
Pharmacokinetic–Pharmacometabolomic Approach in Early-Phase Clinical Trials: A Way Forward for Targeted Therapy in Type 2 Diabetes

 ,
, 
Abstract
:1. Introduction
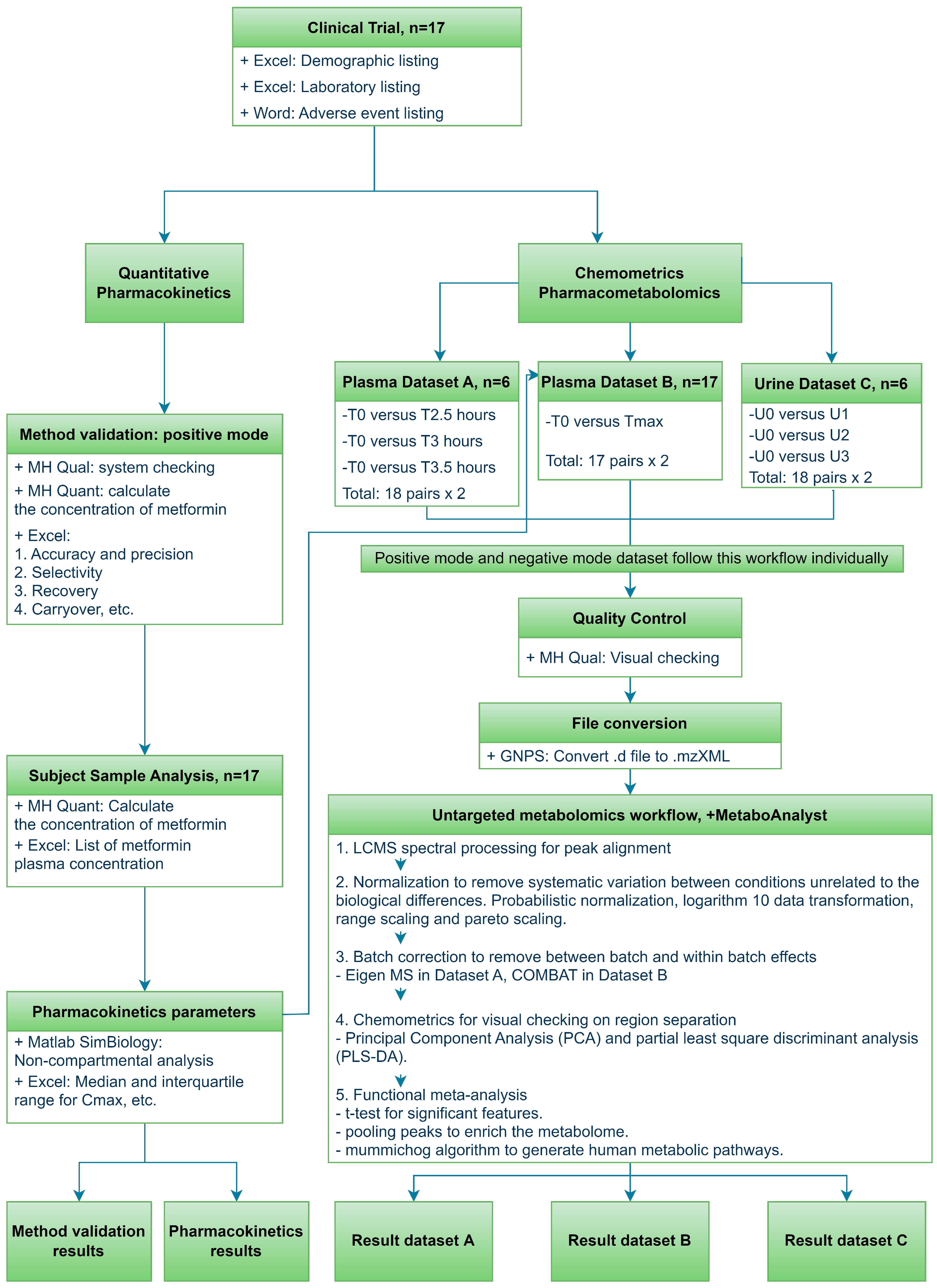
2. Materials and Methods
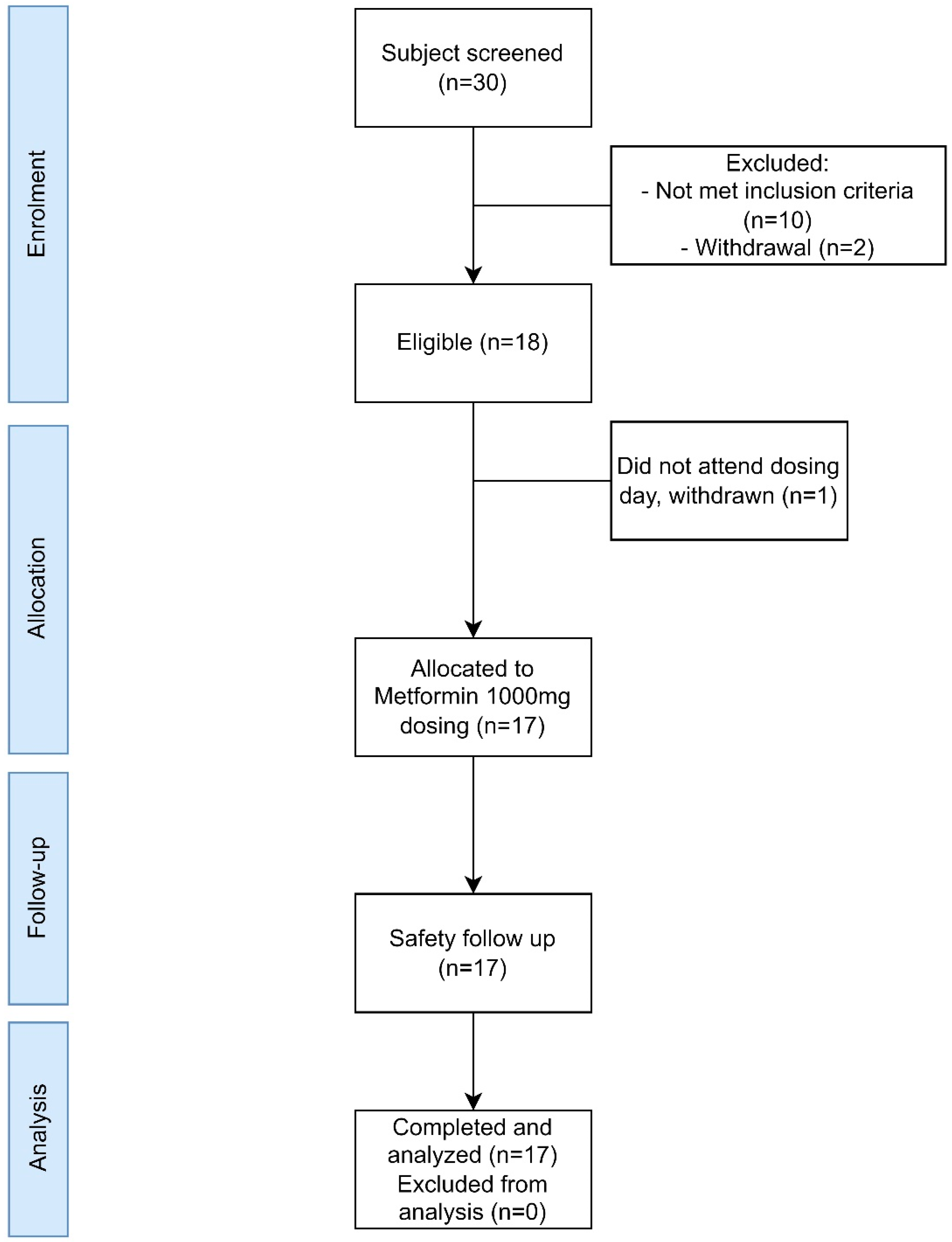
2.1. Study Design
2.2. Bioanalytical
2.3. Method Validation and Statistical Analysis for Pharmacokinetics
2.4. Data Processing and Statistical Method for Untargeted Metabolomics
3. Results
3.1. Clinical Trial Results
3.2. Metformin Analytical Method Validation
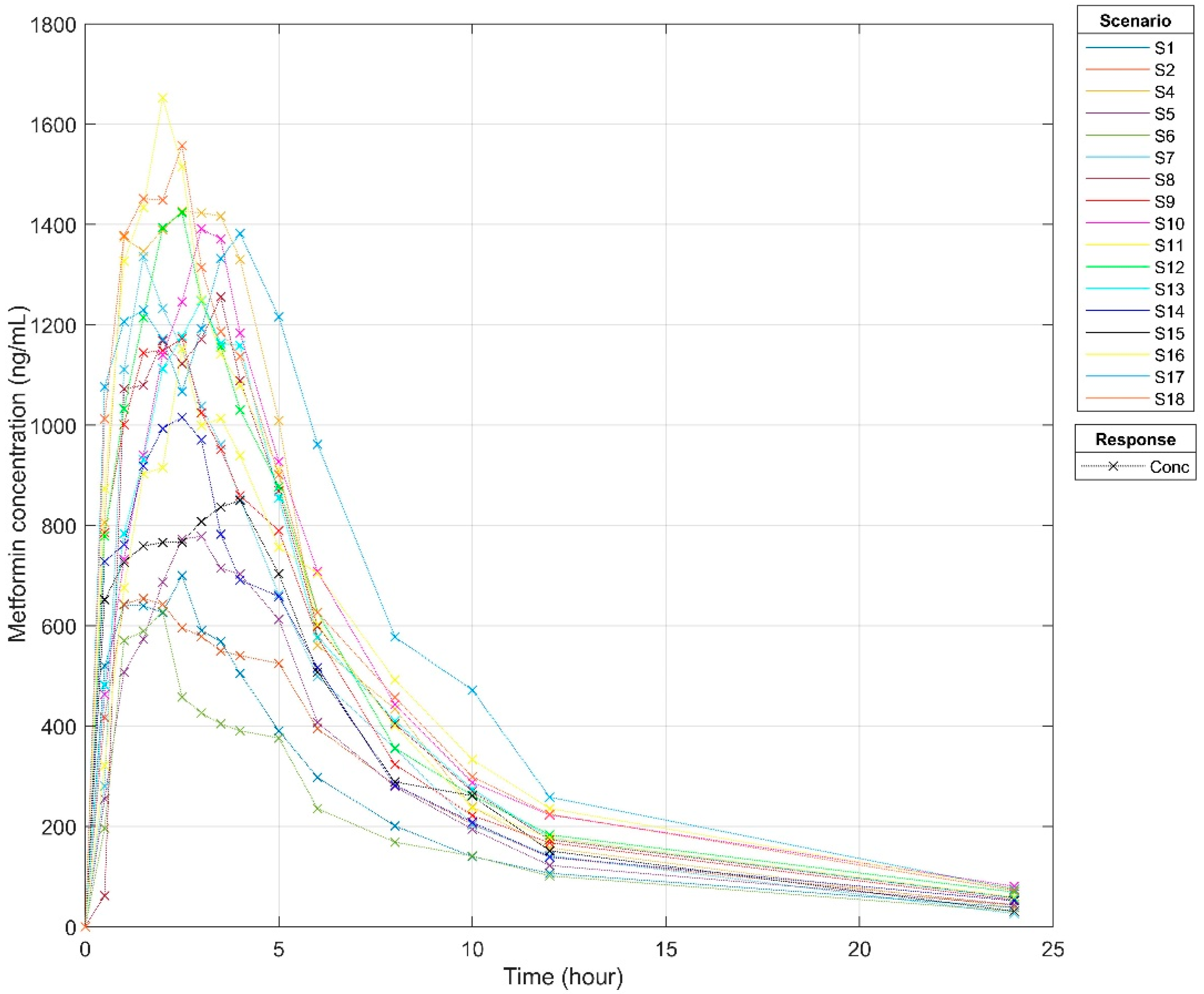
3.3. Pharmacokinetics Profiles
3.4. Metabolomics Analysis of Metformin in Plasma and Urine Samples
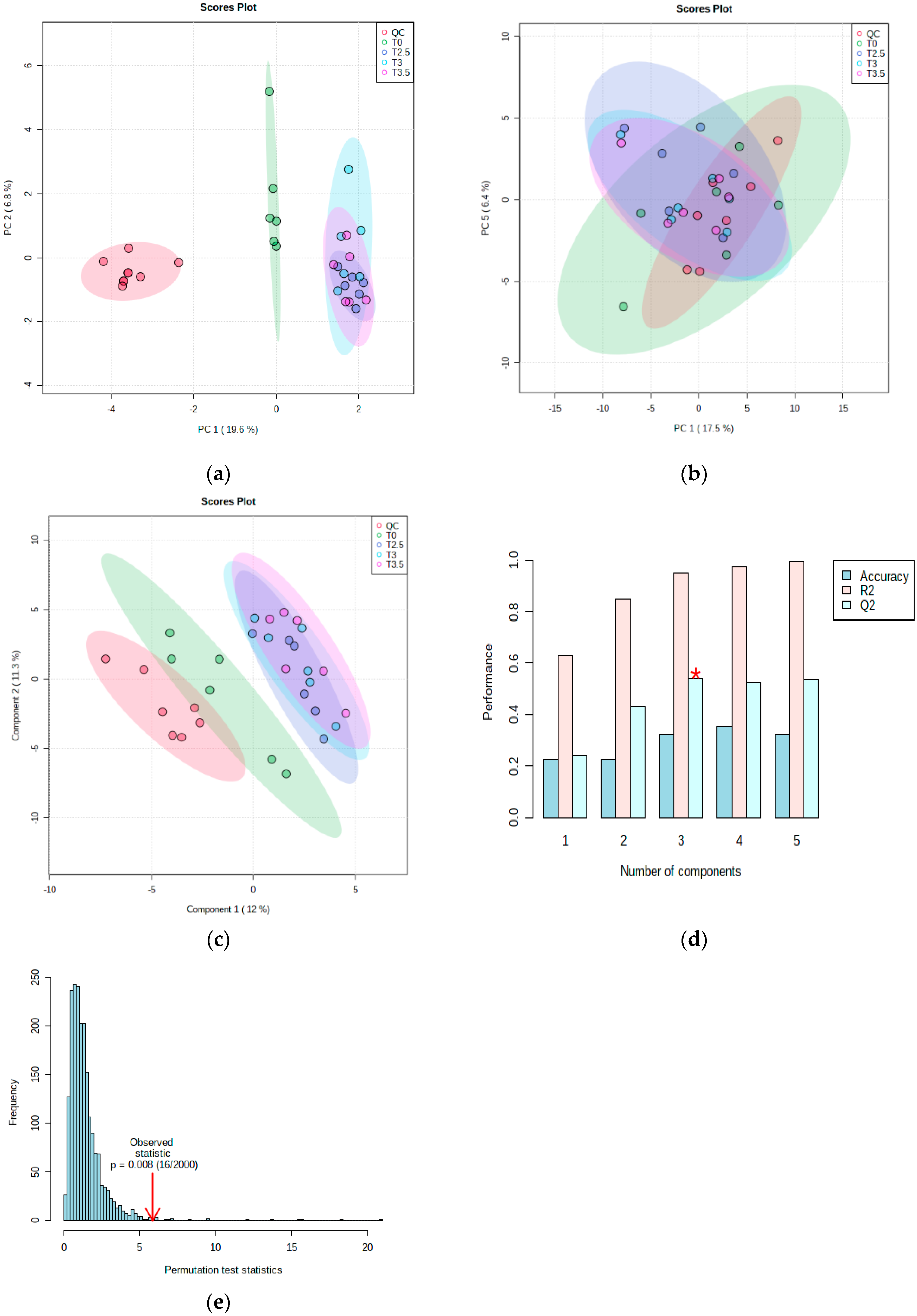
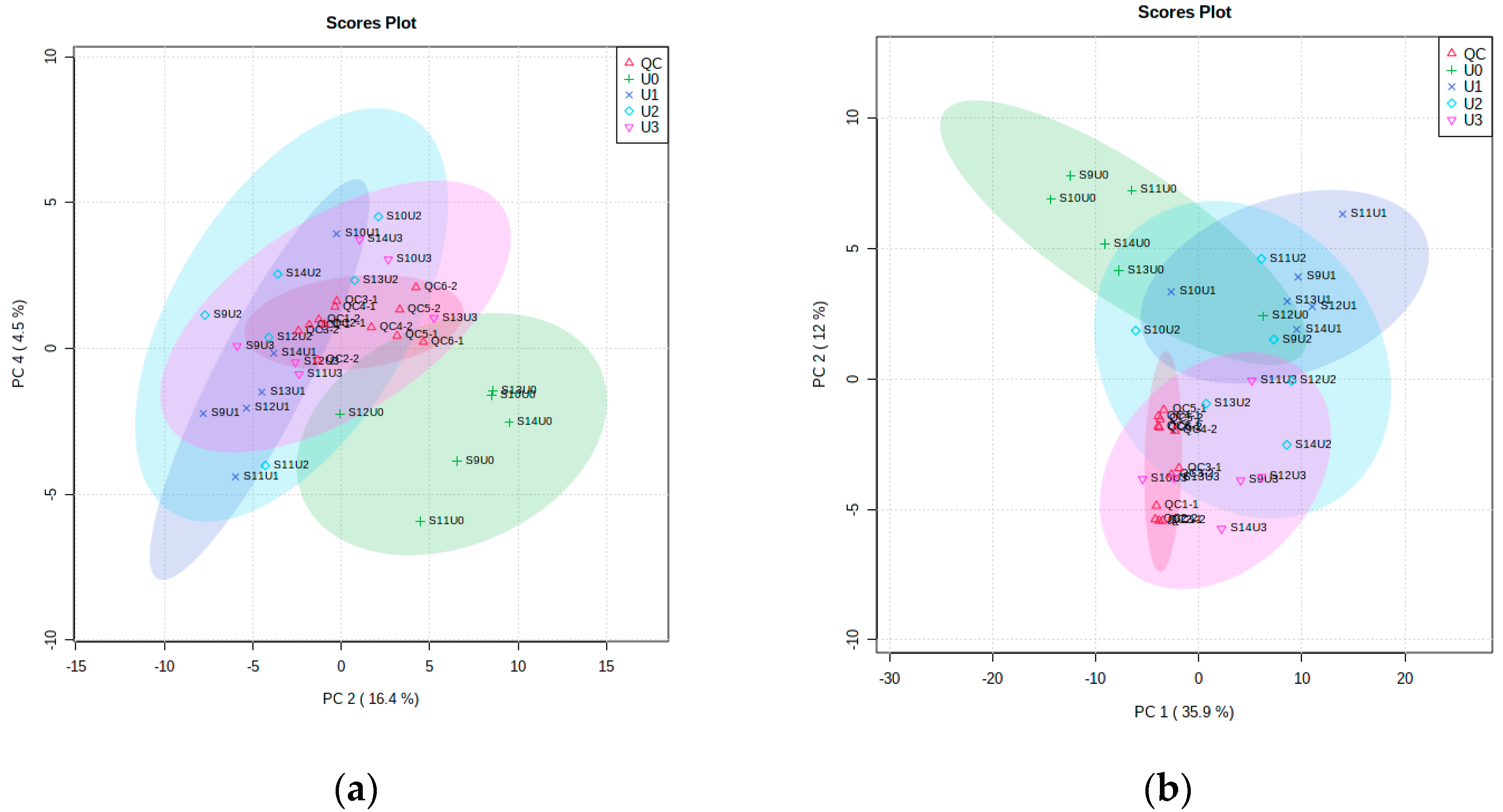
3.4.1. Metabolomic Multivariate Analysis
3.4.2. Metabolomic Functional Pathway Analyses
4. Discussion
4.1. Clinical Trial
4.2. Pharmacokinetics
4.3. Metabolomics in Plasma and Urine Samples
4.3.1. Arginine and Proline Metabolism
4.3.2. Valine, Leucine and Isoleucine Biosynthesis
4.3.3. Glutathione Metabolism
4.3.4. Galactose Metabolism
4.3.5. Tryptophan Metabolism
4.3.6. Retinol Metabolism
4.3.7. Starch and Sucrose Metabolism
4.3.8. Glycosaminoglycan Degradation
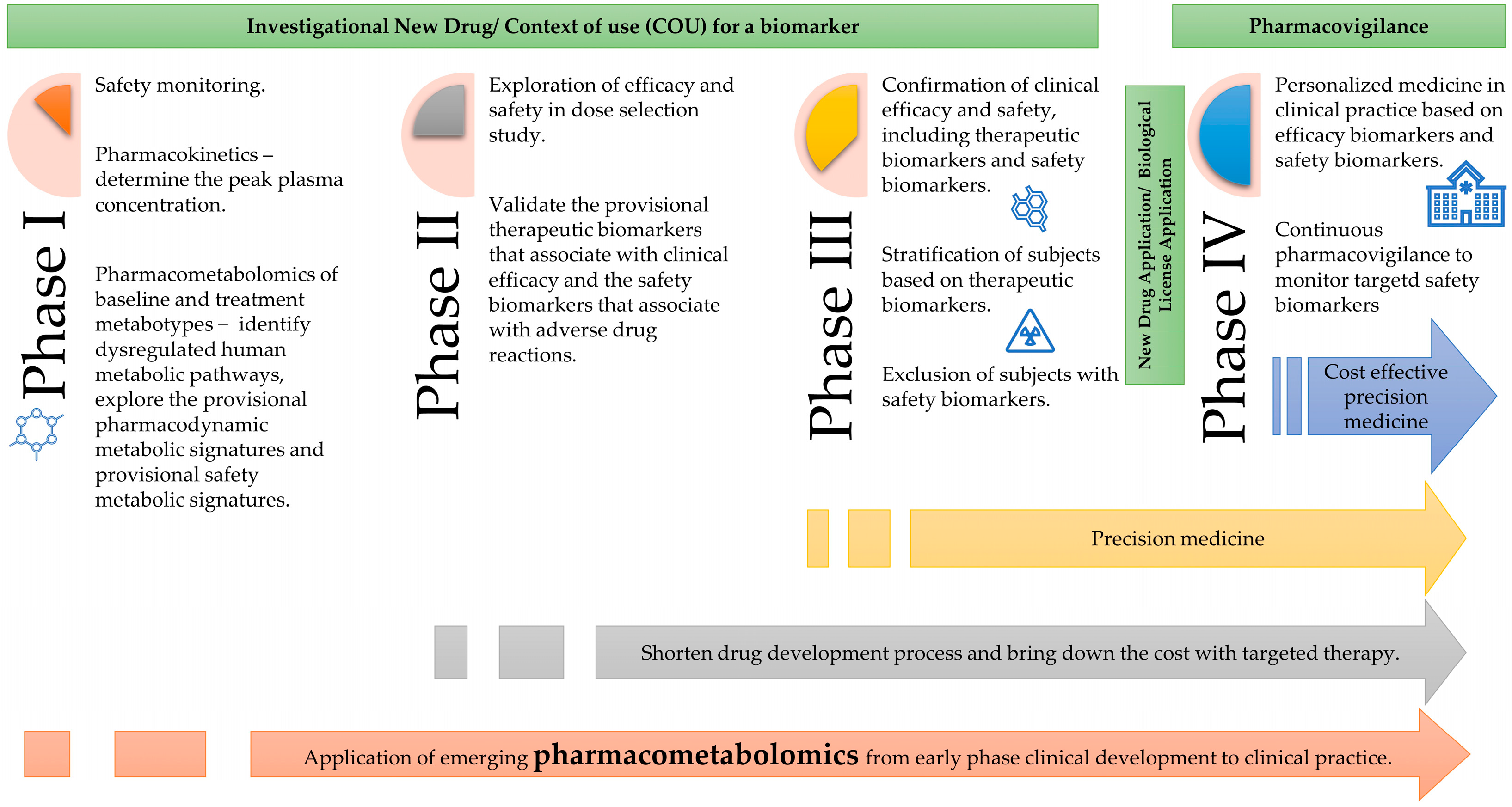
4.4. Application of Pharmacometabolomics in Clinical Drug Development
4.5. Limitation and Future Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| LCMS | liquid chromatography mass spectrometry; |
| Cmax | maximum plasma concentration; |
| SD | standard deviation; |
| AUC | area under the curve; |
| Tmax | time to reach maximum plasma concentration; |
| BCAA | branched-chain amino acid |
References
- Schranner, D.; Kastenmüller, G.; Schönfelder, M.; Römisch-Margl, W.; Wackerhage, H. Metabolite Concentration Changes in Humans After a Bout of Exercise: A Systematic Review of Exercise Metabolomics Studies. Sports Med.—Open 2020, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, S.Z.; Begley, P.; Mullard, G.; Hollywood, K.A.; Bishop, P.N. Introduction to metabolomics and its applications in ophthalmology. Eye 2016, 30, 773–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaddurah-Daouk, R.; Weinshilboum, R.M. Pharmacometabolomics: Implications for clinical pharmacology and systems pharmacology. Clin. Pharmacol. Ther. 2014, 95, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Beger, R.D.; Schmidt, M.A.; Kaddurah-Daouk, R. Current Concepts in Pharmacometabolomics, Biomarker Discovery, and Precision Medicine. Metabolites 2020, 10, 129. [Google Scholar] [CrossRef] [Green Version]
- Kantae, V.; Krekels, E.H.J.; Esdonk, M.J.V.; Lindenburg, P.; Harms, A.C.; Knibbe, C.A.J.; Van der Graaf, P.H.; Hankemeier, T. Integration of pharmacometabolomics with pharmacokinetics and pharmacodynamics: Towards personalized drug therapy. Metab. Off. J. Metab. Soc. 2016, 13, 9. [Google Scholar] [CrossRef] [Green Version]
- Waller, D.G.; Sampson, A.P. 2-Pharmacokinetics. In Medical Pharmacology and Therapeutics, 5th ed.; Waller, D.G., Sampson, A.P., Eds.; Elsevier: Southampton, UK, 2018; pp. 33–62. [Google Scholar]
- Brayfield, A. Martindale: The Complete Drug Reference; Pharmaceutical Press: London, UK, 2017; Volume A. [Google Scholar]
- Gong, L.; Goswami, S.; Giacomini, K.M.; Altman, R.B.; Klein, T.E. Metformin pathways: Pharmacokinetics and pharmacodynamics. Pharmacogenetics Genom. 2012, 22, 820–827. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.W. Metabolomic Approaches to Investigate the Effect of Metformin: An Overview. Int. J. Mol. Sci. 2021, 22, 10275. [Google Scholar] [CrossRef]
- LaMoia, T.E.; Shulman, G.I. Cellular and Molecular Mechanisms of Metformin Action. Endocr. Rev. 2020, 42, 77–96. [Google Scholar] [CrossRef]
- Dahabiyeh, L.A.; Mujammami, M.; Arafat, T.; Benabdelkamel, H.; Alfadda, A.A.; Abdel Rahman, A.M. A Metabolic Pattern in Healthy Subjects Given a Single Dose of Metformin: A Metabolomics Approach. Front. Pharmacol. 2021, 12, 705932. [Google Scholar] [CrossRef]
- Aleidi, S.M.; Dahabiyeh, L.A.; Gu, X.; Al Dubayee, M.; Alshahrani, A.; Benabdelkamel, H.; Mujammami, M.; Li, L.; Aljada, A.; Abdel Rahman, A.M. Obesity Connected Metabolic Changes in Type 2 Diabetic Patients Treated with Metformin. Front. Pharmacol. 2021, 11, 616157. [Google Scholar] [CrossRef]
- Aljofan, M.; Riethmacher, D. Anticancer activity of metformin: A systematic review of the literature. Future Sci. OA 2019, 5, FSO410. [Google Scholar] [CrossRef] [Green Version]
- Ladeiras-Lopes, R.; Fontes-Carvalho, R.; Bettencourt, N.; Sampaio, F.; Gama, V.; Leite-Moreira, A. Novel therapeutic targets of metformin: Metabolic syndrome and cardiovascular disease. Expert. Opin. Ther. Targets 2015, 19, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Eppinga, R.N.; Kofink, D.; Dullaart, R.P.; Dalmeijer, G.W.; Lipsic, E.; van Veldhuisen, D.J.; van der Horst, I.C.; Asselbergs, F.W.; van der Harst, P. Effect of Metformin on Metabolites and Relation with Myocardial Infarct Size and Left Ventricular Ejection Fraction After Myocardial Infarction. Circ. Cardiovasc. Genet. 2017, 10, e001564. [Google Scholar] [CrossRef] [Green Version]
- Burt, T.; Nandal, S. Pharmacometabolomics in Early-Phase Clinical Development. Clin. Transl. Sci. 2016, 9, 128–138. [Google Scholar] [CrossRef]
- BEST (Biomarkers, EndpointS, and Other Tools) Resource. Available online: https://www.ncbi.nlm.nih.gov/books/NBK338448/?report=reader (accessed on 28 April 2022).
- Agilent Technologies. MassHunter METLIN Metabolite PCD/PCDL Quick Start Guide. Available online: https://www.agilent.com/cs/library/usermanuals/Public/G6825-90008_MetlinPCDL_QuickStart.pdf (accessed on 1 July 2021).
- Dunn, W.B.; Wilson, I.D.; Nicholls, A.W.; Broadhurst, D. The importance of experimental design and QC samples in large-scale and MS-driven untargeted metabolomic studies of humans. Bioanalysis 2012, 4, 2249–2264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steuer, A.E.; Brockbals, L.; Kraemer, T. Metabolomic Strategies in Biomarker Research–New Approach for Indirect Identification of Drug Consumption and Sample Manipulation in Clinical and Forensic Toxicology? Front. Chem. 2019, 7, 319. [Google Scholar] [CrossRef] [PubMed]
- USFDA. Bioanalytical Method Validation Guidance for Industry. Available online: https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf (accessed on 4 November 2021).
- Chong, J.; Wishart, D.S.; Xia, J. Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar] [CrossRef]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.-É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef]
- Pang, Z.; Chong, J.; Li, S.; Xia, J. MetaboAnalystR 3.0: Toward an Optimized Workflow for Global Metabolomics. Metabolites 2020, 10, 186. [Google Scholar] [CrossRef]
- Karpievitch, Y.V.; Nikolic, S.B.; Wilson, R.; Sharman, J.E.; Edwards, L.M. Metabolomics data normalization with EigenMS. PLoS ONE 2014, 9, e116221. [Google Scholar] [CrossRef]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Park, Y.; Duraisingham, S.; Strobel, F.H.; Khan, N.; Soltow, Q.A.; Jones, D.P.; Pulendran, B. Predicting network activity from high throughput metabolomics. PLoS Comput. Biol. 2013, 9, e1003123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Conference of Harmonization. Guidance on General Consideration for Clinical Trials. Available online: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E8/Step4/E8_Guideline.pdf (accessed on 22 March 2022).
- Ivy, S.P.; Siu, L.L.; Garrett-Mayer, E.; Rubinstein, L. Approaches to phase 1 clinical trial design focused on safety, efficiency, and selected patient populations: A report from the clinical trial design task force of the National Cancer Institute Investigational Drug Steering Committee. Clin. Cancer Res. 2010, 16, 1726–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mokdad, A.A.; Xie, X.-J.; Zhu, H.; Gerber, D.E.; Heitjan, D.F. Statistical justification of expansion cohorts in phase 1 cancer trials. Cancer 2018, 124, 3339–3345. [Google Scholar] [CrossRef] [Green Version]
- Nyamundanda, G.; Gormley, I.C.; Fan, Y.; Gallagher, W.M.; Brennan, L. MetSizeR: Selecting the optimal sample size for metabolomic studies using an analysis based approach. BMC Bioinform. 2013, 14, 338. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, F.; Scheen, A. Understanding and overcoming metformin gastrointestinal intolerance. Diabetes Obes. Metab. 2017, 19, 473–481. [Google Scholar] [CrossRef]
- Friedrich, C.; Br, T.; Ring, A.; Meinicke, T. Bioequivalence of Glucophage ® (Metformin) Tablets from Europe and the United States Tested in Healthy Volunteers. J. Bioequivalence Bioavailab. 2014, 6, 61–66. [Google Scholar]
- Chow, S.C.; Liu, J. Meta-analysis for bioequivalence review. J. Biopharm. Stat. 1997, 7, 97–111. [Google Scholar] [CrossRef]
- Rotroff, D.M.; Oki, N.O.; Liang, X.; Yee, S.W.; Stocker, S.L.; Corum, D.G.; Meisner, M.; Fiehn, O.; Motsinger-Reif, A.A.; Giacomini, K.M.; et al. Pharmacometabolomic Assessment of Metformin in Non-diabetic, African Americans. Front. Pharm. 2016, 7, 135. [Google Scholar] [CrossRef] [Green Version]
- Menge, B.A.; Schrader, H.; Ritter, P.R.; Ellrichmann, M.; Uhl, W.; Schmidt, W.E.; Meier, J.J. Selective amino acid deficiency in patients with impaired glucose tolerance and type 2 diabetes. Regul. Pept. 2010, 160, 75–80. [Google Scholar] [CrossRef]
- Nie, C.; He, T.; Zhang, W.; Zhang, G.; Ma, X. Branched Chain Amino Acids: Beyond Nutrition Metabolism. Int. J. Mol. Sci. 2018, 19, 954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, W.; Chen, J.; Meng, Y.; Yang, J.; Cui, Q.; Zhou, Y. Metformin Alters Gut Microbiota of Healthy Mice: Implication for Its Potential Role in Gut Microbiota Homeostasis. Front. Microbiol. 2018, 9, 336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safai, N.; Suvitaival, T.; Ali, A.; Spégel, P.; Al-Majdoub, M.; Carstensen, B.; Vestergaard, H.; Ridderstråle, M. Effect of metformin on plasma metabolite profile in the Copenhagen Insulin and Metformin Therapy (CIMT) trial. Diabet. Med. 2018, 35, 944–953. [Google Scholar] [CrossRef] [PubMed]
- Lagman, M.; Ly, J.; Saing, T.; Kaur Singh, M.; Vera Tudela, E.; Morris, D.; Chi, P.-T.; Ochoa, C.; Sathananthan, A.; Venketaraman, V. Investigating the causes for decreased levels of glutathione in individuals with type II diabetes. PLoS ONE 2015, 10, e0118436. [Google Scholar] [CrossRef] [Green Version]
- Lutchmansingh, F.K.; Hsu, J.W.; Bennett, F.I.; Badaloo, A.V.; McFarlane-Anderson, N.; Gordon-Strachan, G.M.; Wright-Pascoe, R.A.; Jahoor, F.; Boyne, M.S. Glutathione metabolism in type 2 diabetes and its relationship with microvascular complications and glycemia. PLoS ONE 2018, 13, e0198626. [Google Scholar] [CrossRef] [Green Version]
- Ewis, S.A.; Abdel-Rahman, M.S. Effect of metformin on glutathione and magnesium in normal and streptozotocin-induced diabetic rats. J. Appl. Toxicol. 1995, 15, 387–390. [Google Scholar] [CrossRef]
- Elekofehinti, O.O.; Ariyo, E.O.; Iwaloye, O.; Obafemi, T.O. Co-administration of metformin and gallic acid modulates JAK/STAT signaling pathway and glutathione metabolism in fructose-fed streptozotocin diabetic Rats. Phytomedicine Plus 2022, 2, 100181. [Google Scholar] [CrossRef]
- Muthukumaran, P.; Thiyagarajan, G.; Arun Babu, R.; Lakshmi, B.S. Raffinose from Costus speciosus attenuates lipid synthesis through modulation of PPARs/SREBP1c and improves insulin sensitivity through PI3K/AKT. Chem. Biol. Interact. 2018, 284, 80–89. [Google Scholar] [CrossRef]
- Elango, D.; Rajendran, K.; Van der Laan, L.; Sebastiar, S.; Raigne, J.; Thaiparambil, N.A.; El Haddad, N.; Raja, B.; Wang, W.; Ferela, A.; et al. Raffinose Family Oligosaccharides: Friend or Foe for Human and Plant Health? Front. Plant Sci. 2022, 13, 829118. [Google Scholar] [CrossRef]
- Oxenkrug, G. Insulin resistance and dysregulation of tryptophan-kynurenine and kynurenine-nicotinamide adenine dinucleotide metabolic pathways. Mol. Neurobiol. 2013, 48, 294–301. [Google Scholar] [CrossRef] [Green Version]
- Oxenkrug, G.F. Increased Plasma Levels of Xanthurenic and Kynurenic Acids in Type 2 Diabetes. Mol. Neurobiol. 2015, 52, 805–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muzik, O.; Burghardt, P.; Yi, Z.; Kumar, A.; Seyoum, B. Successful metformin treatment of insulin resistance is associated with down-regulation of the kynurenine pathway. Biochem. Biophys. Res. Commun. 2017, 488, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Meyramov, G.; Meyramova, A. Diabetogenic Zinc Binding B-Cytotoxic Chemicals: Mechanisms of Action and Methods for Prevention of Diabetes. J. Obes. Eat. Disord. 2016, 2, 100019. [Google Scholar] [CrossRef]
- Basu, T.K.; Tze, W.J.; Leichter, J. Serum vitamin A and retinol-binding protein in patients with insulin-dependent diabetes mellitus. Am. J. Clin. Nutr. 1989, 50, 329–331. [Google Scholar] [CrossRef]
- Martinoli, L.; Di Felice, M.; Seghieri, G.; Ciuti, M.; De Giorgio, L.A.; Fazzini, A.; Gori, R.; Anichini, R.; Franconi, F. Plasma retinol and alpha-tocopherol concentrations in insulin-dependent diabetes mellitus: Their relationship to microvascular complications. Int. J. Vitam. Nutr. Res. 1993, 63, 87–92. [Google Scholar]
- Graham, T.E.; Kahn, B.B. Tissue-specific alterations of glucose transport and molecular mechanisms of intertissue communication in obesity and type 2 diabetes. Horm. Metab. Res. 2007, 39, 717–721. [Google Scholar] [CrossRef] [Green Version]
- Zabetian-Targhi, F.; Mahmoudi, M.J.; Rezaei, N.; Mahmoudi, M. Retinol binding protein 4 in relation to diet, inflammation, immunity, and cardiovascular diseases. Adv. Nutr. 2015, 6, 748–762. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Graham, T.E.; Mody, N.; Preitner, F.; Peroni, O.D.; Zabolotny, J.M.; Kotani, K.; Quadro, L.; Kahn, B.B. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature 2005, 436, 356–362. [Google Scholar] [CrossRef]
- Perumalsamy, S.; Ahmad, W.A.W.; Huri, H.Z. Retinol-Binding Protein-4—A Predictor of Insulin Resistance and the Severity of Coronary Artery Disease in Type 2 Diabetes Patients with Coronary Artery Disease. Biology 2021, 10, 858. [Google Scholar] [CrossRef]
- Hussein, M.M.A.; El-Belbasi, H.I.; Morsy, M.A.; Saadeldin, I.M.; Alshammari, G.M. The synergistic effect of fenretinide and metformin to achieve a decrease in insulin resistance and inflammatory mediators: An in vivo study. All Life 2020, 13, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Electronic Medicines Compendium. Metformin 500 mg Tablets. Available online: https://www.medicines.org.uk/emc/product/594/smpc#gref (accessed on 1 May 2022).
- Jura-Półtorak, A.; Olczyk, P.; Chałas-Lipka, A.; Komosińska-Vassev, K.; Kuźnik-Trocha, K.; Winsz-Szczotka, K.; Ivanova, D.; Kiselova-Kaneva, Y.; Krysik, K.; Telega, A.; et al. Urinary sulphated glycosaminoglycans excretion in obese patients with type 2 diabetes mellitus treated with metformin. Arch. Physiol. Biochem. 2019, 128, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Zhang, Y.; Liu, J.; Lian, X.; Tang, J.; Zhu, F. Pharmacometabonomics: Data processing and statistical analysis. Brief. Bioinform. 2021, 22, bbab138. [Google Scholar] [CrossRef] [PubMed]
- Kaddurah-Daouk, R.; Weinshilboum, R. Metabolomic Signatures for Drug Response Phenotypes: Pharmacometabolomics Enables Precision Medicine. Clin. Pharmacol. Ther. 2015, 98, 71–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mussap, M.; Loddo, C.; Fanni, C.; Fanos, V. Metabolomics in pharmacology—A delve into the novel field of pharmacometabolomics. Expert Rev. Clin. Pharmacol. 2020, 13, 115–134. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical and Demographics, (n = 17) | Screening | Follow-Up |
|---|---|---|
| Ethnic, n (%) | ||
| Malay | 9 (52.9) | |
| Chinese | 5 (29.4) | |
| Indian | 2 (11.8) | |
| Bidayuh | 1 (5.9) | |
| Sex, n (%) | ||
| Male | 17 (100.0) | |
| Age, mean (range), years * | 25 (22–27) | |
| Weight, mean (range), kg * | 63.9 (57.0–74.4) | |
| Height, mean (range), cm * | 166 (165–170) | |
| BMI, mean (range), kg/m2 * | 23.5 (22.1–25.0) | |
| Virology test | ||
| Hepatitis Bs Ag (HbsAg) | Not detected | |
| Hepatitis C antibody (Anti0 HBs) | Not detected | |
| HIV Ag/Ab Combo | Not detected | |
| Biochemistry | ||
| Sodium (mmol/L) * | 140.0 (138.0–142.0) | 139.0 (138.0–139.0) |
| Potassium (mmol/L) * | 4.4 (4.2–4.6) | 4.0 (3.9–4.1) |
| Chloride (mmol/L) * | 103.0 (102.0–105.0) | 103.0 (103.0–104.0) |
| Total CO2 (mmol/L) * | 30.0 (30.0–31.0) | 29.0 (28.0–30.0) |
| Anion Gap (mmol/L) * | 11.0 (10.0–12.0) | 10.0 (9.0–11.0) |
| Urea (mmol/L) * | 4.9 (3.6–5.4) | 4.1 (4.0–4.8) |
| Creatinine (µmol/L) * | 82.0 (75.0–85.0) | 87.0 (83.0–93.0) |
| Liver function test | ||
| Albumin (g/L) * | 44.0 (42.0–44.0) | 39.0 (38.0–40.0) |
| Total bilirubin (µmol/L) * | 18.0 (14.0–20.0) | 11.0 (7.0–14.0) |
| Alkaline phosphatase (u/L) * | 72.0 (63.0–80.0) | 71.0 (65.0–86.0) |
| Alanine aminotransferase (u/l) * | 21.0 (17.0–26.0) | 19.0 (16.0–32.0) |
| Gamma GT (u/L) * | 19.0 (12.0–25.0) | 17.0 (12.0–22.0) |
| Complete blood count * | ||
| Hemoglobin (g/L) * | 160.0 (156.0–166.0) | 143.0 (140.0–148.0) |
| Hematocrit (l/L) * | 0.49 (0.47–0.49) | 0.43 (0.42–0.44) |
| Red blood cell (1012/L) * | 5.5 (5.4–5.9) | 5.1 (5.0–5.2) |
| Mean corpuscular volume (fl) * | 85.0 (82.0–88.0) | 85.0 (84.0–87.0) |
| Mean corpuscular hemoglobin (pg) * | 28.6 (27.2–29.7) | 28.7 (27.7–29.1) |
| Mean corpuscular hemoglobin concentration (g/L) * | 333.0 (327.0–342.0) | 335.0 (329.0–346.0) |
| Red cell distribution width (%) * | 12.2 (12.1–13.4) | 12.3 (12.2–12.5) |
| White blood cell (109/L) * | 6.8 (5.7–7.1) | 6.8 (6.2–8.2) |
| Platelet (109/L) * | 275.0 (247.0–319.0) | 273.0 (237.0–297.0) |
| Parameter | Results | |||
|---|---|---|---|---|
| Between run accuracy | LLOQ 106.71%, LQC 96.05%, MQC 99.95%, HQC 93.97% | |||
| Between run precision | LQC 3.88, MQC 5.56, HQC 7.67 | |||
| Within run accuracy | LLOQ | LQC | MQC | HQC |
| Batch 1 | 111.17% | 101.40% | 102.67% | 90.68% |
| Batch 2 | 99.82% | 92.45% | 96.23% | 89.61% |
| Batch 3 | 109.15% | 94.31% | 100.95% | 101.63% |
| Within run precision | LLOQ | LQC | MQC | HQC |
| Batch 1 | 0.74 | 0.68 | 3.31 | 2.89 |
| Batch 2 | 1.82 | 2.87 | 1.62 | 1.94 |
| Batch 3 | 5.47 | 3.42 | 2.85 | 3.47 |
| Selectivity | No peak was observed at the metformin retention time for six biological batches. | |||
| Recovery | 88.58%, %CV9.85 | |||
| Carryover | No carry over is observed after 10 alternating injections of blank plasma and HQC. | |||
| Stability | LQC CV | HQC CV | ||
| Bench top room temperature (6 h) | −0.11 | −0.09 | ||
| Three freeze-thaw cycles | −0.11 | −0.23 | ||
| Auto-sampler | −0.15 | −0.12 | ||
| Long-term (3 months) | 0.00 | −0.18 | ||
| Parameter | Median (Interquartile Range) |
|---|---|
| C_max (ng/mL) | 1248 (849–1391) |
| T_max (h) | 2.5 (2.5–3.0) |
| AUC0_infinity (ng*h/mL) | 9510 (7313–10,411) |
| AUC_0–24 (ng*h/mL) | 8955 (7099–10,020) |
| T_half (h) | 6.8 (5.5–7.0) |
| CL (mL/min) * | 1884 (32.3) |
| Human Metabolic Pathways (Pathway Total Metabolites in KEGG) | Dataset A, n = 6 | Dataset B, n = 17 | U0–U1, n = 6 | Compound with Significant Hits (p-Value ≤ 0.05) |
|---|---|---|---|---|
| Total Hit (Significant Hit Number, p-Value ≤ 0.05) | ||||
| Arginine and proline metabolism (37) | 24 (4) | 31 (3) | 28 (5) | L-Proline A; D-Proline A; S-Adenosylmethioninamine AB; N-Acetylputrescine AB; Creatine BU; Gamma-Aminobutyric acid U; 4-AminobutyraldehydeU; L-4-Hydroxyglutamate semialdehyde U; L-Glutamic acidU |
| Glycine, serine and threonine metabolism (30) | 17 (2) | 23 (1) | 21 (2) | Betaine aldehyde A; Glyceric acid A; Choline B; Creatine BU; Dimethylglycine U |
| Steroid hormone biosynthesis (85) | 83 (12) | 84 (1) | 84 (3) | Cholesterol A; 20a,22b-Dihydroxycholesterol A; 17alpha,20alpha-Dihydroxycholesterol A; Dehydroepiandrosterone A; Cortisol; 17a,21-Dihydroxy-5b-pregnane-3,11,20-trione A; Testosterone A; Etiocholanedione A; Androstanedione A; 18-Hydroxycorticosterone A; 11-Dehydrocorticosterone A; Tetrahydrocortisol A; Testosterone glucuronide A; Estrone glucuronide A; Estriol-16-Glucuronide AB; 11b-HydroxyprogesteroneU; 11b-Hydroxyandrost-4-ene-3,17-dione U; 2-Methoxyestrone U; 2-Methoxyestradiol U; 19-Hydroxyandrost-4-ene-3,17-dione U; 19-Oxoandrost-4-ene-3,17-dione U; 19-Oxotestosterone U; Cholesterol sulfate U; 16a-Hydroxyandrost-4-ene-3,17-dione U; Adrenosterone U |
| Glutathione metabolism (19) | 11 (2) | 13 (1) | 10 (1) | Aminopropylcadaverine AB; Trypanothione disulfide A; L-Glutamic acid U |
| Galactose metabolism (27) | 24 (2) | 26 (1) | 25 (1) | D-Gal alpha 1->6D-Gal alpha 1->6D-Glucose AB; Raffinose AB; D-Galactose U; Alpha-D-Glucose U; D-Galactose U; D-Glucose U; D Fructose U; D-Mannose U; myo-Inositol U |
| Starch and sucrose metabolism (13) | 13 (1) | 13 (1) | 12(1) | Dextrin AB; D-Fructose U; D-Glucose U |
| Metabolism of xenobiotics by cytochrome P450 (68) | 40 (5) | 54 (1) | 49 (3) | Glutathione episulfonium ion ABU; 2-(S-Glutathionyl)acetyl chloride A; Trichloroethanol glucuronide A; S-(2-Chloroacetyl)glutathione A; (1R)-Hydroxy-(2R)-glutathionyl-1,2-dihydronaphthalene A; alpha-[3-[(Hydroxymethyl)nitrosoamino]propyl]-3-pyridinemethanol U; 1-(Methylnitrosoamino)-4-(3-pyridinyl)-1,4-butanediol U |
| Ubiquinone and other terpenoid-quinone biosynthesis (9) | 9 (4) | 9 (1) | 9 (1) | Vitamin K1 AB; Vitamin K2 A; Menaquinol A; Vitamin K1 2,3-epoxide A; 2,3-Epoxymenaquinone U |
| Cysteine and methionine metabolism (33) | 22 (1) | 28 (1) | 25(1) | S-Adenosylmethioninamine AB; L-Alpha-aminobutyric acid U |
| Tryptophan metabolism (41) | 23 (1) | 33 (1) | 36(1) | L-Tryptophan B; 5-Hydroxy-N-formylkynurenine A; 5-Hydroxy-L-tryptophanU |
| Aminoacyl-tRNA biosynthesis (22) | 14 (1) | 19 (1) | 16 (2) | L-Proline A; L-Tryptophan B; L-Isoleucine U; L-Leucine U; L-Glutamic acid U |
| Riboflavin metabolism (4) | 2 (1) | 3 (1) | - | Riboflavin AB |
| Retinol metabolism (16) | 16 (1) | 16 (1) | - | B-Carotene B; Retinoyl b-glucuronide AB |
| Glycerophospholipid metabolism (13) | 7 (1) | 12 (1) | - | Acetylcholine A; Choline B |
| Human Metabolic Pathways | Pathway Total Metabolites/Total Metabolites Hit (Significant Metabolites Hit, p ≤ 0.005) | Compound with Significant Hits (p-Value ≤ 0.05) | ||
|---|---|---|---|---|
| U0 vs. U1 | U0 vs. U2 | U0 vs. U3 | ||
| Arginine and proline metabolism (37) | 28 (5) | 26 (2) | - | Creatine U1U2; Gamma-Aminobutyric acid U1; 4-Aminobutyraldehyde U1; L-4-Hydroxyglutamate semialdehyde U1U2; L-Glutamic acid U1U2 |
| Glycine, serine and threonine metabolism (30) | 21 (2) | 22 (1) | 22 (1) | Creatine U1U2; Dimethylglycine U1; L-2-Amino-3-oxobutanoic acid U3 |
| Glycosaminoglycan degradation (21) | 9 (2) | - | - | (GalNAc)2 (GlcA)1 (S)1 U1; (GlcA)2 (GlcNAc)1 (S)2 U1; DWA-2 U1 |
| Drug metabolism—cytochrome P450 (43) | 38 (4) | - | - | Alcophosphamide U1; Codeine-6-glucuronide U1; Citalopram N-oxide U1; L-alpha-Acetyl-N,N-dinormethadol U1 |
| Butanoate metabolism (15) | 9 (2) | 9 (1) | - | 2-Hydroxyglutarate U1; Gamma-Aminobutyric acid U1; L-Glutamic acid U1U2 |
| Arginine biosynthesis (14) | 10 (1) | 9 (1) | - | L-Glutamic acid U1U2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tee, K.B.; Ibrahim, L.; Hashim, N.M.; Saiman, M.Z.; Zakaria, Z.H.; Huri, H.Z. Pharmacokinetic–Pharmacometabolomic Approach in Early-Phase Clinical Trials: A Way Forward for Targeted Therapy in Type 2 Diabetes. Pharmaceutics 2022, 14, 1268. https://doi.org/10.3390/pharmaceutics14061268
Tee KB, Ibrahim L, Hashim NM, Saiman MZ, Zakaria ZH, Huri HZ. Pharmacokinetic–Pharmacometabolomic Approach in Early-Phase Clinical Trials: A Way Forward for Targeted Therapy in Type 2 Diabetes. Pharmaceutics. 2022; 14(6):1268. https://doi.org/10.3390/pharmaceutics14061268
Chicago/Turabian StyleTee, Khim Boon, Luqman Ibrahim, Najihah Mohd Hashim, Mohd Zuwairi Saiman, Zaril Harza Zakaria, and Hasniza Zaman Huri. 2022. "Pharmacokinetic–Pharmacometabolomic Approach in Early-Phase Clinical Trials: A Way Forward for Targeted Therapy in Type 2 Diabetes" Pharmaceutics 14, no. 6: 1268. https://doi.org/10.3390/pharmaceutics14061268