
Methods of Liposomes Preparation: Formation and Control Factors of Versatile Nanocarriers for Biomedical and Nanomedicine Application


Abstract
:1. Introduction
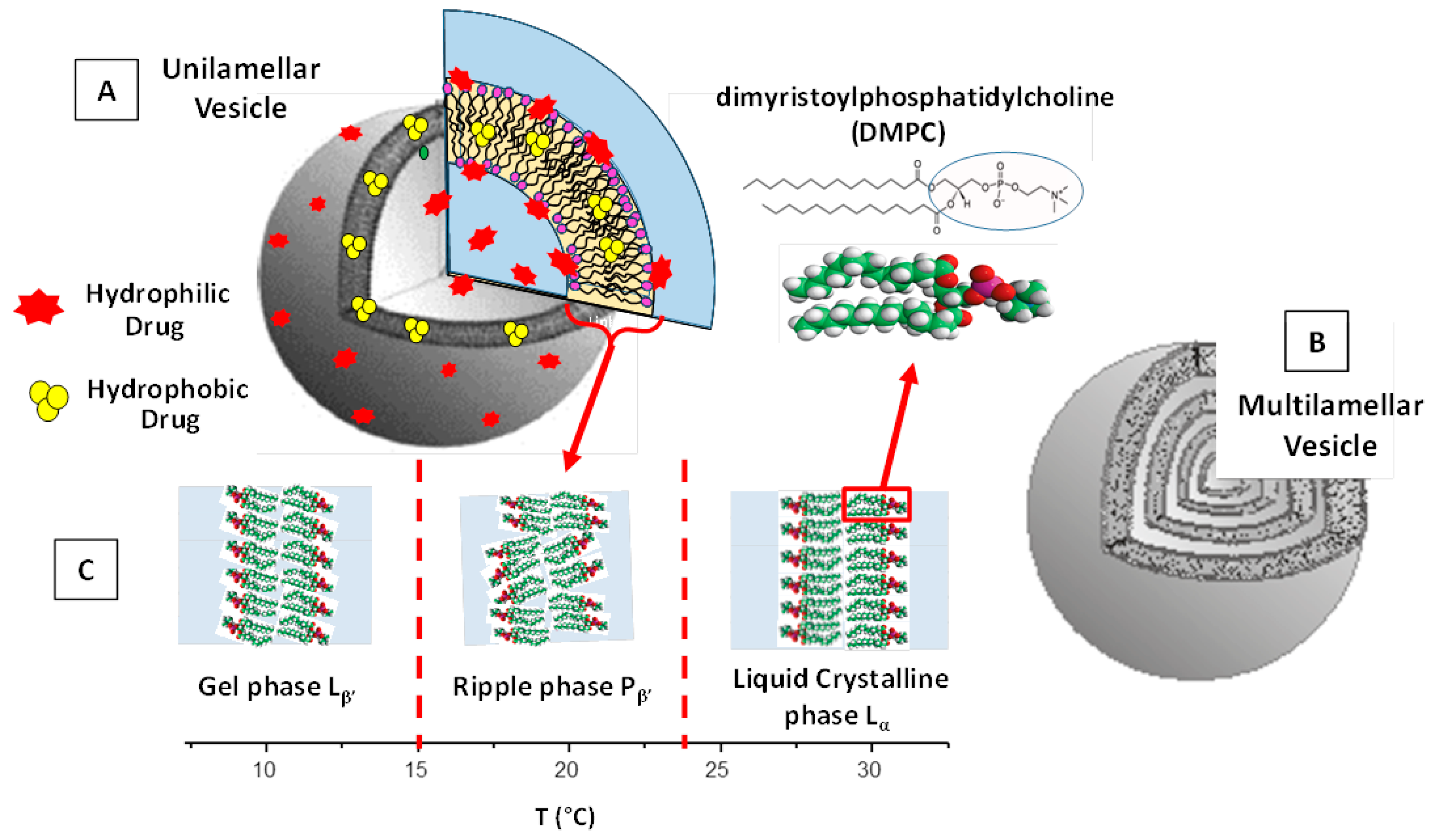
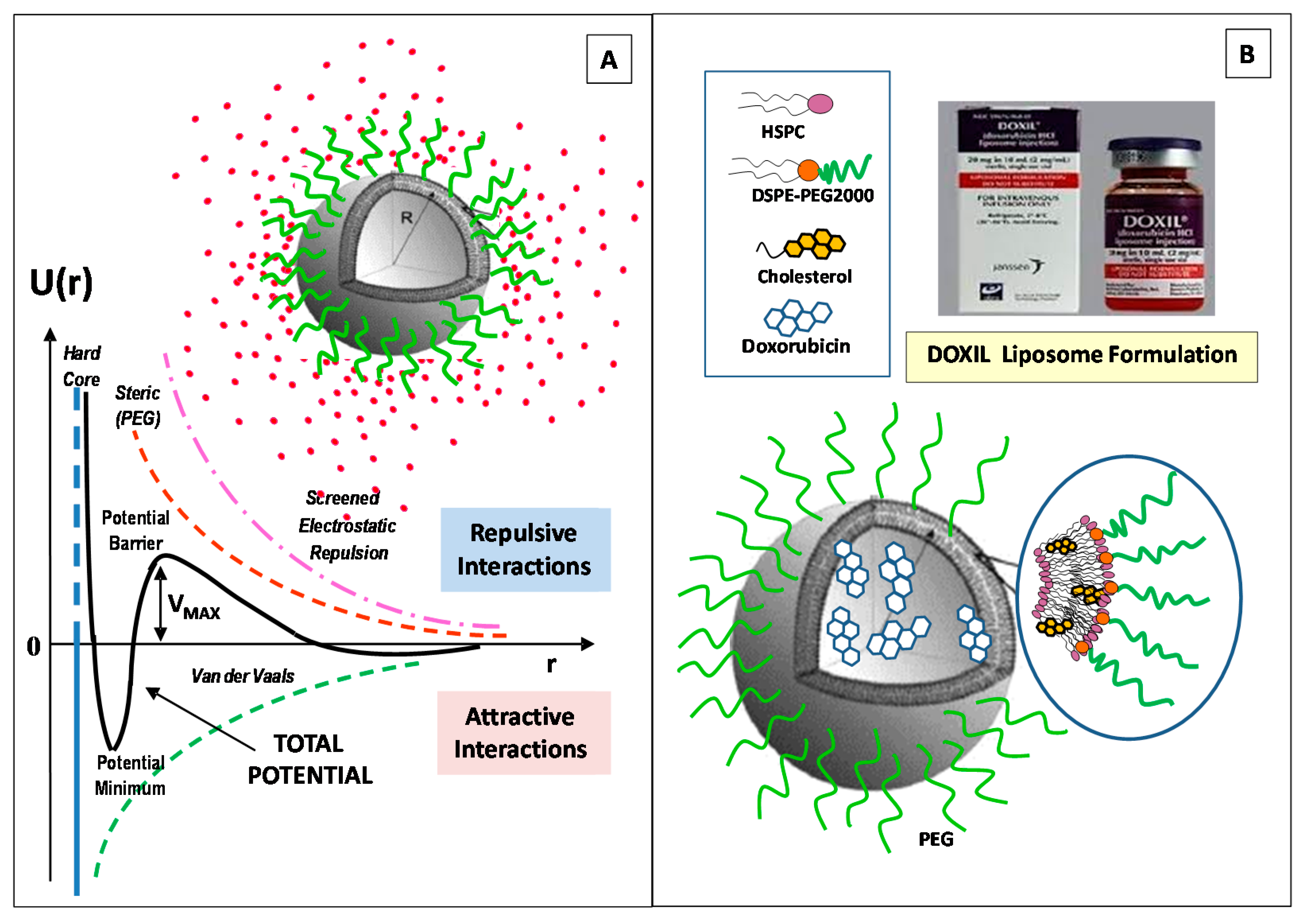
2. Structural Features and Main Control Factors of Liposomes
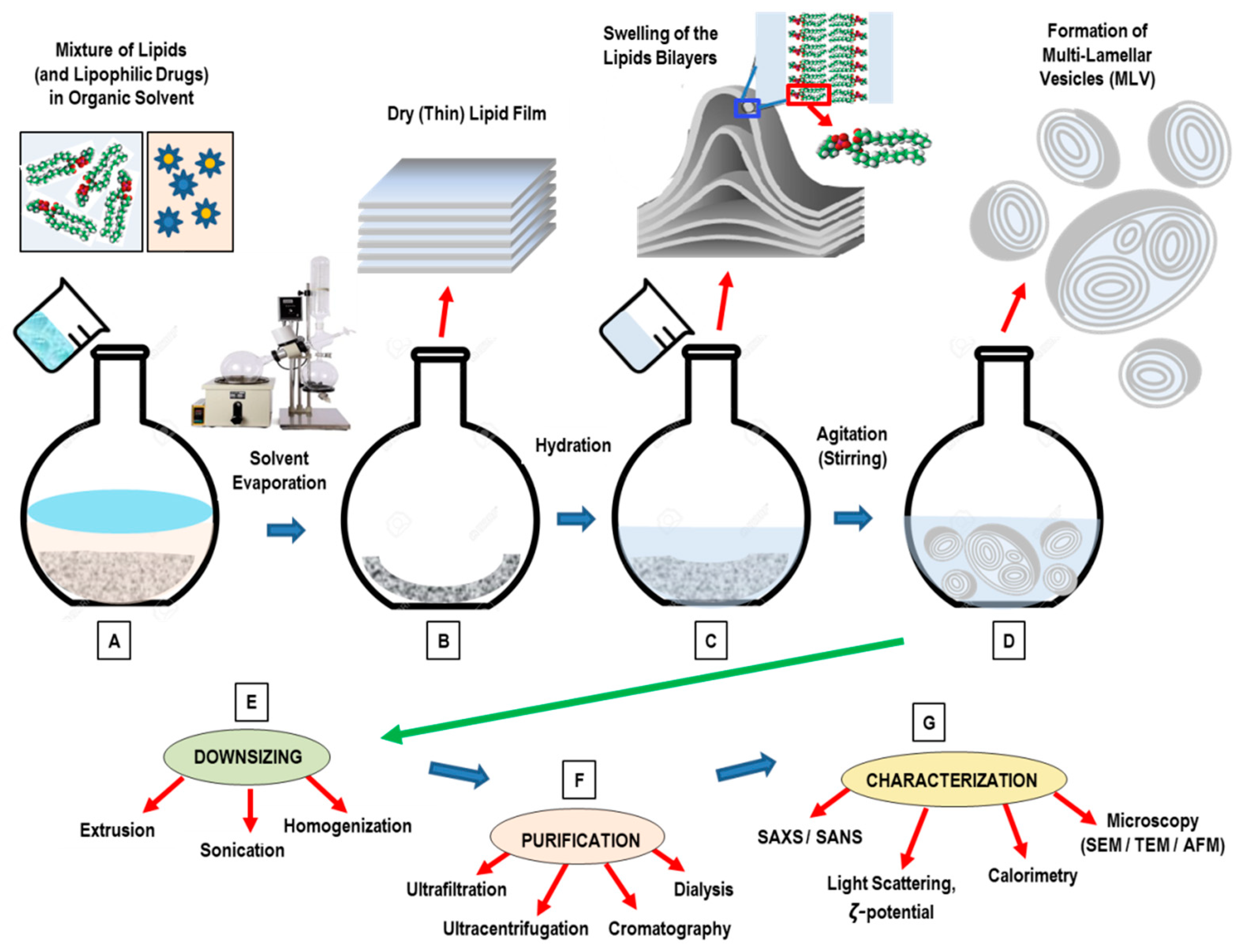
3. Conventional Methods for the Preparation of Liposomes
- Dissolution of lipids in an organic solvent;
- Drying-down of the resultant lipidic solution from the organic solvent;
- Hydrating the lipid with an aqueous media (followed by agitation/stirring);
- Downsizing (and/or change in lamellarity);
- Post-formation processing (purification, sterilization);
- Characterization of the final nanoformulation product.
3.1. Thin-Film Hydration (TFH) Method (Bangham Method)
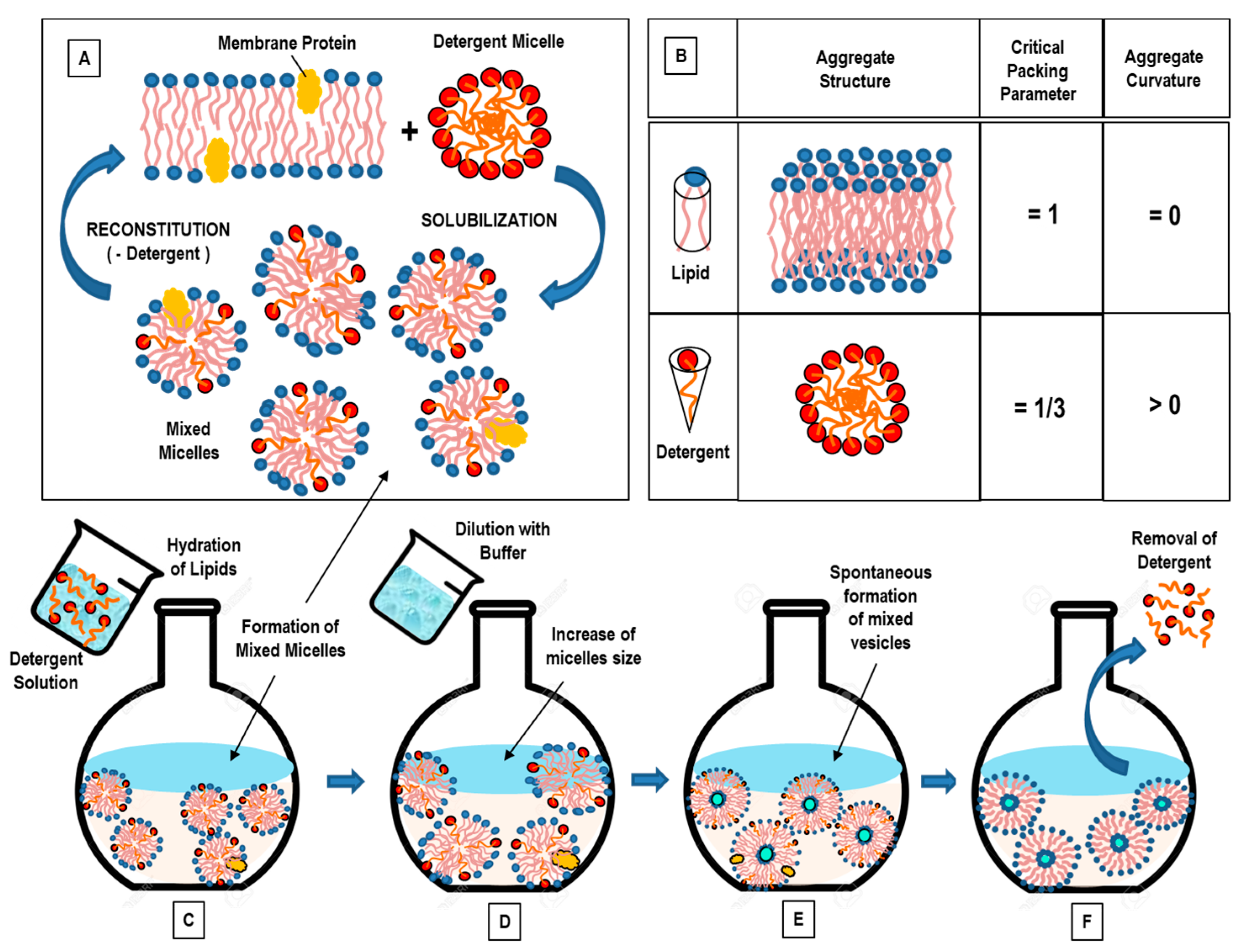
3.2. Detergent Removal (Depletion) Method
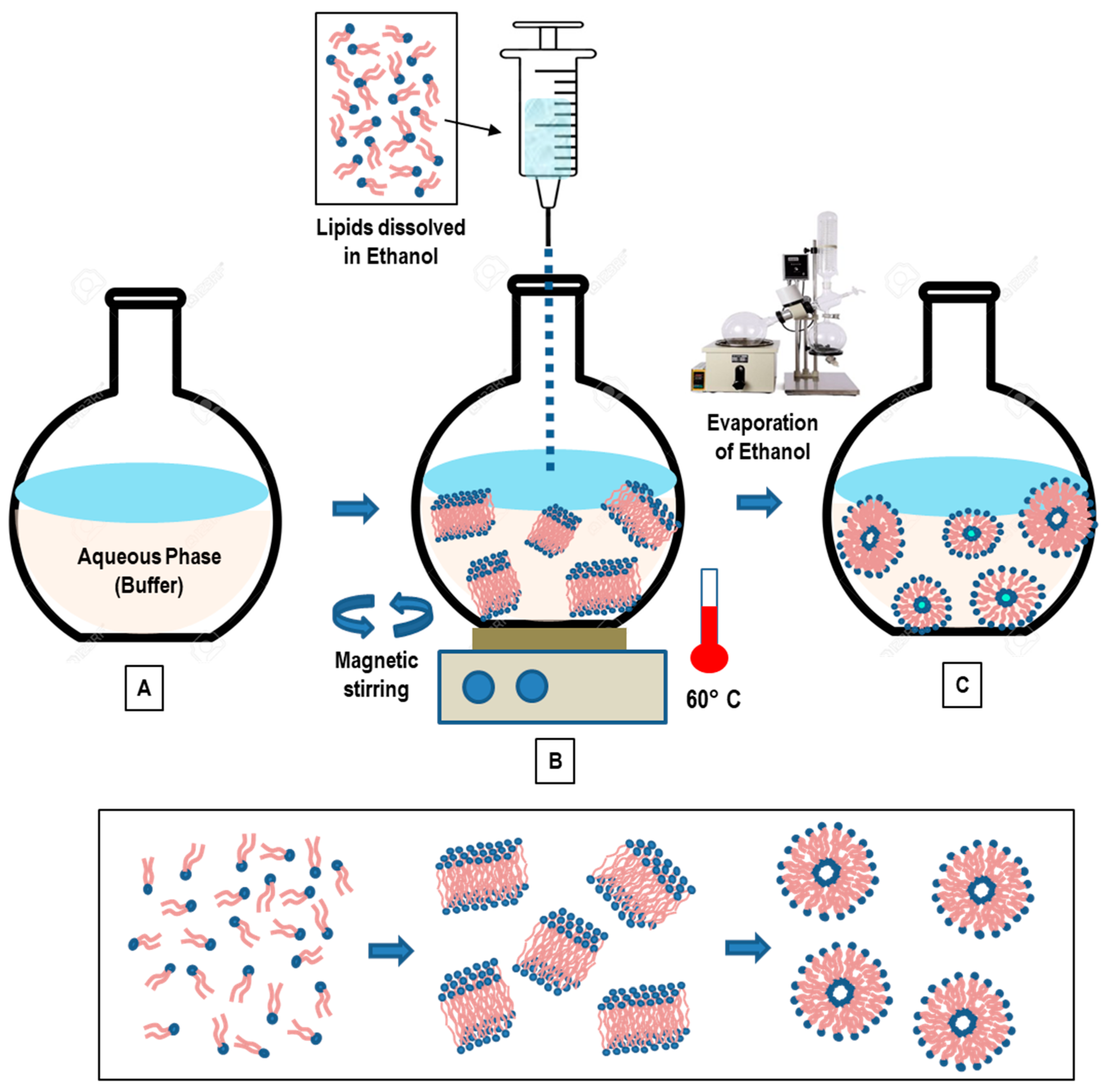
3.3. Solvent Injection Method
3.3.1. Ethanol Injection Method
3.3.2. Ether Injection Method
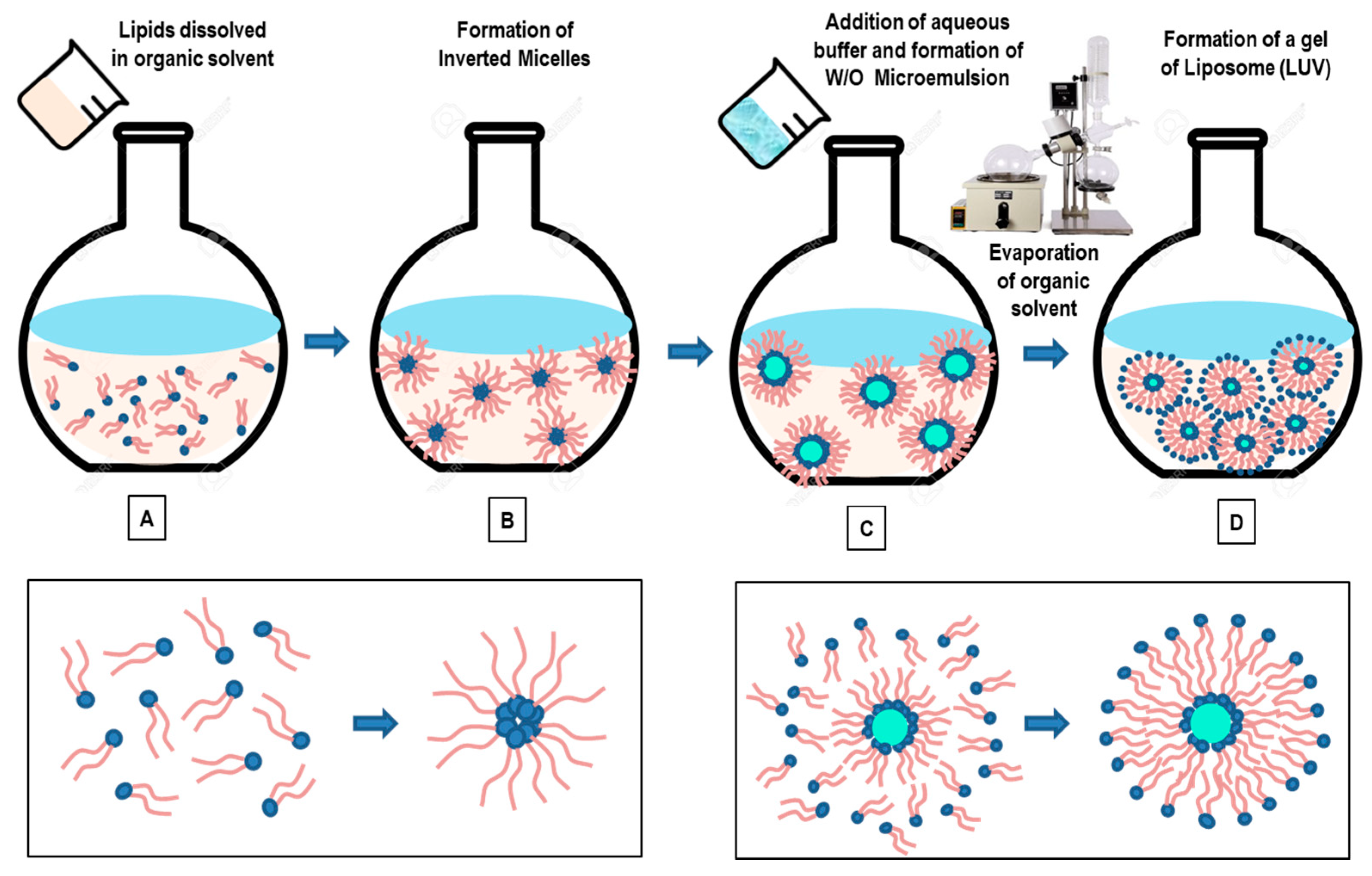
3.4. Reverse-Phase Evaporation Method
4. Downsizing and Post-Formation Processing
4.1. Sonication Method
4.2. Extrusion Method
4.3. High-Pressure Homogenization Method
5. Novel Technologies for Liposome Preparation
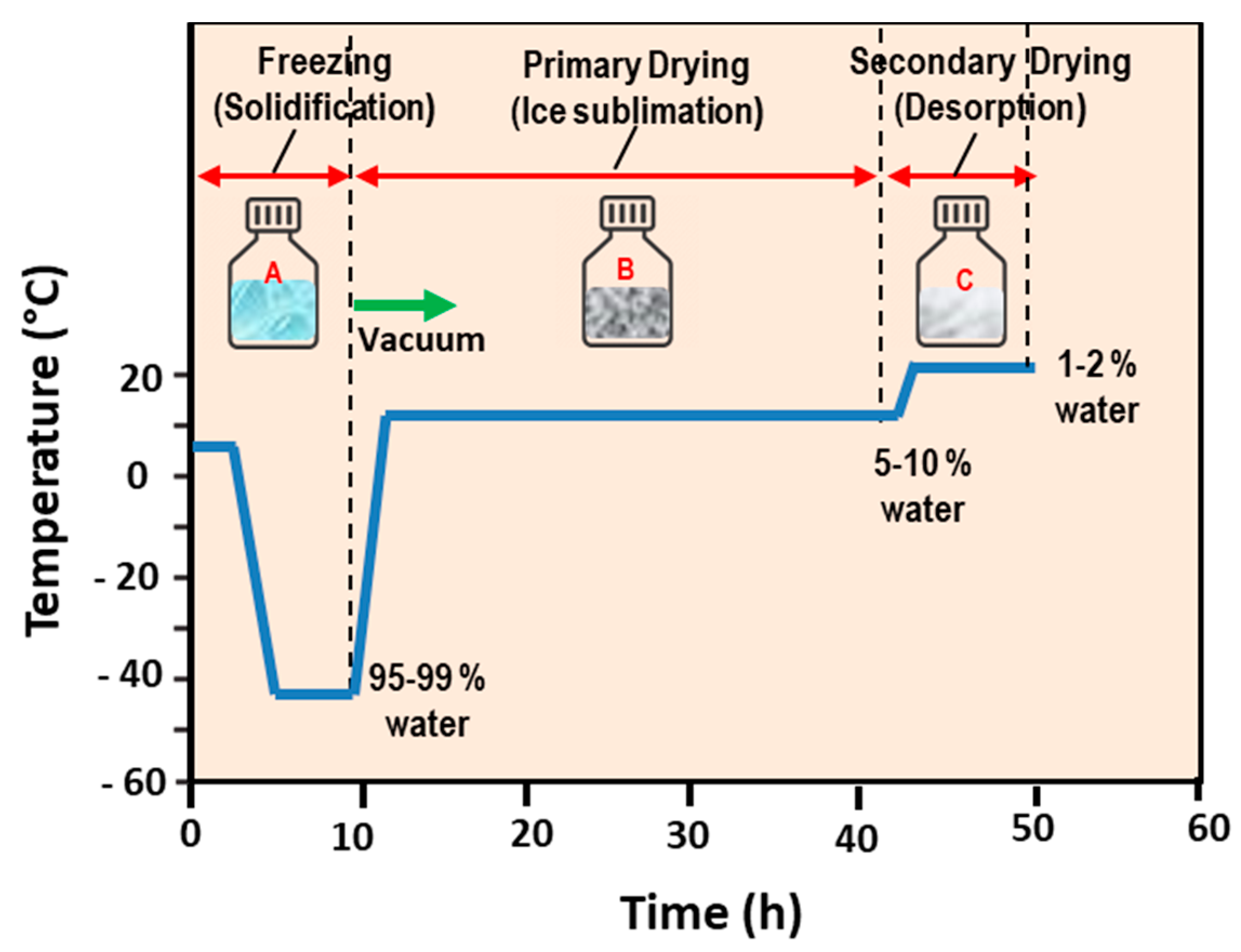
5.1. Freeze-Drying (Lyophilization) Method
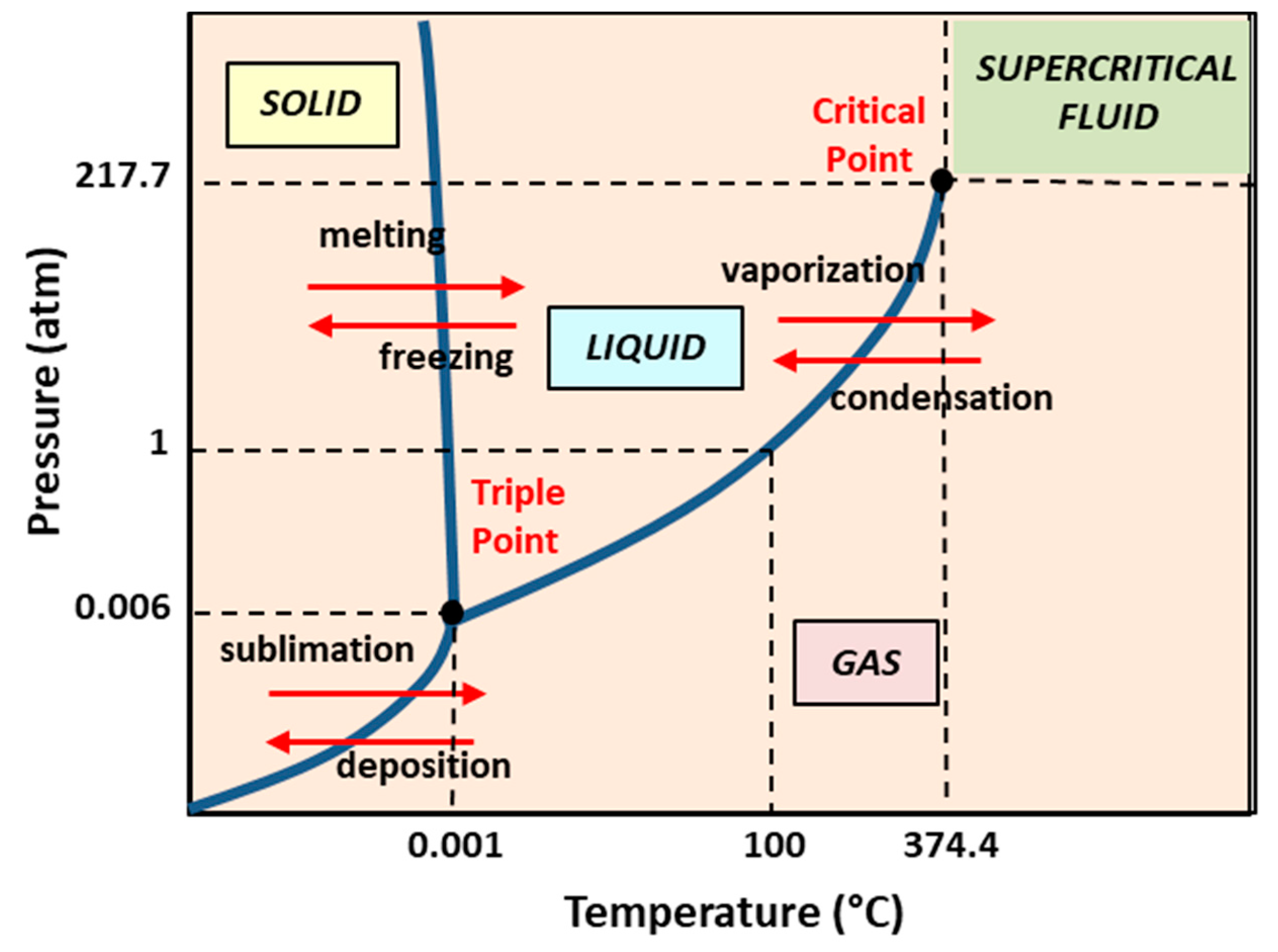
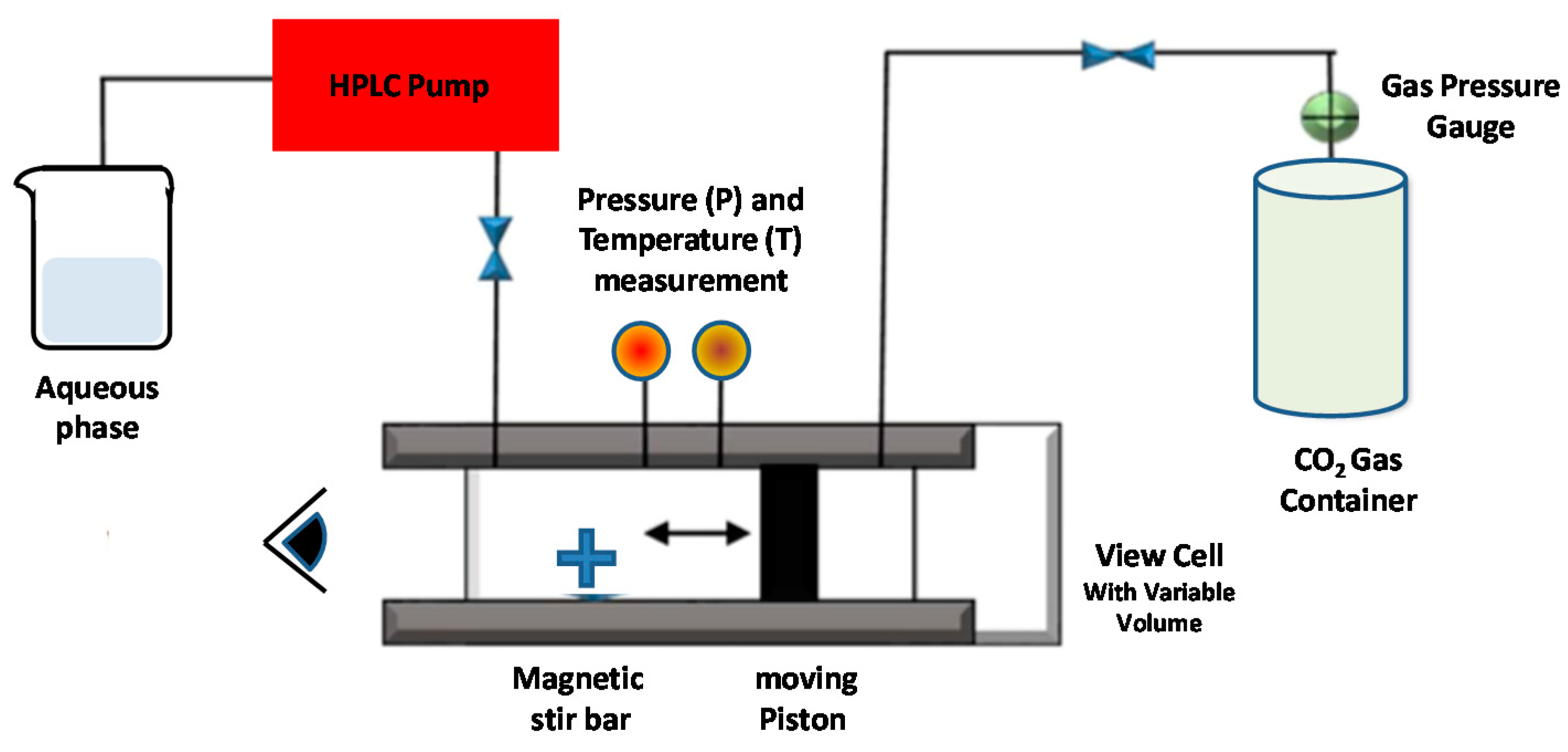
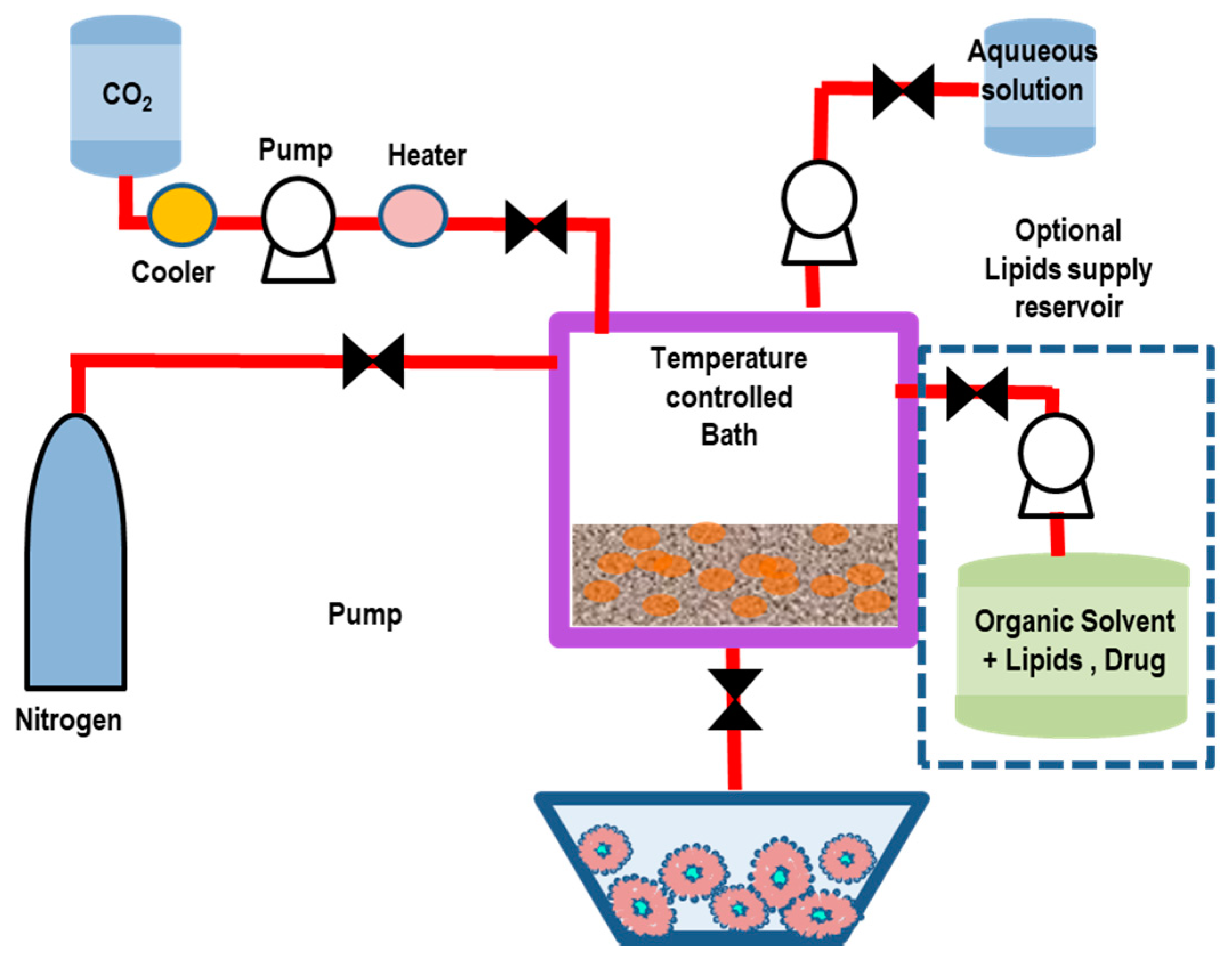
5.2. Dense Gas Technology: Supercritical Fluid-Assisted Methods
5.2.1. Supercritical Reverse-Phase Evaporation (SC-RPE) Method
5.2.2. Supercritical Anti-Solvent (SAS) Method
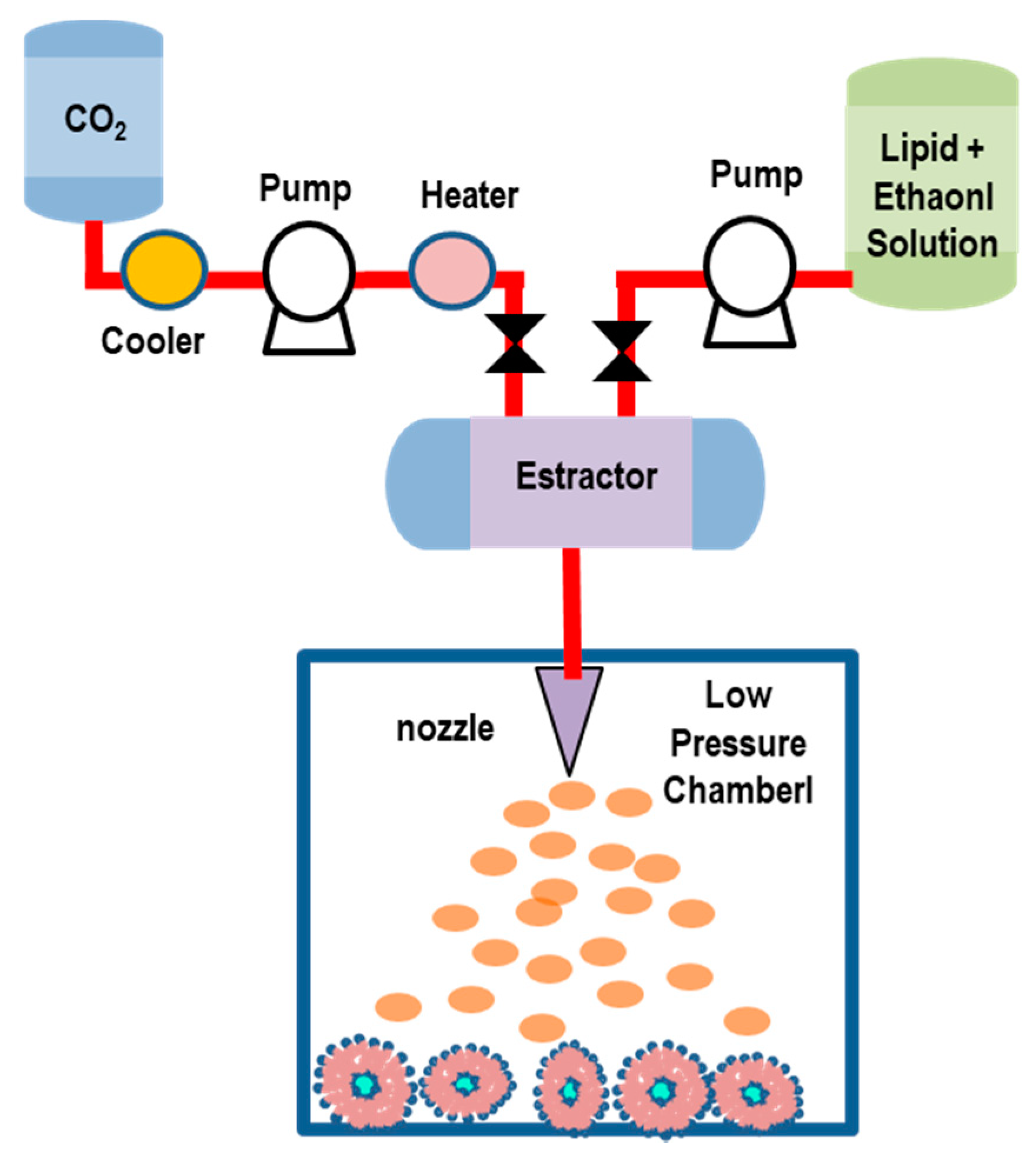
5.2.3. Rapid Expansion of a Supercritical Solution (RESS) Method
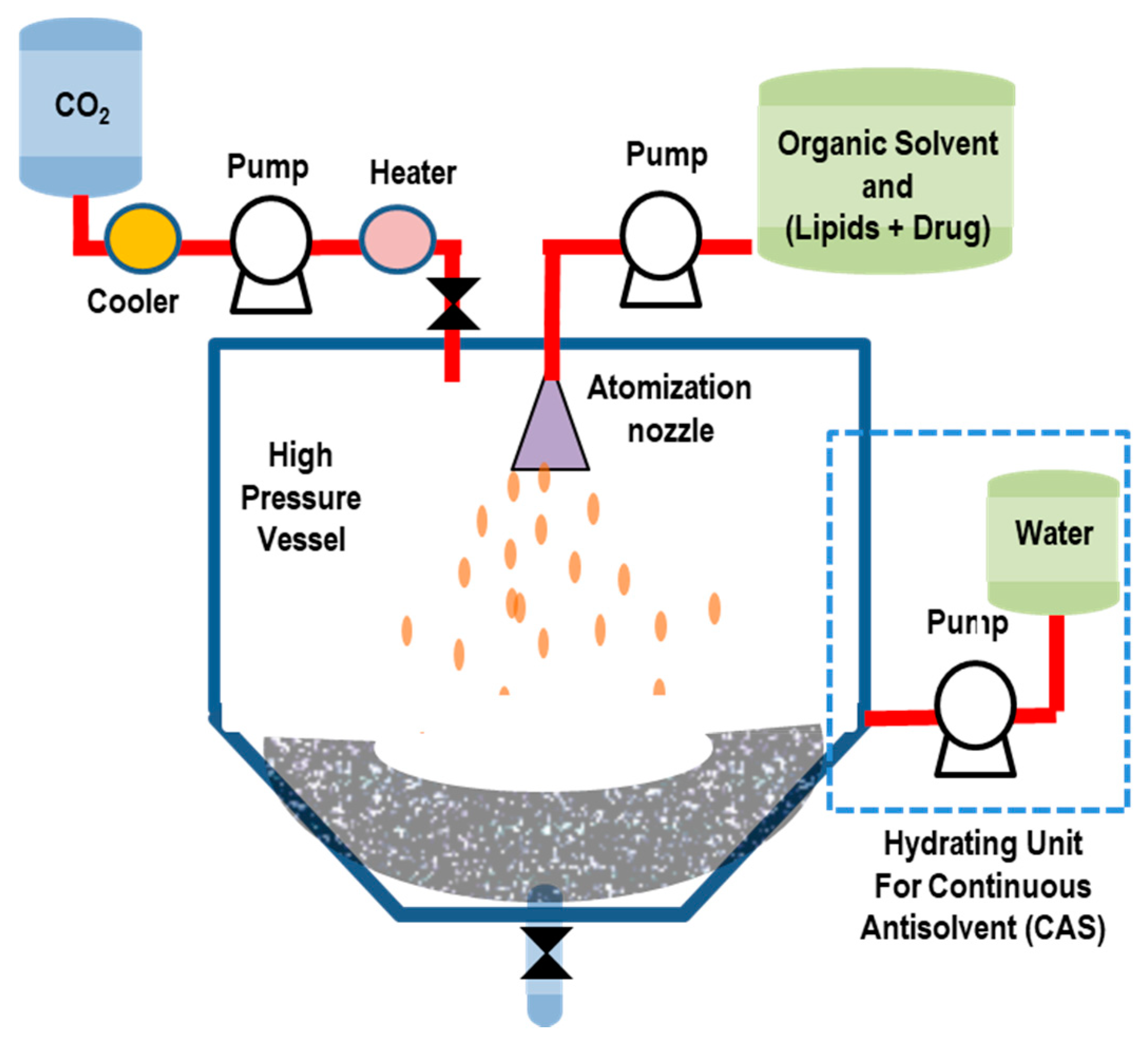
5.2.4. Supercritical-Assisted Liposome Formation (SuperLip) Method
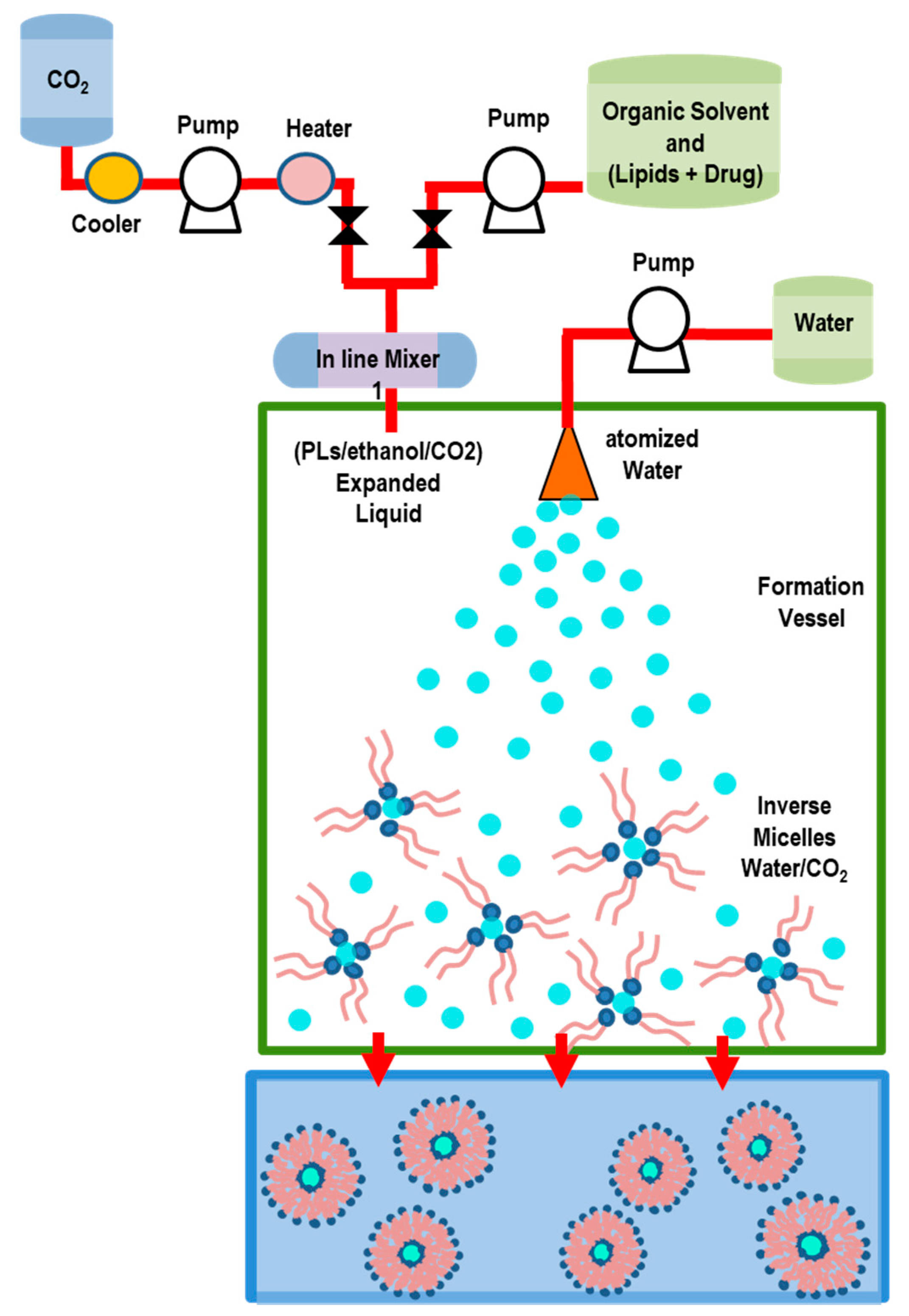
5.2.5. Depressurization of an Expanded Liquid Organic Solution into Aqueous Suspension (DELOS) Method
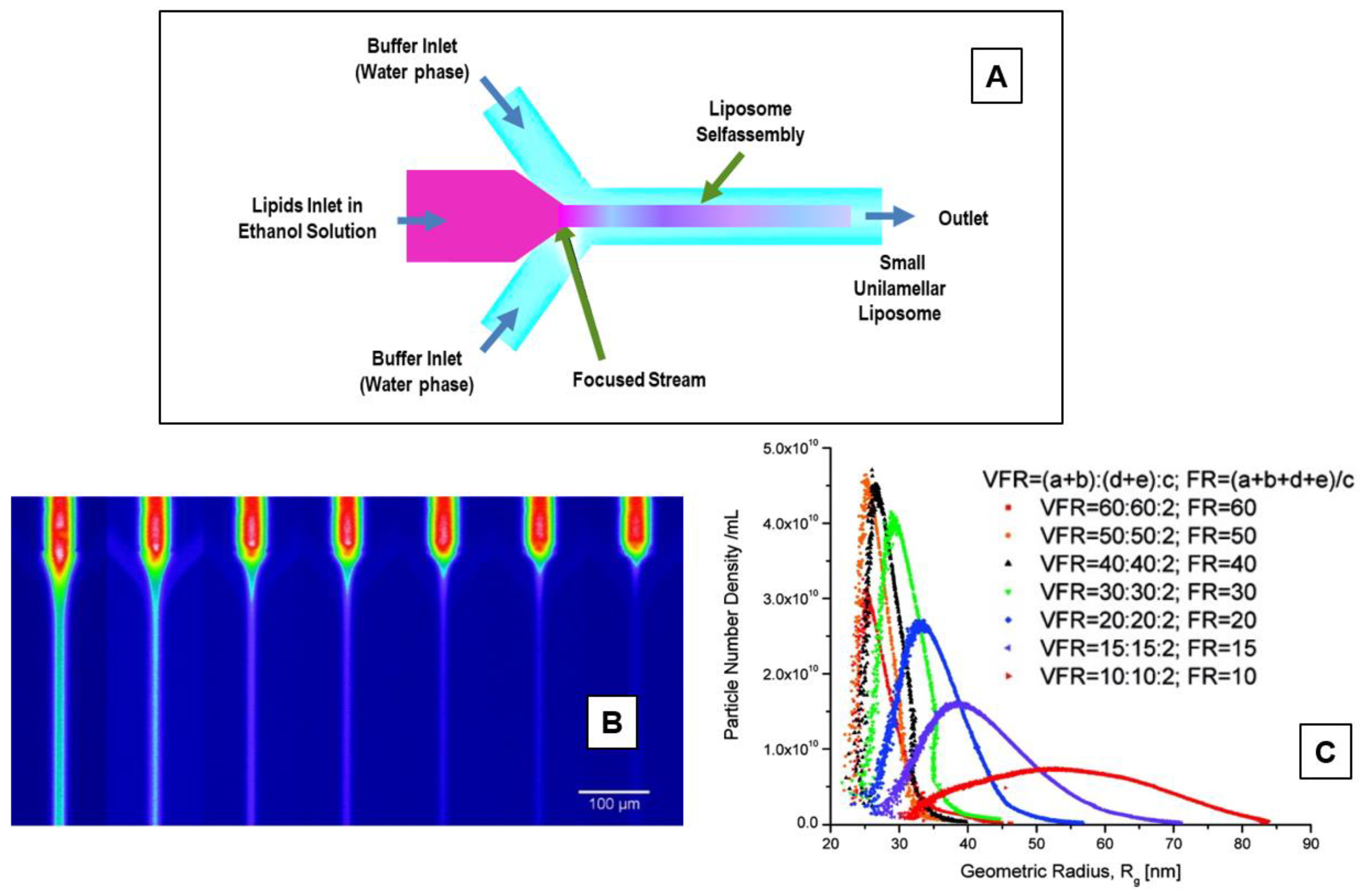
5.3. Microfluidic (Channel) Methods
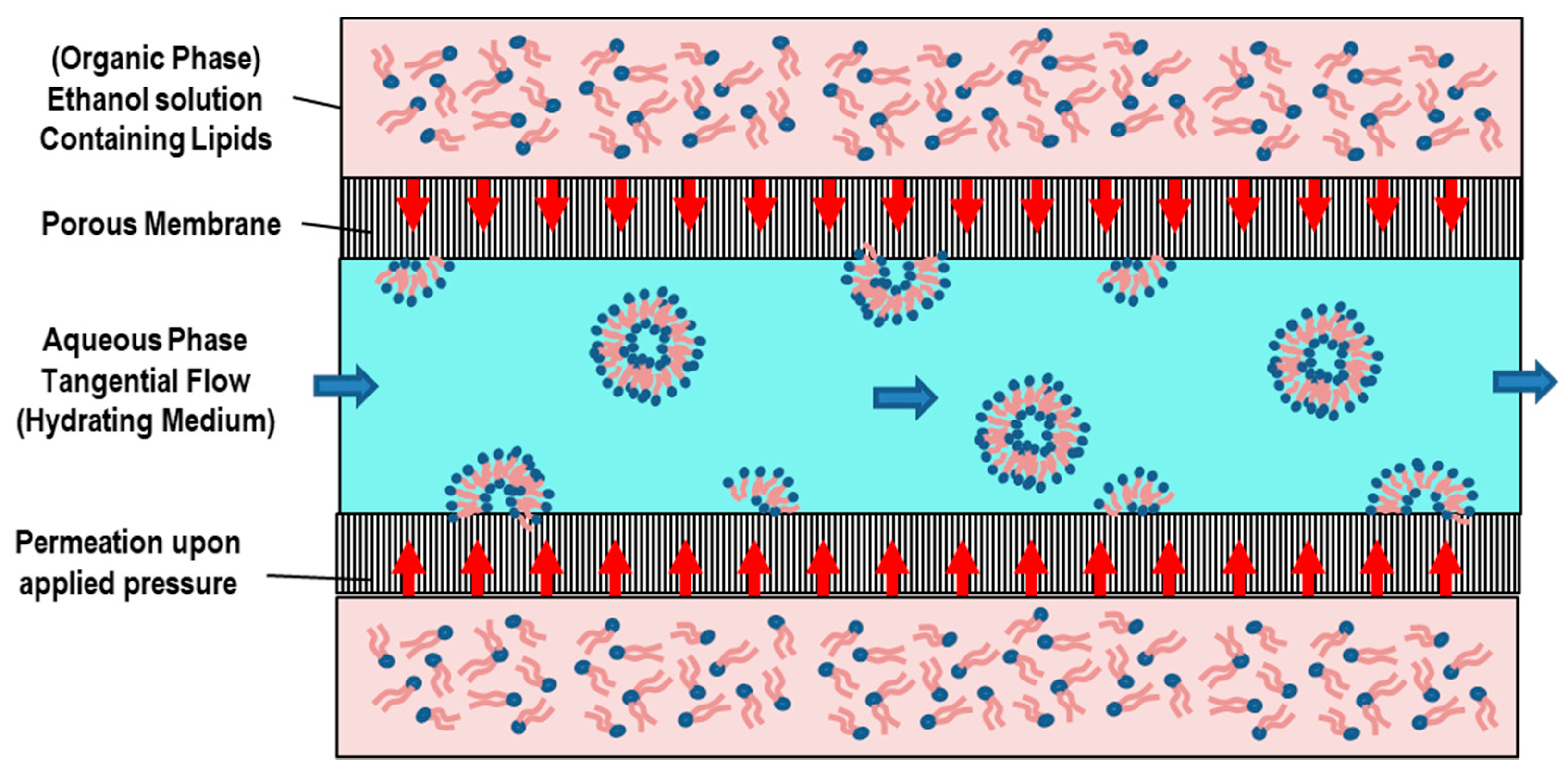
5.4. Membrane Contactor Method
6. Drug Loading in Liposome Nanoformulations
7. Post-Formation Processing of Liposomes
7.1. Purification of Liposomes Nanoformulations
7.2. Sterilization of Liposomes
7.3. Microfluidic Lab-on-Chip Nanodevices for the Combined Formation, Drug Loading, and Purification of Liposomes
Micro- and Nanofabrication Techniques
8. Characterization Methods of Liposome Nanocarriers
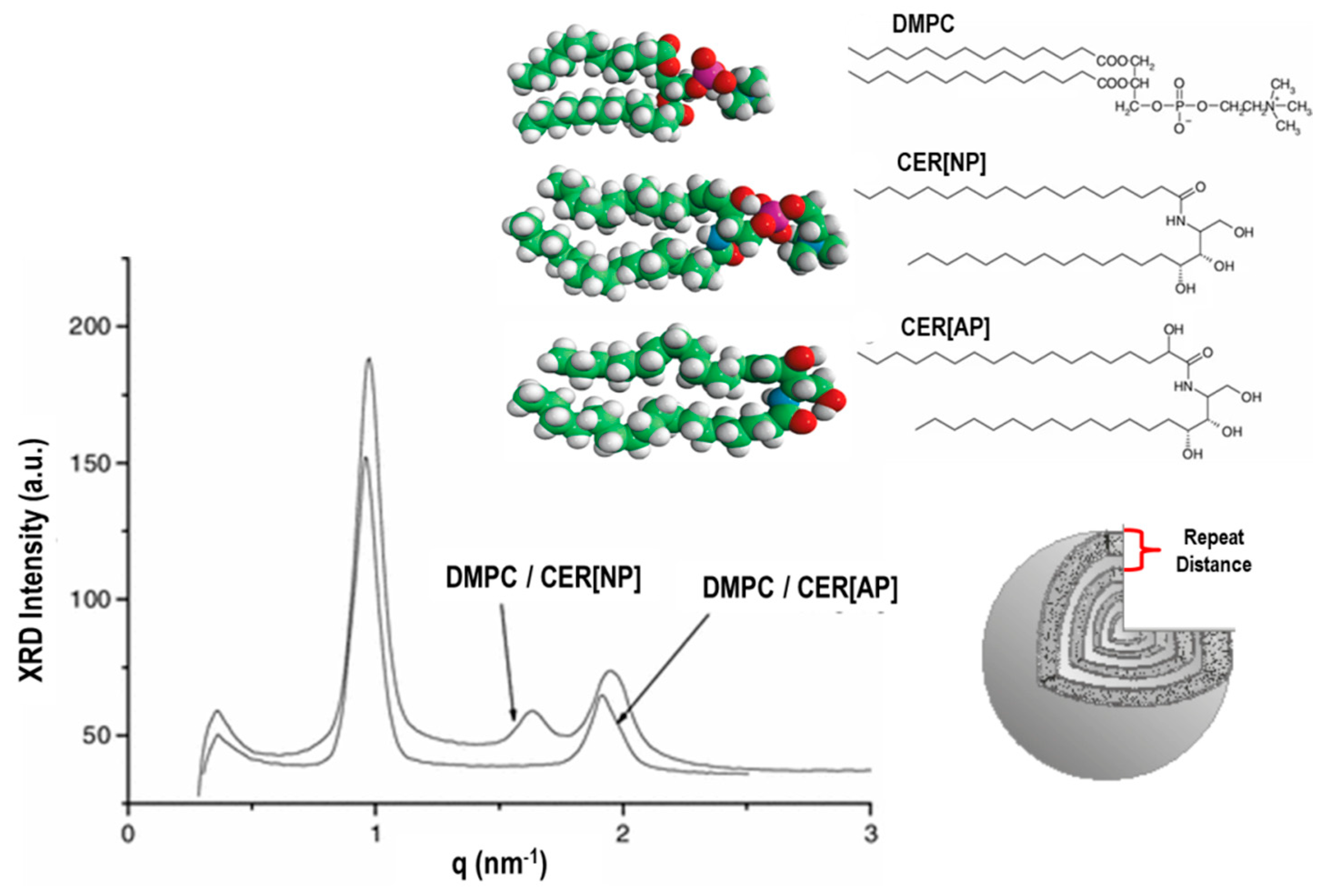
8.1. Small-Angle X-ray/Neutron Scattering (SAXS/SANS) and Diffraction Techniques
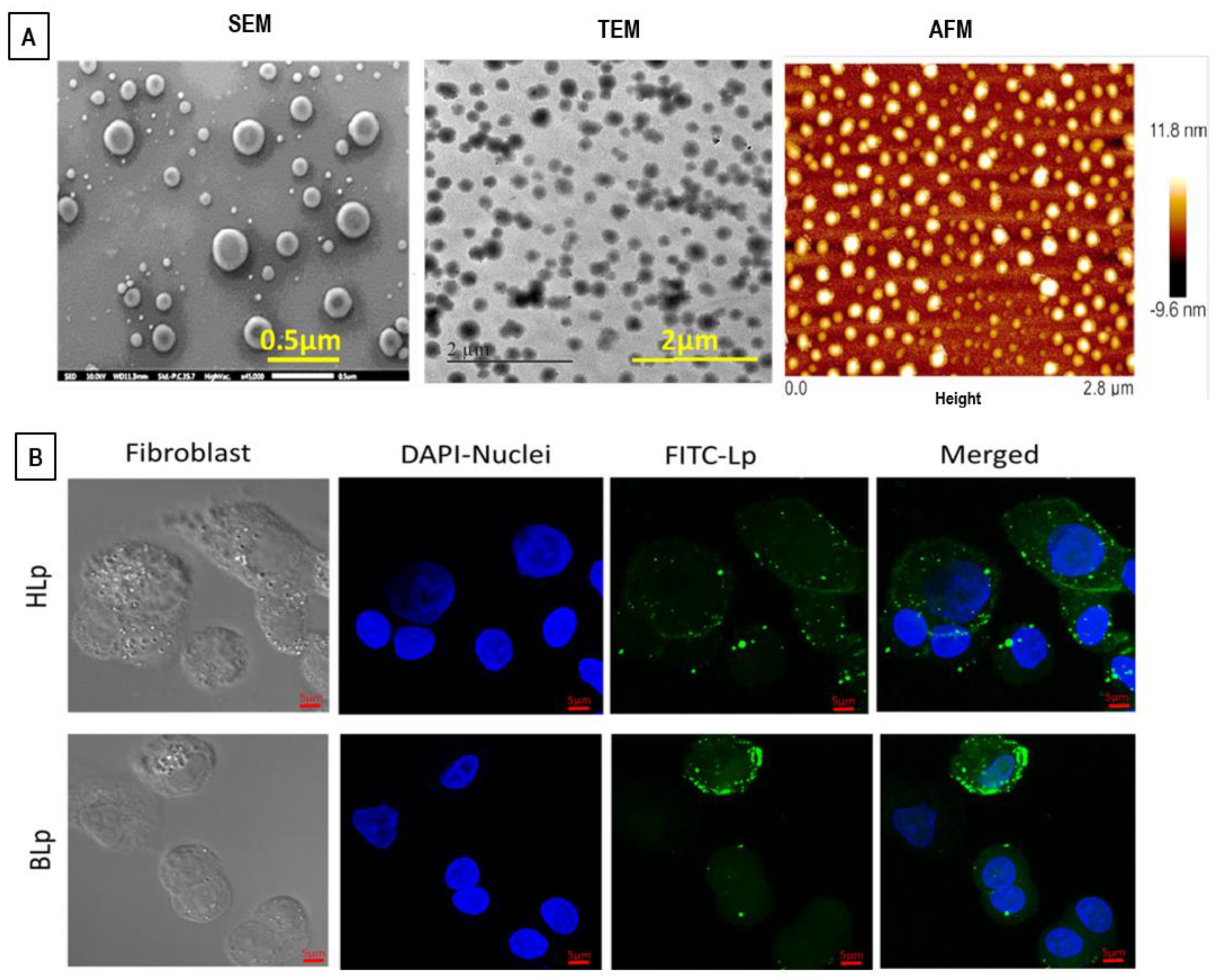
8.2. Electron Microscopy and Atomic Force Microscopy (AFM) Techniques
8.3. Light (Fluorescence and Confocal) Microscopy Techniques
8.4. Dynamic Light Scattering Technique
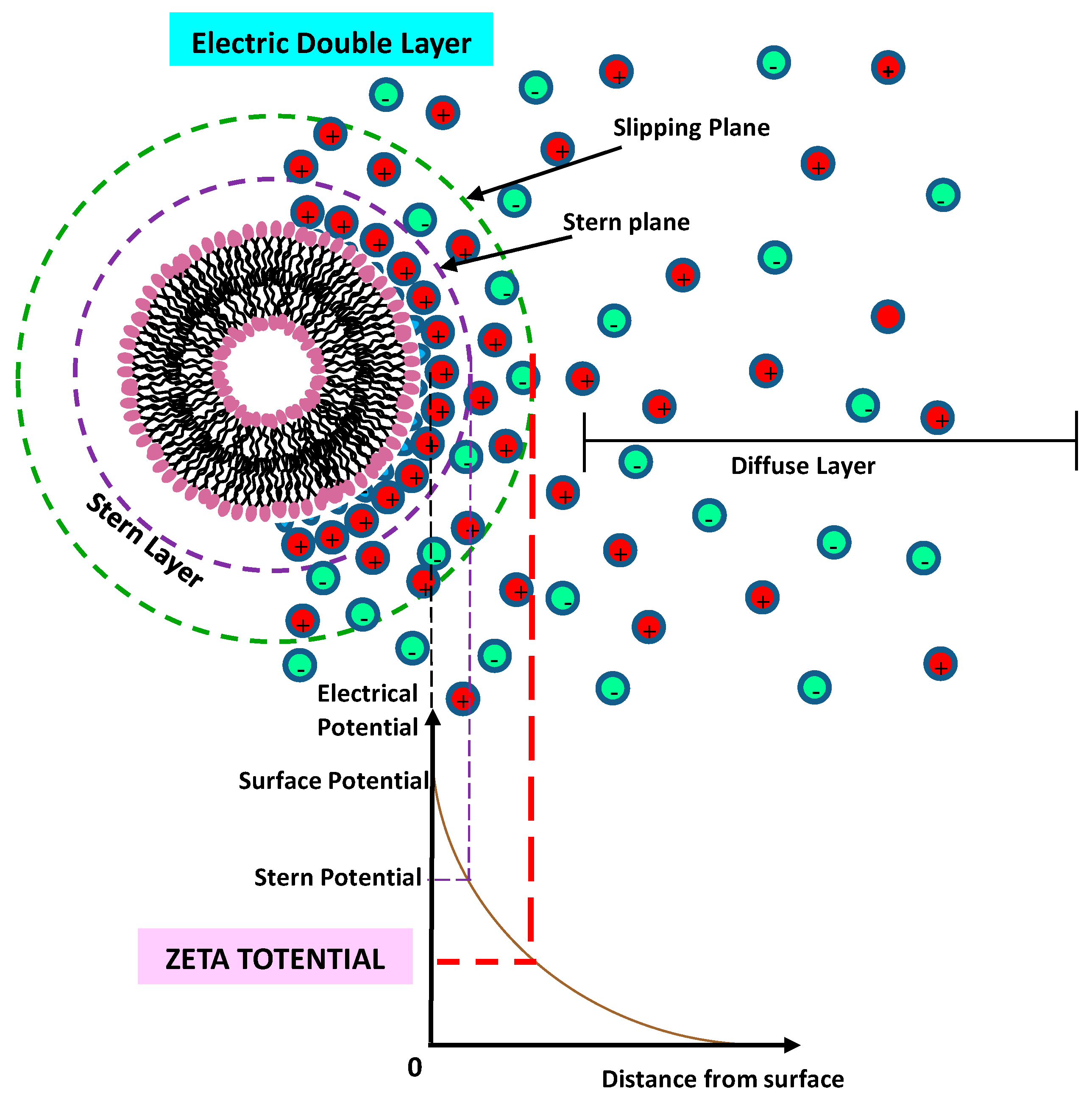
8.5. Zeta (ζ) Potential Technique
8.6. Other Complementary Characterization Techniques
9. Technology Transfer and Regulatory Perspectives
- -
- the assessment of the benefit/risk of the liposomal systems;
- -
- definition of the critical quality attributes of the final product;
- -
- guide for the formulation studies in the early stages of development (designing preclinical/clinical trials);
- -
- support to the pharmaceutical quality control system and sustaining the post-marketing variations.
The Quality by Design (QbD) Method
- (1)
- A definition of the quality target product profile (QTPP), based on some specific properties of the product, in order to ensure the desired quality, safety, and efficacy of the drug product, and by considering the critical factors of the administration (such as specific route, dosage, bioavailability, strength, and stability);
- (2)
- The identification of the main factors, such as the critical quality attributes (CQAs) of the targeted product, the critical material attributes (CMAs), and the critical process parameters (CPPs), which are related to the selected production method;
- (3)
- The identification of the risk assessment (RA) through a science-based process that ranks the parameters’ impact on the CQAs of the product, and identifies (between them) the critical material attributes (CMAs) and critical process parameters (CPPs);
- (4)
- The development of a design space (DS), i.e., a study of the combination and interaction (in a mathematical form) of the input variables (such as the CMAs and CPPs) and their impact on CQAs and the process parameters (to ensure desired product quality). To establish the design space with a minimum number of experiments, the Design of Experiments (DoE) strategy is used;
- (5)
- The definition (and implementation) of a control strategy (CS), with the aim of stimulating the continuous products’ improvement;
- (6)
- A life cycle management.
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bozzuto, G.; Molinari, A.A. Liposomes as nanomedical devices. Int. J. Nanomed. 2015, 10, 975–999. [Google Scholar] [CrossRef] [Green Version]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliver. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, D.; Calandra, P.; Barreca, D.; Magazù, S.; Kiselev, M.A. Soft interaction in liposome nanocarriers for therapeutic drug delivery. Nanomaterials 2016, 6, 125. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-K. Liposomes for Enhanced Bioavailability of Water-Insoluble Drugs: In Vivo Evidence and Recent Approaches. Pharmaceutics 2020, 12, 264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewert, K.K.; Scodeller, P.; Simón-Gracia, L.; Steffes, V.M.; Wonder, E.A.; Teesalu, T.; Safinya, C.R. Cationic Liposomes as Vectors for Nucleic Acid and Hydrophobic Drug Therapeutics. Pharmaceutics 2021, 13, 1365. [Google Scholar] [CrossRef]
- Boons, G.-J. Liposomes Modified by Carbohydrate Ligands Can Target B-Cells for the Treatment of B-Cell Lymphomas. Expert Rev. Vaccines 2010, 9, 1251–1256. [Google Scholar] [CrossRef] [Green Version]
- Kumar Giri, T.; Giri, A.; Kumar Barman, T.; Maity, S. Nanoliposome is a Promising Carrier of Protein and Peptide Biomolecule for the Treatment of Cancer. Anti-Cancer Agents Med. Chem. 2016, 16, 816–831. [Google Scholar] [CrossRef]
- Blanken, D.; Foschepoth, D.; Serrão, A.C.; Danelon, C. Genetically controlled membrane synthesis in liposomes. Nat. Commun. 2020, 11, 4317. [Google Scholar] [CrossRef]
- Xia, Y.; Xu, C.; Zhang, X.; Ning, P.; Wang, Z.; Tian, J.; Chen, X. Liposome-based probes for molecular imaging: From basic research to the bedside. Nanoscale 2019, 11, 5822–5838. [Google Scholar] [CrossRef]
- Shao, X.R.; Wei, X.Q.; Zhang, S.; Fu, N.; Lin, Y.-F.; Cai, X.-X. Effects of Micro-environmental pH of Liposome on Chemical Stability of Loaded Drug. Nanoscale Res. Lett. 2017, 12, 504. [Google Scholar] [CrossRef] [Green Version]
- Lombardo, D.; Calandra, P.; Caccamo, M.T.; Magazù, S.; Kiselev, M.A. Colloidal stability of liposomes. AIMS Mater. Sci. 2019, 6, 200–213. [Google Scholar] [CrossRef]
- Pattni, B.S.; Chupin, V.V.; Torchilin, V.P. New Developments in Liposomal Drug Delivery. Chem. Rev. 2015, 115, 10938–10966. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Da Silva, C.G.; Van der Maaden, K.; Ossendorp, F.; Cruz, L.J. Liposome-Based Drug Delivery Systems in Cancer Immunotherapy. Pharmaceutics 2020, 12, 1054. [Google Scholar] [CrossRef]
- Salehi, B.; Calina, D.; Docea, A.O.; Koirala, N.; Aryal, S.; Lombardo, D.; Pasqua, L.; Taheri, Y.; Salgado Castillo, C.M.; Martorell, M.; et al. Curcumin’s Nanomedicine Formulations for Therapeutic Application in Neurological Diseases. J. Clin. Med. 2020, 9, 430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ba, S.; Qiao, M.; Jia, L.; Zhang, J.; Zhao, X.; Hu, H.; Chen, D. Construction of Hierarchical-Targeting pH-Sensitive Liposomes to Reverse Chemotherapeutic Resistance of Cancer Stem-like Cells. Pharmaceutics 2021, 13, 1205. [Google Scholar] [CrossRef]
- Bobo, D.; Robinson, K.J.; Islam, J.; Thurecht, K.J.; Corrie, S.R. Nanoparticle-based medicines: A review of FDA approved materials and clinical trials to date. Pharm. Res 2016, 33, 2373–2387. [Google Scholar] [CrossRef]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Lamichhane, N.; Udayakumar, T.S.; D’Souza, W.D.; Simone, C.B., 2nd; Raghavan, S.R.; Polf, J.; Mahmood, J. Liposomes: Clinical Applications and Potential for Image-Guided Drug Delivery. Molecules 2018, 23, 288. [Google Scholar] [CrossRef] [Green Version]
- Noble, G.T.; Stefanick, J.F.; Ashley, J.D.; Kiziltepe, T.; Bilgicer, B. Ligand-targeted liposome design: Challenges and fundamental considerations. Trends Biotechnol. 2014, 32, 32–45. [Google Scholar] [CrossRef]
- Dutta, S.; Moses, J.A.; Anandharamakrishnan, C. Encapsulation of Nutraceutical Ingredients in Liposomes and Their Potential for Cancer Treatment. Nutr. Cancer 2018, 70, 1184–1198. [Google Scholar] [CrossRef]
- Subramani, T.; Ganapathyswamy, H. An overview of liposomal nano-encapsulation techniques and its applications in food and nutraceutical. J. Food Sci. Technol. 2020, 57, 3545–3555. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Im, H. Theranostics Based on Liposome: Looking Back and Forward. Nucl. Med. Mol. Imaging 2019, 53, 242–246. [Google Scholar] [CrossRef]
- Seleci, M.; Ag Seleci, D.; Scheper, T.; Stahl, F. Theranostic Liposome-Nanoparticle Hybrids for Drug Delivery and Bioimaging. Int. J. Mol. Sci. 2017, 18, 1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.; Gao, J.; Ding, Y.; Lu, Y.; Wei, Q.; Cui, D.; Fan, J.; Li, X.; Zhu, E.; Lu, Y.; et al. Multi-Functional Liposome: A Powerful Theranostic Nano-Platform Enhancing Photodynamic Therapy. Adv. Sci. 2021, 8, e2100876. [Google Scholar] [CrossRef]
- Fahmy, S.A.; Azzazy, H.M.E.-S.; Schaefer, J. Liposome Photosensitizer Formulations for Effective Cancer Photodynamic Therapy. Pharmaceutics 2021, 13, 1345. [Google Scholar] [CrossRef] [PubMed]
- Fobian, S.-F.; Cheng, Z.; ten Hagen, T.L.M. Smart Lipid-Based Nanosystems for Therapeutic Immune Induction against Cancers: Perspectives and Outlooks. Pharmaceutics 2022, 14, 26. [Google Scholar] [CrossRef]
- Sackmann, E. Physical basis of self-organization and function of membranes: Physics of vesicles. In Handbook of Biological Physics; Lipowsky, R., Sackmann, E., Eds.; Elsevier: Amsterdam, The Netherlands, 1995; Volume 1, pp. 213–303. [Google Scholar]
- Katsaras, J.; Gutberlet, T. Lipid Bilayers. Structure and Interactions; Springer: Berlin/Heidelberg, Germany, 2000. [Google Scholar]
- Helfrich, W. Elastic properties of lipid bilayers: Theory and possible experiments. Z. Nat. C 1973, 28, 693–703. [Google Scholar] [CrossRef]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef] [Green Version]
- Israelachvili, J.N. Physics of Amphiphiles: Micelles, Vesicles and Microemulsions; Degiorgio, V., Corti, M., Eds.; North-Holland: Amsterdam, The Netherlands, 1985. [Google Scholar]
- Israelachvili, J.; Wennerström, H. Role of hydration and water structure in biological and colloidal interactions. Nature 1996, 379, 219–225. [Google Scholar] [CrossRef]
- Lombardo, D.; Munaò, M.; Calandra, P.; Pasqua, L.; Caccamo, M.T. Evidence of pre-micellar aggregates in water solution of amphiphilic PDMS-PEO block copolymer. Phys. Chem. Chem. Phys. 2019, 21, 11983–11991. [Google Scholar] [CrossRef]
- Ishida, T.; Harashima, H.; Kiwada, H. Liposome clearance. Biosci. Rep. 2012, 22, 197–224. [Google Scholar] [CrossRef] [PubMed]
- Kraft, J.C.; Freeling, J.P.; Wang, Z.; Ho, R.J.Y. Emerging research and clinical development trends of liposome and lipid nanoparticle drug delivery systems. J. Pharm. Sci. 2014, 103, 29–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merino, M.; Zalba, S.; Garrido, M.J. Immunoliposomes in clinical oncology: State of the art and future perspectives. J. Control. Release 2018, 275, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Luzzati, V.; Tardieu, A. Lipid Phases: Structure and Structural Transitions. Annu. Rev. Phys. Chem. 1974, 25, 79–94. [Google Scholar] [CrossRef]
- Nagle, J.F.; Tristram-Nagle, S. Structure of lipid bilayers. Biochim. Biophys. Acta. 2000, 1469, 159–195. [Google Scholar] [CrossRef] [Green Version]
- Lesieur, P.; Kiselev, M.A.; Barsukov, L.I.; Lombardo, D. Temperature-induced micelle to vesicle transition: Kinetic effects in the DMPC/NaC system. J. Appl. Cryst. 2000, 33, 623–627. [Google Scholar] [CrossRef] [Green Version]
- Kiselev, M.A.; Lombardo, D. Structural characterization in mixed lipid membrane systems by neutron and X-ray scattering. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 3700–3717. [Google Scholar] [CrossRef]
- Rawicz, W.; Olbrich, K.C.; Mcintosh, T.; Needham, D.; Evans, E. Effect of Chain Length and Unsaturation on Elasticity of Lipid Bilayers. Biophys. J. 2000, 79, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Cullis, P.R.; De Kruijff, B. Lipid polymorphism and the functional roles of lipids in biological membranes. Biochim. Biophys. Acta BBA Rev. Biomembr. 1979, 559, 399–420. [Google Scholar] [CrossRef]
- Mouritsen, O.G. Lipids, curvature, and nano-medicine. Eur. J. Lipid Sci. Technol. 2011, 113, 1174–1187. [Google Scholar] [CrossRef] [Green Version]
- Lombardo, D.; Kiselev, M.A.; Magazu, S.; Calandra, P. Amphiphiles Self-Assembly: Basic Concepts and Future Perspectives of Supramolecular Approaches. Adv. Cond. Matter Phys. 2015, 2015, 151683. [Google Scholar] [CrossRef] [Green Version]
- Kashapov, R.; Gaynanova, G.; Gabdrakhmanov, D.; Kuznetsov, D.; Pavlov, R.; Petrov, K.; Zakharova, L.; Sinyashin, O. Self-Assembly of Amphiphilic Compounds as a Versatile Tool for Construction of Nanoscale Drug Carriers. Int. J. Mol. Sci. 2020, 21, 6961. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, D.; Longo, A.; Darcy, R.; Mazzaglia, A. Structural properties of nonionic cyclodextrin colloids in water. Langmuir 2004, 20, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, K.; Kolli, H.B.; Killingmoe Christensen, M.; Bore, S.L.; Diezemann, G.; Gauss, J.; Milano, G.; Lund, R.; Cascella, M. Supramolecular packing drives morphological transitions of charged surfactant micelles. Angew. Chem. Int. Ed. 2020, 59, 18591–18598. [Google Scholar] [CrossRef]
- Mallamace, F.; Beneduci, R.; Gambadauro, P.; Lombardo, D.; Chen, S.H. Glass and percolation transitions in dense attractive micellar system. Phys. A Stat. Mech. Appl. 2001, 302, 202–219. [Google Scholar] [CrossRef]
- Chen, S.H.; Mallamace, F.; Faraone, A.; Gambadauro, P.; Lombardo, D.; Chen, W.R. Observation of a re-entrant kinetic glass transition in a micellar system with temperature-dependent attractive interaction. Eur. Phys. J. E Soft Matter 2002, 9, 283–286. [Google Scholar] [CrossRef]
- Nakhaei, P.; Margiana, R.; Bokov, D.O.; Abdelbasset, W.K.; Kouhbanani, M.A.J.; Varma, R.S.; Marofi, F.; Jarahian, M.; Beheshtkhoo, N. Liposomes: Structure, Biomedical Applications, and Stability Parameters With Emphasis on Cholesterol. Front. Bioeng. Biotechnol. 2021, 9, 705886. [Google Scholar] [CrossRef]
- Lee, S.-C.; Lee, K.-E.; Kim, J.-J.; Lim, S.-H. The Effect of Cholesterol in the Liposome Bilayer on the Stabilization of Incorporated Retinol. J. Liposome Res. 2005, 15, 157–166. [Google Scholar] [CrossRef]
- Kaddah, S.; Khreich, N.; Kaddah, F.; Charcosset, C.; Greige-Gerges, H. Cholesterol modulates the liposome membrane fluidity and permeability for a hydrophilic molecule. Food Chem. Toxicol. 2018, 113, 40–48. [Google Scholar] [CrossRef]
- Hernández-Caselles, T.; Villalaín, J.; Gómez-Fernández, J.C. Influence of liposome charge and composition on their interaction with human blood serum proteins. Mol. Cell. Biochem. 1993, 120, 119–126. [Google Scholar] [CrossRef]
- Jobin, M.L.; Alves, I.D. On the importance of electrostatic interactions between cell penetrating peptides and membranes: A pathway toward tumor cell selectivity? Biochimie 2014, 107, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Pöyry, S.; Vattulainen, I. Role of charged lipids in membrane structures—Insight given by simulations. Biochim. Biophys. Acta 2016, 1858, 2322–2333. [Google Scholar] [CrossRef]
- Claessens, M.M.; van Oort, B.F.; Leermakers, F.A.; Hoekstra, F.A.; Cohen Stuart, M.A. Charged lipid vesicles: Effects of salts on bending rigidity, stability, and size. Biophys. J. 2004, 87, 3882–3893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, C.R.; Bondurant, B.; McLean, S.D.; McGovern, K.A.; O’Brien, D.F. Liposome-cell interactions in vitro: Effect of liposome surface charge on the binding and endocytosis of conventional and sterically stabilized liposomes. Biochemistry 1998, 37, 12875–12883. [Google Scholar] [CrossRef] [PubMed]
- Campbell, R.B.; Ying, B.; Kuesters, G.M.; Hemphill, R. Fighting cancer: From the bench to bedside using second generation cationic liposomal therapeutics. J. Pharm. Sci. 2009, 98, 411–429. [Google Scholar] [CrossRef]
- Inglut, C.T.; Sorrin, A.J.; Kuruppu, T.; Vig, S.; Cicalo, J.; Ahmad, H.; Huang, H.C. Immunological and Toxicological Considerations for the Design of Liposomes. Nanomaterials 2000, 10, 190. [Google Scholar] [CrossRef] [Green Version]
- Gref, R.; Lück, M.; Quellec, P.; Marchand, M.; Dellacherie, E.; Harnisch, S.; Blunk, T.; Müller, R.H. ‘Stealth’ corona-core nanoparticles surface modified by polyethylene glycol (PEG): Influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids Surf. B Biointerfaces 2000, 18, 301–313. [Google Scholar] [CrossRef]
- Immordino, M.L.; Dosio, F.; Cattel, L. Stealth liposomes: Review of the basic science, rationale, and clinical applications, existing and potential. Int. J. Nanomed. 2006, 1, 297–315. [Google Scholar]
- Mohamed, M.; Abu Lila, A.S.; Shimizu, T.; Alaaeldin, E.; Hussein, A.; Sarhan, H.A.; Szebeni, J.; Ishida, T. PEGylated liposomes: Immunological responses. Sci. Technol. Adv. Mater. 2019, 20, 710–724. [Google Scholar] [CrossRef] [Green Version]
- Thi, T.T.H.; Pilkington, E.H.; Nguyen, D.H.; Lee, J.S.; Park, K.D.; Truong, N.P. The Importance of Poly(ethylene glycol) Alternatives for Overcoming PEG Immunogenicity in Drug Delivery and Bioconjugation. Polymers 2020, 12, 298. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.; Wang, B.; Sun, Z.; Cheng, J.; Zhao, H.; Wu, K.; Sun, p.; Shen, Q.; Li, M.; Fan, Q. Multifunctional Theranostic Liposomes Loaded with a Hypoxia-Activated Prodrug for Cascade-Activated Tumor Selective Combination Therapy. ACS Appl. Mater. Interfaces 2019, 11, 39410–39423. [Google Scholar] [CrossRef] [PubMed]
- New, R.C.C. Preparation of liposomes. In Liposomes: A Practical Approach; New, R.C.C., Ed.; Oxford University Press: New York, NY, USA, 1990; pp. 33–104. [Google Scholar]
- Zhang, H. Thin-Film Hydration Followed by Extrusion Method for Liposome Preparation. In Liposomes. Methods in Molecular Biology; D’Souza, G., Ed.; Humana Press: New York, NY, USA, 2017; Volume 1522. [Google Scholar] [CrossRef]
- Xiang, B.; De-ying, C. Liposome-Based Drug Delivery Systems. In Biomaterial Engineering; Lu, W.-L., Qi, X.-R., Eds.; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar] [CrossRef]
- Lasch, J.; Weissig, V.; Brandl, M. Preparation of liposomes. In Liposomes: A Practical Approach, 2nd ed.; Torchilin, V., Weissig, V., Eds.; Oxford University Press: New York, NY, USA, 2003; pp. 3–29. [Google Scholar]
- Jeon, M.; Kim, G.; Lee, W.; Im, H.-J. Development of theranostic PEGylated liposomal Au-liposome for effective tumor passive targeting and photothermal therapy. J. Nucl. Med. 2020, 61 (Suppl. 1), 1076. [Google Scholar]
- Wang, G.; Yang, Y.; Yi, D.; Yuan, L.; Yin, P.-H.; Ke, X.; Jun-Jie, W.; Tao, M.-F. Eudragit S100 prepared pH-responsive liposomes-loaded betulinic acid against colorectal cancer in vitro and in vivo. J. Liposome Res. 2021, 13, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Jiskoot, W.; Teerlink, T.; Beuvery, E.C.; Crommelin, D.J. Preparation of liposomes via detergent removal from mixed micelles by dilution. The effect of bilayer composition and process parameters on liposome characteristics. Pharm. Weekbl. Sci. 1986, 8, 259–265. [Google Scholar] [CrossRef]
- Ollivon, M.; Lesieur, S.; Grabielle-Madelmont, C.; Paternostre, M. Vesicle reconstitution from lipid-detergent mixed micelles. Biochim. Biophys. Acta 2000, 1508, 34–50. [Google Scholar] [CrossRef] [Green Version]
- Leng, J.; Egelhaaf, S.U.; Cates, M.E. Kinetics of the micelle-to-vesicle transition: Aqueous lecithin-bile salt mixtures. Biophys. J. 2003, 85, 1624–1646. [Google Scholar] [CrossRef] [Green Version]
- Kiselev, M.A.; Lombardo, D.; Lesieur, P.; Kisselev, A.M.; Borbely, S.; Simonova, T.N.; Barsukov, L.I. Membrane Self Assembly in Mixed DMPC/NaC Systems by SANS. Chem. Phys. 2008, 345, 173–180. [Google Scholar] [CrossRef]
- Kiselev, M.; Janich, M.; Hildebrand, A.; Strunz, P.; Neubert, R.; Lombardo, D. Structural transition in aqueous lipid/bile salt [DPPC/NaDC] supramolecular aggregates: SANS and DLS study. Chem. Phys. 2013, 424, 93–99. [Google Scholar] [CrossRef]
- Niroomand, H.; Venkatesan, G.A.; Sarles, S.A.; Mukherjee, D.; Khomami, B. Lipid-Detergent Phase Transitions during Detergent-Mediated Liposome Solubilization. J. Membr. Biol. 2016, 249, 523–538. [Google Scholar] [CrossRef]
- Juan-Colás, J.; Dresser, L.; Morris, K.; Lagadou, H.; Ward, R.H.; Burns, A.; Tear, S.P.; Johnson, S.D.; Leake, M.C.; Quinn, S.D. The Mechanism of Vesicle Solubilization by the Detergent Sodium Dodecyl Sulfate. Langmuir 2020, 36, 11499–11507. [Google Scholar] [CrossRef]
- Majeed, S.; Ahmad, A.B.; Sehar, U.; Georgieva, E.R. Lipid Membrane Mimetics in Functional and Structural Studies of Integral Membrane Proteins. Membranes 2021, 11, 685. [Google Scholar] [CrossRef]
- Neves, P.; Lopes, S.C.; Sousa, I.; Garcia, S.; Eaton, P.; Gameiro, P. Characterization of membrane protein reconstitution in LUVs of different lipid composition by fluorescence anisotropy. J. Pharm. Biomed. Anal. 2009, 49, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Lasic, D.D. Mechanisms of liposome formation. J. Liposome Res. 1995, 5, 431–441. [Google Scholar] [CrossRef]
- Gouda, A.; Sakr, O.S.; Nasr, M.; Sammour, O.A. Ethanol injection technique for liposomes formulation: An insight into development, influencing factors, challenges and applications. J. Drug Deliv. Sci. Technol. 2020, 61, 102174. [Google Scholar] [CrossRef]
- Jaafar-Maalej, C.; Diab, R.; Andrieu, V.; Elaissari, A.; Fessi, H. Ethanol injection method for hydrophilic and lipophilic drug-loaded liposome preparation. J. Liposome Res. 2009, 20, 228–243. [Google Scholar] [CrossRef]
- Guimarães, D.; Noro, J.; Loureiro, A.; Lager, F.; Renault, G.; Cavaco-Paulo, A.; Nogueira, E. Increased Encapsulation Efficiency of Methotrexate in Liposomes for Rheumatoid Arthritis Therapy. Biomedicines 2020, 8, 630. [Google Scholar] [CrossRef]
- Deamer, D.; Bangham, A.D. Large volume liposomes by an ether vaporization method. Biochim. Biophys. Acta 1976, 443, 629–634. [Google Scholar] [CrossRef]
- DDeamer, D.W. Preparation and properties of ether-injection liposomes. Ann. N. Y. Acad. Sci. 1978, 308, 250–258. [Google Scholar] [CrossRef]
- Hasan, M.; Hamiduzzaman; Jahan, I.; Hasan, A. Asaduzzaman Formulation Development, Characterization and In-Vitro Evaluation of Tamoxifen Loaded Liposomes. J. Pharm. Res. Int. 2020, 32, 64–82. [Google Scholar] [CrossRef]
- Hauschild, S.; Lipprandt, U.; Rumplecker, A.; Borchert, U.; Rank, A.; Schubert, R.; Förster, S. Direct preparation and loading of lipid and polymer vesicles using inkjets. Small 2005, 1, 1177–1180. [Google Scholar] [CrossRef]
- Szoka, F., Jr.; Papahadjopoulos, D. Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse-phase evaporation. Proc. Natl. Acad. Sci. USA 1978, 75, 4194–4198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pidgeon, C.; McNeely, S.; Schmidt, T.; Johnson, J.E. Multilayered vesicles prepared by reverse-phase evaporation: Liposome structure and optimum solute entrapment. Biochemistry 1987, 26, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Shi, N.-Q.; Qi, X.-R. Preparation of Drug Liposomes by Reverse-Phase Evaporation. In Liposome-Based Drug Delivery Systems. Biomaterial Engineering; Lu, W.L., Qi, X.R., Eds.; Springer: Berlin/Heidelberg, Germany, 2021. [Google Scholar] [CrossRef]
- Béalle, G.; Di Corato, R.; Kolosnjaj-Tabi, J.; Dupuis, V.; Clément, O.; Gazeau, F.; Wilhelm, C.; Ménager, C. Ultra Magnetic Liposomes for MR Imaging, Targeting, and Hyperthermia. Langmuir 2012, 28, 11834–11842. [Google Scholar] [CrossRef] [PubMed]
- Do, H.D.; Ménager, C.; Michel, A.; Seguin, J.; Korichi, T.; Dhotel, H.; Marie, C.; Doan, B.-T.; Mignet, N. Development of Theranostic Cationic Liposomes Designed for Image-Guided Delivery of Nucleic Acid. Pharmaceutics 2020, 12, 854. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, Y.-Y.; Chang, Q.; Shu, Q.; Shen, N.; Wang, H.; Xie, Y.; Deng, X. Pressure-Controlled Encapsulation of Graphene Quantum Dots into Liposomes by the Reverse-Phase Evaporation Method. Langmuir 2021, 37, 14096–14104. [Google Scholar] [CrossRef]
- Mendez, R.; Banerjee, S. Sonication-Based Basic Protocol for Liposome Synthesis. Methods Mol. Biol. 2017, 1609, 255–260. [Google Scholar] [CrossRef]
- Olson, F.; Hunt, C.A.; Szoka, F.; Vail, W.; Papahadjopoulos, D. Preparation of liposomes of defined size distribution by extrusion through polycarbonate membranes. Biochim. Biophys. Acta 1979, 557, 9–23. [Google Scholar] [CrossRef]
- Schneider, T.; Sachse, A.; Rößling, G.; Brandl, M. Generation of contrast-carrying liposomes of defined size with a new continuous high pressure extrusion method. Int. J. Pharm. 1995, 117, 1–12. [Google Scholar] [CrossRef]
- Ong, S.S.G.; Chitneni, M.; Lee, K.S.; Ming, L.C.; Yuen, K.H. Evaluation of Extrusion Technique for Nanosizing Liposomes. Pharmaceutics 2016, 8, 36. [Google Scholar] [CrossRef]
- Hamilton, R.L., Jr.; Goerke, J.; Guo, L.S.; Williams, M.C.; Havel, R.J. Unilamellar liposomes made with the French pressure cell: A simple preparative and semiquantitative technique. J. Lipid. Res. 1980, 21, 981–992. [Google Scholar] [CrossRef]
- Brandl, M.; Bachmann, D.; Drechsler, M.; Bauer, K.H. Liposome preparation by a new high pressure homogenizer Gaulin Micron Lab 40. Drug Dev. Ind. Pharm. 1990, 16, 2167–2191. [Google Scholar] [CrossRef]
- Li, C.; Deng, Y. A novel method for the preparation of liposomes: Freeze drying of monophase solutions. J. Pharm. Sci. 2004, 93, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Franzé, S.; Selmin, F.; Samaritani, E.; Minghetti, P.; Cilurzo, F. Lyophilization of Liposomal Formulations: Still Necessary, Still Challenging. Pharmaceutics 2018, 10, 139. [Google Scholar] [CrossRef] [Green Version]
- Kiselev, M.A.; Lesieur, P.; Kisselev, A.M.; Lombardo, D.; Killany, M.; Lesieur, S.; Ollivon, M. A sucrose solutions application to the study of model biological membranes. Nucl. Instrum. Methods Phys. Res. A 2001, 470, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Kiselev, M.; Lesieur, P.; Kisselev, A.M.; Lombardo, D.; Killany, M.; Lesieur, S. Sucrose solutions as prospective medium to study the vesicle structure: SAXS and SANS study. J. Alloys Compd. 2001, 328, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Crowe, J.H.; Spargo, B.J.; Crowe, L.M. Preservation of dry liposomes does not require retention of residual water. Proc. Natl. Acad. Sci. USA 1987, 84, 1537–1540. [Google Scholar] [CrossRef] [Green Version]
- Stark, B.; Pabst, G.; Prassl, R. Long-term stability of sterically stabilized liposomes by freezing and freeze-drying: Effects of cryoprotectants on structure. Eur. J. Pharm. Sci. 2010, 41, 546–555. [Google Scholar] [CrossRef]
- Meure, L.A.; Foster, N.R.; Dehghani, F. Conventional and dense gas techniques for the production of liposomes: A review. AAPS PharmSciTech 2008, 9, 798–809. [Google Scholar] [CrossRef] [Green Version]
- Maja, L.; Željko, K.; Mateja, P. Sustainable technologies for liposome preparation. J. Supercrit. Fluids 2000, 165, 104984. [Google Scholar] [CrossRef]
- Castor, T.P.; Chu, L. Methods and Apparatus for Making Liposomes Containing Hydrophobic Drugs. U.S. Patent 5,776,486, 7 July 1998. [Google Scholar]
- Otake, K.; Imura, T.; Sakai, H.; Abe, M. Development of a new preparation method of liposomes using supercritical carbon dioxide. Langmuir 2001, 17, 3898–3901. [Google Scholar] [CrossRef]
- Imura, T.; Gotoh, T.; Otake, K.; Yoda, S.; Takebayashi, Y.; Yokoyama, S.; Takebayashi, H.; Sakai, H.; Yuasa, M.; Abe, M. Control of physicochemical properties of liposomes using a supercritical reverse phase evaporation method. Langmuir 2003, 19, 2021–2025. [Google Scholar] [CrossRef]
- Otake, K.; Shimomura, T.; Goto, T.; Imura, T.; Furuya, T.; Yoda, S.; Takebayashi, Y.; Sakai, H.; Abe, M. Preparation of liposomes using an improved supercritical reverse phase evaporation method. Langmuir 2006, 22, 2543–2550. [Google Scholar] [CrossRef] [PubMed]
- Aburai, K.; Yagi, N.; Yokoyama, Y.; Okuno, H.; Sakai, K.; Sakai, H.; Sakamoto, K.; Abe, M. Preparation of liposomes modified with lipopeptides using a supercritical carbon dioxide reverse-phase evaporation method. J. Oleo Sci. 2011, 60, 209–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesoin, L.; Crampon, C.; Boutin, O.; Badens, E. Preparation of liposomes using the supercritical anti-solvent (SAS) process and comparison with a conventional method. J. Supercrit. Fluids 2011, 57, 162–174. [Google Scholar] [CrossRef]
- Lesoin, L.; Crampon, C.; Boutin, O.; Badens, E. Development of a continuous dense gas process for the production of liposomes. J. Supercrit. Fluids 2011, 60, 51–62. [Google Scholar] [CrossRef]
- Soh, S.H.; Lee, L.Y. Microencapsulation and Nanoencapsulation Using Supercritical Fluid (SCF) Techniques. Pharmaceutics 2019, 11, 21. [Google Scholar] [CrossRef] [Green Version]
- Türk, M. Particle synthesis by rapid expansion of supercritical solutions (RESS): Current state, further perspectives and needs. J. Aerosol Sci. 2022, 161, 105950. [Google Scholar] [CrossRef]
- Chakravarty, P.; Famili, A.; Nagapudi, K.; Al-Sayah, M.A. Using Supercritical Fluid Technology as a Green Alternative During the Preparation of Drug Delivery Systems. Pharmaceutics 2019, 11, 629. [Google Scholar] [CrossRef] [Green Version]
- Wen, Z.; Liu, B.; Zheng, Z.; You, X.; Pua, Y.; Li, Q. Preparation of liposomes entrapping essential oil from Atractylodes macrocephala Koidz by modified RESS technique. Chem. Eng. Res. Des. 2010, 88, 1102–1107. [Google Scholar] [CrossRef]
- Zhang, Q.; Ou, C.; Ye, S.; Song, X.; Luo, S. Construction of nanoscale liposomes loaded with melatonin via supercritical fluid technology. J. Microencapsul. 2017, 34, 687–698. [Google Scholar] [CrossRef]
- Jiao, Z.; Wang, X.; Han, S.; Zha, X.; Xia, J. Preparation of vitamin C liposomes by rapid expansion of supercritical solution process: Experiments and optimization. J. Drug Deliv. Sci. Technol. 2019, 51, 1–6. [Google Scholar] [CrossRef]
- Trucillo, P.; Campardelli, R.; Reverchon, E. A versatile supercritical assisted process for the one-shot production of liposomes. J. Supercrit. Fluids 2019, 146, 136–143. [Google Scholar] [CrossRef]
- Trucillo, P.; Martino, M.; Reverchon, E. Supercritical Assisted Production of Lutein-Loaded Liposomes and Modelling of Drug Release. Processes 2021, 9, 1162. [Google Scholar] [CrossRef]
- Trucillo, P.; Cardea, S.; Baldino, L.; Reverchon, E. Production of liposomes loaded alginate aerogels using two supercritical CO2 assisted techniques. J. CO2 Util. 2020, 39, 101161. [Google Scholar] [CrossRef]
- Meure, L.A.; Knott, R.; Foster, N.R.; Dehghani, F. The Depressurization of an Expanded Solution into Aqueous Media for the Bulk Production of Liposomes. Langmuir 2009, 25, 326–337. [Google Scholar] [CrossRef]
- Zhao, L.; Temelli, F. Preparation of liposomes using a modified supercritical process via depressurization of liquid phase. J. Supercrit. Fluids 2015, 100, 110–120. [Google Scholar] [CrossRef]
- Boloix, A.; Feiner-Gracia, N.; Köber, M.; Repetto, J.; Pascarella, R.; Soriano, A.; Masanas, M.; Segovia, N.; Vargas-Nadal, G.; Merlo-Mas, J.; et al. Engineering pH-Sensitive Stable Nanovesicles for Delivery of MicroRNA Therapeutics. Small 2021, 18, 2101959. [Google Scholar] [CrossRef]
- Merlo-Mas, J.; Tomsen-Melero, J.; Corchero, J.-L.; González-Mira, E.; Font, A.; Pedersen, J.N.; García-Aranda, N.; Cristóbal-Lecina, E.; Alcaina-Hernando, M.; Mendoza, R.; et al. Application of Quality by Design to the robust preparation of a liposomal GLA formulation by DELOS-susp method. J. Supercrit. Fluids 2021, 173, 105204. [Google Scholar] [CrossRef]
- Van Swaay, D.; Demello, A. Microfluidic methods for forming liposomes. Lab Chip 2013, 13, 752–767. [Google Scholar] [CrossRef]
- Jahn, A.; Vreeland, W.N.; DeVoe, D.L.; Locascio, L.E.; Gaitan, M. Microfluidic directed formation of liposomes of controlled size. Langmuir 2007, 23, 6289–6293. [Google Scholar] [CrossRef]
- Carugo, D.; Bottaro, E.; Owen, J.; Stride, E.; Nastruzzi, C. Liposome production by microfluidics: Potential and limiting factors. Sci. Rep. 2016, 6, 25876. [Google Scholar] [CrossRef] [Green Version]
- Al-Amin, M.D.; Bellato, F.; Mastrotto, F.; Garofalo, M.; Malfanti, A.; Salmaso, S.; Caliceti, P. Dexamethasone Loaded Liposomes by Thin-Film Hydration and Microfluidic Procedures: Formulation Challenges. Int. J. Mol. Sci. 2020, 21, 1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shum, H.C.; Lee, D.; Yoon, I.; Kodger, T.; Weitz, D.A. Double Emulsion Templated Monodisperse Phospholipid Vesicles. Langmuir 2008, 24, 7651–7653. [Google Scholar] [CrossRef] [PubMed]
- Funakoshi, K.; Suzuki, H.; Takeuchi, S. Formation of giant lipid vesiclelike compartments from a planar lipid membrane by a pulsed jet flow. J. Am. Chem. Soc. 2007, 129, 12608–12609. [Google Scholar] [CrossRef] [PubMed]
- Bruna, G.; Carvalho, B.G.; Ceccato, B.T.; Michelon, M.; Han, S.W.; de la Torre, L.G. Advanced Microfluidic Technologies for Lipid Nano-Microsystems from Synthesis to Biological Application. Pharmaceutics 2022, 14, 141. [Google Scholar] [CrossRef]
- Zhang, G.; Sun, J. Lipid in Chips: A Brief Review of Liposomes Formation by Microfluidics. Int. J. Nanomed. 2021, 16, 7391–7416. [Google Scholar] [CrossRef]
- Jaafar-Maalej, C.; Charcosset, C.; Fessi, H. A new method for liposome preparation using a membrane contactor. J. Liposome Res. 2010, 21, 213–220. [Google Scholar] [CrossRef]
- Laouini, A.; Jaafar-Maalej, C.; Sfar, S.; Charcosset, C.; Fessi, H. Liposome preparation using a hollow fiber membrane contactor—Application to spironolactone encapsulation. Int. J. Pharm. 2011, 415, 53–61. [Google Scholar] [CrossRef]
- Laouini, A.; Charcosset, C.; Fessi, H.; Holdich, R.; Vladisavljević, G. Preparation of liposomes: A novel application of microengineered membranes-investigation of the process parameters and application to the encapsulation of vitamin E. RSC Adv. 2013, 3, 4985–4994. [Google Scholar] [CrossRef] [Green Version]
- Delma, K.L.; Lechanteur, A.; Evrard, B.; Semdé, R.; Piel, G. Sterilization methods of liposomes: Drawbacks of conventional methods and perspectives. Int. J. Pharm. 2021, 597, 120271. [Google Scholar] [CrossRef]
- Soares, G.; Learmonth, D.A.; Vallejo, M.; Davila, S.P.; González, P.; Sousa, R.A.; Oliveira, A. Supercritical CO2 technology: The next standard sterilization technique? Mater. Sci. Eng. C 2019, 99, 520–540. [Google Scholar] [CrossRef] [PubMed]
- Hood, R.R.; Vreeland, W.N.; DeVoe, D. Microfluidic remote loading for rapid single-step liposomal drug preparation. Lab Chip 2014, 14, 3359–3367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastner, E.; Verma, V.; Lowry, D.; Perrie, Y. Microfluidic-controlled manufacture of liposomes for the solubilisation of a poorly water soluble drug. Int. J. Pharm. 2015, 485, 122–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimov, N.; Kastner, E.; Hussain, M.; Perrie, Y.; Szita, N. Formation and purification of tailored liposomes for drug delivery using a module-based micro continuous-flow system. Sci. Rep. 2017, 7, 12045. [Google Scholar] [CrossRef] [Green Version]
- Yadav, S.; Sharma, A.K.; Kumar, P. Nanoscale Self-Assembly for Therapeutic Delivery. Front. Bioeng. Biotechnol. 2020, 8, 127. [Google Scholar] [CrossRef]
- Lombardo, D.; Calandra, P.; Pasqua, L.; Magazù, S. Self-Assembly of Organic Nanomaterials and Biomaterials: The Bottom-Up Approach for Functional Nanostructures Formation and Advanced Applications. Materials 2020, 13, 1048. [Google Scholar] [CrossRef] [Green Version]
- Calandra, P.; Caschera, D.; Liveri, V.T.; Lombardo, D. How self-assembly of amphiphilic molecules can generate complexity in the nanoscale. Colloids Surf. A Physicochem. Eng. 2015, 484, 164–183. [Google Scholar] [CrossRef]
- Zemb, T.; Lindner, P. Neutron, X-rays and Light Scattering Methods Applied to Soft Condensed Matter; North-Holland: Amsterdam, The Netherlands, 2002. [Google Scholar]
- Di Cola, E.; Grillo, I.; Ristori, S. Small Angle X-ray and Neutron Scattering: Powerful Tools for Studying the Structure of Drug-Loaded Liposomes. Pharmaceutics 2016, 8, 10. [Google Scholar] [CrossRef]
- Fitter, J.; Gutberlet, T.; Katsaras, J. Neutron scattering in Biology. In Techniques and Applications; Springer: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- Belloni, L. Interacting monodisperse and polydisperse spheres. In Neutron X-ray and Light Scattering; Lindner, P., Zemb, T., Eds.; Elsevier Science Publishers B. V.: New York, NY, USA, 1991. [Google Scholar]
- Schmidt, P.W. Modern Aspects of Small-Angle Scattering; Brumberger, H., Ed.; Kluwer Academic: Dordrecht, The Netherlands, 1995. [Google Scholar]
- Lombardo, D.; Calandra, P.; Kiselev, M.A. Structural Characterization of Biomaterials by Means of Small Angle X-rays and Neutron Scattering (SAXS and SANS), and Light Scattering Experiments. Molecules 2020, 25, 5624. [Google Scholar] [CrossRef]
- Kinnun, J.J.; Scott, H.L.; Ashkar, R.; Katsaras, J. Biomembrane Structure and Material Properties Studied with Neutron Scattering. Front. Chem. 2021, 9, 642851. [Google Scholar] [CrossRef] [PubMed]
- Kiselev, M.A.; Zemlyanaya, E.V.; Ryabova, N.Y.; Hauss, T.; Dante, S.; Lombardo, D. Water distribution function across the curved lipid bilayer: SANS study. Chem. Phys. 2008, 345, 185–190. [Google Scholar] [CrossRef]
- Dorrell, M.; Heberle, F.A.; Katsaras, J.; Lyman, E.; Sodt, A.J. Nanoscale Structure of Lipid Bilayers Revealed by In-Silico and Experimental Small Angle Neutron Scattering. Biophys. J. 2019, 116, 90a. [Google Scholar] [CrossRef] [Green Version]
- Kiselev, M.; Zemlyanaya, E.V.; Ryabova, N.Y.; Hauß, T.; Almásy, L.; Funari, S.S.; Zbytovská, J.; Lombardo, D. Influence of ceramide on the internal structure and hydration of the phospholipid bilayer studied by neutron and X-ray scattering. Appl. Phys. A 2014, 116, 319–325. [Google Scholar] [CrossRef]
- Amenitsch, H.; Bernstorff, S.; Kriechbaum, M.; Lombardo, D.; Mio, H.; Rappolt, M.; Laggner, P. Performance and first results of the ELETTRA high-flux beamline for small-angle X-ray scattering. J. Appl. Crystallogr. 1997, 30, 872–876. [Google Scholar] [CrossRef]
- Narayanan, T.; Sztucki, M.; Van Vaerenbergh, P.; Léonardon, J.; Gorini, J.; Claustre, L.; Sever, F.; Morse, J.; Boesecke, P. A multipurpose instrument for time-resolved ultra-small-angle and coherent X-ray scattering. J. Appl. Crystallogr. 2018, 51, 1511–1524. [Google Scholar] [CrossRef] [Green Version]
- Turco Liveri, V.; Lombardo, D.; Pochylski, M.; Calandra, P. Molecular association of small amphiphiles: Origin of ionic liquid properties in dibutyl phosphate/propylamine binary mixtures. J. Mol. Liq. 2018, 263, 274–281. [Google Scholar] [CrossRef]
- Koch, M.H.; Vachette, P.; Svergun, D.I. Small-angle scattering: A view on the properties, structures and structural changes of biological macromolecules in solution. Q. Rev. Biophys. 2003, 36, 147–227. [Google Scholar] [CrossRef] [Green Version]
- Weyerich, B.; Brunner-Popela, J.; Glatter, O. Small-angle scattering of interacting particles. II. Generalized indirect Fourier transformation under consideration of the effective structure factor for polydisperse systems. J. Appl. Crystallogr. 1999, 32, 197–209. [Google Scholar] [CrossRef]
- Micali, N.; Scolaro, L.M.; Romeo, A.; Lombardo, D.; Lesieur, P.; Mallamace, F. Structural properties of methanol-polyamidoamine dendrimer solutions. Phys. Rev. E 1998, 58, 6229–6235. [Google Scholar] [CrossRef]
- Sachs, J.; Petrache, H.; Woolf, T. Interpretation of small angle X-ray measurements guided by molecular dynamics simulations of lipid bilayers. Chem. Phys. Lipids 2003, 26, 211–223. [Google Scholar] [CrossRef]
- Kucerka, N.; Nagle, J.F.; Sachs, J.N.; Feller, S.E.; Pencer, J.; Jackson, A.; Katsaras, J. Lipid bilayer structure determined by the simultaneous analysis of neutron and X-ray scattering data. Biophys. J. 2008, 95, 2356–2367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardo, D.; Calandra, P.; Bellocco, E.; Laganà, G.; Barreca, D.; Magazù, S.; Wanderlingh, U.; Kiselev, M.A. Effect of anionic and cationic polyamidoamine (PAMAM) dendrimers on a model lipid membrane. Biochim. Biophys. Acta 2016, 1858, 2769–2777. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, D.; Calandra, P.; Magazù, S.; Wanderlingh, U.; Barreca, D.; Pasqua, L.; Kiselev, M.A. Soft nanoparticles charge expression within lipid membranes: The case of amino terminated dendrimers in bilayers vesicles. Colloids Surf. B Biointerfaces 2018, 170, 609–616. [Google Scholar] [CrossRef]
- Lombardo, D.; Calandra, P.; Caccamo, M.T.; Magazù, S.; Pasqua, L.; Kiselev, M.A. Interdisciplinary approaches to the study of biological membranes. AIMS Biophys. 2020, 7, 267. [Google Scholar] [CrossRef]
- Klang, V.; Valenta, C.; Matsko, N.B. Electron microscopy of pharmaceutical systems. Micron 2013, 44, 45–74. [Google Scholar] [CrossRef]
- Bibi, S.; Kaur, R.; Henriksen-Lacey, M.; McNeil, S.E.; Wilkhu, J.; Lattmann, E.; Christensen, D.; Mohammed, A.R.; Perrie, Y. Microscopy imaging of liposomes: From coverslips to environmental SEM. Int. J. Pharm. 2011, 417, 138–150. [Google Scholar] [CrossRef]
- Ruozi, B.; Belletti, D.; Tombesi, A.; Tosi, G.; Bondioli, L.; Forni, F.; Vandelli, M.A. AFM, ESEM, TEM, and CLSM in liposomal characterization: A comparative study. Int. J. Nanomed. 2011, 6, 557–563. [Google Scholar] [CrossRef] [Green Version]
- Helvig, S.; Azmi, I.D.M.; Moghimi, S.M.; Yaghmur, A. Recent advances in cryo-TEM imaging of soft lipid nanoparticles. AIMS Biophys. 2015, 2, 116–130. [Google Scholar] [CrossRef]
- Almgren, M.; Edwards, K.; Karlsson, G.B. Cryo transmission electron microscopy of liposomes and related structures. Colloids Surf. A Physicochem. Eng. 2000, 174, 3–21. [Google Scholar] [CrossRef]
- Spyratou, E.; Mourelatou, E.A.; Makropoulou, M.; Demetzos, C. Atomic force microscopy: A tool to study the structure, dynamics and stability of liposomal drug delivery systems. Expert Opin. Drug Deliv. 2009, 6, 305–317. [Google Scholar] [CrossRef]
- Robson, A.L.; Dastoor, P.C.; Flynn, J.; Palmer, W.; Martin, A.; Smith, D.W.; Woldu, A.; Hua, S. Advantages and Limitations of Current Imaging Techniques for Characterizing Liposome Morphology. Front. Pharmacol. 2018, 9, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouvrais, H.; Pott, T.; Bagatolli, L.A.; Ipsen, J.H.; Meleard, P. Impact of membrane-anchored fluorescent probes on the mechanical properties of lipid bilayers. Biochim. Biophys. Acta 2010, 1798, 1333–1337. [Google Scholar] [CrossRef] [PubMed]
- Mertins, O.; Dimova, R. Insights on the interactions of chitosan with phospholipid vesicles. Part II: Membrane stiffening and pore formation. Langmuir 2013, 29, 14552–14559. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.B.; Davidson, M.W. Confocal laser scanning microscopy. In Fundamentals of Light Microscopy and Electronic Imaging, 2nd ed.; Murphy, D.B., Ed.; John Wiley and Sons: New York, NY, USA, 2012; pp. 265–305. [Google Scholar] [CrossRef]
- Solomon, M.A. Determination of the Subcellular Distribution of Liposomes Using Confocal Microscopy. Methods Mol. Biol. 2017, 1522, 119–130. [Google Scholar] [CrossRef]
- Bruun, K.; Hille, C. Study on intracellular delivery of liposome encapsulated quantum dots using advanced fluorescence microscopy. Sci. Rep. 2019, 9, 10504. [Google Scholar] [CrossRef]
- Sattler, E.C.; Maier, T.; Hoffmann, V.S.; Hegyi, J.; Ruzicka, T.; Berking, C. Noninvasive in vivo detection and quantification of Demodex mites by confocal laser scanning microscopy. Br. J. Dermatol. 2012, 167, 1042–1047. [Google Scholar] [CrossRef]
- Vaghasiya, K.; Sharma, A.; Kumar, K.; Ray, E.; Adlakha, S.; Katare, O.P.; Hota, S.K.; Verma, R.K. Heparin-Encapsulated Metered-Dose Topical “Nano-Spray Gel” Liposomal Formulation Ensures Rapid On-Site Management of Frostbite Injury by Inflammatory Cytokines Scavenging, ACS Biomater. Sci. Eng. 2019, 5, 6617–6631. [Google Scholar] [CrossRef]
- Berne, B.J.; Pecora, R. Dynamic Light Scattering; John Wiley: New York, NY, USA, 1976. [Google Scholar]
- Brown, W. Dynamic Light Scattering: The Method and some Applications; Clarendon Press: Oxford, UK, 1993. [Google Scholar]
- Hunter, R.J. Foundations of Colloid Science; Oxford University Press: New York, NY, USA, 1986; Volume I–II. [Google Scholar]
- McNeil-Watson, F.; Tscharnuter, W.; Miller, J. A new instrument for the measurement of very small electrophoretic mobilities using phase analysis light scattering (PALS). Colloids Surf. A 1998, 140, 53–57. [Google Scholar] [CrossRef]
- Carvalho, P.M.; Felício, M.R.; Santos, N.C.; Gonçalves, S.; Domingues, M.M. Application of Light Scattering Techniques to Nanoparticle Characterization and Development. Front. Chem. 2018, 6, 237. [Google Scholar] [CrossRef]
- Mazzaglia, A.; Angelini, N.; Darcy, R.; Donohue, R.; Lombardo, D.; Micali, N.; Sciortino, M.T.; Villari, V.; Scolaro, L.M. Novel heterotopic colloids of anionic porphyrins entangled in cationic amphiphilic cyclodextrins: Spectroscopic investigation and intracellular delivery. Chemistry 2003, 9, 5762–5769. [Google Scholar] [CrossRef]
- Mazzaglia, A.; Angelini, N.; Lombardo, D.; Micali, N.; Patané, S.; Villari, V.; Scolaro, L.M. Amphiphilic cyclodextrin carriers embedding porphyrins: Charge and size modulation of colloidal stability in heterotopic aggregates. J. Phys. Chem. B 2005, 109, 7258–7265. [Google Scholar] [CrossRef] [PubMed]
- Lebrón, J.A.; López-López, M.; García-Calderón, C.B.; Rosado, I.V.; Balestra, F.R.; Huertas, P.; Rodik, R.V.; Kalchenko, V.I.; Bernal, E.; Moyá, M.L.; et al. Multivalent Calixarene-Based Liposomes as Platforms for Gene and Drug Delivery. Pharmaceutics 2021, 13, 1250. [Google Scholar] [CrossRef] [PubMed]
- Grabielle-Madelmont, C.; Lesieur, S.; Ollivon, M. Characterization of loaded liposomes by size exclusion chromatography. J. Biochem. Biophys. Methods 2003, 56, 189–217. [Google Scholar] [CrossRef]
- Nieh, M.P.; Heberle, F.A.; Katsaras, J. Characterization of Biological Membranes Structure and Dynamics; De Gruyter: Berlin, Germany, 2019. [Google Scholar]
- Pignatello, R.; Musumeci, T.; Basile, L.; Carbone, C.; Puglisi, G. Biomembrane models and drug-biomembrane interaction studies: Involvement in drug design and development. J. Pharm. Bioallied Sci. 2011, 3, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Pignatello, R. Drug-Biomembrane Interaction Studies, the Application of Calorimetric Techniques; Elsevier: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Abraham, T.; Lewis, R.N.; Hodges, R.S.; McElhaney, R.N. Isothermal titration calorimetry studies of the binding of a rationally designed analogue of the antimicrobial peptide gramicidin s to phospholipid bilayer membranes. Biochemistry 2005, 44, 2103–2112. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.M.; Patel, A.B.; de Graaf, R.A.; Behar, K.L. Determination of liposomal encapsulation efficiency using proton NMR spectroscopy. Chem. Phys. Lipids 2004, 127, 113–120. [Google Scholar] [CrossRef]
- Cruciani, O.; Mannina, L.; Sobolev, A.; Cametti, C.; Segre, A. An improved NMR study of liposomes using 1-palmitoyl-2-oleoyl-sn-glycero-3-phospatidylcholine as model. Molecules 2006, 11, 334–344. [Google Scholar] [CrossRef] [Green Version]
- Sainz, V.; Conniot, J.; Matos, A.I.; Peres, C.; Zupanǒiǒ, E.; Moura, L.; Silva, L.C.; Florindo, H.F.; Gaspar, R.S. Regulatory aspects on nanomedicines. Biochem. Biophys. Res. Commun. 2015, 468, 504–510. [Google Scholar] [CrossRef]
- Raval, N.; Maheshwari, R.G.; Kalyane, D.; Youngren-Ortiz, S.R.; Chougule, M.B.; Tekade, R.K. Importance of Physicochemical Characterization of Nanoparticles in Pharmaceutical Product Development; Elsevier BV: Amsterdam, The Netherlands, 2019; pp. 369–400. [Google Scholar] [CrossRef]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Davarani, F.H.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M. Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Németh, Z.; Pallagi, E.; Dobó, D.G.; Csóka, I. A Proposed Methodology for a Risk Assessment-Based Liposome Development Process. Pharmaceutics 2020, 12, 1164. [Google Scholar] [CrossRef]
- European Medicine Agency (EMA). Reflection Paper on the Data Requirements for Intravenous Liposomal Products Developed with Reference to an Innovator Liposomal Product. 2013. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/reflection-paper-data-requirements-intravenous-liposomal-products-developed-reference-innovator_en.pdf (accessed on 18 February 2022).
- Food and Drug Administration (FDA). Liposome Drug Products Chemistry, Manufacturing, and Controls; Human Pharmacokinetics and Bioavailability; and Labeling Documentation Guidance for Industry. 2018. Available online: http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm (accessed on 18 February 2022).
- Sabir, F.; Katona, G.; Pallagi, E.; Dobó, D.G.; Akel, H.; Berkesi, D.; Kónya, Z.; Csóka, I. Quality-by-design-based development of n-propyl-gallate-loaded hyaluronic-acid-coated liposomes for intranasal administration. Molecules 2021, 26, 1429. [Google Scholar] [CrossRef] [PubMed]
- Porfire, A.; Achim, M.; Barbalata, C.; Rus, I.; Tomuta, I.; Cristea, C. Pharmaceutical Development of Liposomes Using the QbD Approach. Liposomes Adv. Perspect 2019, 2019, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Lu, B.; Ma, Q.; Zhang, J.; Liu, R.; Yue, Z.; Xu, C.; Li, Z.; Lin, H. Preparation and characterization of bupivacaine multivesicular liposome: A QbD study about the effects of formulation and process on critical quality attributes. Int. J. Pharm. 2021, 598, 120335. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Khan, M.A.; Burgess, D.J. A quality by design (QbD) case study on liposomes containing hydrophilic API: I. Formulation, processing design and risk assessment. Int. J. Pharm. 2011, 419, 52–59. [Google Scholar] [CrossRef]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Advantages | Disadvantages | Liposomes | Ref. |
|---|---|---|---|---|
| Thin-Film Hydration (Bangham method) | Simple procedure, good encapsulation efficiency (EE) (both with small and large drugs) | Large use of organic solvent (difficult to remove) lower EE for water-soluble drugs, small-scale production, no particle size control, time consuming, sterilization needed | Polydisperse MLVs | [66,67,68] |
| Detergent Removal (Depletion) Method | Simple procedure, good EE (both with small and large drugs) | Need of large amount of organic (and residual) solvent, poor EE (for lipophilic drugs), low final liposome concentrations (low yield), time consuming, sterilization issue | MLVs, LUVs | [71,72,73,74,75,76] |
| Solvent (Ethanol/Ether) Injection | Simple, rapid, reproducible Ether injection gives greater EE. | Removal of ethanol is difficult, as it forms azeotrope with water, difficult to handle biologically active macromolecules in ethanol, possible nozzle blockage (ether system), sterilization issue | SMVs, SUVs | [81,82,83,84,85] |
| Reverse-Phase Evaporation | Simple process, suitable EE | Large quantity of organic solvent, not suitable for fragile (bio-) drugs, time consuming, sterilization issue | MLVs, LUVs | [88,89,90] |
| Novel Technologies | ||||
| Freeze Drying (Lyophilization) | Low organic solvent residue, suitable for large-scale production, prevents the physical degradation of liposomes during storage, increases liposomes’ shelf-life | Time- and energy-consuming, may induce (structural/size) alterations in formed vesicle, loss of encapsulated material, sterilization issue | MLVs, LUVs SMVs, SUVs | [100,101,102,103,104,105] |
| SC Reverse-Phase Evaporation (SC-RPE) | Control of particle size, possible in situ sterilization, low organic solvent (env. friendly), quickly encapsulates both hydrophilic and lipophilic materials, large-scale production | high pressure used, high capital cost, low encapsulation efficiency, low liposome stability | LUVs, MLVs | [109,110,111,112] |
| Supercritical Anti-Solvent (SAS) | Relatively simple (and repeatable), control of particle size, low organic solvent (and residues), in situ sterilization, | High capital cost, low yield and EE, possible aggregation of particles, presence of residual (toxic) solvents in the final product | LUVs, MLVs | [113,114] |
| Rapid Expansion of a Supercritical Solution (RESS) | Control of particle size, possible in situ sterilization, low organic solvent consumption (that can be reused) | low yield and EE, high pressure (up to 250 bar) used, high production cost, poor solubility of (polymer-based) biomaterials in SC-CO2, difficulty of the separation between co-solvents and vesicles during the depressurization process, may involve nozzle blockages | OLMs, MLVs, ULVs | [115,116,117,118,119,120] |
| Supercritical Assisted Liposome Formation (SuperLip) | Continuous and replicable process, encapsulates hydrophilic drugs, high EE, low solvent residue | time-consuming process, requires high pressure, high capital cost | LUVs, MLVs | [121,123] |
| Depressurization of an Expanded Liquid Organic Solution (DELOS) | Simple and rapid process, control of the liposomes size, possibility to obtain small sizes, shape uniformity/homogeneity, and good stability, possibility to reduce sterols use, high EE (hydrophilic drugs) | residual organic solvent, nozzle blockage | LUVs, MLVs | [124,125,126,127] |
| Microfluidic (Micro Hydrodynamic Focusing—MHF), (Microfluidic Droplets—MD), (Pulsed Jet Flow—PJF) | Good particle size control, possibility to upgrade to novel methods for liposome preparation (lab on chip) | organic solvent (difficult to remove), not suitable for bulk production, high cost of microfluidic channels | SUVs, LUVs (for MHF), GUVs (MD), GUVs (PJF) | [128,129,130,131,132,133,134,135] |
| Membrane Contactor | Fast process, high EE, good size (distribution) control, scaling-up abilities (for industry) | possibility of clogging the pores, membrane blockage, high temperature, sterilization issues | MLVs, | [136,137,138] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lombardo, D.; Kiselev, M.A. Methods of Liposomes Preparation: Formation and Control Factors of Versatile Nanocarriers for Biomedical and Nanomedicine Application. Pharmaceutics 2022, 14, 543. https://doi.org/10.3390/pharmaceutics14030543
Lombardo D, Kiselev MA. Methods of Liposomes Preparation: Formation and Control Factors of Versatile Nanocarriers for Biomedical and Nanomedicine Application. Pharmaceutics. 2022; 14(3):543. https://doi.org/10.3390/pharmaceutics14030543
Chicago/Turabian StyleLombardo, Domenico, and Mikhail A. Kiselev. 2022. "Methods of Liposomes Preparation: Formation and Control Factors of Versatile Nanocarriers for Biomedical and Nanomedicine Application" Pharmaceutics 14, no. 3: 543. https://doi.org/10.3390/pharmaceutics14030543