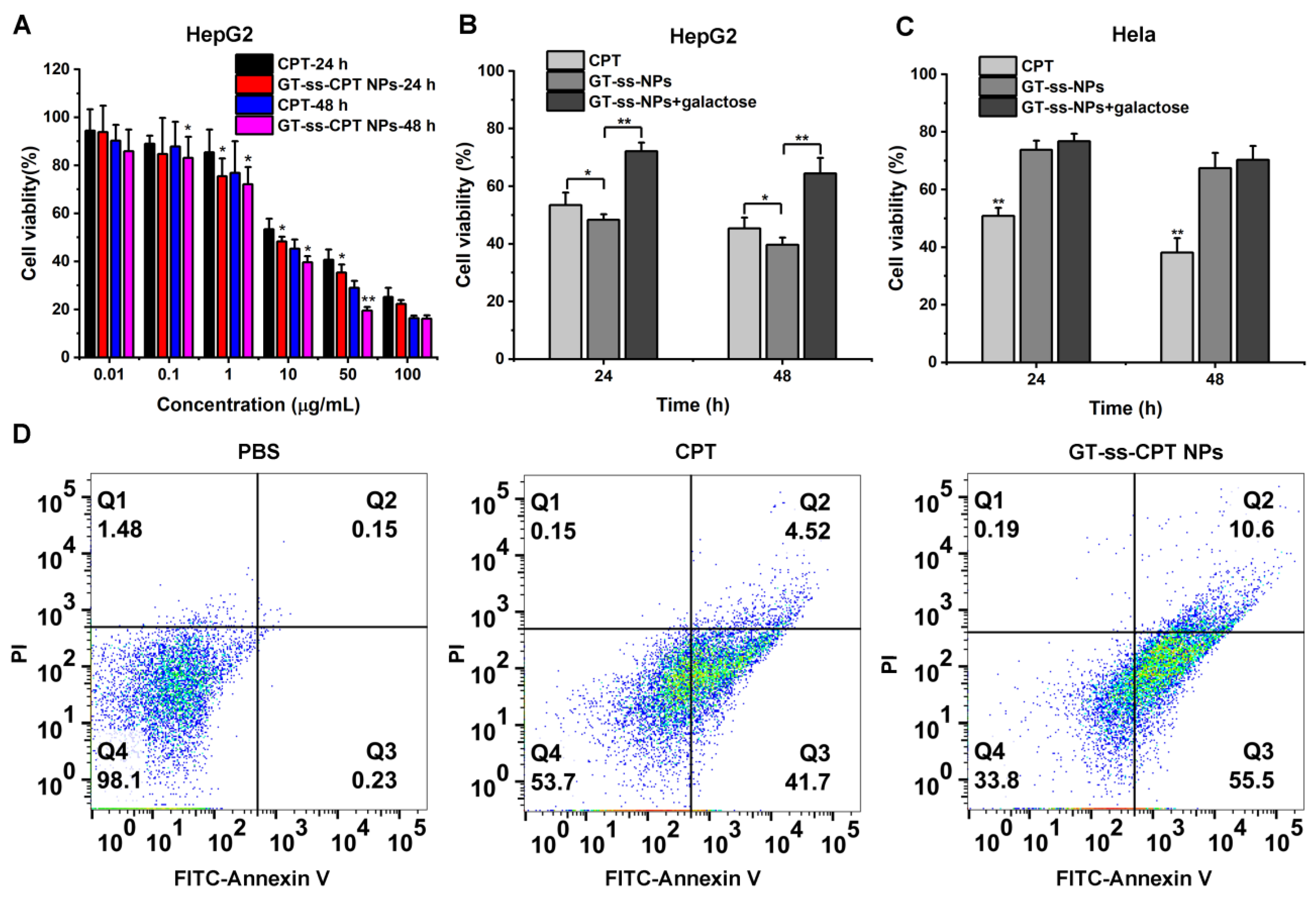
3.4. In Vitro Cytotoxicity and Cell Apoptosis
3.5. Hemolysis
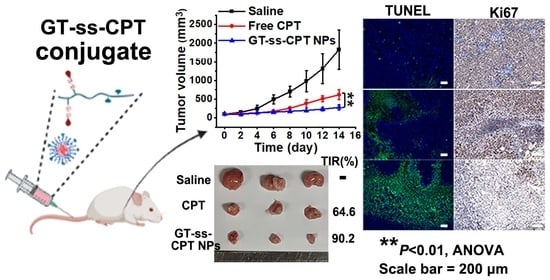
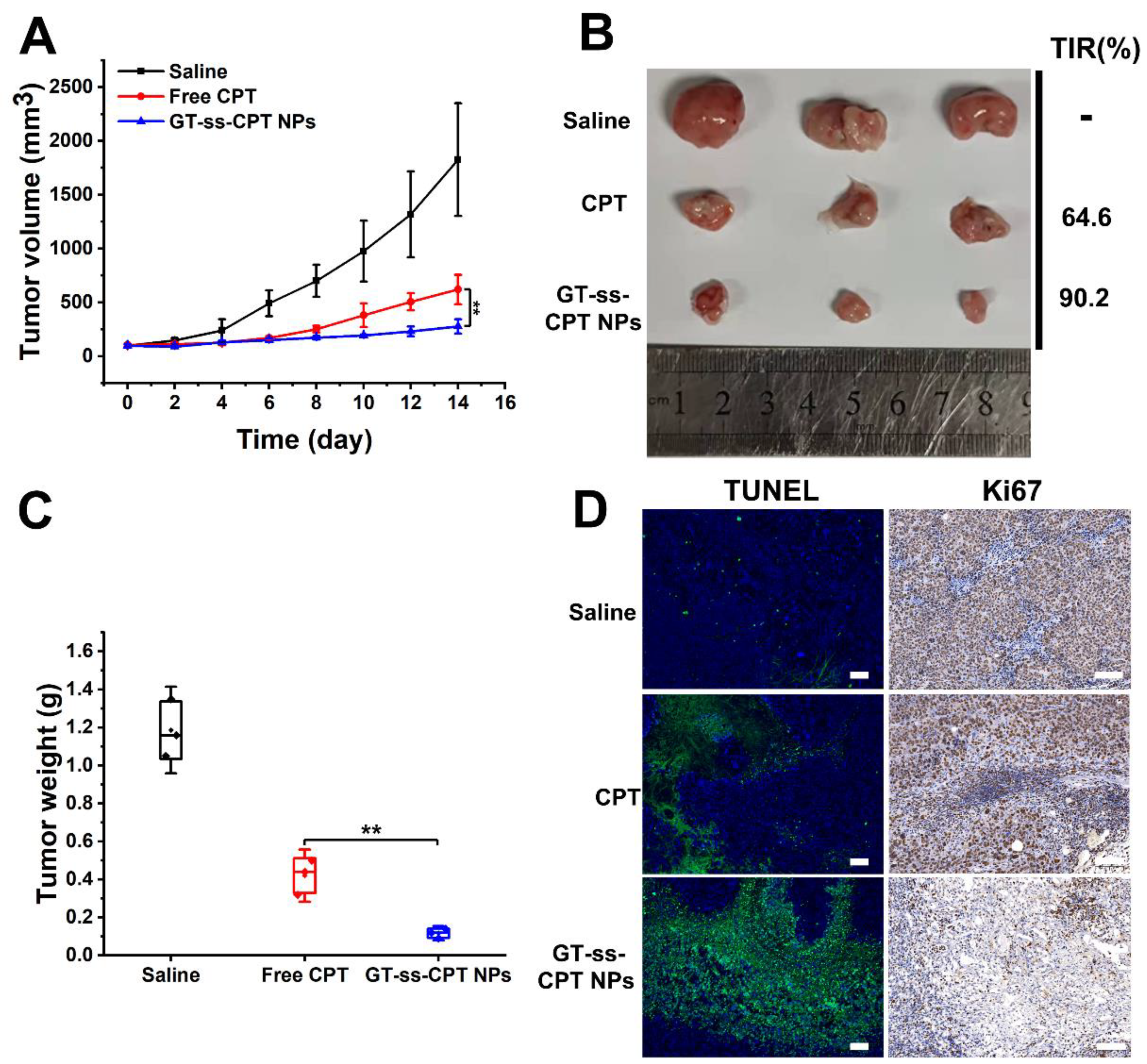
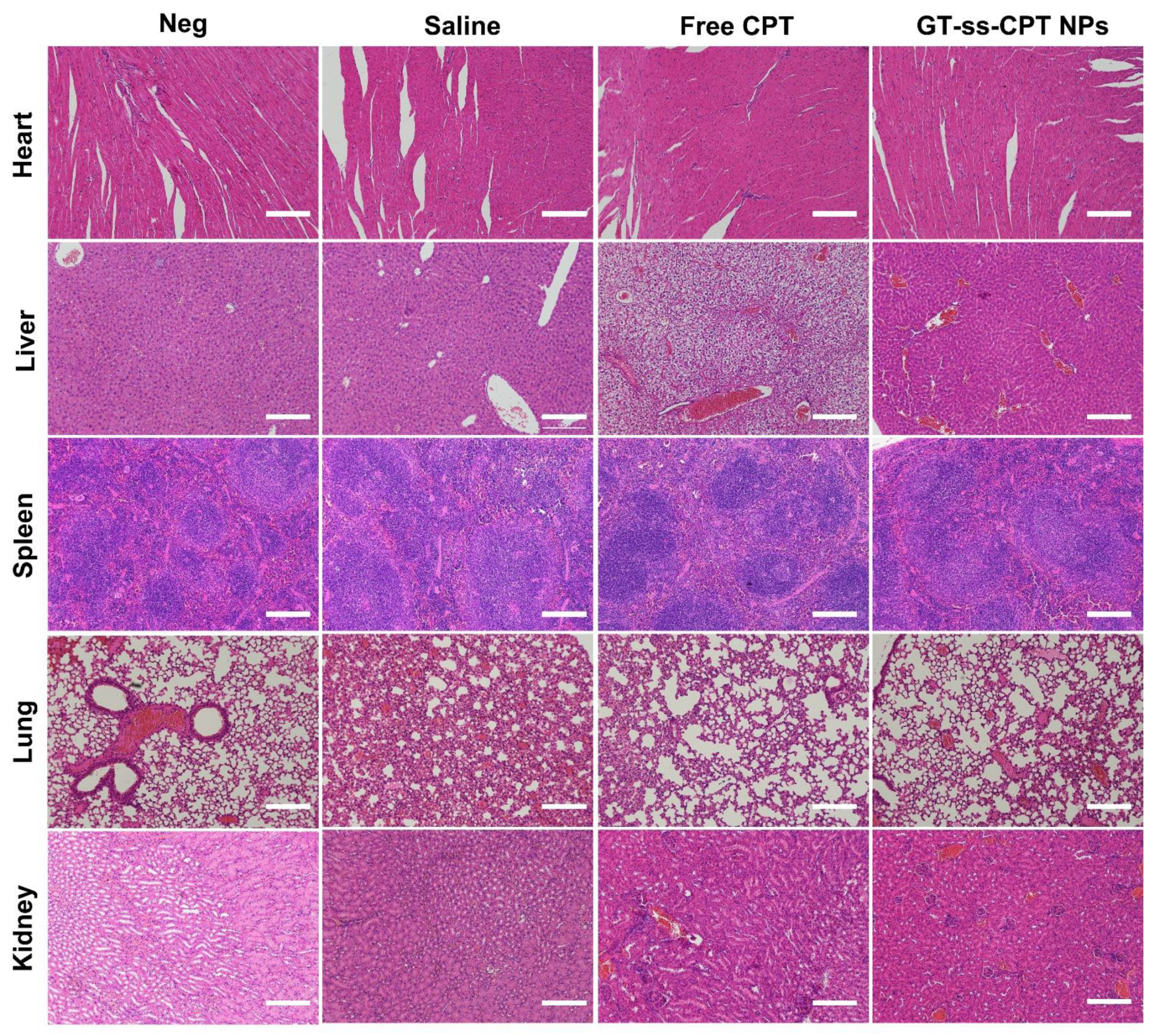
3.6. In Vivo Antitumor Efficacy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, G.; Sun, B.; Li, Y.; Luo, C.; He, Z.; Sun, J. Small-Molecule Prodrug Nanoassemblies: An Emerging Nanoplatform for Anticancer Drug Delivery. Small 2021, 17, e2101460. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Ma, Y.; Li, L. The application of prodrug-based nano-drug delivery strategy in cancer combination therapy. Colloid. Surf. B Biointerfaces 2016, 146, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Taresco, V.; Alexander, C.; Singh, N.; Pearce, A.K. Stimuli-Responsive Prodrug Chemistries for Drug Delivery. Adv. Therapeut. 2018, 1, 1800030. [Google Scholar] [CrossRef] [Green Version]
- Anwanwan, D.; Singh, S.K.; Singh, S.; Saikam, V.; Singh, R. Challenges in liver cancer and possible treatment approaches. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188314. [Google Scholar] [CrossRef]
- D’Souza, A.A.; Devarajan, P.V. Asialoglycoprotein receptor mediated hepatocyte targeting—Strategies and applications. J. Control. Release 2015, 203, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Zhong, J.; Zhao, M.; Tang, Y.; Han, N.; Hua, L.; Xu, T.; Wang, C.; Zhu, B. Galactose-modified selenium nanoparticles for targeted delivery of doxorubicin to hepatocellular carcinoma. Drug Deliv. 2019, 26, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Yu, P.; Chen, Y.; Sun, B.; Dong, P.; Zhu, T.; Meng, X. N-acetylgalactosamine-decorated nanoliposomes for targeted delivery of paclitaxel to hepatocellular carcinoma. Eur. J. Med. Chem. 2021, 222, 113605. [Google Scholar] [CrossRef]
- Lu, L.; Li, B.; Lin, C.; Li, K.; Liu, G.; Xia, Z.; Luo, Z.; Cai, K. Redox-responsive amphiphilic camptothecin prodrug nanoparticles for targeted liver tumor therapy. J. Mater. Chem. B 2020, 8, 3918–3928. [Google Scholar] [CrossRef]
- Khdair, A.; Hamad, I.; Alkhatib, H.; Bustanji, Y.; Mohammad, M.; Tayem, R.; Aiedeh, K. Modified-chitosan nanoparticles: Novel drug delivery systems improve oral bioavailability of doxorubicin. Eur. J. Pharm. Sci. 2016, 93, 38–44. [Google Scholar] [CrossRef]
- Pramanik, A.; Laha, D.; Pramanik, P.; Karmakar, P. A novel drug “copper acetylacetonate” loaded in folic acid-tagged chitosan nanoparticle for efficient cancer cell targeting. J. Drug Target 2014, 22, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Li, N.M.; Gao, P.; Yang, S.; Ning, Q.; Huang, W.; Li, Z.P.; Ye, P.J.; Xiang, L.; He, D.X.; et al. In vitro and in vivo evaluation of macromolecular prodrug GC-FUA based nanoparticle for hepatocellular carcinoma chemotherapy. Drug Deliv. 2017, 24, 459–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, L.; Tang, C.; Yin, C.H. Effect of binding affinity for siRNA on the in vivo antitumor efficacy of polyplexes. Biomaterials 2013, 34, 5317–5327. [Google Scholar] [CrossRef]
- Han, L.; Tang, C.; Yin, C. Enhanced antitumor efficacies of multifunctional nanocomplexes through knocking down the barriers for siRNA delivery. Biomaterials 2015, 44, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhao, S.; Meng, F.; Wang, H.; Sun, L.; Li, G.; Gao, F.; Chen, F. Nrf2 Down-Regulation by Camptothecin Favors Inhibiting Invasion, Metastasis and Angiogenesis in Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 661157. [Google Scholar] [CrossRef]
- Thomas, C.J.; Rahier, N.J.; Hecht, S.M. Camptothecin: Current perspectives. Bioorg. Med. Chem. 2004, 12, 1585–1604. [Google Scholar] [CrossRef]
- Slichenmyer, W.J.; Rowinsky, E.K.; Donehower, R.C.; Kaufmann, S.H. The current status of camptothecin analogues as antitumor agents. J. Natl. Cancer Inst. 1993, 85, 271–291. [Google Scholar] [CrossRef]
- Wen, Y.; Wang, Y.; Liu, X.; Zhang, W.; Xiong, X.; Han, Z.; Liang, X. Camptothecin-based nanodrug delivery systems. Cancer Biol. Med. 2017, 14, 363–370. [Google Scholar] [CrossRef] [Green Version]
- Hershberger, K.K.; Gauger, A.J.; Bronstein, L.M. Utilizing Stimuli Responsive Linkages to Engineer and Enhance Polymer Nanoparticle-Based Drug Delivery Platforms. ACS Appl. Bio Mater. 2021, 4, 4720–4736. [Google Scholar] [CrossRef]
- Sun, H.; Zhang, Y.; Zhong, Z. Reduction-sensitive polymeric nanomedicines: An emerging multifunctional platform for targeted cancer therapy. Adv. Drug Deliv. Rev. 2018, 132, 16–32. [Google Scholar] [CrossRef]
- Hou, C.; Ma, N.; Shen, Z.; Chi, G.; Chao, S.; Pei, Y.; Chen, L.; Lu, Y.; Pei, Z. A GSH-Responsive Nanoprodrug System Based on Self-Assembly of Lactose Modified Camptothecin for Targeted Drug Delivery and Combination Chemotherapy. Int. J. Nanomed. 2020, 15, 10417–10424. [Google Scholar] [CrossRef]
- Chen, Z.; He, N.; Chen, M.; Zhao, L.; Li, X. Tunable conjugation densities of camptothecin on hyaluronic acid for tumor targeting and reduction-triggered release. Acta Biomater. 2016, 43, 195–207. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, M.; Luo, X.; Zhang, H.; Liu, C.; Li, H.; Li, X. Tuning multiple arms for camptothecin and folate conjugations on star-shaped copolymers to enhance glutathione-mediated intracellular drug delivery. Polym. Chem. 2015, 6, 2192–2203. [Google Scholar] [CrossRef]
- Chen, D.; Huang, Y.; Xu, S.; Jiang, H.; Wu, J.; Jin, X.; Zhu, X. Self-Assembled Polyprodrug Amphiphile for Subcutaneous Xenograft Tumor Inhibition with Prolonged Acting Time In Vivo. Macromol. Biosci. 2017, 17, 1700174. [Google Scholar] [CrossRef]
- Zhou, H.Y.; Zhang, Y.P.; Zhang, W.F.; Chen, X.G. Biocompatibility and characteristics of injectable chitosan-based thermosensitive hydrogel for drug delivery. Carbohyd. Polym. 2011, 83, 1643–1651. [Google Scholar] [CrossRef]
- Kong, F.; Tang, C.; Yin, C. Benzylguanidine and Galactose Double-Conjugated Chitosan Nanoparticles with Reduction Responsiveness for Targeted Delivery of Doxorubicin to CXCR 4 Positive Tumors. Bioconjug. Chem. 2020, 31, 2446–2455. [Google Scholar] [CrossRef]
- Lu, N.; Xi, L.; Zha, Z.; Wang, Y.; Han, X.; Ge, Z. Acid-responsive endosomolytic polymeric nanoparticles with amplification of intracellular oxidative stress for prodrug delivery and activation. Biomater. Sci. 2021, 9, 4613–4629. [Google Scholar] [CrossRef]
- Pramanik, A.; Xu, Z.; Shamsuddin, S.H.; Khaled, Y.S.; Ingram, N.; Maisey, T.; Tomlinson, D.; Coletta, P.L.; Jayne, D.; Hughes, T.A.; et al. Affimer Tagged Cubosomes: Targeting of Carcinoembryonic Antigen Expressing Colorectal Cancer Cells Using In Vitro and In Vivo Models. ACS Appl. Mater. Interfaces 2022, 14, 11078–11091. [Google Scholar] [CrossRef]
- Jia, Y.Y.; Zhang, J.J.; Zhang, Y.X.; Wang, W.; Li, C.; Zhou, S.Y.; Zhang, B.L. Construction of redox-responsive tumor targeted cisplatin nano-delivery system for effective cancer chemotherapy. Int. J. Pharm. 2020, 580, 119190. [Google Scholar] [CrossRef]
- Tahvilian, R.; Tajani, B.; Sadrjavadi, K.; Fattahi, A. Preparation and characterization of pH-sensitive camptothecin-cis-aconityl grafted chitosan oligosaccharide nanomicelles. Int. J. Biol. Macromol. 2016, 92, 795–802. [Google Scholar] [CrossRef]
- Ma, P.; Sun, Y.; Chen, J.; Li, H.; Zhu, H.; Gao, X.; Bi, X.; Zhang, Y. Enhanced anti-hepatocarcinoma efficacy by GLUT1 targeting and cellular microenvironment-responsive PAMAM-camptothecin conjugate. Drug Deliv. 2018, 25, 153–165. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Zhao, Y.; Liu, Y.; Chang, X.; Chen, C.; Zhao, Y. Cellular uptake, intracellular trafficking, and cytotoxicity of nanomaterials. Small 2011, 7, 1322–1337. [Google Scholar] [CrossRef]
- He, Y.; Xu, Y.; Huang, Y.; Quang, H.; Xia, X.; Zhao, H.; Xiao, Y.; Lu, W.; Yu, J. Redox sensitive nano-capsules self-assembled from hyaluronic acid-hydroxychloroquine conjugates for CD44-targeted delivery of hydroxychloroquine to combat breast cancer metastasis in vitro and in vivo. Colloid. Surf. B Biointerfaces 2022, 210, 112249. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yuan, J.; Luo, X.; Chen, M.; Chen, Z.; Zhao, Y.; Li, X. Folate-decorated and reduction-sensitive micelles assembled from amphiphilic polymer-camptothecin conjugates for intracellular drug delivery. Mol. Pharm. 2014, 11, 4258–4269. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Bremner, D.H.; Ye, Y.; Lou, J.; Niu, S.; Zhu, L.M. A dual-prodrug nanoparticle based on chitosan oligosaccharide for enhanced tumor-targeted drug delivery. Colloid. Surf. A 2021, 619, 126512. [Google Scholar] [CrossRef]
- Podsiedlik, M.; Markowicz-Piasecka, M.; Sikora, J. Erythrocytes as model cells for biocompatibility assessment, cytotoxicity screening of xenobiotics and drug delivery. Chem. Biol. Interact. 2020, 332, 109305. [Google Scholar] [CrossRef] [PubMed]
- International Organization for Standardization. Biological Evaluation of Medical Devices—Part 4: Selection of Tests for Interactions with Blood; International Organization for Standardization: Geneva, Switzerland, 2017; pp. 10993–10994. Available online: https://www.iso.org/standard/63448.html (accessed on 16 June 2021).
- Hentze, H.; Latta, M.; Kunstle, G.; Dhakshinamoorthy, S.; Ng, P.Y.; Porter, A.G.; Wendel, A. Topoisomerase inhibitor camptothecin sensitizes mouse hepatocytes in vitro and in vivo to TNF-mediated apoptosis. Hepatology 2004, 39, 1311–1320. [Google Scholar] [CrossRef]
- Malhotra, S.; Dumoga, S.; Joshi, A.; Mohanty, S.; Singh, N. Polymeric micelles coated with hybrid nanovesicles enhance the therapeutic potential of the reversible topoisomerase inhibitor camptothecin in a mouse model. Acta Biomater. 2021, 121, 579–591. [Google Scholar] [CrossRef]
- Qu, Y.; Chu, B.; Wei, X.; Lei, M.; Hu, D.; Zha, R.; Zhong, L.; Wang, M.; Wang, F.; Qian, Z. Redox/pH dual-stimuli responsive camptothecin prodrug nanogels for “on-demand” drug delivery. J. Control. Release 2019, 296, 93–106. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, C.; Qin, J.; Liu, X.; Kong, F. Galactosed and Reduction-Responsive Nanoparticles Assembled from Trimethylchitosan–Camptothecin Conjugates for Enhanced Hepatocellular Carcinoma Therapy. Pharmaceutics 2022, 14, 1315. https://doi.org/10.3390/pharmaceutics14071315
Fu C, Qin J, Liu X, Kong F. Galactosed and Reduction-Responsive Nanoparticles Assembled from Trimethylchitosan–Camptothecin Conjugates for Enhanced Hepatocellular Carcinoma Therapy. Pharmaceutics. 2022; 14(7):1315. https://doi.org/10.3390/pharmaceutics14071315
Chicago/Turabian StyleFu, Chen, Jingcan Qin, Xinlong Liu, and Fei Kong. 2022. "Galactosed and Reduction-Responsive Nanoparticles Assembled from Trimethylchitosan–Camptothecin Conjugates for Enhanced Hepatocellular Carcinoma Therapy" Pharmaceutics 14, no. 7: 1315. https://doi.org/10.3390/pharmaceutics14071315