
Physiologically Based Pharmacokinetic Modelling and Simulation to Predict the Plasma Concentration Profile of Doxorubicin



Abstract
:
1. Introduction
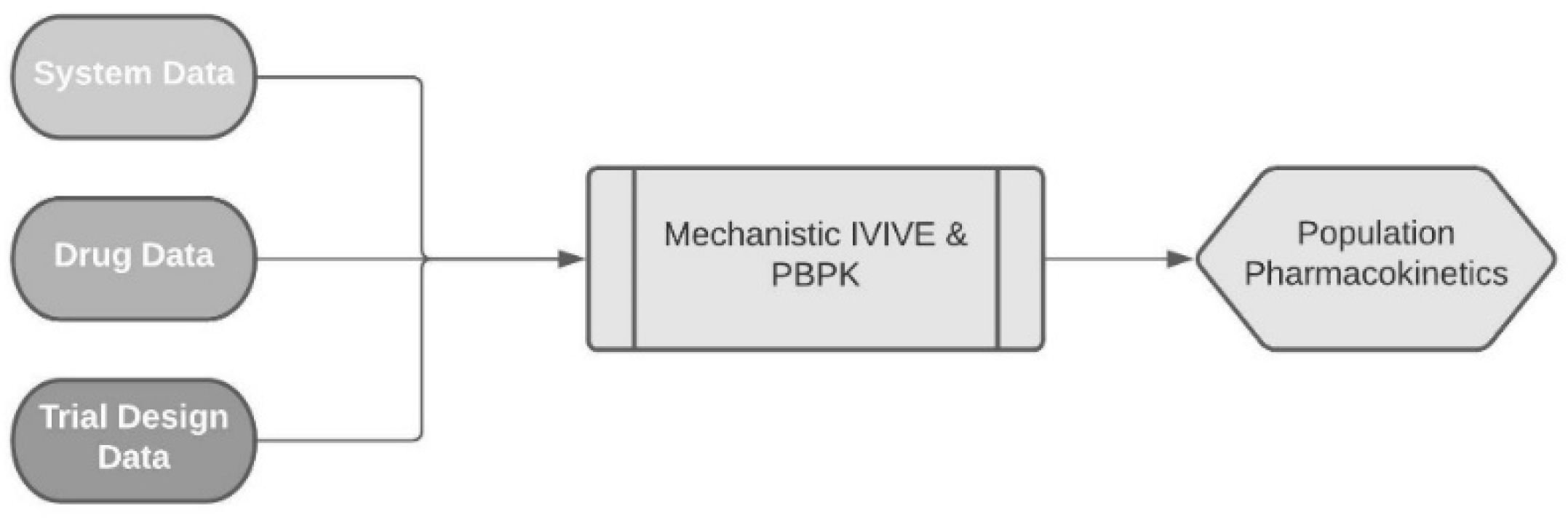
2. Materials and Methods
2.1. Clinical Studies Used
2.2. Software
2.3. Virtual Population Characteristics (System Data)
2.4. Development of DOX PBPK Model (Drug Data)
2.4.1. Calculating DOX Renal Clearance
Method A
Method B
2.4.2. Calculating DOX Hepatic Clearance
Calculating the Hepatic Blood Flow for Each Patient i
Calculating Unbound Fraction in Blood
Calculating Hepatic Blood Clearance
Calculating Intrinsic Hepatic Clearance
Separating Hepatic Clearance to Hepatic Metabolic Clearance and Biliary Excretion
Correction of Intrinsic Biliary Clearance for In Vitro System Scaling Factors
Calculating Intrinsic Metabolic Clearance
- The enzymes that participate in the primary metabolic pathway, which is the two-electron reduction, mainly aldoketoreductases (AKRs) and carbonylreductases (CBRs) are located in the cytoplasm and are expected to be found in the cytoplasmic fraction after centrifugation.
- The enzymes that participate in the secondary metabolic pathway, which is the one-electron reduction, are located mainly in mitochondria and sarcoplasmic reticulum and are expected to be found in the microsomal fraction after centrifugation.
- The enzymes that participate in the minor metabolic pathway, which is the deglycosylation, are not specified, contribute only 1–2% of the total metabolism and thus their contribution can be attributed per 106 hepatocytes.
2.4.3. Selecting a Distribution Model for DOX
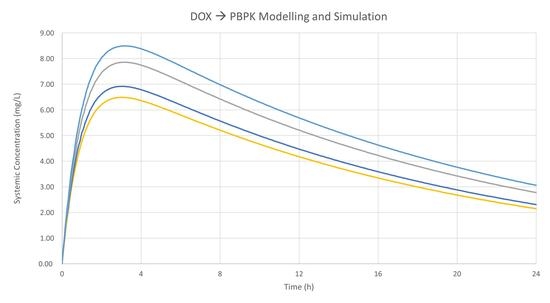
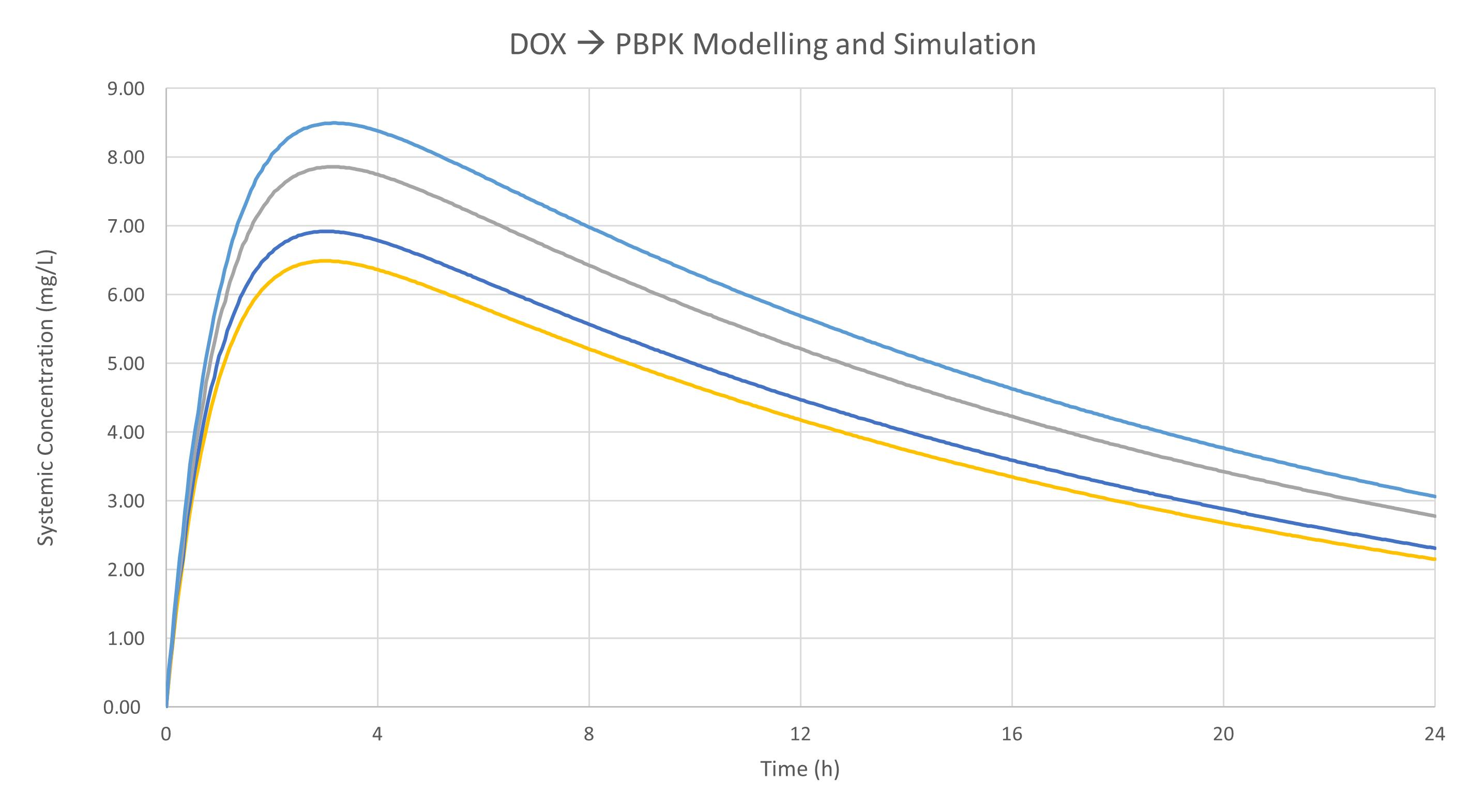
2.4.4. Generating DOX Models

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | CLR (L/h) | CLmet | Distribution Model | P.E. Kp Scalar | P.E. Vsac (L/Kg) | P.E. Qsac (L/h) |
|---|---|---|---|---|---|---|
| 1 | 7.04 | HEP | mPBPK | NA | 31.5495 | 151.3618 |
| 2 | 7.04 | HEP | fPBPK | 5.3119 | NA | NA |
| 3 | 7.04 | DIST | mPBPK | NA | 31.3833 | 212.3364 |
| 4 | 7.04 | DIST | fPBPK | 5.3119 | NA | NA |
| 5 | 8.67 | HEP | mPBPK | NA | 31.2603 | 237.3875 |
| 6 | 8.67 | HEP | fPBPK | 5.237 | NA | NA |
| 7 | 8.67 | DIST | mPBPK | NA | 31.3827 | 211.6487 |
| 8 | 8.67 | DIST | fPBPK | 5.3119 | NA | NA |
2.5. Virtual Patient Demographics for the Development of the Model Based on the Training Dataset (Clinical Settings Data)
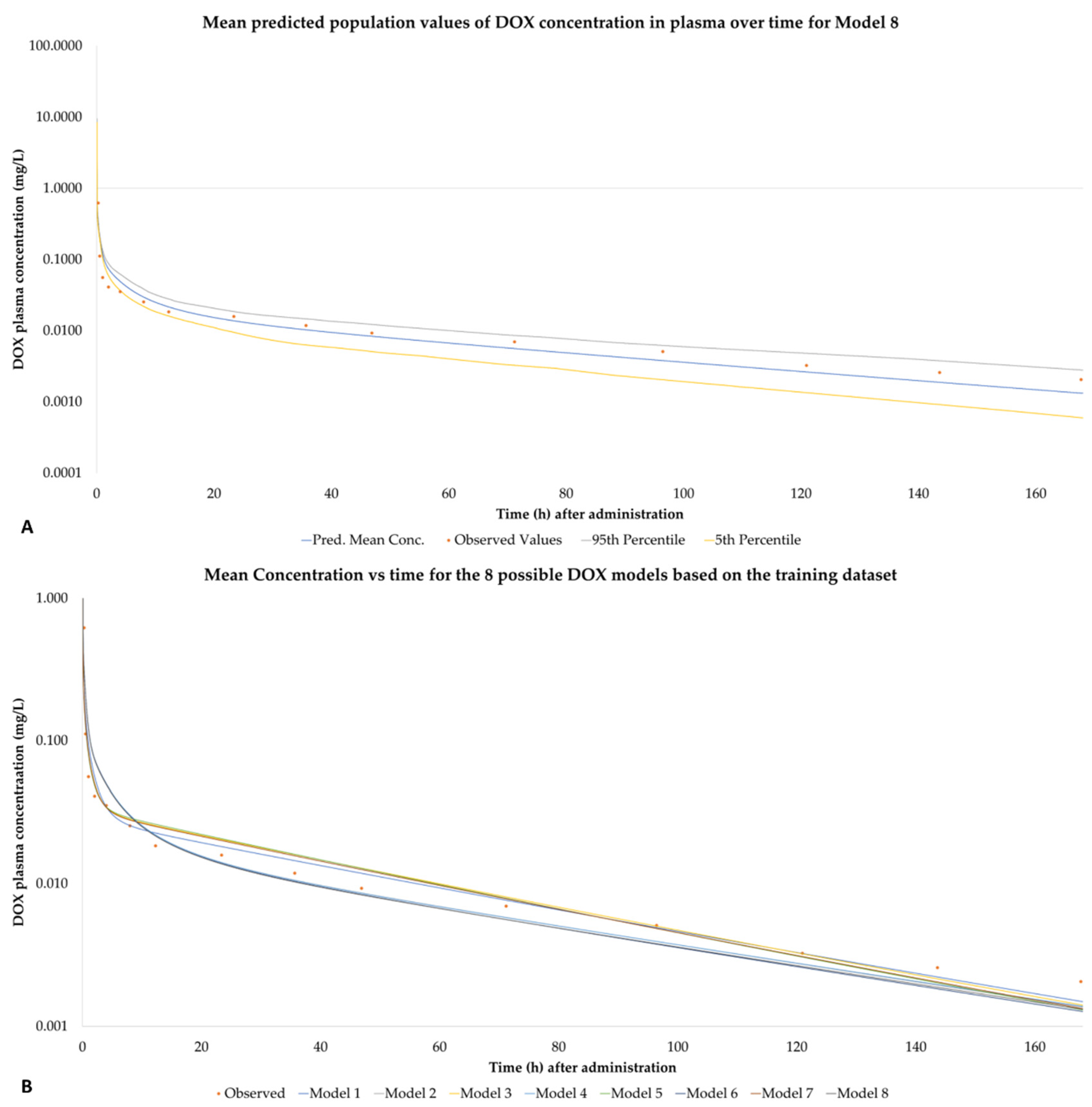
2.6. Results Based on Training Dataset
2.7. Observed Concentration Values for the Trainind Dataset
3. Results-Validation of Models
3.1. Patient Demographics for the Development of the Model Based on the Validation Dataset
3.2. Administration and Sample Retrieval
3.3. Analytical Method and Pharmacokinetic Analysis
3.4. Clinical Settings of Virtual Patients for Validation Dataset
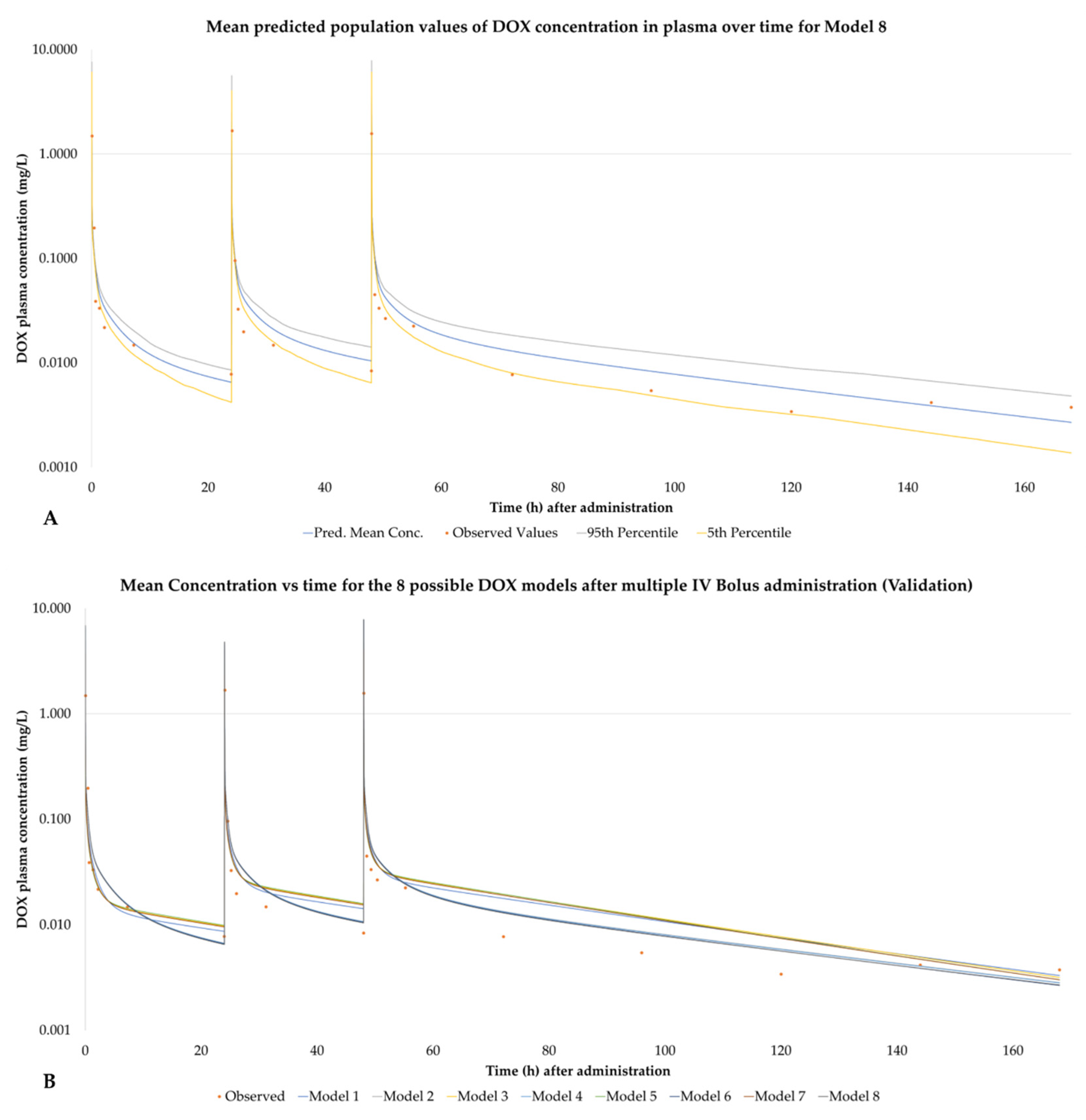
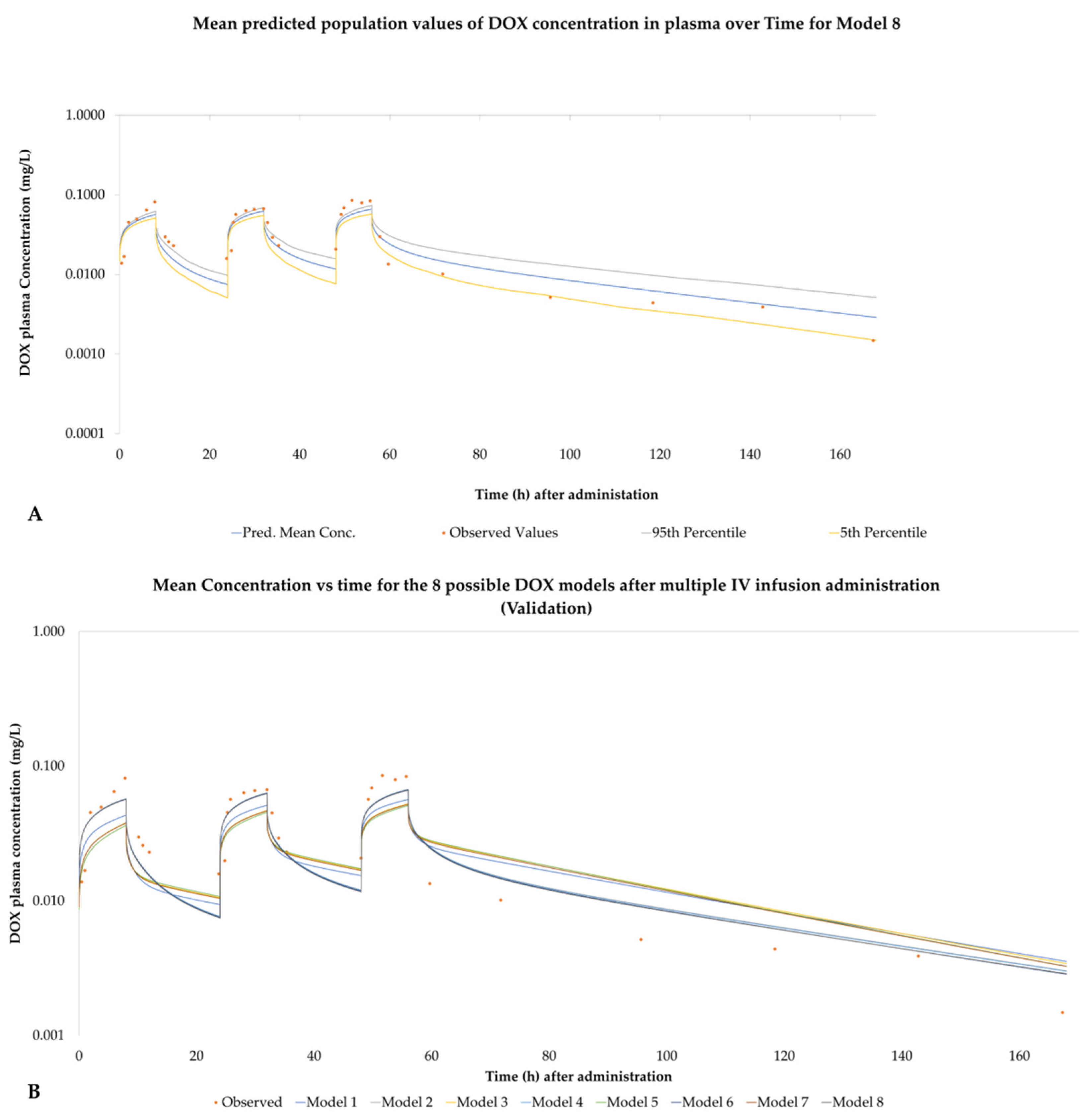
3.5. PBPK Models Performance
3.6. Observed Concentration Values for the Validation Dataset
4. Discussion
4.1. Discussion of the Performance of the Different DOX Models
4.1.1. Model Performance Based on Training Dataset
4.1.2. Model Performance Based on IV Bolus Validation
4.1.3. Model Performance Based on IV Infusion Validation
4.2. Limitations of the Models Based on Procedure
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weiss, R.B. The anthracyclines: Will we ever find a better doxorubicin? Semin. Oncol. 1992, 19, 670–686. [Google Scholar] [PubMed]
- Carvalho, C.; Santos, R.; Cardoso, S.; Correia, S.; Oliveira, P.; Santos, M.; Moreira, P. Doxorubicin: The Good, the Bad and the Ugly Effect. Curr. Med. Chem. 2009, 16, 3267–3285. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, K.; Zhang, J.; Honbo, N.; Karliner, J.S. Doxorubicin Cardiomyopathy. Cardiology 2010, 115, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Whirl-Carrillo, M.; Huddart, R.; Gong, L.; Sangkuhl, K.; Thorn, C.F.; Whaley, R.; Klein, T.E. An Evidence-Based Framework for Evaluating Pharmacogenomics Knowledge for Personalized Medicine. Clin. Pharmacol. Ther. 2021, 110, 563–572. [Google Scholar] [CrossRef]
- Whirl-Carrillo, M.; McDonagh, E.M.; Hebert, J.M.; Gong, L.; Sangkuhl, K.; Thorn, C.F.; Altman, R.B.; Klein, T.E. Pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther. 2012, 92, 414–417. [Google Scholar] [CrossRef]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharm. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef]
- Meiners, B.; Shenoy, C.; Zordoky, B.N. Clinical and preclinical evidence of sex-related differences in anthracycline-induced cardiotoxicity. Biol. Sex. Differ. 2018, 9, 38. [Google Scholar] [CrossRef]
- Barenholz, Y. Doxil®—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 2013, 65, 157–170. [Google Scholar] [CrossRef]
- Yuan, D.; He, H.; Wu, Y.; Fan, J.; Cao, Y. Physiologically Based Pharmacokinetic Modeling of Nanoparticles. J. Pharm. Sci. 2019, 108, 58–72. [Google Scholar] [CrossRef] [Green Version]
- Silverman, L.; Barenholz, Y. Effect of major tumor metabolites on release of doxorubicin from Doxil–implications for precision nano-medicine. Precis. Nanomed. 2020, 3, 699–708. [Google Scholar] [CrossRef]
- Rostami-Hodjegan, A. Reverse Translation in PBPK and QSP: Going Backwards in Order to Go Forward with Confidence. Clin. Pharmacol. Ther. 2018, 103, 224–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, H.M.; Rowland-Yeo, K. Basic Concepts in Physiologically Based Pharmacokinetic Modeling in Drug Discovery and Development. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rowland, M.; Peck, C.; Tucker, G. Physiologically-Based Pharmacokinetics in Drug Development and Regulatory Science. Annu. Rev. Pharm. Toxicol. 2011, 51, 45–73. [Google Scholar] [CrossRef]
- Vizirianakis, I.S.; Miliotou, A.N.; Mystridis, G.A.; Andriotis, E.G.; Andreadis, I.I.; Papadopoulou, L.C.; Fatouros, D.G. Tackling pharmacological response heterogeneity by PBPK modeling to advance precision medicine productivity of nanotechnology and genomics therapeutics. Expert Rev. Precis. Med. Drug Dev. 2019, 4, 139–151. [Google Scholar] [CrossRef]
- Vizirianakis, I.S.; Mystridis, G.A.; Avgoustakis, K.; Fatouros, D.G.; Spanakis, M. Enabling personalized cancer medicine decisions: The challenging pharmacological approach of PBPK models for nanomedicine and pharmacogenomics (Review). Oncol. Rep. 2016, 35, 1891–1904. [Google Scholar] [CrossRef] [Green Version]
- Dubbelboer, I.R.; Lilienberg, E.; Sjogren, E.; Lennernas, H. A Model-Based Approach To Assessing the Importance of Intracellular Binding Sites in Doxorubicin Disposition. Mol. Pharm. 2017, 14, 686–698. [Google Scholar] [CrossRef]
- Gustafson, D.L.; Rastatter, J.C.; Colombo, T.; Long, M.E. Doxorubicin pharmacokinetics: Macromolecule binding, metabolism, and excretion in the context of a physiologic model. J. Pharm. Sci. 2002, 91, 1488–1501. [Google Scholar] [CrossRef]
- Hanke, N.; Teifel, M.; Moj, D.; Wojtyniak, J.G.; Britz, H.; Aicher, B.; Sindermann, H.; Ammer, N.; Lehr, T. A physiologically based pharmacokinetic (PBPK) parent-metabolite model of the chemotherapeutic zoptarelin doxorubicin-integration of in vitro results, Phase I and Phase II data and model application for drug-drug interaction potential analysis. Cancer Chemother. Pharmacol. 2018, 81, 291–304. [Google Scholar] [CrossRef]
- He, H.; Liu, C.; Wu, Y.; Zhang, X.; Fan, J.; Cao, Y. A Multiscale Physiologically-Based Pharmacokinetic Model for Doxorubicin to Explore Its Mechanisms of Cytotoxicity and Cardiotoxicity in Human Physiological Contexts. Pharm. Res. 2018, 35, 174. [Google Scholar] [CrossRef]
- Jamei, M.; Dickinson, G.L.; Rostami-Hodjegan, A. A Framework for Assessing Inter-individual Variability in Pharmacokinetics Using Virtual Human Populations and Integrating General Knowledge of Physical Chemistry, Biology, Anatomy, Physiology and Genetics: A Tale of ‘Bottom-Up’ vs ‘Top-Down’ Recognition of Covariates. Drug Metab. Pharm. Acokine. 2009, 24, 53–75. [Google Scholar] [CrossRef]
- Camaggi, C.M.; Comparsi, R.; Strocchi, E.; Testoni, F.; Angelelli, B.; Pannuti, F. Epirubicin and doxorubicin comparative metabolism and pharmacokinetics. A cross-over study. Cancer Chemother. Pharmacol. 1988, 21, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Speth, P.A.; Linssen, P.C.; Boezeman, J.B.; Wessels, H.M.; Haanen, C. Cellular and plasma adriamycin concentrations in long-term infusion therapy of leukemia patients. Cancer Chemother. Pharmacol. 1987, 20, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Shebley, M.; Sandhu, P.; Emami Riedmaier, A.; Jamei, M.; Narayanan, R.; Patel, A.; Peters, S.A.; Reddy, V.P.; Zheng, M.; de Zwart, L.; et al. Physiologically Based Pharmacokinetic Model Qualification and Reporting Procedures for Regulatory Submissions: A Consortium Perspective. Clin. Pharmacol. Ther. 2018, 104, 88–110. [Google Scholar] [CrossRef]
- Davies, D.F.; Shock, N.W. Age changes in glomerular filtration rate, effective renal plasma flow, and tubular excretory capacity in adult males. J. Clin. Investig. 1950, 29, 496–507. [Google Scholar] [CrossRef] [Green Version]
- Wright, J.G.; Boddy, A.V.; Highley, M.; Fenwick, J.; McGill, A.; Calvert, A.H. Estimation of glomerular filtration rate in cancer patients. Br. J. Cancer 2001, 84, 452–459. [Google Scholar] [CrossRef] [Green Version]
- Drugbank.ca. Doxorubicin. Available online: https://www.drugbank.ca/drugs/DB00997 (accessed on 20 December 2021).
- Barter, Z.E.; Bayliss, M.K.; Beaune, P.H.; Boobis, A.R.; Carlile, D.J.; Edwards, R.J.; Houston, J.B.; Lake, B.G.; Lipscomb, J.C.; Pelkonen, O.R.; et al. Scaling factors for the extrapolation of in vivo metabolic drug clearance from in vitro data: Reaching a consensus on values of human microsomal protein and hepatocellularity per gram of liver. Curr. Drug Metab. 2007, 8, 33–45. [Google Scholar] [CrossRef]
- Barter, Z.E.; Chowdry, J.E.; Harlow, J.R.; Snawder, J.E.; Lipscomb, J.C.; Rostami-Hodjegan, A. Covariation of human microsomal protein per gram of liver with age: Absence of influence of operator and sample storage may justify interlaboratory data pooling. Drug Metab. Dispos. 2008, 36, 2405–2409. [Google Scholar] [CrossRef] [Green Version]
- Wagner, C.; Pan, Y.; Hsu, V.; Sinha, V.; Zhao, P. Predicting the Effect of CYP3A Inducers on the Pharmacokinetics of Substrate Drugs Using Physiologically Based Pharmacokinetic (PBPK) Modeling: An Analysis of PBPK Submissions to the US FDA. Clin. Pharm. 2016, 55, 475–483. [Google Scholar] [CrossRef]
- Abduljalil, K.; Cain, T.; Humphries, H.; Rostami-Hodjegan, A. Deciding on success criteria for predictability of pharmacokinetic parameters from in vitro studies: An analysis based on in vivo observations. Drug Metab. Dispos. 2014, 42, 1478–1484. [Google Scholar] [CrossRef]
- Guest, E.J.; Aarons, L.; Houston, J.B.; Rostami-Hodjegan, A.; Galetin, A. Critique of the two-fold measure of prediction success for ratios: Application for the assessment of drug-drug interactions. Drug Metab. Dispos. 2011, 39, 170–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawar, S.; Shevalkar, G.; Vavia, P. Glucosamine-anchored doxorubicin-loaded targeted nano-niosomes: Pharmacokinetic, toxicity and pharmacodynamic evaluation. J. Drug Target. 2016, 24, 730–743. [Google Scholar]
- Johnson, T.N.; Tucker, G.T.; Tanner, M.S.; Rostami-Hodjegan, A. Changes in liver volume from birth to adulthood: A meta-analysis. Liver Transpl. 2005, 11, 1481–1493. [Google Scholar]
- Heinemann, A.; Wischhusen, F.; Püschel, K.; Rogiers, X. Standard liver volume in the Caucasian population. Liver Transpl. Surg. 1999, 5, 366–368. [Google Scholar]




| Dose (mg/m2) | Administration | N. 1 | Women (%) | Age (Years) | Weight (kg) | BSA (m2) | Dataset | Reference |
|---|---|---|---|---|---|---|---|---|
| 60 | Single IV bolus | 8 | NA | 57.63 ± 9.28 (42–72) | 69.15 ± 14.86 (45.9–90.0) | 1.72 ± 0.18 (1.40–1.92) | Training | [22] |
| 30 | Q24 h × 3 IV bolus | 7 | 44.5% 2 | 44 ± 17 2 (17–67) | NA | NA | Validation | [23] |
| 30 | Q24 h × 3 IV infusion over 8 h. | 4 | 44.5% 2 | 44 ± 17 2 (17–67) | NA | NA | Validation | [23] |
| Physicochemical Property | Value | Comments/References |
| MW (g/mol) | 543.51 | Calculated |
| LogPo:w | 1.27 | [23] |
| Drug Type | Ampholyte | [24] |
| pKa 1 | 9.53 | [22] |
| pKa 2 | 8.94 | [22] |
| B:P Ratio | 1.15 | [25] see limitations section |
| fu,p | 0.25 | drugbank.ca (accessed on 20 December 2021) Supplementary Materials Section S2 |
| Elimination | Value (CV%) | Comments/References |
| Renal Clearance L/h | 7.04 ± 2.10 (29.8%) 8.67 ± 2.85(32.86%) | Method A Method B |
| Biliary excretion μL/min/106 cells | 24.80 ± 10.89 (43.93%) | - |
| Metabolic Clearance | - | - |
| μL/min/106 cells (HEP) | 30.38 ± 13.34(43.93%) | Method C |
| μL/min/mg protein (HLM) | 86.29 ± 38.12(44.17%) | - |
| μL/min/mg protein (HLC) | 38.74 ± 17.01(43.93%) | - |
| Distribution of clearance | See Section 2.4.2 | Method D |
| Distribution | Value | Comments/References |
| Minimal PBPK | - | Method E |
| Vss (L/kg) Vsac (L/kg) Qsac (L/h) | 31.923 P.E. tool P.E. | U.I. See Section 2.4.4 See Section 2.4.4 |
| Full PBPK | - | Method F |
| Vss (L/kg) Kp Scalar | 34.831 P.E. tool | Predicted by Method 3 See Section 2.4.4 |
| System | Prediction | CLuint Measuring Unit | Scaling Factor 1 | Scaling Factor 2 |
|---|---|---|---|---|
| Hepatocytes | Metabolic clearance Biliary excretion | μL/min per 106 cells | HPGL | Liver weight |
| Cytosolic fraction | Metabolic clearance | μL/min per mg of protein | CPPGL | |
| Microsomal fraction | Metabolic clearance | μL/min per mg of protein | MPPGL |
| Gender | Through Hepatic Artery | Through Portal Vein | Total |
|---|---|---|---|
| Males | 6.5% | 19.0% | 25.5% |
| Females | 6.5% | 21.5% | 28.0% |
| Average for both genders | 26.75% | ||
| Way of Elimination | Percentage |
|---|---|
| fbile (%) | 40% |
| fmet (%) | 49% |
| fe (%) | 11% |
| Pathway | Approximate Relative Contribution | CLint | Measuring Unit | CV % |
|---|---|---|---|---|
| HLM | 37% | 31.929 | μL/min/mg | 44.17% |
| HLC | 60% | 23.241 | μL/min/mg | 43.93% |
| HEP | 3% | 0.911 | μL/min/106 cells | 43.93% |
| Parameters | Cmax (mg/L) | AUC0–168 (mg∙h/L) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Model | CLR (L/h) | CLmet | Dist. * | Pred. | Obs. | Diff. | Pred. | Obs. | Diff. |
| 1 | 7.04 | HEP | mPBPK | 6.085 | 5.474 | 11.2% | 1.976 | 1.939 | 1.90% |
| 2 | 7.04 | HEP | fPBPK | 9.048 | 65.3% | 1.942 | 0.16% | ||
| 3 | 7.04 | DIST | mPBPK | 4.963 | −9.3% | 1.970 | 1.59% | ||
| 4 | 7.04 | DIST | fPBPK | 9.048 | 65.3% | 1.940 | 0.07% | ||
| 5 | 8.67 | HEP | mPBPK | 4.582 | −16.3% | 1.950 | 0.56% | ||
| 6 | 8.67 | HEP | fPBPK | 9.027 | 64.9% | 1.909 | −1.56% | ||
| 7 | 8.67 | DIST | mPBPK | 4.957 | −9.4% | 1.934 | −0.24% | ||
| 8 | 8.67 | DIST | fPBPK | 9.022 | 64.8% | 1.904 | −1.79% | ||
| Therapeutic Scheme | Dose | N. of Patients | Cmax (ng/mL) | AUC0–120 (mg × h/L) | Vss (L) |
|---|---|---|---|---|---|
| IV bolus | 30 mg/m2 Q24 h × 3 | 7 | 1640 ± 470 (9980 at 90 s) | 2.3 ± 0.5 | 1450 ± 84 |
| 8 h infusion | 30 mg/m2 Q24 h × 3 | 4 | 85 ± 50 | 2.0 ±1.3 |
| Parameters | Cmax (mg/L) | AUC0–120 (mg∙h/L) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Model | CLR (L/h) | CLmet | Dist. * | Pred. | Obs. | Diff. | Pred. | Obs. | Diff. |
| 1 | 7.04 | HEP | mPBPK | 4.331 | 9.980 | −56.6% | 2.542 | 2.300 | 10.51% |
| 2 | 7.04 | HEP | fPBPK | 6.896 | −30.9% | 2.574 | 11.89% | ||
| 3 | 7.04 | DIST | mPBPK | 3.787 | −62.1% | 2.560 | 11.32% | ||
| 4 | 7.04 | DIST | fPBPK | 6.896 | −30.9% | 2.563 | 11.45% | ||
| 5 | 8.67 | HEP | mPBPK | 3.560 | −64.3% | 2.528 | 9.90% | ||
| 6 | 8.67 | HEP | fPBPK | 6.884 | −31.0% | 2.538 | 10.33% | ||
| 7 | 8.67 | DIST | mPBPK | 3.784 | −62.1% | 2.522 | 9.64% | ||
| 8 | 8.67 | DIST | fPBPK | 6.882 | −31.0% | 2.521 | 9.59% | ||
| Parameters | Cmax (μg/L) | AUC0–120 (mg∙h/L) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Model | CLR (L/h) | CLmet | Dist. * | Pred. | Obs. | Diff. | Pred. | Obs. | Diff. |
| 1 | 7.04 | HEP | mPBPK | 32.316 | 85.000 | −62.0% | 2.518 | 2.000 | 25.91% |
| 2 | 7.04 | HEP | fPBPK | 34.521 | −59.4% | 2.566 | 28.31% | ||
| 3 | 7.04 | DIST | mPBPK | 30.411 | −64.2% | 2.532 | 26.62% | ||
| 4 | 7.04 | DIST | fPBPK | 34.441 | −59.5% | 2.556 | 27.79% | ||
| 5 | 8.67 | HEP | mPBPK | 29.439 | −65.4% | 2.502 | 25.08% | ||
| 6 | 8.67 | HEP | fPBPK | 34.223 | −59.7% | 2.531 | 26.56% | ||
| 7 | 8.67 | DIST | mPBPK | 30.080 | −64.6% | 2.494 | 24.71% | ||
| 8 | 8.67 | DIST | fPBPK | 34.077 | −59.9% | 2.513 | 25.67% | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mystridis, G.A.; Batzias, G.C.; Vizirianakis, I.S. Physiologically Based Pharmacokinetic Modelling and Simulation to Predict the Plasma Concentration Profile of Doxorubicin. Pharmaceutics 2022, 14, 541. https://doi.org/10.3390/pharmaceutics14030541
Mystridis GA, Batzias GC, Vizirianakis IS. Physiologically Based Pharmacokinetic Modelling and Simulation to Predict the Plasma Concentration Profile of Doxorubicin. Pharmaceutics. 2022; 14(3):541. https://doi.org/10.3390/pharmaceutics14030541
Chicago/Turabian StyleMystridis, George A., Georgios C. Batzias, and Ioannis S. Vizirianakis. 2022. "Physiologically Based Pharmacokinetic Modelling and Simulation to Predict the Plasma Concentration Profile of Doxorubicin" Pharmaceutics 14, no. 3: 541. https://doi.org/10.3390/pharmaceutics14030541