
Impact of Pharmacokinetic and Pharmacodynamic Properties of Monoclonal Antibodies in the Management of Psoriasis


 , ,
, ,
Abstract
:1. Introduction
1.1. Pathophysiology of Psoriasis
1.2. Clinical Endpoints of Psoriasis

2. Pharmacokinetic/Pharmacodynamic Properties of Monoclonal Antibodies in Psoriasis
2.1. Pharmacokinetic Properties
2.2. Pharmacodynamic Properties
3. Monoclonal Antibody Approved for Psoriasis
4. Population Pharmacokinetic/Pharmacodynamic Models for Monoclonal Antibodies in Psoriasis
4.1. Adalimumab
4.2. Golimumab
4.3. Ustekinumab
4.4. Secukinumab
4.5. Ixekizumab
4.6. Brodalumab
4.7. Guselkumab
4.8. Tildrakizumab
4.9. Risankizumab
5. Therapeutic Drug Monitoring of Monoclonal Antibodies in Psoriasis

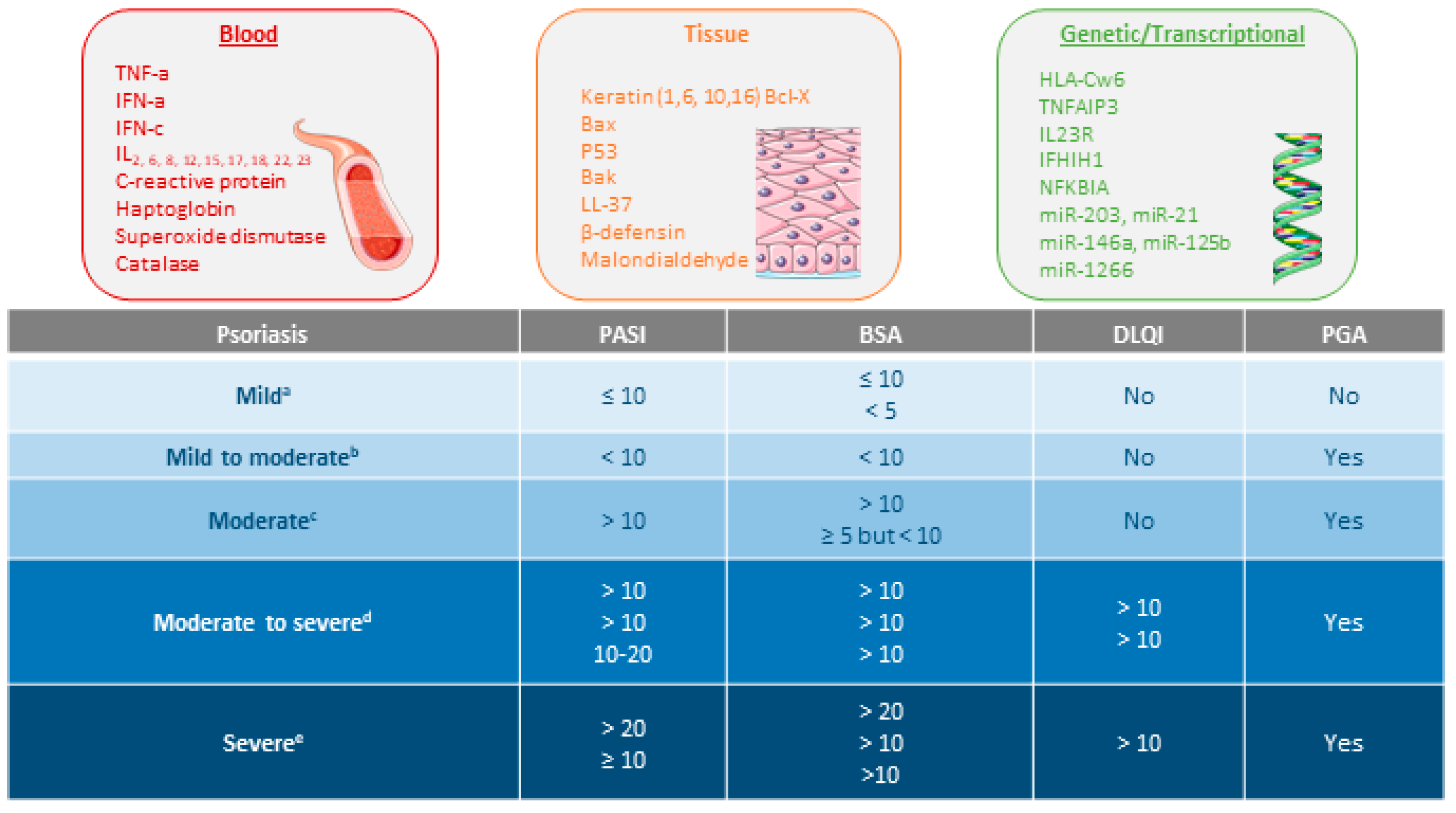{kind=link}
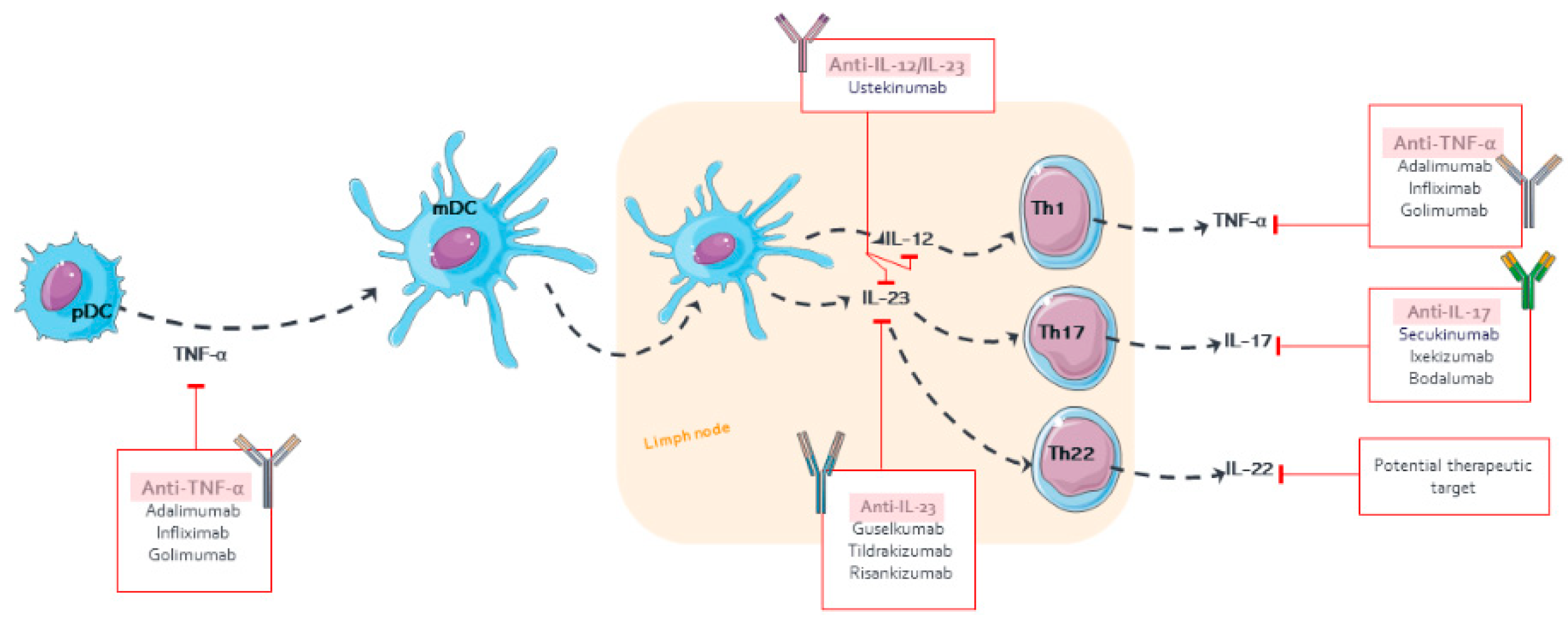{kind=link}
| Drug | Incidence of ADA, % | Ctrough, μm/mL (Response) | Therapeutic Range |
|---|---|---|---|
| Etanercept | 0.0–18.3 [49,50,126,135,136,137] | NA | NA |
| Adalimumab | 6.5–45.0 [51,123,126,138,139] | 3.51 [113], 7.84 [123], 9.7 [138] (PASI75) | 3.51–7.0 [113] |
| Infliximab | 5.4–54.2 [52,53,123,140,141,142,143,144,145,146] | 0.92 [123] (PASI75) | NA |
| 3.16 [146] (PASI50) | |||
| Ustekinumab | 3.5–6.0 [57,132,147,148,149,150,151] | NA | NA |
| Secukinumab | 0.3–0.4 [58] | 33.2 [128] (PASI ≤ 2) | NA |
| Ixekizumab | 9.0–13.4 [152] | NA | NA |
6. Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADA | Antidrug antibodies |
| Alb | Albumin |
| ALK | Alkaline phosphatase |
| AUC | Area under the plasma concentration–time curve |
| BID | Twice a day |
| BIW | Twice a week |
| BSA | Body Surface Area |
| BW | Body weight |
| Cavg | Average concentration |
| Cmax | Maximum concentration |
| CMT | Compartment |
| CL | Clearance |
| CL/F | Apparent clearance |
| Cr | Creatinine |
| CrCL | Creatinine clearance |
| CRP | Baseline C-reactive protein level |
| Ctrough | Minimum trough concentration |
| D | Day |
| DB | Diabetes |
| DLQI | Dermatologic Life Quality Index |
| DNA | Deoxyribonucleic acid |
| EC50 | Serum drug concentration causing 50% of the maximum effect |
| Emax | Maximum drug effect |
| E–R | Exposure–response |
| Fc | Fragment crystallizable |
| FcγR | Fcγ receptor |
| FcRn | Neonatal Fc receptor |
| γ | Hill’s coefficient |
| hs-CRP | High-sensitivity C-reactive protein |
| HTA | Hypertension |
| IC50 | Median serum drug concentration causing 50% of the maximum inhibitory effect |
| IFN | Interferon |
| IIV | Inter-individual variability |
| IL | Interleukin |
| IM | Intramuscular |
| IV | Intravenous |
| ka | Absorption rate constant |
| Km | Michaelis–Menten constant |
| kin | Formation rate of psoriatic skin lesions |
| kout | Remission rate of psoriatic skin lesion |
| LE | Linear elimination |
| mAb | Monoclonal antibody |
| mDC | Myeloid dendritic cell |
| MM | Mixture model |
| MTX | Past methotrexate use |
| NF- κB | Nuclear factor kappa B |
| NLE | Nonlinear elimination |
| NR | Non-responder patients |
| OR | Oral |
| PASI | Psoriasis Area Severity Index |
| PASI75-W12 | PASI75 responder status at the week 12 primary |
| PGA | Physician Global Assessment |
| PDE4 | Phosphodiesterase-4 |
| PD | Pharmacodynamic |
| PK | Pharmacokinetics |
| PK/PD | Pharmacokinetics/pharmacodymamics |
| PLBM | Maximum placebo effect |
| PP | Palmoplantar psoriasis. |
| PsA | Psoriatic arthritis |
| Pso | Psoriasis |
| Q | Intercompartmental transfer clearance |
| QD | Every day |
| QW | Every week |
| Q2W | Once every 2 weeks |
| Q4W | Once every 4 weeks |
| Q8W | Once every 8 weeks |
| Q12W | Once every 12 weeks |
| R | Responder patients |
| RSE | Relative standard error |
| SC | Subcutaneous |
| SM | Single model |
| SMDs | Small-molecule drugs |
| sPGA | Static Physician Global Assessment |
| TMDD | Target mediated drug disposition |
| TDM | Therapeutic drug monitoring |
| TID | Three times a day |
| Th | T helper lymphocytes |
| TNF-α | Tumor necrosis factor alpha |
| URI | Upper respiratory infection |
| VC | Central volume of distribution |
| V/F | Apparent volume of distribution |
| Vmax | maximal velocity for nonlinear elimination |
| VPz | Peripheral volume of distribution |
| W | Week |
References
- Di Meglio, P.; Villanova, F.; Nestle, F.O. Psoriasis. Cold Spring Harb. Perspect. Med. 2014, 4, a015354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parisi, R.; Iskandar, I.Y.K.; Kontopantelis, E.; Augustin, M.; Griffiths, C.E.M.; Ashcroft, D.M. National, regional, and worldwide epidemiology of psoriasis: Systematic analysis and modelling study. BMJ 2020, 369, m1590. [Google Scholar] [CrossRef] [PubMed]
- Menter, A.; Gottlieb, A.; Feldman, S.R.; Van Voorhees, A.S.; Leonardi, C.L.; Gordon, K.B.; Lebwohl, M.; Koo, J.Y.; Elmets, C.A.; Korman, N.J. Guidelines of care for the management of psoriasis and psoriatic arthritis: Section 1. Overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J. Am. Acad. Dermatol. 2008, 58, 826–850. [Google Scholar] [CrossRef] [PubMed]
- Schadler, E.D.; Ortel, B.; Mehlis, S.L. Biologics for the primary care physician: Review and treatment of psoriasis. Dis. Mon. DM 2018, 65, 51–90. [Google Scholar] [CrossRef] [PubMed]
- Todke, P.; Shah, V.H. Psoriasis: Implication to disease and therapeutic strategies, with an emphasis on drug delivery approaches. Int. J. Dermatol. 2018, 57, 1387–1402. [Google Scholar] [CrossRef] [PubMed]
- Salomon, J.; Szepietowski, J.C.; Proniewicz, A. Psoriatic nails: A prospective clinical study. J. Cutan. Med. Surg. 2003, 7, 317–321. [Google Scholar] [CrossRef]
- Mahil, S.K.; Capon, F.; Barker, J.N. Genetics of psoriasis. Dermatol. Clin. 2015, 33, 1–11. [Google Scholar] [CrossRef]
- Zeng, J.; Luo, S.; Huang, Y.; Lu, Q. Critical role of environmental factors in the pathogenesis of psoriasis. J. Dermatol. 2017, 44, 863–872. [Google Scholar] [CrossRef] [Green Version]
- Nestle, F.O.; Kaplan, D.H.; Barker, J. Psoriasis. N. Engl. J. Med. 2009, 361, 496–509. [Google Scholar] [CrossRef]
- Perera, G.K.; Di Meglio, P.; Nestle, F.O. Psoriasis. Annu. Rev. Pathol. 2012, 7, 385–422. [Google Scholar] [CrossRef]
- Kim, J.; Krueger, J.G. The immunopathogenesis of psoriasis. Dermatol. Clin. 2015, 33, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Greb, J.E.; Goldminz, A.M.; Elder, J.T.; Lebwohl, M.G.; Gladman, D.D.; Wu, J.J.; Mehta, N.N.; Finlay, A.Y.; Gottlieb, A.B. Psoriasis. Nat. Rev. Dis. Primers 2016, 2, 16082. [Google Scholar] [CrossRef]
- Armstrong, A.W.; Read, C. Pathophysiology, Clinical Presentation, and Treatment of Psoriasis: A Review. JAMA 2020, 323, 1945–1960. [Google Scholar] [CrossRef]
- Conrad, C.; Gilliet, M. Psoriasis: From Pathogenesis to Targeted Therapies. Clin. Rev. Allergy Immunol. 2018, 54, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Rashmi, R.; Rao, K.S.; Basavaraj, K.H. A comprehensive review of biomarkers in psoriasis. Clin. Exp. Dermatol. 2009, 34, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Villanova, F.; Di Meglio, P.; Nestle, F.O. Biomarkers in psoriasis and psoriatic arthritis. Ann. Rheum. Dis. 2013, 72 (Suppl. S2), ii104–ii110. [Google Scholar] [CrossRef] [PubMed]
- Aydin, B.; Arga, K.Y.; Karadag, A.S. Omics-Driven Biomarkers of Psoriasis: Recent Insights, Current Challenges, and Future Prospects. Clin. Cosmet. Investig. Dermatol. 2020, 13, 611–625. [Google Scholar] [CrossRef]
- Spuls, P.I.; Lecluse, L.L.; Poulsen, M.L.; Bos, J.D.; Stern, R.S.; Nijsten, T. How good are clinical severity and outcome measures for psoriasis?: Quantitative evaluation in a systematic review. J. Investig. Dermatol. 2010, 130, 933–943. [Google Scholar] [CrossRef] [Green Version]
- Bożek, A.; Reich, A. The reliability of three psoriasis assessment tools: Psoriasis area and severity index, body surface area and physician global assessment. Adv. Clin. Exp. Med. 2017, 26, 851–856. [Google Scholar] [CrossRef] [Green Version]
- Mrowietz, U.; Kragballe, K.; Reich, K.; Spuls, P.; Griffiths, C.; Nast, A.; Franke, J.; Antoniou, C.; Arenberger, P.; Balieva, F. Definition of treatment goals for moderate to severe psoriasis: A European consensus. Arch. Dermatol. Res. 2011, 303, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Menter, A.; Korman, N.J.; Elmets, C.A.; Feldman, S.R.; Gelfand, J.M.; Gordon, K.B.; Gottlieb, A.; Koo, J.Y.; Lebwohl, M.; Leonardi, C.L.; et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: Section 6. Guidelines of care for the treatment of psoriasis and psoriatic arthritis: Case-based presentations and evidence-based conclusions. J. Am. Acad. Dermatol. 2011, 65, 137–174. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. CHMP/EWP/2454/02 Corr—Guideline on Clinical Investigation of Medicinal Products Indicated for the Treatment of Psoriasis. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-clinical-investigation-medicinal-products-indicated-treatment-psoriasis_en.pdf (accessed on 23 September 2021).
- Daudén, E.; Puig, L.; Ferrándiz, C.; Sánchez-Carazo, J.L.; Hernanz-Hermosa, J.M. Consensus document on the evaluation and treatment of moderate-to-severe psoriasis: Psoriasis Group of the Spanish Academy of Dermatology and Venereology. J. Eur. Acad. Dermatol. Venereol. JEADV 2016, 30 (Suppl. S2), 1–18. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.H.; Yiu, Z.Z.; Bale, T.; Burden, A.D.; Coates, L.C.; Edwards, W.; MacMahon, E.; Mahil, S.; McGuire, A.; Murphy, R.; et al. British Association of Dermatologists guidelines for biologic therapy for psoriasis 2020—A rapid update. Br. J. Dermatol. 2020, 183, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Ryman, J.T.; Meibohm, B. Pharmacokinetics of monoclonal antibodies. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 576–588. [Google Scholar] [CrossRef] [PubMed]
- KÖHler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Mould, D.R.; Green, B. Pharmacokinetics and pharmacodynamics of monoclonal antibodies. BioDrugs 2010, 24, 23–39. [Google Scholar] [CrossRef]
- Regazzi, M.; Joseè, G.; Molinaro, M. Monoclonal Antibody Monitoring: Clinically Relevant Aspects, A Systematic Critical Review. Ther. Drug Monit. 2019, 42, 45–56. [Google Scholar] [CrossRef]
- Zhao, L.; Shang, E.Y.; Sahajwalla, C.G. Application of pharmacokinetics–pharmacodynamics/clinical response modeling and simulation for biologics drug development. J. Pharm. Sci. 2012, 101, 4367–4382. [Google Scholar] [CrossRef]
- Thomas, V.A.; Balthasar, J.P. Understanding Inter-Individual Variability in Monoclonal Antibody Disposition. Antibodies 2019, 8, 56. [Google Scholar] [CrossRef] [Green Version]
- Posner, J.; Barrington, P.; Brier, T.; Datta-Mannan, A. Monoclonal Antibodies: Past, Present and Future. In Concepts and Principles of Pharmacology: 100 Years of the Handbook of Experimental Pharmacology; Barrett, J.E., Page, C.P., Michel, M.C., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 81–141. [Google Scholar]
- Lobo, E.D.; Hansen, R.J.; Balthasar, J.P. Antibody pharmacokinetics and pharmacodynamics. J. Pharm. Sci. 2004, 93, 2645–2668. [Google Scholar] [CrossRef]
- Wang, W.; Wang, E.Q.; Balthasar, J.P. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin. Pharmacol. Ther. 2008, 84, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Zinner, R.G.; Glisson, B.S.; Fossella, F.V.; Pisters, K.M.; Kies, M.S.; Lee, P.M.; Massarelli, E.; Sabloff, B.; Fritsche, H.A., Jr.; Ro, J.Y.; et al. Trastuzumab in combination with cisplatin and gemcitabine in patients with Her2-overexpressing, untreated, advanced non-small cell lung cancer: Report of a phase II trial and findings regarding optimal identification of patients with Her2-overexpressing disease. Lung Cancer 2004, 44, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Gaudreault, J.; Shiu, V.; Bricarello, A.; Christian, B.J.; Zuch, C.L.; Mounho, B. Concomitant administration of bevacizumab, irinotecan, 5-fluorouracil, and leucovorin: Nonclinical safety and pharmacokinetics. Int. J. Toxicol. 2005, 24, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Ettlinger, D.E.; Mitterhauser, M.; Wadsak, W.; Ostermann, E.; Farkouh, A.; Schueller, J.; Czejka, M. In vivo disposition of irinotecan (CPT-11) and its metabolites in combination with the monoclonal antibody cetuximab. Anticancer Res. 2006, 26, 1337–1341. [Google Scholar] [PubMed]
- Xu, L.; Zuch, C.L.; Lin, Y.S.; Modi, N.B.; Lum, B.L. Pharmacokinetics and safety of bevacizumab administered in combination with cisplatin and paclitaxel in cynomolgus monkeys. Cancer Chemother. Pharmacol. 2008, 61, 607–614. [Google Scholar] [CrossRef]
- Wright, A.; Sato, Y.; Okada, T.; Chang, K.; Endo, T.; Morrison, S. In vivo trafficking and catabolism of IgG1 antibodies with Fc associated carbohydrates of differing structure. Glycobiology 2000, 10, 1347–1355. [Google Scholar] [CrossRef] [Green Version]
- Israel, E.J.; Wilsker, D.F.; Hayes, K.C.; Schoenfeld, D.; Simister, N.E. Increased clearance of IgG in mice that lack beta 2-microglobulin: Possible protective role of FcRn. Immunology 1996, 89, 573–578. [Google Scholar] [CrossRef]
- Junghans, R.P.; Anderson, C.L. The protection receptor for IgG catabolism is the beta2-microglobulin-containing neonatal intestinal transport receptor. Proc. Natl. Acad. Sci. USA 1996, 93, 5512–5516. [Google Scholar] [CrossRef] [Green Version]
- Liu, L. Pharmacokinetics of monoclonal antibodies and Fc-fusion proteins. Protein Cell 2018, 9, 15–32. [Google Scholar] [CrossRef]
- Dirks, N.L.; Meibohm, B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin. Pharm. 2010, 49, 633–659. [Google Scholar] [CrossRef]
- Lucas, A.T.; Robinson, R.; Schorzman, A.N.; Piscitelli, J.A.; Razo, J.F.; Zamboni, W.C. Pharmacologic Considerations in the Disposition of Antibodies and Antibody-Drug Conjugates in Preclinical Models and in Patients. Antibodies 2019, 8, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friberg, L.E.; Henningsson, A.; Maas, H.; Nguyen, L.; Karlsson, M.O. Model of chemotherapy-induced myelosuppression with parameter consistency across drugs. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2002, 20, 4713–4721. [Google Scholar] [CrossRef] [PubMed]
- Dayneka, N.L.; Garg, V.; Jusko, W.J. Comparison of four basic models of indirect pharmacodynamic responses. J. Pharmacokinet. Biopharm. 1993, 21, 457–478. [Google Scholar] [CrossRef]
- Molinelli, E.; Campanati, A.; Ganzetti, G.; Offidani, A. Biologic Therapy in Immune Mediated Inflammatory Disease: Basic Science and Clinical Concepts. Curr. Drug Saf. 2016, 11, 35–43. [Google Scholar] [CrossRef]
- Lebwohl, M. Psoriasis. Ann. Intern. Med. 2018, 168, Itc49–Itc64. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.B.; Lebwohl, M.G. Review of safety and efficacy of approved systemic psoriasis therapies. Int. J. Dermatol. 2019, 58, 649–658. [Google Scholar] [CrossRef]
- Leonardi, C.L.; Powers, J.L.; Matheson, R.T.; Goffe, B.S.; Zitnik, R.; Wang, A.; Gottlieb, A.B. Etanercept as monotherapy in patients with psoriasis. N. Engl. J. Med. 2003, 349, 2014–2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papp, K.A.; Tyring, S.; Lahfa, M.; Prinz, J.; Griffiths, C.E.; Nakanishi, A.M.; Zitnik, R.; van de Kerkhof, P.C.; Melvin, L. A global phase III randomized controlled trial of etanercept in psoriasis: Safety, efficacy, and effect of dose reduction. Br. J. Dermatol. 2005, 152, 1304–1312. [Google Scholar] [CrossRef]
- Menter, A.; Tyring, S.K.; Gordon, K.; Kimball, A.B.; Leonardi, C.L.; Langley, R.G.; Strober, B.E.; Kaul, M.; Gu, Y.; Okun, M.; et al. Adalimumab therapy for moderate to severe psoriasis: A randomized, controlled phase III trial. J. Am. Acad. Dermatol. 2008, 58, 106–115. [Google Scholar] [CrossRef]
- Menter, A.; Feldman, S.R.; Weinstein, G.D.; Papp, K.; Evans, R.; Guzzo, C.; Li, S.; Dooley, L.T.; Arnold, C.; Gottlieb, A.B. A randomized comparison of continuous vs. intermittent infliximab maintenance regimens over 1 year in the treatment of moderate-to-severe plaque psoriasis. J. Am. Acad. Dermatol. 2007, 56, 31.e1–31.e15. [Google Scholar] [CrossRef]
- Reich, K.; Nestle, F.O.; Papp, K.; Ortonne, J.P.; Evans, R.; Guzzo, C.; Li, S.; Dooley, L.T.; Griffiths, C.E. Infliximab induction and maintenance therapy for moderate-to-severe psoriasis: A phase III, multicentre, double-blind trial. Lancet 2005, 366, 1367–1374. [Google Scholar] [CrossRef]
- Gottlieb, A.B.; Blauvelt, A.; Thaçi, D.; Leonardi, C.L.; Poulin, Y.; Drew, J.; Peterson, L.; Arendt, C.; Burge, D.; Reich, K. Certolizumab pegol for the treatment of chronic plaque psoriasis: Results through 48 weeks from 2 phase 3, multicenter, randomized, double-blinded, placebo-controlled studies (CIMPASI-1 and CIMPASI-2). J. Am. Acad. Dermatol. 2018, 79, 302–314.e306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kavanaugh, A.; McInnes, I.; Mease, P.; Krueger, G.G.; Gladman, D.; Gomez-Reino, J.; Papp, K.; Zrubek, J.; Mudivarthy, S.; Mack, M.; et al. Golimumab, a new human tumor necrosis factor alpha antibody, administered every four weeks as a subcutaneous injection in psoriatic arthritis: Twenty-four-week efficacy and safety results of a randomized, placebo-controlled study. Arthritis Rheum. 2009, 60, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, C.L.; Kimball, A.B.; Papp, K.A.; Yeilding, N.; Guzzo, C.; Wang, Y.; Li, S.; Dooley, L.T.; Gordon, K.B. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet 2008, 371, 1665–1674. [Google Scholar] [CrossRef]
- Papp, K.A.; Langley, R.G.; Lebwohl, M.; Krueger, G.G.; Szapary, P.; Yeilding, N.; Guzzo, C.; Hsu, M.C.; Wang, Y.; Li, S.; et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2). Lancet 2008, 371, 1675–1684. [Google Scholar] [CrossRef]
- Langley, R.G.; Elewski, B.E.; Lebwohl, M.; Reich, K.; Griffiths, C.E.; Papp, K.; Puig, L.; Nakagawa, H.; Spelman, L.; Sigurgeirsson, B.; et al. Secukinumab in plaque psoriasis—Results of two phase 3 trials. N. Engl. J. Med. 2014, 371, 326–338. [Google Scholar] [CrossRef] [Green Version]
- Gordon, K.B.; Blauvelt, A.; Papp, K.A.; Langley, R.G.; Luger, T.; Ohtsuki, M.; Reich, K.; Amato, D.; Ball, S.G.; Braun, D.K.; et al. Phase 3 Trials of Ixekizumab in Moderate-to-Severe Plaque Psoriasis. N. Engl. J. Med. 2016, 375, 345–356. [Google Scholar] [CrossRef]
- Griffiths, C.E.; Reich, K.; Lebwohl, M.; van de Kerkhof, P.; Paul, C.; Menter, A.; Cameron, G.S.; Erickson, J.; Zhang, L.; Secrest, R.J.; et al. Comparison of ixekizumab with etanercept or placebo in moderate-to-severe psoriasis (UNCOVER-2 and UNCOVER-3): Results from two phase 3 randomised trials. Lancet 2015, 386, 541–551. [Google Scholar] [CrossRef]
- Papp, K.A.; Reich, K.; Paul, C.; Blauvelt, A.; Baran, W.; Bolduc, C.; Toth, D.; Langley, R.G.; Cather, J.; Gottlieb, A.B.; et al. A prospective phase III, randomized, double-blind, placebo-controlled study of brodalumab in patients with moderate-to-severe plaque psoriasis. Br. J. Dermatol. 2016, 175, 273–286. [Google Scholar] [CrossRef]
- Lebwohl, M.; Strober, B.; Menter, A.; Gordon, K.; Weglowska, J.; Puig, L.; Papp, K.; Spelman, L.; Toth, D.; Kerdel, F.; et al. Phase 3 Studies Comparing Brodalumab with Ustekinumab in Psoriasis. N. Engl. J. Med. 2015, 373, 1318–1328. [Google Scholar] [CrossRef]
- Blauvelt, A.; Papp, K.A.; Griffiths, C.E.; Randazzo, B.; Wasfi, Y.; Shen, Y.K.; Li, S.; Kimball, A.B. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: Results from the phase III, double-blinded, placebo- and active comparator-controlled VOYAGE 1 trial. J. Am. Acad. Dermatol. 2017, 76, 405–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reich, K.; Armstrong, A.W.; Foley, P.; Song, M.; Wasfi, Y.; Randazzo, B.; Li, S.; Shen, Y.K.; Gordon, K.B. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: Results from the phase III, double-blind, placebo- and active comparator-controlled VOYAGE 2 trial. J. Am. Acad. Dermatol. 2017, 76, 418–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reich, K.; Warren, R.B.; Iversen, L.; Puig, L.; Pau-Charles, I.; Igarashi, A.; Ohtsuki, M.; Falqués, M.; Harmut, M.; Rozzo, S.; et al. Long-term efficacy and safety of tildrakizumab for moderate-to-severe psoriasis: Pooled analyses of two randomized phase III clinical trials (reSURFACE 1 and reSURFACE 2) through 148 weeks. Br. J. Dermatol. 2020, 182, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Blauvelt, A.; Leonardi, C.L.; Gooderham, M.; Papp, K.A.; Philipp, S.; Wu, J.J.; Igarashi, A.; Flack, M.; Geng, Z.; Wu, T.; et al. Efficacy and Safety of Continuous Risankizumab Therapy vs. Treatment Withdrawal in Patients With Moderate to Severe Plaque Psoriasis: A Phase 3 Randomized Clinical Trial. JAMA Dermatol. 2020, 156, 649–658. [Google Scholar] [CrossRef] [Green Version]
- Gisondi, P.; Del Giglio, M.; Girolomoni, G. Treatment Approaches to Moderate to Severe Psoriasis. Int. J. Mol. Sci. 2017, 18, 2427. [Google Scholar] [CrossRef] [Green Version]
- Boehncke, W.-H. Etiology and pathogenesis of psoriasis. Rheum. Dis. Clin. 2015, 41, 665–675. [Google Scholar] [CrossRef]
- Gordon, K.B.; Langley, R.G.; Leonardi, C.; Toth, D.; Menter, M.A.; Kang, S.; Heffernan, M.; Miller, B.; Hamlin, R.; Lim, L.; et al. Clinical response to adalimumab treatment in patients with moderate to severe psoriasis: Double-blind, randomized controlled trial and open-label extension study. J. Am. Acad. Dermatol. 2006, 55, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, N.M.; Nader, A.M.; Noertersheuser, P.; Okun, M.; Awni, W.M. Impact of immunogenicity on pharmacokinetics, efficacy and safety of adalimumab in adult patients with moderate to severe chronic plaque psoriasis. J. Eur. Acad. Dermatol. Venereol. JEADV 2016, 31, 490–497. [Google Scholar] [CrossRef]
- Xu, Z.; Vu, T.; Lee, H.; Hu, C.; Ling, J.; Yan, H.; Baker, D.; Beutler, A.; Pendley, C.; Wagner, C.; et al. Population pharmacokinetics of golimumab, an anti-tumor necrosis factor-alpha human monoclonal antibody, in patients with psoriatic arthritis. J. Clin. Pharm. 2009, 49, 1056–1070. [Google Scholar] [CrossRef]
- Gottlieb, A.; Menter, A.; Mendelsohn, A.; Shen, Y.K.; Li, S.; Guzzo, C.; Fretzin, S.; Kunynetz, R.; Kavanaugh, A. Ustekinumab, a human interleukin 12/23 monoclonal antibody, for psoriatic arthritis: Randomised, double-blind, placebo-controlled, crossover trial. Lancet 2009, 373, 633–640. [Google Scholar] [CrossRef]
- Zhu, Y.; Hu, C.; Lu, M.; Liao, S.; Marini, J.C.; Yohrling, J.; Yeilding, N.; Davis, H.M.; Zhou, H. Population pharmacokinetic modeling of ustekinumab, a human monoclonal antibody targeting IL-12/23p40, in patients with moderate to severe plaque psoriasis. J. Clin. Pharm. 2009, 49, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.W.; Mendelsohn, A.; Pendley, C.; Davis, H.M.; Zhou, H. Population pharmacokinetics of ustekinumab in patients with active psoriatic arthritis. Int. J. Clin. Pharmacol. Ther. 2010, 48, 830–846. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Hu, C.; Zhu, Y.; Lu, M.; Liao, S.; Yeilding, N.; Davis, H.M. Population-based exposure-efficacy modeling of ustekinumab in patients with moderate to severe plaque psoriasis. J. Clin. Pharm. 2010, 50, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Zhou, H. An improved approach for confirmatory phase III population pharmacokinetic analysis. J. Clin. Pharm. 2008, 48, 812–822. [Google Scholar] [CrossRef] [Green Version]
- Pan, S.; Tsakok, T.; Dand, N.; Lonsdale, D.O.; Loeff, F.C.; Bloem, K.; de Vries, A.; Baudry, D.; Duckworth, M.; Mahil, S.; et al. Using Real-World Data to Guide Ustekinumab Dosing Strategies for Psoriasis: A Prospective Pharmacokinetic-Pharmacodynamic Study. Clin. Transl. Sci. 2020, 13, 400–409. [Google Scholar] [CrossRef]
- Hu, C.; Zhang, J.; Zhou, H. Confirmatory analysis for phase III population pharmacokinetics. Pharm. Stat. 2011, 10, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Szapary, P.O.; Mendelsohn, A.M.; Zhou, H. Latent variable indirect response joint modeling of a continuous and a categorical clinical endpoint. J. Pharmacokinet. Pharmacodyn. 2014, 41, 335–349. [Google Scholar] [CrossRef]
- Hueber, W.; Patel, D.D.; Dryja, T.; Wright, A.M.; Koroleva, I.; Bruin, G.; Antoni, C.; Draelos, Z.; Gold, M.H.; Durez, P.; et al. Effects of AIN457, a fully human antibody to interleukin-17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci. Transl. Med. 2010, 2, 52ra72. [Google Scholar] [CrossRef] [Green Version]
- Bruin, G.; Loesche, C.; Nyirady, J.; Sander, O. Population pharmacokinetic modeling of secukinumab in patients with moderate to severe psoriasis. J. Clin. Pharmacol. 2017, 57, 876–885. [Google Scholar] [CrossRef] [Green Version]
- Rich, P.; Sigurgeirsson, B.; Thaci, D.; Ortonne, J.P.; Paul, C.; Schopf, R.E.; Morita, A.; Roseau, K.; Harfst, E.; Guettner, A.; et al. Secukinumab induction and maintenance therapy in moderate-to-severe plaque psoriasis: A randomized, double-blind, placebo-controlled, phase II regimen-finding study. Br. J. Dermatol. 2013, 168, 402–411. [Google Scholar] [CrossRef]
- Reich, K.; Papp, K.A.; Matheson, R.T.; Tu, J.H.; Bissonnette, R.; Bourcier, M.; Gratton, D.; Kunynetz, R.A.; Poulin, Y.; Rosoph, L.A.; et al. Evidence that a neutrophil-keratinocyte crosstalk is an early target of IL-17A inhibition in psoriasis. Exp. Dermatol. 2015, 24, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.A.; Langley, R.G.; Sigurgeirsson, B.; Abe, M.; Baker, D.R.; Konno, P.; Haemmerle, S.; Thurston, H.J.; Papavassilis, C.; Richards, H.B. Efficacy and safety of secukinumab in the treatment of moderate-to-severe plaque psoriasis: A randomized, double-blind, placebo-controlled phase II dose-ranging study. Br. J. Dermatol. 2013, 168, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Paul, C.; Lacour, J.P.; Tedremets, L.; Kreutzer, K.; Jazayeri, S.; Adams, S.; Guindon, C.; You, R.; Papavassilis, C. Efficacy, safety and usability of secukinumab administration by autoinjector/pen in psoriasis: A randomized, controlled trial (JUNCTURE). J. Eur. Acad. Dermatol. Venereol. JEADV 2015, 29, 1082–1090. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, C.; Matheson, R.; Zachariae, C.; Cameron, G.; Li, L.; Edson-Heredia, E.; Braun, D.; Banerjee, S. Anti–Interleukin-17 Monoclonal Antibody Ixekizumab in Chronic Plaque Psoriasis. N. Engl. J. Med. 2012, 366, 1190–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tham, L.-S.; Tang, C.-C.; Choi, S.-L.; Satterwhite, J.H.; Cameron, G.S.; Banerjee, S. Population exposure-response model to support dosing evaluation of ixekizumab in patients with chronic plaque psoriasis. J. Clin. Pharm. 2014, 54, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Chigutsa, E.; Velez de Mendizabal, N.; Chua, L.; Heathman, M.; Friedrich, S.; Jackson, K.; Reich, K. Exposure-Response Modeling to Characterize the Relationship Between Ixekizumab Serum Drug Concentrations and Efficacy Responses at Week 12 in Patients with Moderate to Severe Plaque Psoriasis. J. Clin. Pharm. 2018, 58, 1489–1500. [Google Scholar] [CrossRef]
- Papp, K.A.; Reid, C.; Foley, P.; Sinclair, R.; Salinger, D.H.; Williams, G.; Dong, H.; Krueger, J.G.; Russell, C.B.; Martin, D.A. Anti-IL-17 receptor antibody AMG 827 leads to rapid clinical response in subjects with moderate to severe psoriasis: Results from a phase I, randomized, placebo-controlled trial. J. Investig. Dermatol. 2012, 132, 2466–2469. [Google Scholar] [CrossRef] [Green Version]
- Endres, C.J.; Salinger, D.H.; Köck, K.; Gastonguay, M.R.; Martin, D.A.; Klekotka, P.; Nirula, A.; Gibbs, M.A. Population pharmacokinetics of brodalumab in healthy adults and adults with psoriasis from single and multiple dose studies. J. Clin. Pharm. 2014, 54, 1230–1238. [Google Scholar] [CrossRef]
- Timmermann, S.; Hall, A. Population pharmacokinetics of brodalumab in patients with moderate to severe plaque psoriasis. Basic Clin. Pharm. Toxicol. 2019, 125, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Salinger, D.H.; Endres, C.J.; Martin, D.A.; Gibbs, M.A. A semi-mechanistic model to characterize the pharmacokinetics and pharmacodynamics of brodalumab in healthy volunteers and subjects with psoriasis in a first-in-human single ascending dose study. Clin. Pharmacol. Drug Dev. 2014, 3, 276–283. [Google Scholar] [CrossRef]
- Papp, K.A.; Leonardi, C.; Menter, A.; Ortonne, J.P.; Krueger, J.G.; Kricorian, G.; Aras, G.; Li, J.; Russell, C.B.; Thompson, E.H.; et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N. Engl. J. Med. 2012, 366, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Randazzo, B.; Sharma, A.; Zhou, H. Improvement in latent variable indirect response modeling of multiple categorical clinical endpoints: Application to modeling of guselkumab treatment effects in psoriatic patients. J. Pharmacokinet. Pharmacodyn. 2017, 44, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Gordon, K.B.; Duffin, K.C.; Bissonnette, R.; Prinz, J.C.; Wasfi, Y.; Li, S.; Shen, Y.K.; Szapary, P.; Randazzo, B.; Reich, K. A Phase 2 Trial of Guselkumab versus Adalimumab for Plaque Psoriasis. N. Engl. J. Med. 2015, 373, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Yao, Z.; Chen, Y.; Randazzo, B.; Zhang, L.; Xu, Z.; Sharma, A.; Zhou, H. A comprehensive evaluation of exposure-response relationships in clinical trials: Application to support guselkumab dose selection for patients with psoriasis. J. Pharmacokinet. Pharmacodyn. 2018, 45, 523–535. [Google Scholar] [CrossRef]
- Yao, Z.; Hu, C.; Zhu, Y.; Xu, Z.; Randazzo, B.; Wasfi, Y.; Chen, Y.; Sharma, A.; Zhou, H. Population Pharmacokinetic Modeling of Guselkumab, a Human IgG1λ Monoclonal Antibody Targeting IL-23, in Patients with Moderate to Severe Plaque Psoriasis. J. Clin. Pharm. 2018, 58, 613–627. [Google Scholar] [CrossRef]
- Khalilieh, S.; Hodsman, P.; Xu, C.; Tzontcheva, A.; Glasgow, S.; Montgomery, D. Pharmacokinetics of Tildrakizumab (MK-3222), an Anti-IL-23 Monoclonal Antibody, After Intravenous or Subcutaneous Administration in Healthy Subjects. Basic Clin. Pharm. Toxicol. 2018, 123, 294–300. [Google Scholar] [CrossRef] [Green Version]
- Jauslin, P.; Kulkarni, P.; Li, H.; Vatakuti, S.; Hussain, A.; Wenning, L.; Kerbusch, T. Population-Pharmacokinetic Modeling of Tildrakizumab (MK-3222), an Anti-Interleukin-23-p19 Monoclonal Antibody, in Healthy Volunteers and Subjects with Psoriasis. Clin. Pharm. 2019, 58, 1059–1068. [Google Scholar] [CrossRef]
- Zandvliet, A.; Glasgow, S.; Horowitz, A.; Montgomery, D.; Marjason, J.; Mehta, A.; Xu, C.; van Vugt, M.; Khalilieh, S. Tildrakizumab, a novel anti-IL-23 monoclonal antibody, is unaffected by ethnic variability in Caucasian, Chinese, and Japanese subjects. Int. J. Clin. Pharmacol. Ther. 2015, 53, 139–146. [Google Scholar] [CrossRef]
- Khalilieh, S.; Hussain, A.; Montgomery, D.; Levine, V.; Shaw, P.M.; Bodrug, I.; Mekokishvili, L.; Bailey-Smith, C.; Glasgow, X.S.; Cheng, A.; et al. Effect of tildrakizumab (MK-3222), a high affinity, selective anti-IL23p19 monoclonal antibody, on cytochrome P450 metabolism in subjects with moderate to severe psoriasis. Br. J. Clin. Pharm. 2018, 84, 2292–2302. [Google Scholar] [CrossRef] [Green Version]
- Papp, K.; Thaçi, D.; Reich, K.; Riedl, E.; Langley, R.G.; Krueger, J.G.; Gottlieb, A.B.; Nakagawa, H.; Bowman, E.P.; Mehta, A.; et al. Tildrakizumab (MK-3222), an anti-interleukin-23p19 monoclonal antibody, improves psoriasis in a phase IIb randomized placebo-controlled trial. Br. J. Dermatol. 2015, 173, 930–939. [Google Scholar] [CrossRef]
- Reich, K.; Papp, K.A.; Blauvelt, A.; Tyring, S.K.; Sinclair, R.; Thaçi, D.; Nograles, K.; Mehta, A.; Cichanowitz, N.; Li, Q.; et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): Results from two randomised controlled, phase 3 trials. Lancet 2017, 390, 276–288. [Google Scholar] [CrossRef]
- Kerbusch, T.; Li, H.; Wada, R.; Jauslin, P.M.; Wenning, L. Exposure-response characterisation of tildrakizumab in chronic plaque psoriasis: Pooled analysis of 3 randomised controlled trials. Br. J. Clin. Pharm. 2020, 86, 1795–1806. [Google Scholar] [CrossRef] [PubMed]
- Suleiman, A.A.; Minocha, M.; Khatri, A.; Pang, Y.; Othman, A.A. Population Pharmacokinetics of Risankizumab in Healthy Volunteers and Subjects with Moderate to Severe Plaque Psoriasis: Integrated Analyses of Phase I–III Clinical Trials. Clin. Pharmacokinet. 2019, 58, 1309–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, J.G.; Ferris, L.K.; Menter, A.; Wagner, F.; White, A.; Visvanathan, S.; Lalovic, B.; Aslanyan, S.; Wang, E.E.; Hall, D.; et al. Anti-IL-23A mAb BI 655066 for treatment of moderate-to-severe psoriasis: Safety, efficacy, pharmacokinetics, and biomarker results of a single-rising-dose, randomized, double-blind, placebo-controlled trial. J. Allergy Clin. Immunol. 2015, 136, 116–124.e117. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.A.; Blauvelt, A.; Bukhalo, M.; Gooderham, M.; Krueger, J.G.; Lacour, J.P.; Menter, A.; Philipp, S.; Sofen, H.; Tyring, S.; et al. Risankizumab versus Ustekinumab for Moderate-to-Severe Plaque Psoriasis. N. Engl. J. Med. 2017, 376, 1551–1560. [Google Scholar] [CrossRef]
- Ohtsuki, M.; Fujita, H.; Watanabe, M.; Suzaki, K.; Flack, M.; Huang, X.; Kitamura, S.; Valdes, J.; Igarashi, A. Efficacy and safety of risankizumab in Japanese patients with moderate to severe plaque psoriasis: Results from the SustaIMM phase 2/3 trial. J. Dermatol. 2019, 46, 686–694. [Google Scholar] [CrossRef] [Green Version]
- Suleiman, A.A.; Khatri, A.; Oberoi, R.K.; Othman, A.A. Exposure–Response Relationships for the Efficacy and Safety of Risankizumab in Japanese Subjects with Psoriasis. Clin. Pharmacokinet. 2019, 59, 575–589. [Google Scholar] [CrossRef]
- Khatri, A.; Eckert, D.; Oberoi, R.; Suleiman, A.; Pang, Y.; Cheng, L.; Othman, A.A. Pharmacokinetics of Risankizumab in Asian Healthy Subjects and Patients with Moderate to Severe Plaque Psoriasis, Generalized Pustular Psoriasis, and Erythrodermic Psoriasis. J. Clin. Pharm. 2019, 59, 1656–1668. [Google Scholar] [CrossRef] [Green Version]
- Khatri, A.; Suleiman, A.A.; Polepally, A.R.; Othman, A.A. Exposure–Response Relationships for Efficacy and Safety of Risankizumab in Phase II and III Trials in Psoriasis Patients. Clin. Pharmacol. Ther. 2019, 107, 378–387. [Google Scholar] [CrossRef] [Green Version]
- Gordon, K.B.; Strober, B.; Lebwohl, M.; Augustin, M.; Blauvelt, A.; Poulin, Y.; Papp, K.A.; Sofen, H.; Puig, L.; Foley, P.; et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): Results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet 2018, 392, 650–661. [Google Scholar] [CrossRef]
- Menting, S.P.; Coussens, E.; Pouw, M.F.; van den Reek, J.M.; Temmerman, L.; Boonen, H.; de Jong, E.M.; Spuls, P.I.; Lambert, J. Developing a Therapeutic Range of Adalimumab Serum Concentrations in Management of Psoriasis: A Step Toward Personalized Treatment. JAMA Dermatol. 2015, 151, 616–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ternant, D.; Ducourau, E.; Fuzibet, P.; Vignault, C.; Watier, H.; Lequerré, T.; Le Loët, X.; Vittecoq, O.; Goupille, P.; Mulleman, D.; et al. Pharmacokinetics and concentration-effect relationship of adalimumab in rheumatoid arthritis. Br. J. Clin. Pharm. 2015, 79, 286–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papamichael, K.; Vogelzang, E.H.; Lambert, J.; Wolbink, G.; Cheifetz, A.S. Therapeutic drug monitoring with biologic agents in immune mediated inflammatory diseases. Expert Rev. Clin. Immunol. 2019, 15, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Liau, M.M.; Oon, H.H. Therapeutic drug monitoring of biologics in psoriasis. Biol. Targets Ther. 2019, 13, 127–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewski, J.; Poniedziałek, B.; Rzymski, P.; Adamski, Z. Factors affecting response to biologic treatment in psoriasis. Dermatol. Ther. 2014, 27, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Imamura, C.K. Therapeutic drug monitoring of monoclonal antibodies: Applicability based on their pharmacokinetic properties. Drug Metab. Pharm. 2019, 34, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Watson, I.; Potter, J.; Yatscoff, R.; Fraser, A.; Himberg, J.J.; Wenk, M. Editorial. Ther. Drug Monit. 1997, 19, 125. [Google Scholar] [CrossRef]
- Vermeire, S.; Dreesen, E.; Papamichael, K.; Dubinsky, M.C. How, When, and for Whom Should We Perform Therapeutic Drug Monitoring? Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2020, 18, 1291–1299. [Google Scholar] [CrossRef]
- Schots, L.; Grine, L.; Soenen, R.; Lambert, J. Dermatologists on the medical need for therapeutic drug monitoring of biologics in psoriasis: Results of a structured survey. J. Dermatol. Treat. 2020, 15, 1–9. [Google Scholar] [CrossRef]
- Wilkinson, N.; Tsakok, T.; Dand, N.; Bloem, K.; Duckworth, M.; Baudry, D.; Pushpa-Rajah, A.; Griffiths, C.E.M.; Reynolds, N.J.; Barker, J.; et al. Defining the Therapeutic Range for Adalimumab and Predicting Response in Psoriasis: A Multicenter Prospective Observational Cohort Study. J. Investig. Dermatol. 2019, 139, 115–123. [Google Scholar] [CrossRef]
- Takahashi, H.; Tsuji, H.; Ishida-Yamamoto, A.; Iizuka, H. Plasma trough levels of adalimumab and infliximab in terms of clinical efficacy during the treatment of psoriasis. J. Dermatol. 2013, 40, 39–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syversen, S.W.; Goll, G.L.; Jørgensen, K.K.; Olsen, I.C.; Sandanger, Ø.; Gehin, J.E.; Warren, D.J.; Sexton, J.; Mørk, C.; Jahnsen, J.; et al. Therapeutic drug monitoring of infliximab compared to standard clinical treatment with infliximab: Study protocol for a randomised, controlled, open, parallel-group, phase IV study (the NOR-DRUM study). Trials 2020, 21, 13. [Google Scholar] [CrossRef] [Green Version]
- Syversen, S.W.; Jørgensen, K.K.; Goll, G.L.; Brun, M.K.; Sandanger, Ø.; Bjørlykke, K.H.; Sexton, J.; Olsen, I.C.; Gehin, J.E.; Warren, D.J.; et al. Effect of Therapeutic Drug Monitoring vs. Standard Therapy During Maintenance Infliximab Therapy on Disease Control in Patients With Immune-Mediated Inflammatory Diseases: A Randomized Clinical Trial. JAMA 2021, 326, 2375–2384. [Google Scholar] [CrossRef] [PubMed]
- Mahil, S.K.; Arkir, Z.; Richards, G.; Lewis, C.M.; Barker, J.N.; Smith, C.H. Predicting treatment response in psoriasis using serum levels of adalimumab and etanercept: A single-centre, cohort study. Br. J. Dermatol. 2013, 169, 306–313. [Google Scholar] [CrossRef] [Green Version]
- Carrascosa, J.M.; Toro Montecinos, M.; Ballescá, F.; Teniente Serra, A.; Martínez Cáceres, E.; Ferrándiz, C. Correlation between trough serum levels of adalimumab and absolute PASI score in a series of patients with psoriasis. J. Dermatol. Treat. 2018, 29, 140–144. [Google Scholar] [CrossRef]
- Soenen, R.; Meulewaeter, E.; Grine, L.; Van den Berghe, N.; Brouwers, E.; Speeckaert, R.; Lanssens, S.; Temmerman, L.; Lambert, J.; Gils, A. Defining a Minimal Effective Serum Trough Concentration of Secukinumab in Psoriasis: A Step toward Personalized Therapy. J. Investig. Dermatol. 2019, 139, 2232–2235.e2231. [Google Scholar] [CrossRef] [Green Version]
- Menting, S.P.; van den Reek, J.M.; Baerveldt, E.M.; de Jong, E.M.; Prens, E.P.; Lecluse, L.L.; Wolbink, G.J.; Van der Kleij, D.; Spuls, P.I.; Rispens, T. The correlation of clinical efficacy, serum trough levels and antidrug antibodies in ustekinumab-treated patients with psoriasis in a clinical-practice setting. Br. J. Dermatol. 2015, 173, 855–857. [Google Scholar] [CrossRef] [PubMed]
- Toro-Montecinos, M.; Ballescá, F.; Ferrandiz, C.; Teniente-Serra, A.; Martinez-Caceres, E.; Carrascosa, J.M. Usefulness and correlation with clinical response of serum ustekinumab levels measured at 6 weeks versus 12 weeks. J. Dermatol. Treat. 2019, 30, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Van den Berghe, N.; De Keyser, E.; Soenen, R.; Meuleman, L.; Lanssens, S.; Gils, A.; Lambert, J. Clinical response correlates with 4-week postinjection ustekinumab concentrations in patients with moderate-to-severe psoriasis. Br. J. Dermatol. 2019, 182, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Tsakok, T.; Wilson, N.; Dand, N.; Loeff, F.C.; Bloem, K.; Baudry, D.; Duckworth, M.; Pan, S.; Pushpa-Rajah, A.; Standing, J.F.; et al. Association of Serum Ustekinumab Levels With Clinical Response in Psoriasis. JAMA Dermatol. 2019, 155, 1235–1243. [Google Scholar] [CrossRef]
- Reich, K.; Jackson, K.; Ball, S.; Garces, S.; Kerr, L.; Chua, L.; Muram, T.M.; Blauvelt, A. Ixekizumab Pharmacokinetics, Anti-Drug Antibodies, and Efficacy through 60 Weeks of Treatment of Moderate to Severe Plaque Psoriasis. J. Investig. Dermatol. 2018, 138, 2168–2173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, Y.; Khatri, A.; Suleiman, A.A.; Othman, A.A. Clinical Pharmacokinetics and Pharmacodynamics of Risankizumab in Psoriasis Patients. Clin. Pharm. 2019, 59, 311–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyring, S.; Gordon, K.B.; Poulin, Y.; Langley, R.G.; Gottlieb, A.B.; Dunn, M.; Jahreis, A. Long-term safety and efficacy of 50 mg of etanercept twice weekly in patients with psoriasis. Arch. Dermatol. 2007, 143, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Gordon, K.B.; Gottlieb, A.B.; Leonardi, C.L.; Elewski, B.E.; Wang, A.; Jahreis, A.; Zitnik, R. Clinical response in psoriasis patients discontinued from and then reinitiated on etanercept therapy. J. Dermatol. Treat. 2006, 17, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, C.; Strober, B.; Gottlieb, A.B.; Elewski, B.E.; Ortonne, J.P.; van de Kerkhof, P.; Chiou, C.F.; Dunn, M.; Jahreis, A. Long-term safety and efficacy of etanercept in patients with psoriasis: An open-label study. J. Drugs Dermatol. JDD 2010, 9, 928–937. [Google Scholar]
- Lecluse, L.L.; Driessen, R.J.; Spuls, P.I.; de Jong, E.M.; Stapel, S.O.; van Doorn, M.B.; Bos, J.D.; Wolbink, G.J. Extent and clinical consequences of antibody formation against adalimumab in patients with plaque psoriasis. Arch. Dermatol. 2010, 146, 127–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asahina, A.; Nakagawa, H.; Etoh, T.; Ohtsuki, M. Adalimumab in Japanese patients with moderate to severe chronic plaque psoriasis: Efficacy and safety results from a Phase II/III randomized controlled study. J. Dermatol. 2010, 37, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, A.B.; Evans, R.; Li, S.; Dooley, L.T.; Guzzo, C.A.; Baker, D.; Bala, M.; Marano, C.W.; Menter, A. Infliximab induction therapy for patients with severe plaque-type psoriasis: A randomized, double-blind, placebo-controlled trial. J. Am. Acad. Dermatol. 2004, 51, 534–542. [Google Scholar] [CrossRef]
- Adişen, E.; Aral, A.; Aybay, C.; Gürer, M.A. Anti-infliximab antibody status and its relation to clinical response in psoriatic patients: A pilot study. J. Dermatol. 2010, 37, 708–713. [Google Scholar] [CrossRef]
- Hoffmann, J.H.; Hartmann, M.; Enk, A.H.; Hadaschik, E.N. Autoantibodies in psoriasis as predictors for loss of response and anti-infliximab antibody induction. Br. J. Dermatol. 2011, 165, 1355–1358. [Google Scholar] [CrossRef]
- Torii, H.; Nakagawa, H. Long-term study of infliximab in Japanese patients with plaque psoriasis, psoriatic arthritis, pustular psoriasis and psoriatic erythroderma. J. Dermatol. 2011, 38, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, A.B.; Kalb, R.E.; Blauvelt, A.; Heffernan, M.P.; Sofen, H.L.; Ferris, L.K.; Kerdel, F.A.; Calabro, S.; Wang, J.; Kerkmann, U.; et al. The efficacy and safety of infliximab in patients with plaque psoriasis who had an inadequate response to etanercept: Results of a prospective, multicenter, open-label study. J. Am. Acad. Dermatol. 2012, 67, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Zisapel, M.; Zisman, D.; Madar-Balakirski, N.; Arad, U.; Padova, H.; Matz, H.; Maman-Sarvagyl, H.; Kaufman, I.; Paran, D.; Feld, J.; et al. Prevalence of TNF-α blocker immunogenicity in psoriatic arthritis. J. Rheumatol. 2015, 42, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Herszényi, K.; Jókai, H.; Rencz, F.; Brodszky, V.; Nagy, E.; Holló, P. Antidrug antibody formation during tumor necrosis factor α inhibitor treatment of severe psoriatic patients in the real-life practice. Postepy Dermatol. Alergol. 2019, 36, 589–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kauffman, C.L.; Aria, N.; Toichi, E.; McCormick, T.S.; Cooper, K.D.; Gottlieb, A.B.; Everitt, D.E.; Frederick, B.; Zhu, Y.; Graham, M.A.; et al. A phase I study evaluating the safety, pharmacokinetics, and clinical response of a human IL-12 p40 antibody in subjects with plaque psoriasis. J. Investig. Dermatol. 2004, 123, 1037–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, G.G.; Langley, R.G.; Leonardi, C.; Yeilding, N.; Guzzo, C.; Wang, Y.; Dooley, L.T.; Lebwohl, M. A human interleukin-12/23 monoclonal antibody for the treatment of psoriasis. N. Engl. J. Med. 2007, 356, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, C.E.; Strober, B.E.; van de Kerkhof, P.; Ho, V.; Fidelus-Gort, R.; Yeilding, N.; Guzzo, C.; Xia, Y.; Zhou, B.; Li, S.; et al. Comparison of ustekinumab and etanercept for moderate-to-severe psoriasis. N. Engl. J. Med. 2010, 362, 118–128. [Google Scholar] [CrossRef]
- Tsai, T.F.; Ho, J.C.; Song, M.; Szapary, P.; Guzzo, C.; Shen, Y.K.; Li, S.; Kim, K.J.; Kim, T.Y.; Choi, J.H.; et al. Efficacy and safety of ustekinumab for the treatment of moderate-to-severe psoriasis: A phase III, randomized, placebo-controlled trial in Taiwanese and Korean patients (PEARL). J. Dermatol. Sci. 2011, 63, 154–163. [Google Scholar] [CrossRef]
- Kimball, A.B.; Papp, K.A.; Wasfi, Y.; Chan, D.; Bissonnette, R.; Sofen, H.; Yeilding, N.; Li, S.; Szapary, P.; Gordon, K.B. Long-term efficacy of ustekinumab in patients with moderate-to-severe psoriasis treated for up to 5 years in the PHOENIX 1 study. J. Eur. Acad. Dermatol. Venereol. JEADV 2013, 27, 1535–1545. [Google Scholar] [CrossRef]
- Blauvelt, A.; Langley, R.; Leonardi, C.; Gordon, K.; Luger, T.; Ohtsuki, M.; Nickoloff, B.; Kerr, L. Ixekizumab, a novel anti-IL-17A antibody, exhibits low immunogenicity during long-term treatment in patients with psoriasis. J. Am. Acad. Dermatol. 2016, 74, AB258. [Google Scholar] [CrossRef]
- Agrawal, M.; Spencer, E.A.; Colombel, J.F.; Ungaro, R.C. Approach to the Management of Recently Diagnosed Inflammatory Bowel Disease Patients: A User’s Guide for Adult and Pediatric Gastroenterologists. Gastroenterology 2021, 161, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Cartron, G.; Dacheux, L.; Salles, G.; Solal-Celigny, P.; Bardos, P.; Colombat, P.; Watier, H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood 2002, 99, 754–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellor, J.D.; Brown, M.P.; Irving, H.R.; Zalcberg, J.R.; Dobrovic, A. A critical review of the role of Fc gamma receptor polymorphisms in the response to monoclonal antibodies in cancer. J. Hematol. Oncol. 2013, 6, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feuerstein, J.D.; Nguyen, G.C.; Kupfer, S.S.; Falck-Ytter, Y.; Singh, S. American Gastroenterological Association Institute Guideline on Therapeutic Drug Monitoring in Inflammatory Bowel Disease. Gastroenterology 2017, 153, 827–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]

| Drug | Structure | Mechanism of Action | Route | Dosing Regimen | PASI75 (%) | PASI90 (%) | PASI100 (%) | Side Effect |
|---|---|---|---|---|---|---|---|---|
| Etanercept | Fusion protein | TNF-α receptor binding | SC | 25 mg BIW or 50 mg QW | W12: 49 [49,50] | W12: 21–22 [49,50] | Infections, malignancies, and heart failure | |
| Adalimumab | Monoclonal antibody | TNF-α binding | SC | 80 mg LD + 40 mg Q2W | W16: 71 [51] | W16: 37 [51] | W16: 14 [51] | URI, nasopharyngitis, sinusitis, non-melanoma skin cancer, and heart failure thromboembolic events |
| Infliximab | Monoclonal antibody | TNF-α binding | SC | 5 mg/kg induction at W 0, 2 and 6, then Q8W | W10: 75.5 [52]–80 [53] W50: 54.5 [52]–61 [53] | W10: 57 [53] W50: 34.3 [52]–45 [53] | URI, headache, fatigue, and squamous cell or basal cell cancers | |
| Certolizumab pegol | Pegylated antigen-binding fragment | TNF-α binding | SC | 200 or 400 mg Q2W | W16: 76–83 [54] W48: 81–87 [54] | W16: 44–55 [54] W48: 60–62 [54] | W16: 13–19 [54] W48: 24–38 [54] | URI, nasopharyngitis, and cell carcinoma |
| Golimumab | Monoclonal antibody | TNF-α binding | SC | 50 or 100 mg Q4W | W14: 58 [55] W24: 66 [55] | W14: 24 [55] W24: 32 [55] | Infections and cutaneous squamous cell carcinoma | |
| Ustekinumab | Monoclonal antibody | IL-12 and IL-23 binding | SC | 45 or 90 mg at W 0 & 4, then Q12W | W12: 66.4–75.7 [56,57] | W12: 36.7–50.9 [56,57] | W12: 12.5–50.9 [56,57] | URI, nasopharyngitis, headache, and arthralgia |
| Secukinumab | Monoclonal antibody | IL-17A binding | SC | 300 mg Ws 0–4, then Q4W | W12: 81.6–77.1 [58] | W12: 54.2–59.2 [58] W52: 58 [59] | W12: 24.1–28.6 [58] W52: 39.2 [58] | Nasopharyngitis, headache, and diarrhea during induction |
| Ixekizumab | Monoclonal antibody | IL-17A binding | SC | 160 mg W 0, 80 mg Q2W for 3 months, then Q4W | W12: 77.5 [60] W60: 83 [60] | W12: 64.6 [59] W12: 59.7 [60] W60: 73 [61] | W12: 33.6 [59] W12: 30.8 [60] W60: 55 [60] | URI, nasopharyngitis, and headache |
| Brodalumab | Monoclonal antibody | IL-17A binding | SC | 210 mg Q1W for 3 Ws, then Q2W | W12: 83.3 [61]–86.3 [62] W52: 80 [62] | W12: 69 [63] –70.3 [61] W52: 73–75 [62] | W12: 36.7–44.4 [62] w52: 53–56 [62] | URI, nasopharyngitis, headache, and arthralgia |
| Guselkumab | Monoclonal antibody | IL-23 binding | SC | 100 mg at W0 & 4, then Q8W | W16: 86.5 [64]–91.2 [63] W48: 87.8 [63] | W16: 70 [63]–73.3 [63] W48: 76.3 [63] | W16: 34.1 [64]–37.4 [63] W48: 47.4 [63] | URI and nasopharyngitis |
| Tildrakizumab | Monoclonal antibody | IL-23 binding | SC | 100 or 200 mg at W0 & 4, then Q12W | W12: 61–66 [65] | W12: 35–39 [65] | W12: 12–42 [65] | Nasopharyngitis |
| Risankizumab | Monoclonal antibody | IL-23 binding | SC | 150 mg at W0 & 4, then Q12W | W16:88.7 [66] W52:92.8 [66] | W16: 73.2 [66] W52: 85.6 [66] | W16: 47.2 [66] W52: 60 [66] | URI, nasopharyngitis, and headache |
| Drug | Study (Phase) | Disease | Dose Regimens | Subjects (Samples) | PD Endpoint (Samples) |
|---|---|---|---|---|---|
| Adalimumab | M02-528 (II) [69] | Pso | 40 mg Q2W/QW (SC) [70] | 827 P [70] | PASI, PASI75 [70] |
| REVEAL (III) [51] | |||||
| Golimumab | GO-REVEAL (III) [55] | PsA | 50 or 100 Q4W (SC) [71] | 337 P (2029) [71] | |
| Ustekinumab | NCT00267956 (II) [72] | PsA | 90 mg, W 0–3, 12, and 16 (SC) [73] | 130 P (1594) [73] | |
| PHOENIX 1 (III) [56] | Pso | 45 or 90 mg, W 0, 4, then Q12W (SC) [74,75,76,77] | 1937 P (9938) [74] | ||
| PHOENIX 2 (III) [57] | 1312 P [75] | PASI (11624) [75] | |||
| PSUMMIT I (III) [76] PSUMMIT II (III) [78] | 925 P (2837) [79] | PASI (3429), ACR (8561) [79] | |||
| BSTOP/PSORTD [77] | 491 P (797) [77] | PASI (1590) [77] | |||
| Secukinumab | Hueber et al. (I) [80] | Pso | 25–300 mg, then Q4W (SC) [81] | 1233 P [81] | Total IL-17 [81] |
| Rich et al. (II) [82] | |||||
| Reich et al. (II) [83] | |||||
| Papp et al. (II) [84] | 1–10 mg/kg, sD (IV) [81] | ||||
| ERASURE & FIXTURE (III) [58] | |||||
| JUNCTURE (III) [85] | |||||
| Ixekizumab | I1F-MC-RHAJ (II) [86] | Pso | 10–150 mg, W 0, 2, 4, then Q4W (SC) [87] | 115 P (651) [87] | PASI (2096), PASI75 [87] |
| UNCOVER-1 (III) [59] | |||||
| UNCOVER-2 (III) [60] | 80 mg Q2W/Q4W (SC) [88] | 2888 P (2097) [88] | PASI, PASI75, sPGA [88] | ||
| UNCOVER-3 (III) [60] | |||||
| Brodalumab | NCT00867100 (I) [89] | Pso | 7–700 mg, sD (SC, IV) [90,91] | 57 HV, 25 P [92] | PASI [92] |
| NCT01937260 (I) | 140 or 210 mg, sD (SC) [91] | ||||
| NCT00975637 (II) [93] | 70–280 mg first D, W 1, 2, then Q2W/Q4W (SC) [90,91,92] | 196 P (1526) [90] | |||
| AMAGINE-1 (III) [61] | 140 and/or 210 mg, W 0, 1, 2, then Q2W (SC) [91] | ||||
| AMAGINE-2 (III) [62] | 140 and/or 210 mg, W 0, 1, 2, then Q2W/Q4W/Q8W (SC) [91] | 622 P (7725) [91] | PASI (2220), PGA (2456) [94] | ||
| AMAGINE-3 (III) [62] | |||||
| Guselkumab | X-PLORE (II) [95] | Pso | 1.5 mg Q12W (SC) [94] | 238 P (2014) [94] | PASI (17580), PGA (18986) [96] |
| 15 mg Q8W (SC) [94] | |||||
| VOYAGE 1 (III) [63] | 50 mg Q12W (SC) [94,95,96,97] | 1459 P (13031) [96] | |||
| 100 mg Q8W (SC) [94] | |||||
| VOYAGE 2 (III) [64] | 200 mg Q12W (SC) [94] | 1454 P (13014) [97] | |||
| Tildrakizumab | P05776 (I) [98] | Pso | 0.1, 0.5, 3 and 10 mg/kg, sD (IV) [98] | 31 HV (340) [99] | |
| 50 or 200 mg, sD (SC) [98,99] | |||||
| P06306 (I) [100] | 10 mg/kg, sD (IV) [100] | 53 HV (648) [99] | |||
| 50–400 mg, sD (SC) [99,100] | |||||
| P009 (I) [101] | 200 mg, sD (SC) [99,101] | 19 P (309) [99,101] | |||
| P05495 (IIb) [102] | 5–200 W 0, 4, then Q12W (SC) [99,102] | 349 P (4679) [99,102] | |||
| reSURFACE 1 (III) [103] | 100 or 200 W 0, 4, then Q12W (SC) [99,103,104] | 763 P (6329) [99] | PASI75, 90 and 100 [104] | ||
| reSURFACE 2 (III) [103] | 883 P (5016) [99] | ||||
| Risankizumab | NCT02596217/M16-513 (I) [105] | Pso | 18–1200 mg, sD (SC) [105] | 1899 HV and P (13123) [105] | |
| NCT01577550/1311.1 (I) [106] | 0.01–5 mg/kg, sD (IV), 0.25–1 mg/kg, sD (SC) [105] | ||||
| NCT02054481/1311.2 (II) [107] | |||||
| NCT03000075 (II/III) [108] | 18 mg sD (SC), 90 or 180 W 0, 4, 16 (SC) [105,109] | 2095 P [110] | PASI75, 90 and 100, sPGA [110,111] | ||
| NCT03022045 (III) [109] | |||||
| UltIMMa-1 (III) [112] | 75 or 150 mg W 0, 4, then Q12W (SC) [110] | 1903 P [110] | |||
| UltIMMa-2 (III) [112] | |||||
| NCT02672852/IMMhance (III) [105] | 150 mg W 0, 4, then Q12W or 150 mg Q12W (SC) [105,108,109,112] | 1732 P [111] | |||
| NCT02694523/IMMvent (III) [105] |
| mAb | Model | PK Parameters | Covariates (Parameters) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ka, 1/d (%RSE) | ka IIV, %CV (%RSE) | CL, L/d (%RSE) | CL IIV, %CV (%RSE) | VC, L (%RSE) | VC IIV, %CV (%RSE) | Q, L/d (%RSE) | Q IIV % (%RSE) | VP, L (%RSE) | VP IIV, %CV (%RSE) | Km, µg/mL (%RSE) | Vmax, mg/day (%RSE) | Vmax, IIV, %CV (%RSE) | |||
| Adalimumab [70] | 1-CMT, LE | 0.625 (28.8) | 0.586 (3.8) | 62 (8.6) | 11.4 (5.6) | 43.6 (31.5) | BW and study (CL/F) | ||||||||
| BW and study (V/F) | |||||||||||||||
| Golimumab [71] | 1-CMT, LE | 0.908 | 1.38 | 37.6 | 24.9 | 37.9 | BW, ADA, CRP, and smoking (CL/F) | ||||||||
| BW (V/F) | |||||||||||||||
| Ustekinumab [74,75] | 1-CMT, LE | 0.354 (16.2) | 0 (fixed) | 0.465 (2.0) | 41.0 (3.0) | 15.7 (2.0) | 33.2 (3.9) | BW, DB, ADA, Alb, CrCL, ALK, and sex (CL/F) | |||||||
| BW, DB, and race (V/F) | |||||||||||||||
| Ustekinumab [77] | 1-CMT, LE | 0.23 (16.1) | 0.44 (6.7) | 44.7 (10.3) | 10.2 (8.2) | 36.5 (28.9) | Cr and ADA (CL/F) | ||||||||
| BW (V/F) | |||||||||||||||
| Secukinumab [81] | 2-CMT, LE | 0.18 (3.6) | 35 | 0.19 (1.9) | 32 | 3.61 (2.6) | 30 | 0.39 (4.6) | 2.87 (1.9) | 18 | BW (CL) | ||||
| BW (VC) | |||||||||||||||
| Brodalumab [92] | 2-CMT, LE & NLE | 0.255 (10.2) | 75.1 (27.8) | 0.28 | 3.9 (5.1) | 29 (12.8) | 1.01 | 2.89 | 0.01 (fixed) | 4.39 (7.4) | 31.5 (6.9) | ||||
| Brodalumab [92] | 2-CMT, LE & NLE | 0.236 (0.64) | 57.9 (14) | 0.223 (0.62) | 69.2 (13) | 4.62 (0.90) | 69.6 (19) | 0.697 (13) | 15 (fixed) | 1.84 (0.61) | 85.6 (17) | 0.02 (fixed) | 5.16 (2.0) | 37.8 (12) | BW and age (CL) |
| BW and age (VC) | |||||||||||||||
| BW and age (Vmax) | |||||||||||||||
| Brodalumab [91] | 2-CMT, LE & NLE | 0.300 (2.8) | 62.6 | 0.155 (0.20) | 57.5 | 4.68 (0.99) | 25.5 | 0.328 (5.34) | 91 | 2.41 (3.08) | 189 | 0.02 (fixed) | 6.07 (0.53) | 2-CMT, LE & NLE | BW (CL) |
| BW(VC) | |||||||||||||||
| BW(Vmax) | |||||||||||||||
| Guselkumab [94] | 1-CMT, LE | 4.93 (4.9) | 0.567 (3.7) | 30.7 (23.3) | 14.3 (3.4) | 22.9 (26.0) | |||||||||
| Guselkumab [97] | 1-CMT, LE | 1.11 (14.1) | 129 (22.9) | 0.516 (1.19) | 35.6 (6.54) | 13.5 (1.08) | 28.0 (9.85) | BW and DB (CL/F) | |||||||
| BW (V/F) | |||||||||||||||
| Tildrakizumab [99] | 1-CMT, LE | 0.458 (6.8) | 68 (17) | 0.297 (1.1) | 29 (5.9) | 10.7 (1.1) | 21 (15) | Age, BW, Alb, sex, race, and ethnicity (CL) | |||||||
| Age, BW, and sex (VC) | |||||||||||||||
| Risankizumab [105] | 2-CMT, LE | 0.229 (4.8) | 63 (5.5) | 0.243 (1.8) | 24 (3.6) | 4.86 (3.8) | 34 (6.6) | 0.656 (3.7) | 4.25 (2.0) | BW, Alb, Cr, hs-CRP, and ADA ≥ 128 (CL) | |||||
| Risankizumab [109] | 2-CMT, LE | 0.230 (4.8) | 3.36 | 0.244 (1.8) | 5.5 | 4.87 (3.8) | 11.1 | 0.648 (3.7) | 4.25 (2.0) | BW, Alb, Cr, and hs-CRP (CL) | |||||
| BW (VC) | |||||||||||||||
| BW (VP) | |||||||||||||||
| mAb | Model | PD Parameters | Covariates (Parameters) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| kin, PASI units/d (%RSE) | kin IIV, %CV (%RSE) | kout, 1/d (%RSE) | kout IIV, %CV (%RSE) | Emax (%RSE) | γ | EC50 or IC50 µg/mL (%RSE) | EC50 or IC50 IIV, %CV (%RSE) | |||
| Ustekinumab [75] | Indirect response | 0.615 (2.5) | 60 (6.1) | 0.0313 (1.9) | 54 (4.7) | 0.929 (0.2) | 0.606 (3.4) | 283 (7.2) | HTA, MTX (IC50) | |
| Sex (kin) | ||||||||||
| Smoking (kout) | ||||||||||
| Ustekinumab [77] | SM: 15.5 (4.4) | SM: 0.02 (6.9) | SM: 43.6 (7.3) | SM: 43.6 (7.3) | 1 (fixed) | SM: 0.14 (15.0) | SM: 148.3 (9.5) | |||
| MM: 15.8 (4.2) | MM: 0.02 (7.3) | MM: 41.4 (7.6) | MM: 41.4 (7.6) | MM 1: 0.07 (17.3) MM 2: 1.21 (22.2) | MM: 42.7 (58.2) | |||||
| Ixekizumab [87] | Indirect response | 0.89 | 0.0564 | 1 (fixed) | 0.776 (13.5) | R: 0.97 (60.3) NR: 1.46 (35.1) | R: 1660 (22.8) NR: 581 (44.2) | PASI75-W12 (EC50) | ||
| Ixekizumab [88] | Logistic regression (sPGA) | 5.27 (3.6) | 0.184 (34) | CRP (EC50) | ||||||
| BW, PP (Emax) | ||||||||||
| Logistic regression (PASI75, 90, and 100) | 6.02 (4)/5.54 (5)/5.73 (11) | 0.354 (6)/0.268 (4)/0.169 (8) | BW, PP, baseline PASI (Emax) | |||||||
| Brodalumab [92] | Indirect response | 0.862 (40.1) | 0.06389 | 1 (fixed) | 2.86 (49.7) | 136 (19.8) | ||||
| Guselkumab [94] | Latent variable indirect response (joint model) | 0.0212 (5.69) | 6.24 (4.93) | 0.066 (21.3) | ||||||
| Guselkumab [96] | 0.0212 (1.96) | 5.35 (1.54) | 0.038 (6.22) | BW (IC50, kout) | ||||||
| Tildrakizumab [104] | Logistic regression (PASI75, 90 and 100) | PASI75: 62.16 | PASI75: 0.36 | |||||||
| PASI90: 37.89 | PASI90: 0.46 | |||||||||
| PASI100: 14.63 | PASI100: 0.55 | |||||||||
| Indirect response | 1 | 0.25 (4.8) | 183 | |||||||
| Risankizumab [109] | Logistic regression (sPGA) | sPGA: 0.431 (31.6) | sPGA: 0.916 (1.90) | hs-CRP (EC50) | ||||||
| Logistic regression (PASI75, 90, and 100) | PASI75: 0.939 (1.21) | PASI75: 0.203 (34.3) | ||||||||
| PASI90: 0.812 (2.44) | PASI90: 0.812 (2.44) | |||||||||
| PASI100: 0.642 (9.49) | PASI100: 0.642 (9.49) | |||||||||
| Risankizu-mab [109] | Logistic regression (sPGA) | sPGA: 0.916 | sPGA: 0.431 | |||||||
| Logistic regression (PASI75, 90, and 100) | PASI75: 0.939 | PASI75: 0.203 | ||||||||
| PASI90: 0.812 | PASI90: 0.441 | |||||||||
| PASI100: 0.642 | PASI100: 2.36 | |||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Fernández, K.; Mangas-Sanjuán, V.; Merino-Sanjuán, M.; Martorell-Calatayud, A.; Mateu-Puchades, A.; Climente-Martí, M.; Gras-Colomer, E. Impact of Pharmacokinetic and Pharmacodynamic Properties of Monoclonal Antibodies in the Management of Psoriasis. Pharmaceutics 2022, 14, 654. https://doi.org/10.3390/pharmaceutics14030654
Rodríguez-Fernández K, Mangas-Sanjuán V, Merino-Sanjuán M, Martorell-Calatayud A, Mateu-Puchades A, Climente-Martí M, Gras-Colomer E. Impact of Pharmacokinetic and Pharmacodynamic Properties of Monoclonal Antibodies in the Management of Psoriasis. Pharmaceutics. 2022; 14(3):654. https://doi.org/10.3390/pharmaceutics14030654
Chicago/Turabian StyleRodríguez-Fernández, Karine, Víctor Mangas-Sanjuán, Matilde Merino-Sanjuán, Antonio Martorell-Calatayud, Almudena Mateu-Puchades, Mónica Climente-Martí, and Elena Gras-Colomer. 2022. "Impact of Pharmacokinetic and Pharmacodynamic Properties of Monoclonal Antibodies in the Management of Psoriasis" Pharmaceutics 14, no. 3: 654. https://doi.org/10.3390/pharmaceutics14030654