
PEGylated Chitosan Nanoparticles Encapsulating Ascorbic Acid and Oxaliplatin Exhibit Dramatic Apoptotic Effects against Breast Cancer Cells

 ,
,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Preparation of the NPs
2.2.2. Characterization of the Prepared NP
2.2.3. Quantification Method
2.2.4. Determination of Entrapment Efficiency % (EE) of Ascorbic Acid and Oxaliplatin in Chitosan Nanoparticles
2.2.5. Release Study
2.2.6. Cell Viability Assay
Cell Culture
MTT Assay
2.2.7. Flow Cytometry and Cell Apoptosis Assay
3. Results and Discussion
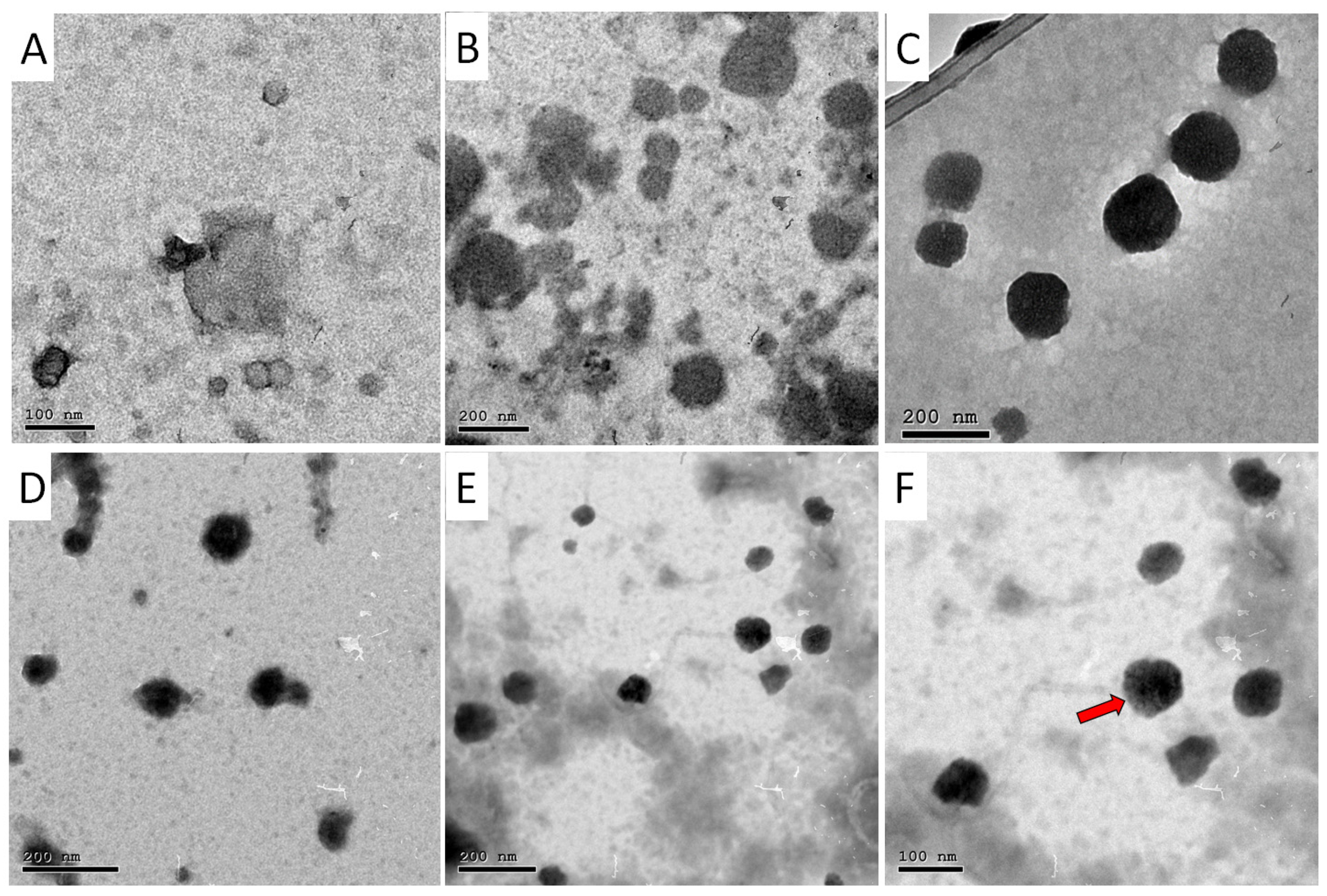
3.1. Hydrodynamic Diameter, Polydispersity Index (PDI), ζ-Potential, Entrapment Efficiency (EE%) and Morphology
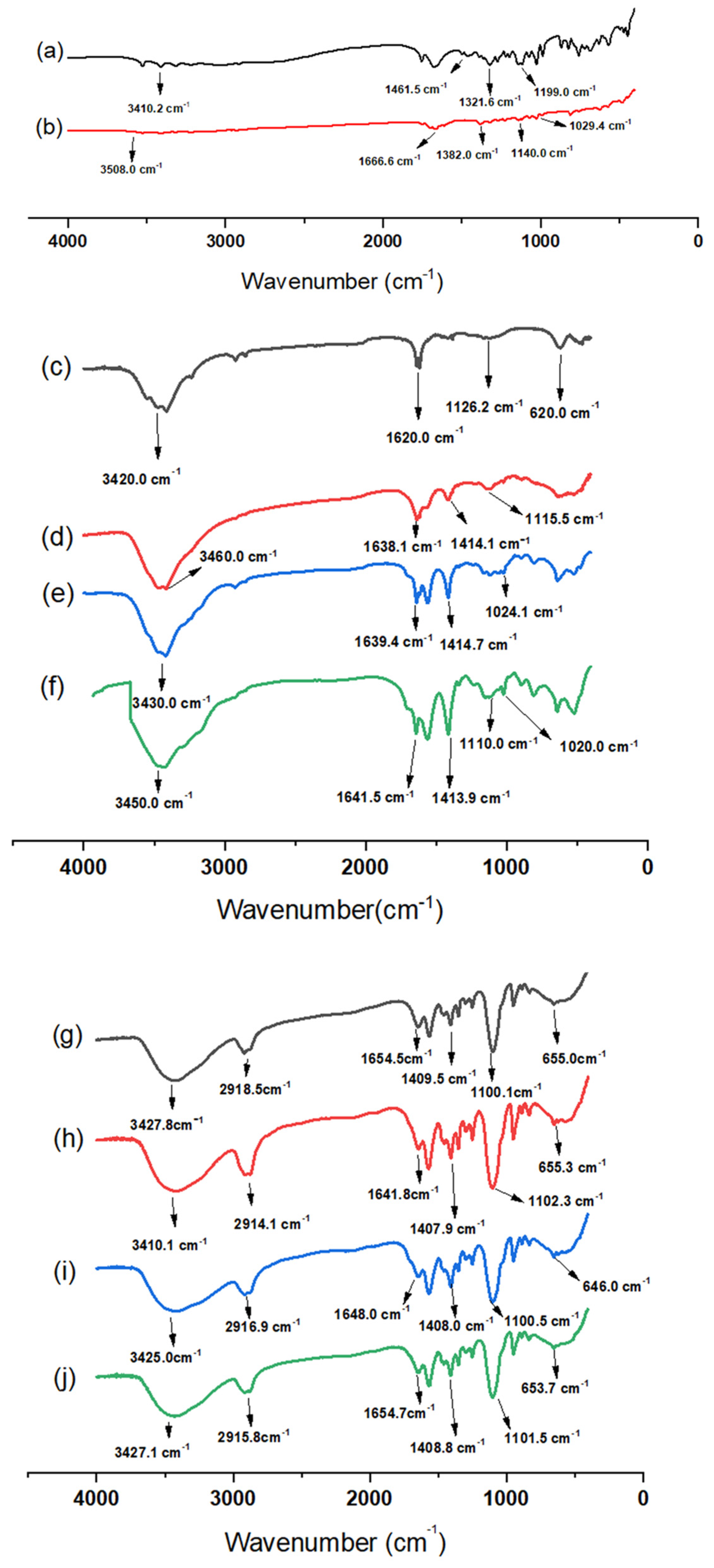
3.2. Fourier-Transform Infrared Spectroscopy (FTIR)
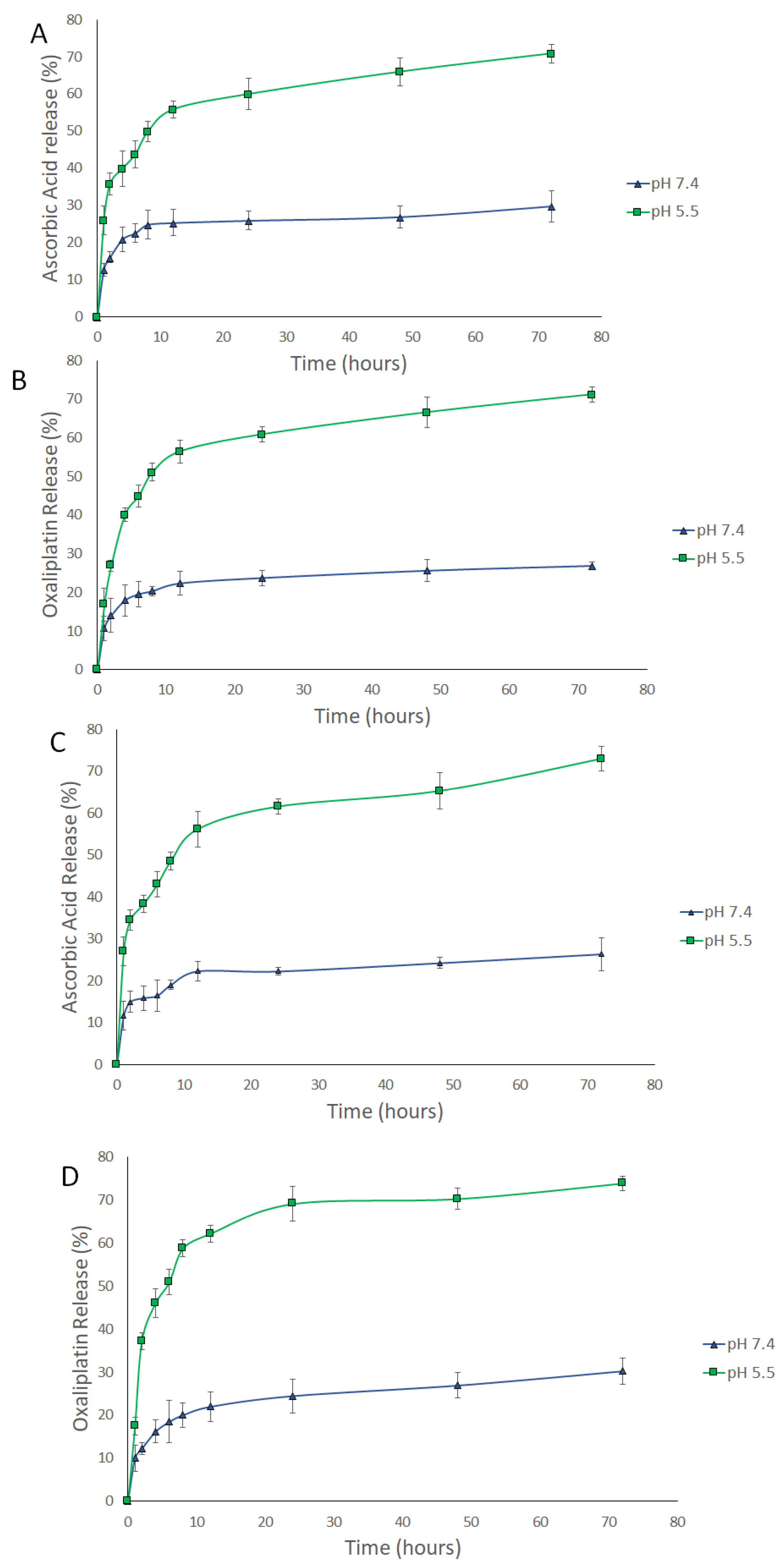
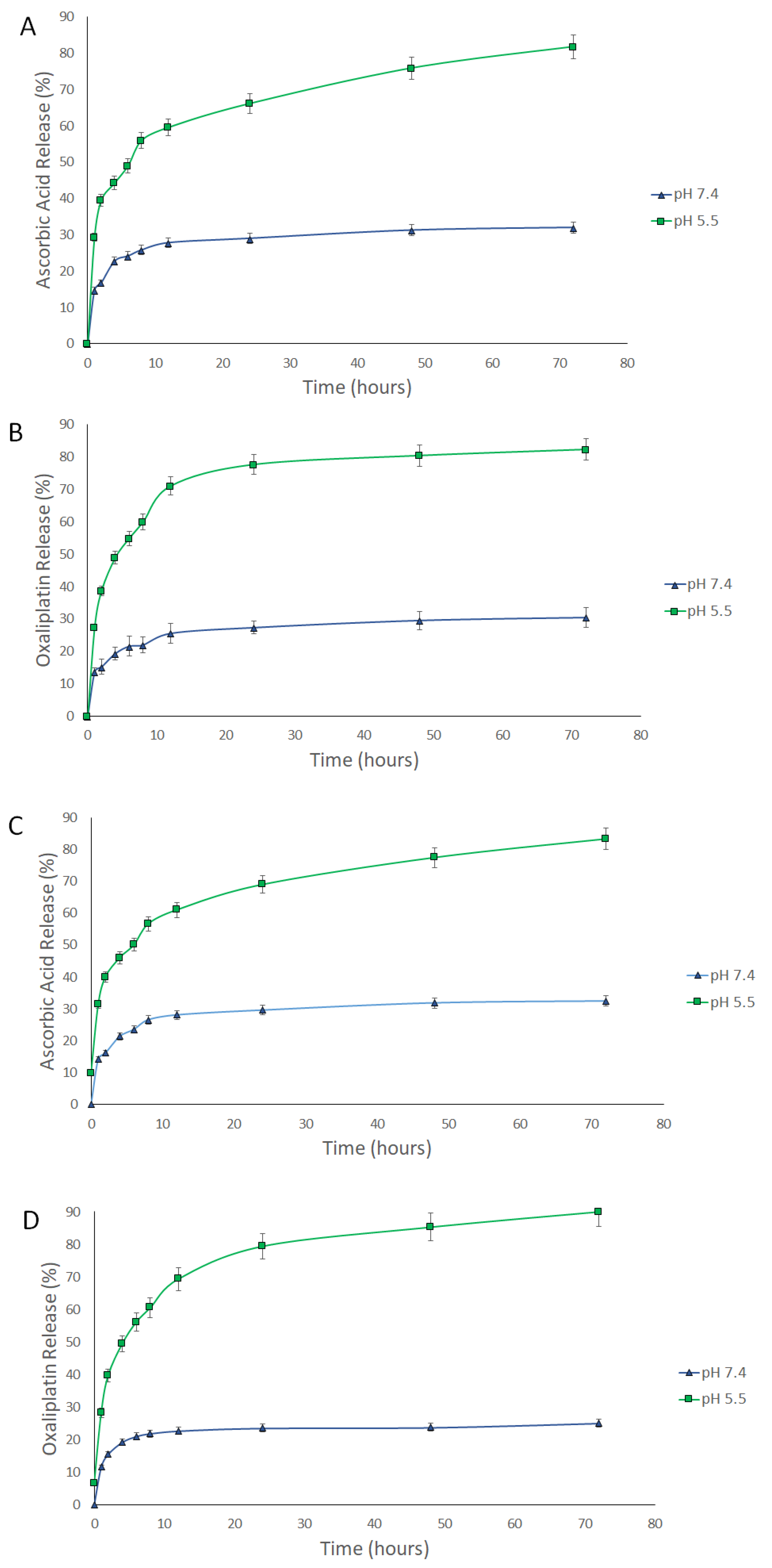
3.3. Release Study
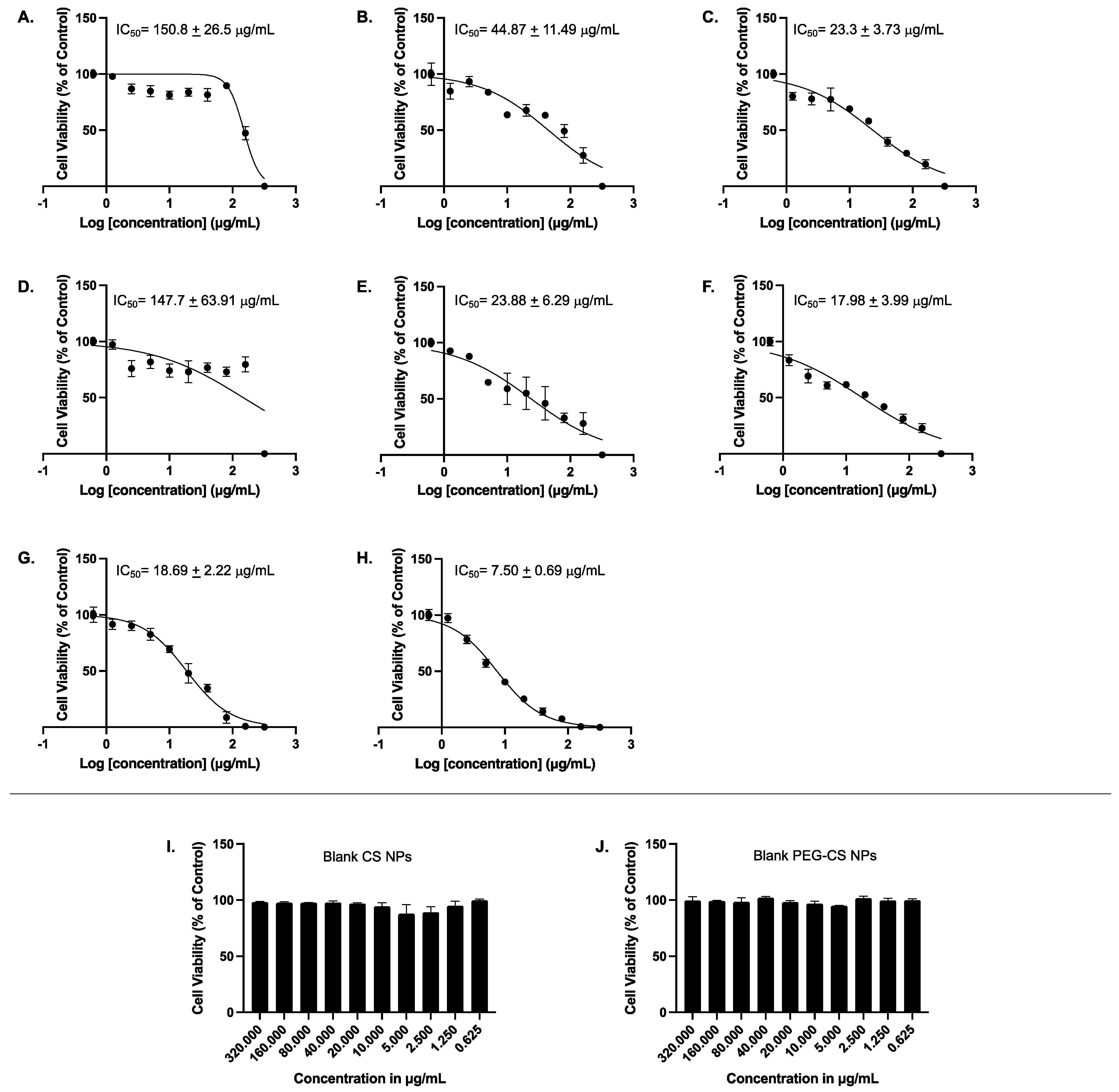
3.4. Cell Viability Assay
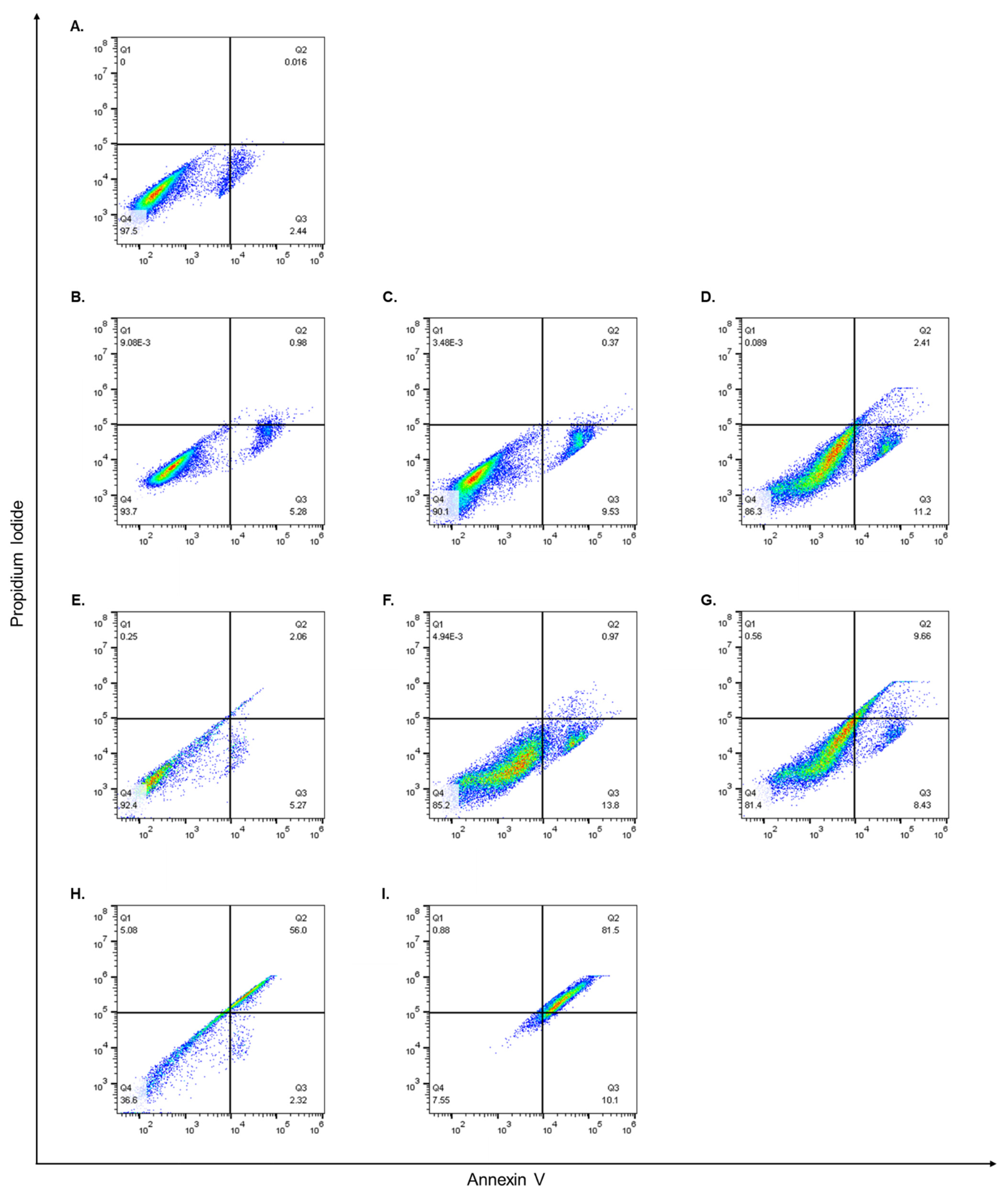
3.5. Flow Cytometry and Cell Apoptosis Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahmy, S.A.; Brüßler, J.; Alawak, M.; El-Sayed, M.M.H.; Bakowsky, U.; Shoeib, T. Chemotherapy Based on Supramolecular Chemistry: A Promising Strategy in Cancer Therapy. Pharmaceutics 2019, 11, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahmy, S.A.; Ponte, F.; Abd El-Rahman, M.K.; Russo, N.; Sicilia, E.; Shoeib, T. Investigation of the host-guest complexation between 4-sulfocalix[4]arene and nedaplatin for potential use in drug delivery. Spectrochim. Acta Part. A 2018, 193, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Fahmy, S.A.; Ponte, F.; Sicilia, E.; Azzazy, H.M.E. Experimental and Computational Investigations of Carboplatin Supramolecular Complexes. ACS Omega 2020, 5, 31456–31466. [Google Scholar] [CrossRef] [PubMed]
- Ritacco, I.; Al Assy, M.; Abd El-Rahman, M.K.; Fahmy, S.A.; Russo, N.; Shoeib, T.; Sicilia, E. Hydrolysis in Acidic Environment and Degradation of Satraplatin: A Joint Experimental and Theoretical Investigation. Inorg. Chem. 2017, 56, 6013–6026. [Google Scholar] [CrossRef] [PubMed]
- Fahmy, S.A.; Ponte, F.; Fawzy, I.M.; Sicilia, E.; Bakowsky, U.; Azzazy, H.M.E. Host-Guest Complexation of Oxaliplatin and Para-Sulfonatocalix[n]Arenes for Potential Use in Cancer Therapy. Molecules 2020, 25, 5926. [Google Scholar] [CrossRef] [PubMed]
- Arango, D.; Wilson, A.J.; Shi, Q.; Corner, G.A.; Aranes, M.J.; Nicholas, C.; Lesser, M.; Mariadason, J.M.; Augenlicht, L.H. Molecular mechanisms of action and prediction of response to oxaliplatin in colorectal cancer cells. Br. J. Cancer 2004, 91, 1931–1946. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Jain, S.K.; Ganesh, N.; Barve, J.; Beg, A.M. Design and development of ligand-appended polysaccharidic nanoparticles for the delivery of oxaliplatin in colorectal cancer. Nanomedicine 2010, 6, 179–190. [Google Scholar] [CrossRef]
- Comella, P.; Casaretti, R.; Sandomenico, C.; Avallone, A.; Franco, L. Role of oxaliplatin in the treatment of colorectal cancer. Ther. Clin. Risk Manag. 2009, 5, 229–238. [Google Scholar] [CrossRef] [Green Version]
- Kalidhindi, R.S.; Tummala, S.; Kumar, M.N.S.; Kuppusamy, G.; Prakash, A.; Mulukutla, S. Preparation, Physicochemical Characterization and In Vitro Evaluation of Oxaliplatin Solid Lipid Nanoparticles for the Treatment of Colorectal Cancer. Indo. Am. J. Pharm. Res. 2014, 4, 3579–3587. [Google Scholar]
- Fahmy, S.A.; Ponte, F.; Fawzy, I.M.; Sicilia, E.; Azzazy, H.M.E. Betaine host–guest complexation with a calixarene receptor: Enhanced in vitro anticancer effect. RSC Adv. 2021, 11, 24673–24680. [Google Scholar] [CrossRef]
- Fahmy, S.A.; Issa, M.Y.; Saleh, B.M.; Meselhy, M.R.; Azzazy, H.M.E. Peganum harmala Alkaloids Self-Assembled Supramolecular Nanocapsules with Enhanced Antioxidant and Cytotoxic Activities. ACS Omega 2021, 6, 11954–11963. [Google Scholar] [CrossRef]
- Fahmy, S.A.; Mahdy, N.K.; Al Mulla, H.; ElMeshad, A.N.; Issa, M.Y.; Azzazy, H.M.E. PLGA/PEG Nanoparticles Loaded with Cyclodextrin-Peganum harmala Alkaloid Complex and Ascorbic Acid with Promising Antimicrobial Activities. Pharmaceutics 2022, 14, 142. [Google Scholar] [CrossRef] [PubMed]
- El-Shafie, S.; Fahmy, S.A.; Ziko, L.; Elzahed, N.; Shoeib, T.; Kakarougkas, A. Encapsulation of Nedaplatin in Novel PEGylated Liposomes Increases Its Cytotoxicity and Genotoxicity against A549 and U2OS Human Cancer Cells. Pharmaceutics 2020, 12, 863. [Google Scholar] [CrossRef] [PubMed]
- Fahmy, S.; Brüßler, J.; Ponte, F.; El-Rahman, M.; Russo, N.; Sicilia, E.; Bakowsky, U.; Shoeib, T. A study on the physicochemical properties and cytotoxic activity of p -sulfocalix[4]arene-nedaplatin complex. J. Phys. Conf. Ser. 2019, 1310, 012011. [Google Scholar] [CrossRef]
- Fahmy, S.; Mamdouh, W. Garlic oil–loaded PLGA nanoparticles with controllable size and shape and enhanced antibacterial activities. J. Appl. Polym. Sci. 2018, 135, 46133. [Google Scholar] [CrossRef]
- Golombek, S.K.; May, J.N.; Theek, B.; Appold, L.; Drude, N.; Kiessling, F.; Lammers, T. Tumor targeting via EPR: Strategies to enhance patient responses. Adv. Drug Deliv. Rev. 2018, 130, 17–38. [Google Scholar] [CrossRef]
- Silva, M.M.; Calado, R.; Marto, J.; Bettencourt, A.; Almeida, A.J.; Goncalves, L.M.D. Chitosan Nanoparticles as a Mucoadhesive Drug Delivery System for Ocular Administration. Mar. Drugs 2017, 15, 370. [Google Scholar] [CrossRef] [Green Version]
- Singla, A.K.; Chawla, M. Chitosan: Some pharmaceutical and biological aspects—An update. J. Pharm. Pharmacol. 2001, 53, 1047–1067. [Google Scholar] [CrossRef]
- Baghdan, E.; Pinnapireddy, S.R.; Strehlow, B.; Engelhardt, K.H.; Schafer, J.; Bakowsky, U. Lipid coated chitosan-DNA nanoparticles for enhanced gene delivery. Int J. Pharm. 2018, 535, 473–479. [Google Scholar] [CrossRef]
- Adhikari, H.S.; Yadav, P.N. Anticancer Activity of Chitosan, Chitosan Derivatives, and Their Mechanism of Action. Int. J. Biomater. 2018, 2018, 2952085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Lee, S.; Kim, J.-H.; Park, K.; Kim, K.; Kwon, I.C. Polymeric nanomedicine for cancer therapy. Prog. Polym. Sci. 2008, 33, 113–137. [Google Scholar] [CrossRef]
- Padayatty, S.J.; Levine, M. Reevaluation of ascorbate in cancer treatment: Emerging evidence, open minds and serendipity. J. Am. Coll. Nutr. 2000, 19, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Krishna, M.C.; Mitchell, J.B.; Corpe, C.P.; Buettner, G.R.; Shacter, E.; Levine, M. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: Action as a pro-drug to deliver hydrogen peroxide to tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 13604–13609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, A.S.; Marques, C.R.; Encarnação, J.C.; Abrantes, A.M.; Marques, I.A.; Laranjo, M.; Oliveira, R.; Casalta-Lopes, J.E.; Gonçalves, A.C.; Sarmento-Ribeiro, A.B.; et al. Ascorbic Acid Chemosensitizes Colorectal Cancer Cells and Synergistically Inhibits Tumor Growth. Front. Physiol. 2018, 23, 911. [Google Scholar] [CrossRef] [Green Version]
- Kurbacher, C.M.; Wagner, U.; Kolster, B.; Andreotti, P.E.; Krebs, D.; Bruckner, H.W. Ascorbic acid (vitamin C) improves the antineoplastic activity of doxorubicin, cisplatin, and paclitaxel in human breast carcinoma cells in vitro. Cancer Lett. 1996, 103, 183–189. [Google Scholar] [CrossRef]
- Wu, C.W.; Liu, H.C.; Yu, Y.L.; Hung, Y.T.; Wei, C.W.; Yiang, G.T. Combined treatment with vitamin C and methotrexate inhibits triple-negative breast cancer cell growth by increasing H2O2 accumulation and activating caspase-3 and p38 pathways. Oncol. Rep. 2017, 37, 2177–2184. [Google Scholar] [CrossRef] [Green Version]
- Calvo, P.; Remunan-Lopez, C.; Vila-Jato, J.L.; Alonso, M.J. Chitosan and chitosan/ethylene oxide-propylene oxide block copolymer nanoparticles as novel carriers for proteins and vaccines. Pharm. Res. 1997, 14, 1431–1436. [Google Scholar] [CrossRef]
- Abdel-Hafez, S.M.; Hathout, R.M.; Sammour, O.A. Tracking the transdermal penetration pathways of optimized curcumin-loaded chitosan nanoparticles via confocal laser scanning microscopy. Int. J. Biol. Macromol. 2018, 108, 753–764. [Google Scholar] [CrossRef]
- Pinnapireddy, S.R.; Duse, L.; Strehlow, B.; Schafer, J.; Bakowsky, U. Composite liposome-PEI/nucleic acid lipopolyplexes for safe and efficient gene delivery and gene knockdown. Colloids Surf. B Biointerfaces 2017, 158, 93–101. [Google Scholar] [CrossRef]
- Fahmy, S.A.; Fawzy, I.M.; Saleh, B.M.; Issa, M.Y.; Bakowsky, U.; Azzazy, H.M.E. Green Synthesis of Platinum and Palladium Nanoparticles Using Peganum harmala L. Seed Alkaloids: Biological and Computational Studies. Nanomaterials 2021, 11, 965. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-X.; Li, T.; Peng, S.; Tao, D. Supramolecular nanocapsules from the self-assembly of amphiphilic calixarene as a carrier for paclitaxel. New J. Chem. 2016, 40, 9923–9929. [Google Scholar] [CrossRef]
- Qiu, L.; Jing, N.; Jin, Y. Preparation and in vitro evaluation of liposomal chloroquine diphosphate loaded by a transmembrane pH-gradient method. Int. J. Pharm. 2008, 361, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Rao, K.; Roome, T.; Aziz, S.; Razzak, A.; Abbas, G.; Imran, M.; Jabri, T.; Gul, J.; Hussain, M.; Sikandar, B.; et al. Bergenin loaded gum xanthan stabilized silver nanoparticles suppress synovial inflammation through modulation of the immune response and oxidative stress in adjuvant induced arthritic rats. J. Mater. Chem. B 2018, 6, 4486–4501. [Google Scholar] [CrossRef]
- Imran, M.; Shah, M.R.; Ullah, F.; Ullah, S.; Sadiq, A.; Ali, I.; Ahmed, F.; Nawaz, W. Double-tailed acyl glycoside niosomal nanocarrier for enhanced oral bioavailability of Cefixime. Artif. Cells Nanomed. Biotechnol. 2017, 45, 1440–1451. [Google Scholar] [CrossRef]
- Zhang, X.G.; Teng, D.Y.; Wu, Z.M.; Wang, X.; Wang, Z.; Yu, D.M.; Li, C.X. PEG-Grafted Chitosan Nanoparticles as an Injectable Carrier for Sustained Protein Release. J. Mater. Sci. Mater. Med. 2008, 19, 3525–3533. [Google Scholar] [CrossRef]
- Pawlak, A.; Mucha, M. Thermogravimetric and FTIR studies of chitosan blends. Thermochimica Acta 2003, 396, 153–166. [Google Scholar] [CrossRef]
- Panicker, C.Y.; Varghese, H.T.; Philip, D. FT-IR, FT-Raman and SERS spectra of Vitamin C. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2006, 65, 802–804. [Google Scholar] [CrossRef]
- Tummala, S.; Gowthamarajan, K.; Satish Kumar, M.N.; Wadhwani, A. Oxaliplatin immuno hybrid nanoparticles for active targeting: An approach for enhanced apoptotic activity and drug delivery to colorectal tumors. Drug Deliv. 2016, 23, 1773–1787. [Google Scholar] [CrossRef] [Green Version]
- Azzazy, H.M.E.; Fahmy, S.A.; Mahdy, N.K.; Meselhy, M.R.; Bakowsky, U. Chitosan-Coated PLGA Nanoparticles Loaded with Peganum harmala Alkaloids with Promising Antibacterial and Wound Healing Activities. Nanomaterials 2021, 11, 2438. [Google Scholar] [CrossRef]
- Boonsongrit, Y.; Mueller, B.W.; Mitrevej, A. Characterization of drug-chitosan interaction by 1H NMR, FTIR and isothermal titration calorimetry. Eur. J. Pharm. Biopharm. 2008, 69, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Chieng, B.W.; Ibrahim, N.; Yunus, W.; Hussein, M. Effects of Graphene Nanopletelets on Poly(Lactic Acid)/Poly(Ethylene Glycol) Polymer Nanocomposites. Polymers 2013, 6, 93–104. [Google Scholar] [CrossRef] [Green Version]
- Sarwar, M.; Ghaffar, A.; Islam, A.; Yasmeen, F.; Oluz, Z.; Cetindag, E.; Duran, H.; Qaiser, A. Controlled drug release behavior of metformin hydrogen chloride from biodegradable films based on chitosan/poly(ethylene glycol) methyl ether blend. Arab. J. Chem. 2017, 13, 393–403. [Google Scholar] [CrossRef]
- Rim, H.P.; Min, K.H.; Lee, H.J.; Jeong, S.Y.; Lee, S.C. pH-Tunable Calcium Phosphate Covered Mesoporous Silica Nanocontainers for Intracellular Controlled Release of Guest Drugs. Angew. Chem. Int. Ed. 2011, 50, 8853–8857. [Google Scholar] [CrossRef]
- Aydin, R.S.T.; Pulat, M. 5-Fluorouracil Encapsulated Chitosan Nanoparticles for pH-Stimulated Drug Delivery: Evaluation of Controlled Release Kinetics. J. Nanomater. 2012, 2012, 313961. [Google Scholar] [CrossRef]
- Agnihotri, S.A.; Mallikarjuna, N.N.; Aminabhavi, T.M. Recent advances on chitosan-based micro- and nanoparticles in drug delivery. J. Control. Release 2004, 100, 5–28. [Google Scholar] [CrossRef]
- Vijayakurup, V.; Thulasidasan, A.T.; Shankar, G.M.; Retnakumari, A.P.; Nandan, C.D.; Somaraj, J.; Antony, J.; Alex, V.V.; Vinod, B.S.; Liju, V.B.; et al. Chitosan Encapsulation Enhances the Bioavailability and Tissue Retention of Curcumin and Improves its Efficacy in Preventing B[a]P-induced Lung Carcinogenesis. Cancer Prev. Res. 2019, 12, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Ma, L.; Liu, Y.; Xu, F.; Nie, J.; Ma, G. Design and characterization of antitumor drug paclitaxel-loaded chitosan nanoparticles by W/O emulsions. Int. J. Biol. Macromol. 2012, 50, 438–443. [Google Scholar] [CrossRef]
- Xiao, B.; Ma, P.; Ma, L.; Chen, Q.; Si, X.; Walter, L.; Merlin, D. Effects of tripolyphosphate on cellular uptake and RNA interference efficiency of chitosan-based nanoparticles in Raw 264.7 macrophages. J. Colloid Interface Sci. 2017, 490, 520–528. [Google Scholar] [CrossRef] [Green Version]
- Parhi, P.; Mohanty, C.; Sahoo, S.K. Nanotechnology-based combinational drug delivery: An emerging approach for cancer therapy. Drug Discov. Today 2012, 17, 1044–1052. [Google Scholar] [CrossRef]
- Espey, M.G.; Chen, P.; Chalmers, B.; Drisko, J.; Sun, A.Y.; Levine, M.; Chen, Q. Pharmacologic ascorbate synergizes with gemcitabine in preclinical models of pancreatic cancer. Free Radic. Biol. Med. 2011, 50, 1610–1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilloteaux, J.; Jamison, J.M.; Neal, D.; Summers, J.L. Synergistic antitumor cytotoxic actions of ascorbate and menadione on human prostate (DU145) cancer cells in vitro: Nucleus and other injuries preceding cell death by autoschizis. Ultrastruct. Pathol. 2014, 38, 116–140. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.E.; Hahm, E.; Bae, S.; Kang, J.S.; Lee, W.J. The enhanced tumor inhibitory effects of gefitinib and L-ascorbic acid combination therapy in non-small cell lung cancer cells. Oncol. Lett. 2017, 14, 276–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Chapman, J.; Levine, M.; Polireddy, K.; Drisko, J.; Chen, Q. High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci. Transl. Med. 2014, 6, 222ra18. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Man, C.; Wang, H.; Lu, X.; Ma, Q.; Cai, Y.; Ma, W. PEGylated multi-walled carbon nanotubes for encapsulation and sustained release of oxaliplatin. Pharm. Res. 2013, 30, 412–423. [Google Scholar] [CrossRef]
- Crowley, L.C.; Marfell, B.J.; Scott, A.P.; Waterhouse, N.J. Quantitation of Apoptosis and Necrosis by Annexin V Binding, Propidium Iodide Uptake, and Flow Cytometry. Cold Spring Harb. Protoc. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Geske, F.J.; Nelson, A.C.; Lieberman, R.; Strange, R.; Sun, T.; Gerschenson, L.E. DNA repair is activated in early stages of p53-induced apoptosis. Cell Death Differ. 2000, 7, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Van der Mark, V.A.; Elferink, R.P.; Paulusma, C.C. P4 ATPases: Flippases in health and disease. Int. J. Mol. Sci. 2013, 14, 7897–7922. [Google Scholar] [CrossRef] [Green Version]
- Pires, A.S.; Marques, C.R.; Encarnacao, J.C.; Abrantes, A.M.; Mamede, A.C.; Laranjo, M.; Goncalves, A.C.; Sarmento-Ribeiro, A.B.; Botelho, M.F. Ascorbic acid and colon cancer: An oxidative stimulus to cell death depending on cell profile. Eur. J. Cell Biol. 2016, 95, 208–218. [Google Scholar] [CrossRef]
- Zanardelli, M.; Micheli, L.; Nicolai, R.; Failli, P.; Ghelardini, C.; Di Cesare Mannelli, L. Different apoptotic pathways activated by oxaliplatin in primary astrocytes vs. colo-rectal cancer cells. Int. J. Mol. Sci. 2015, 16, 5386–5399. [Google Scholar] [CrossRef] [Green Version]
- Igney, F.H.; Krammer, P.H. Death and anti-death: Tumour resistance to apoptosis. Nat. Rev. Cancer 2002, 2, 277–288. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Diameter (nm) | PDI | ζ-Potential (mV) | EE(%) | |
|---|---|---|---|---|---|
| AA | OX | ||||
| CS NPs | 290.30 ± 7.45 | 0.41 ± 0.009 | +27.60 ± 1.48 | - | - |
| AA/CS NPs | 157.20 ± 2.40 | 0.31 ± 0.04 | +22.02 ± 1.50 | 75.49 ± 1.94 | - |
| OX/CS NPs | 188.10 ± 9.70 | 0.25 ± 0.06 | +22.58 ± 1.85 | - | 78.73 ± 2.17 |
| AA-OX/CS NPs | 261.10 ± 9.19 | 0.43 ± 0.03 | +40.40 ± 2.71 | 77.52 ± 2.35 | 79.25 ± 3.93 |
| Samples | Diameter (nm) | PDI | ζ-Potential (mV) | EE (%) | |
|---|---|---|---|---|---|
| AA | OX | ||||
| PEG-CS NPs | 273.30 ± 2.69 | 0.30 ± 0.07 | +15.73 ± 0.98 | - | - |
| AA/PEG-CS NPS | 152.20 ± 2.40 | 0.31 ± 0.04 | +21.84 ± 1.54 | 90.92 ± 1.19 | - |
| OX/PEG-CS NPs | 156.60 ± 4.82 | 0.24 ± 0.03 | +21.31 ± 1.78 | - | 94.11 ± 1.98 |
| AA-OX/PEG CS NPs | 176.00 ± 4.21 | 0.26 ± 0.04 | +28.23 ± 0.93 | 91.84 ± 1.03 | 95.30 ± 1.49 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fahmy, S.A.; Ramzy, A.; Mandour, A.A.; Nasr, S.; Abdelnaser, A.; Bakowsky, U.; Azzazy, H.M.E.-S. PEGylated Chitosan Nanoparticles Encapsulating Ascorbic Acid and Oxaliplatin Exhibit Dramatic Apoptotic Effects against Breast Cancer Cells. Pharmaceutics 2022, 14, 407. https://doi.org/10.3390/pharmaceutics14020407
Fahmy SA, Ramzy A, Mandour AA, Nasr S, Abdelnaser A, Bakowsky U, Azzazy HME-S. PEGylated Chitosan Nanoparticles Encapsulating Ascorbic Acid and Oxaliplatin Exhibit Dramatic Apoptotic Effects against Breast Cancer Cells. Pharmaceutics. 2022; 14(2):407. https://doi.org/10.3390/pharmaceutics14020407
Chicago/Turabian StyleFahmy, Sherif Ashraf, Asmaa Ramzy, Asmaa A. Mandour, Soad Nasr, Anwar Abdelnaser, Udo Bakowsky, and Hassan Mohamed El-Said Azzazy. 2022. "PEGylated Chitosan Nanoparticles Encapsulating Ascorbic Acid and Oxaliplatin Exhibit Dramatic Apoptotic Effects against Breast Cancer Cells" Pharmaceutics 14, no. 2: 407. https://doi.org/10.3390/pharmaceutics14020407