
Design, Synthesis and In Vitro Investigation of Cabozantinib-Based PROTACs to Target c-Met Kinase
 ,
,  , , , , and
, , , , and
Abstract
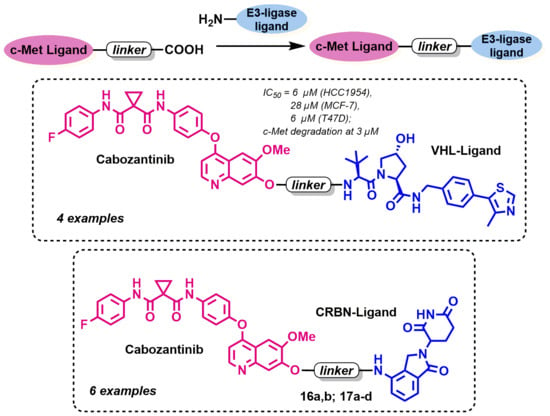
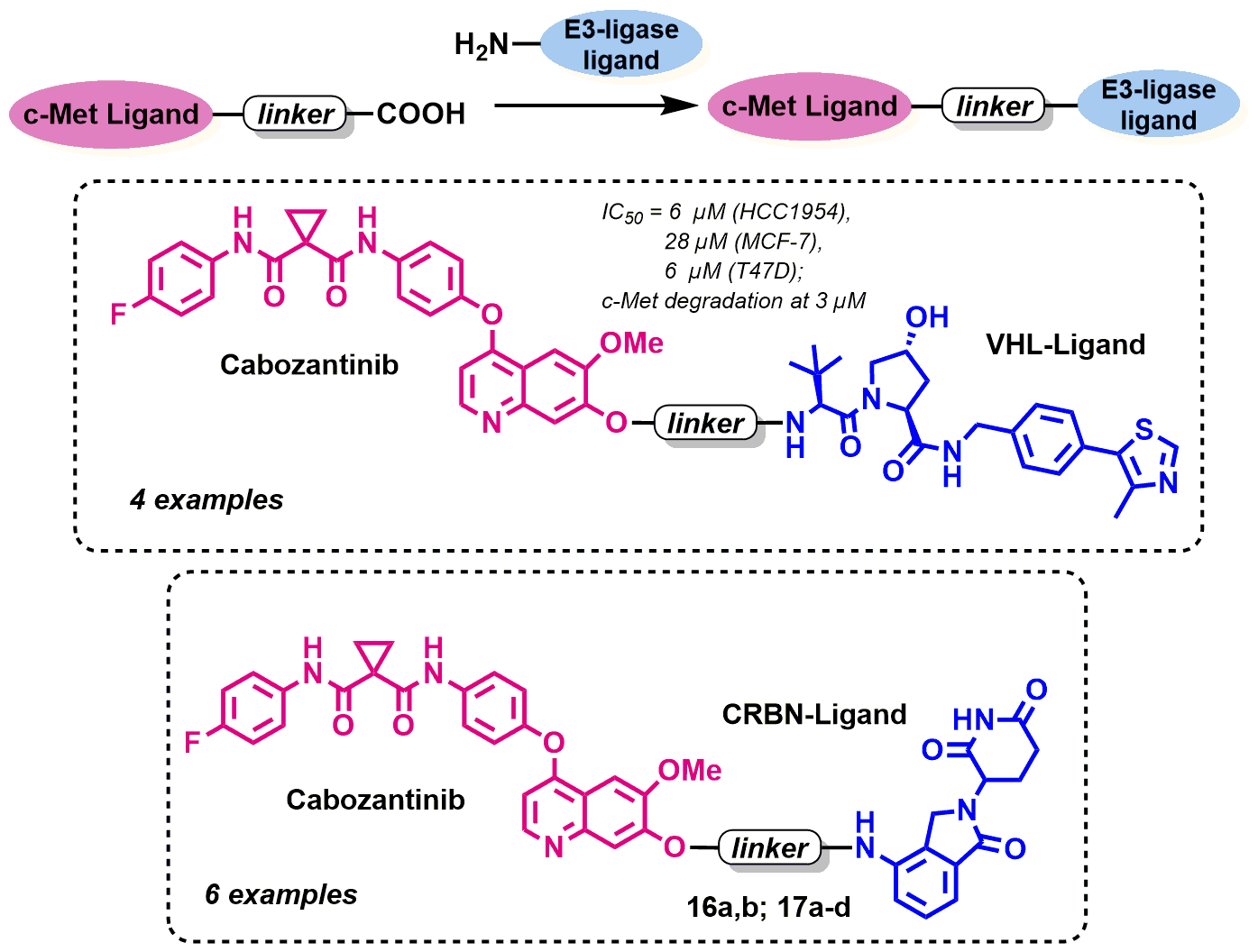
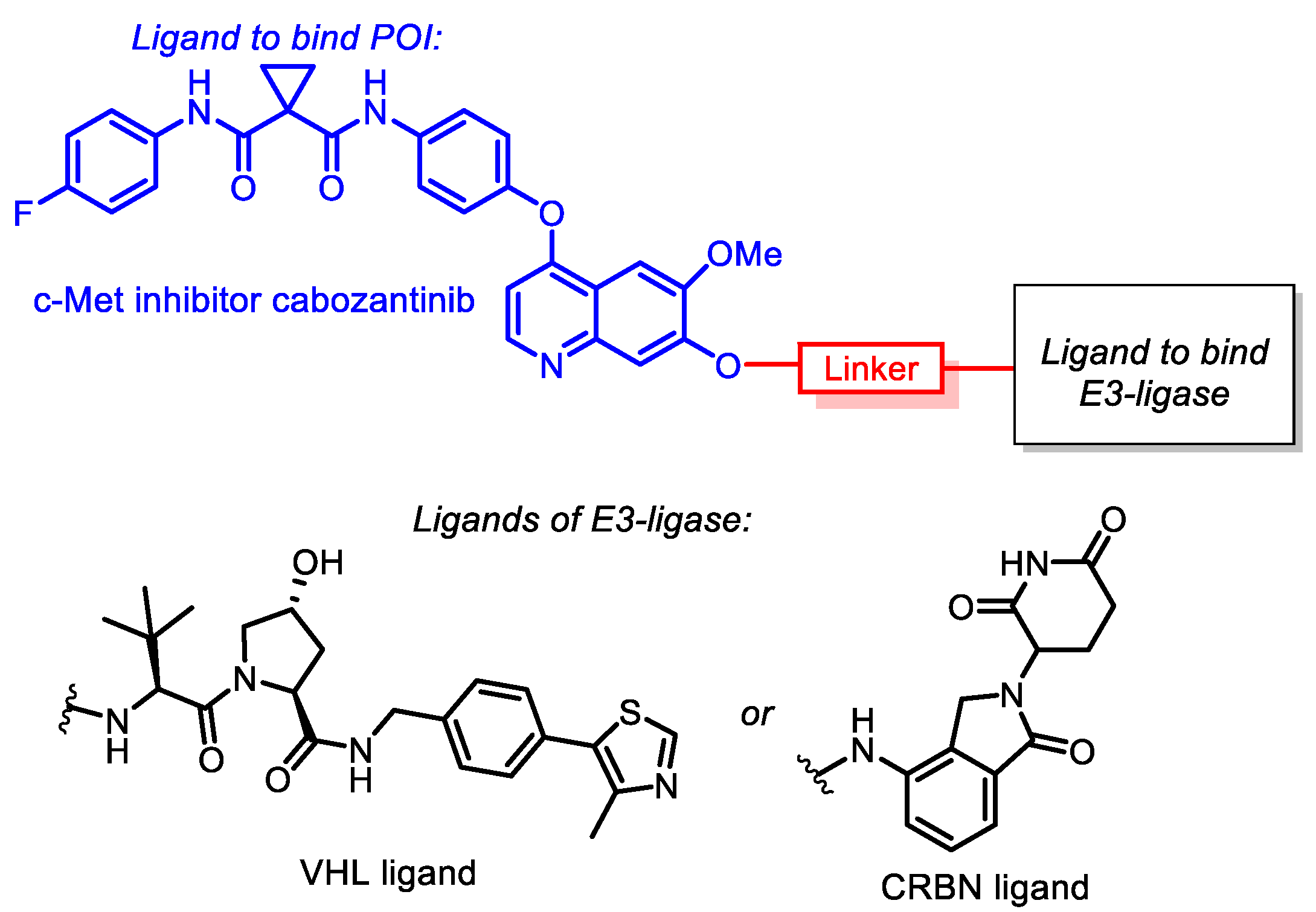
:
1. Introduction
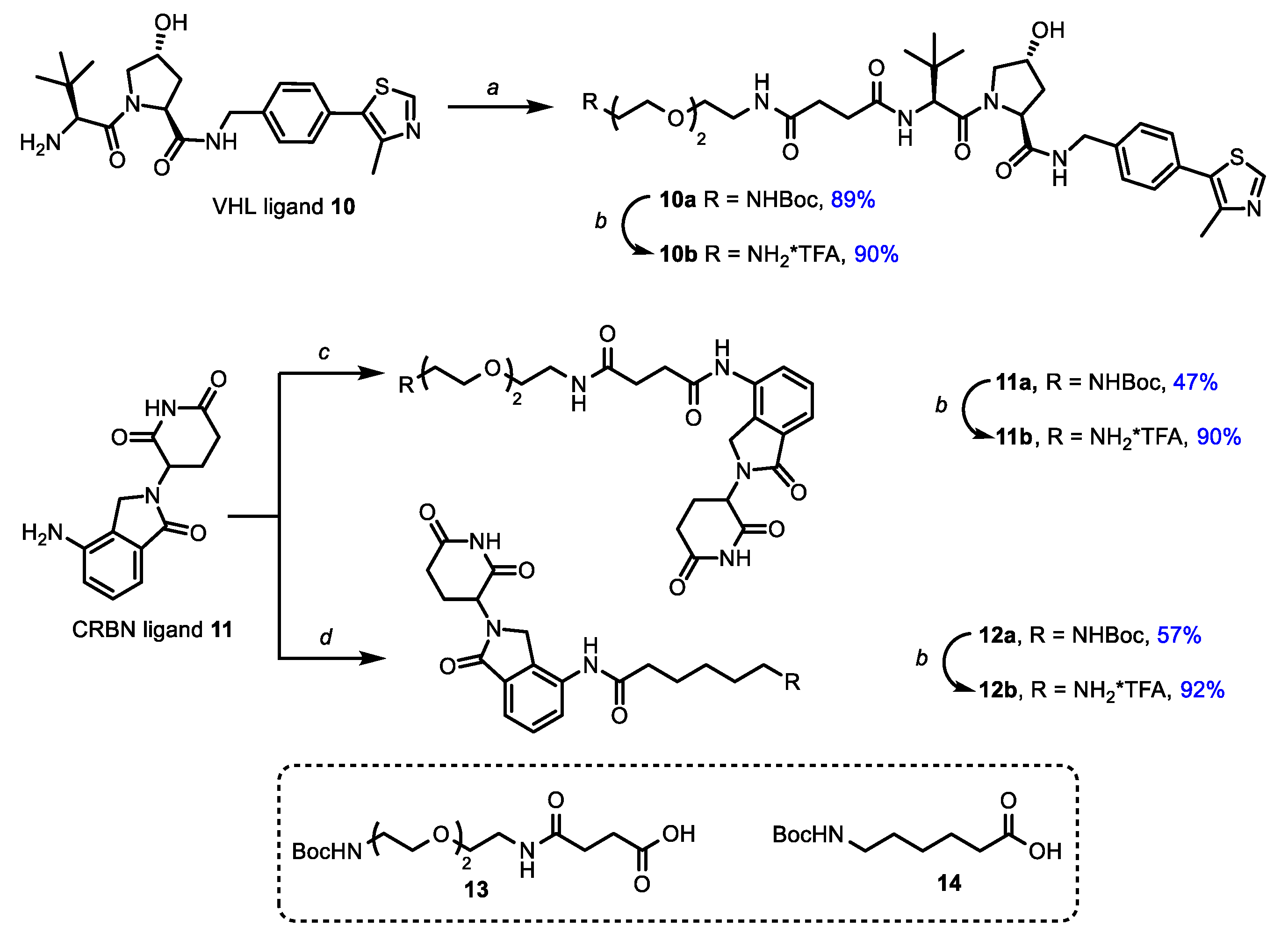
2. Materials and Methods
2.1. Chemistry
2.2. Biological Research
2.2.1. Cell Culture Screening and Assessment of the Antiproliferative Effects
2.2.2. Immunoblotting
3. Results and Discussion
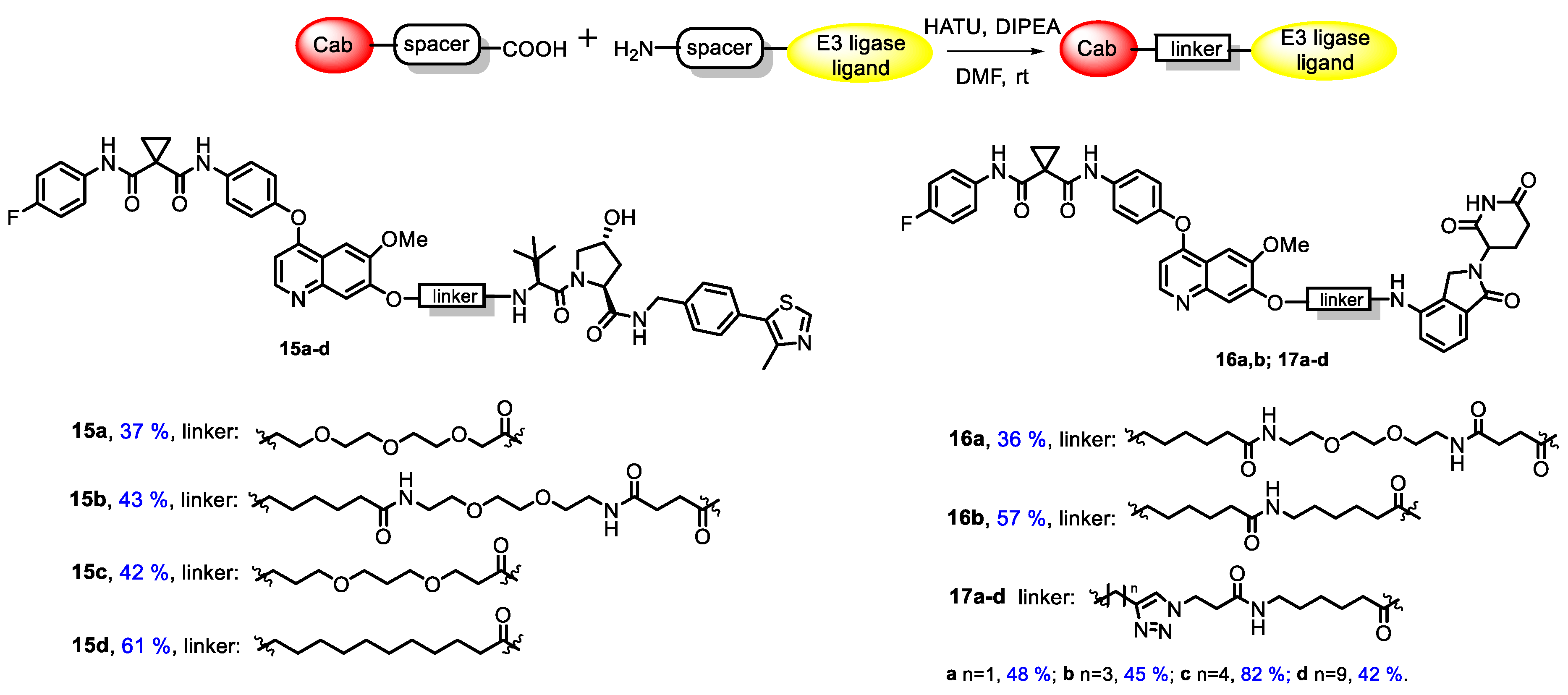
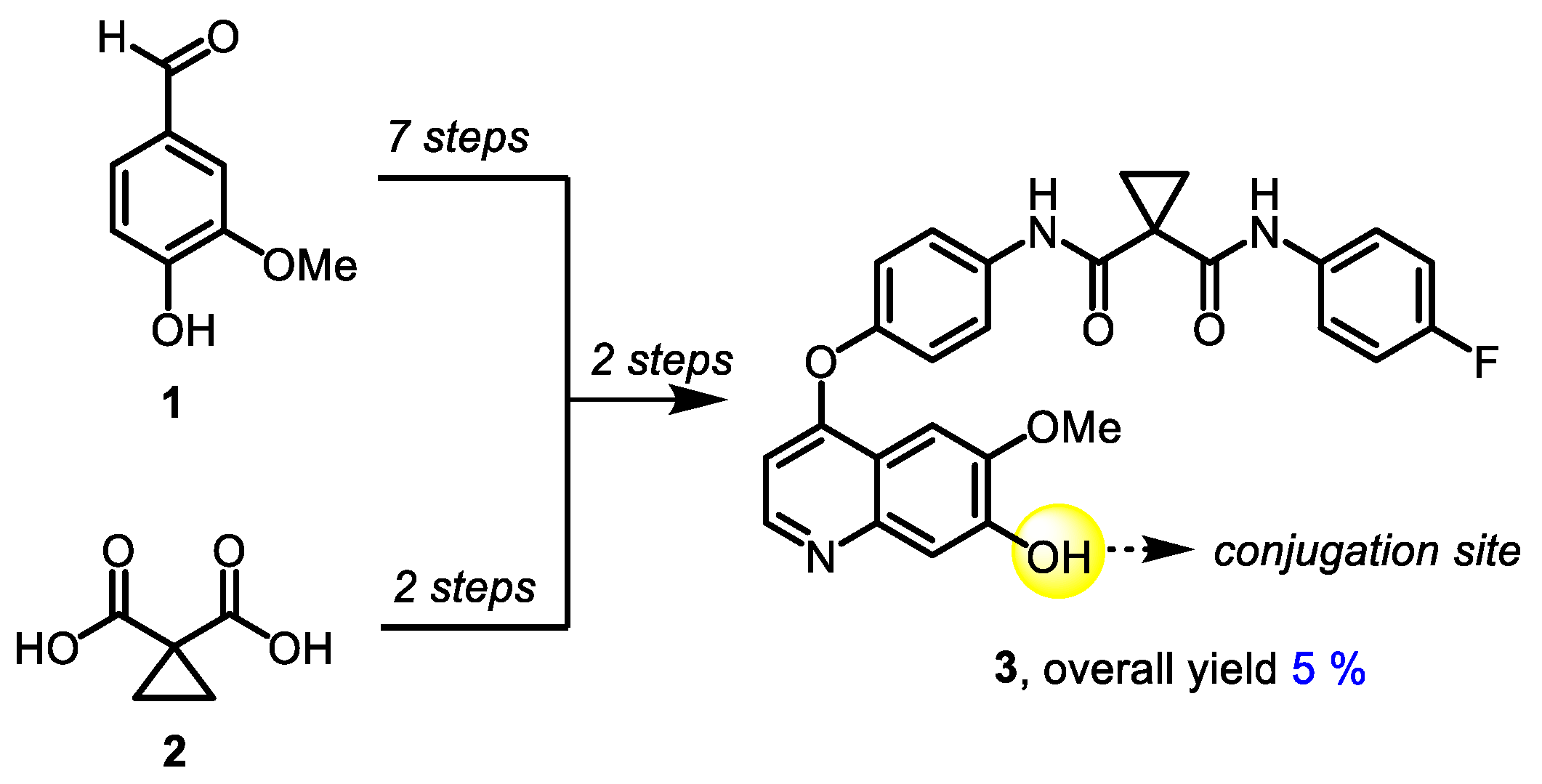
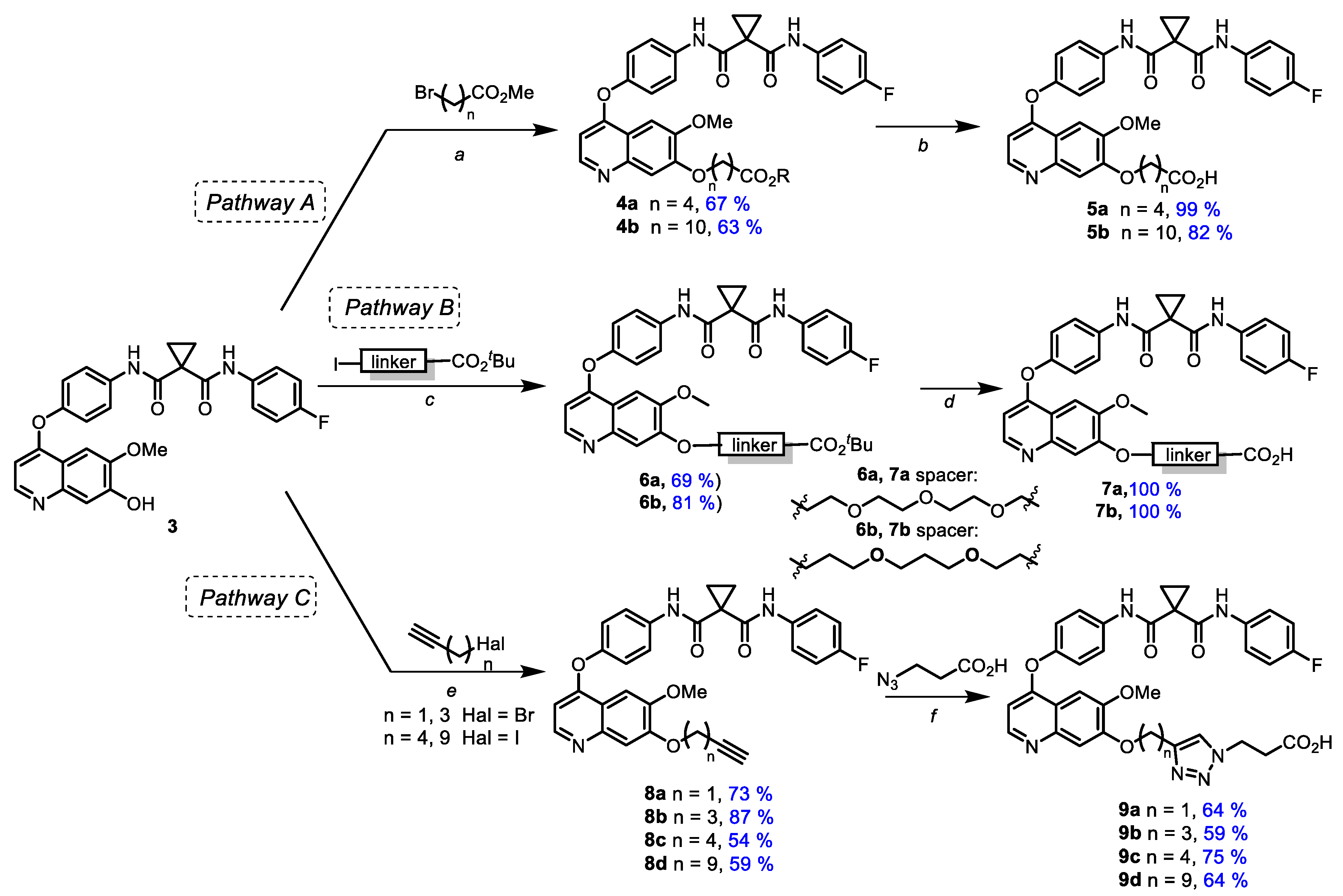
3.1. Chemistry
3.2. Biology
3.2.1. Cancer Cell Line Screening and Antiproliferative Evaluation
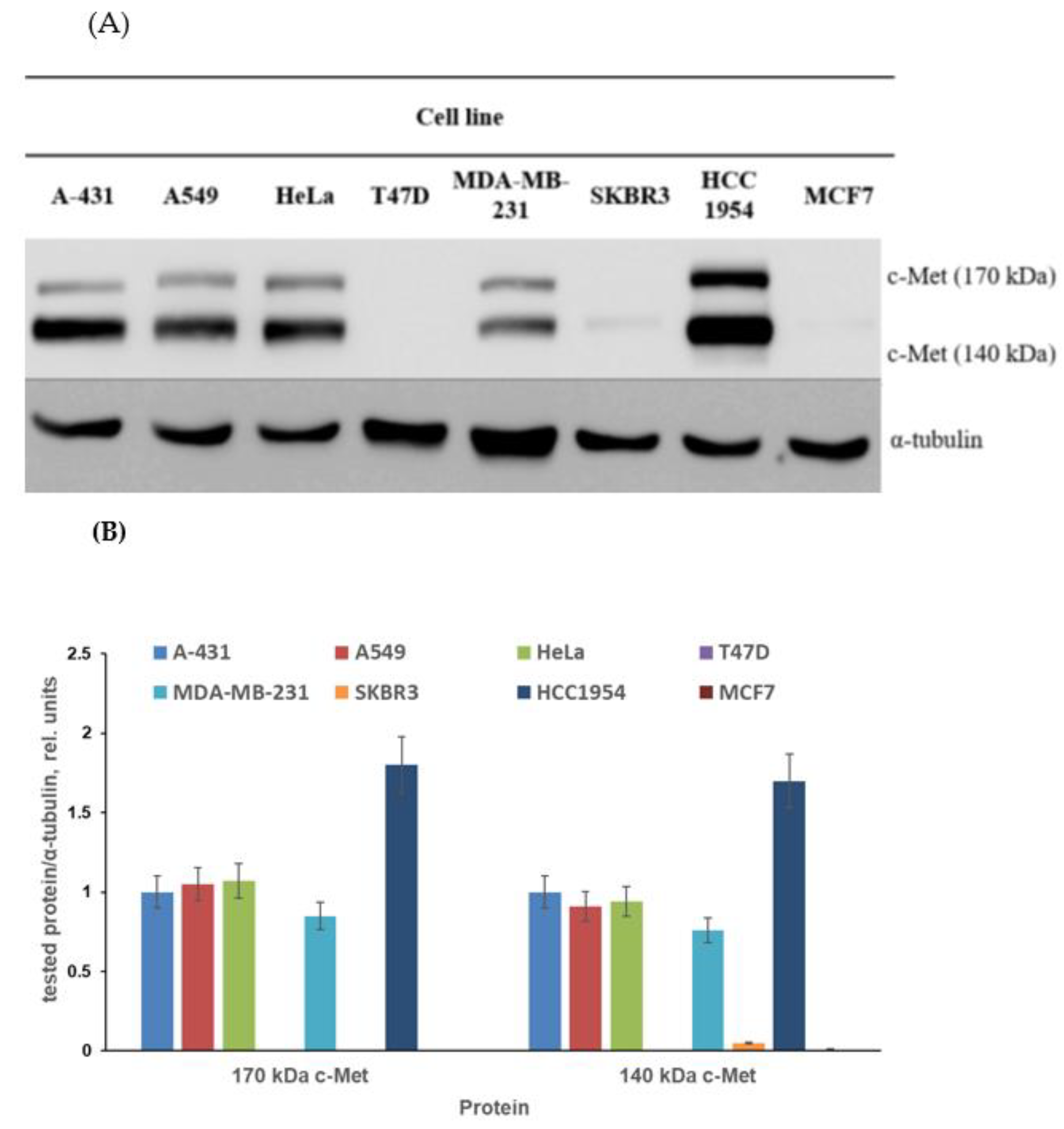
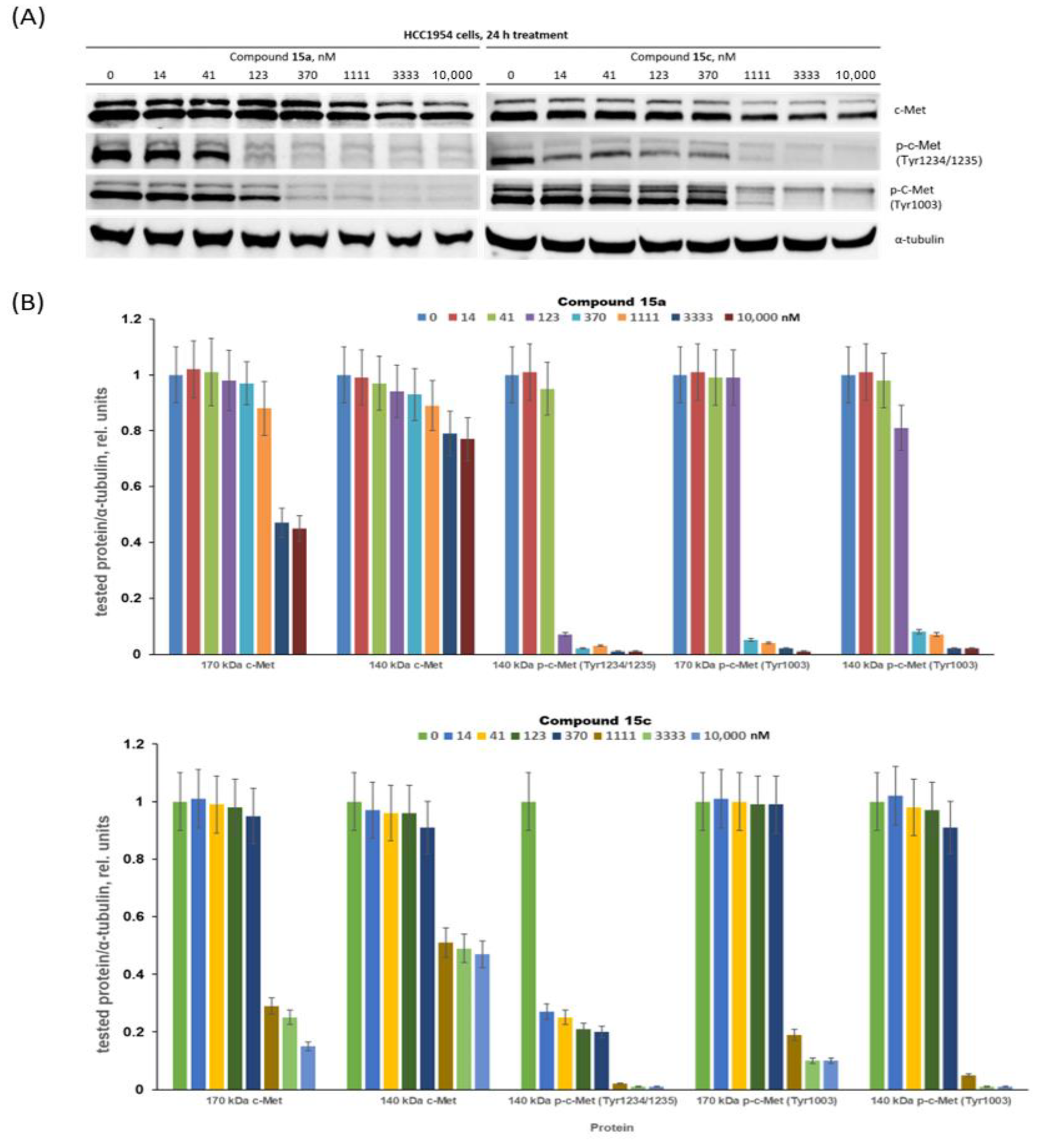
3.2.2. Assessment of c-Met Inhibition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Garber, K. The PROTAC Gold Rush. Nat. Biotechnol. 2022, 40, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric Molecules That Target Proteins to the Skp1–Cullin–F Box Complex for Ubiquitination and Degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC Targeted Protein Degraders: The Past Is Prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Benowitz, A.B.; Jones, K.L.; Harling, J.D. The Therapeutic Potential of PROTACs. Expert Opin. Ther. Pat. 2021, 31, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, X.; Lv, D.; Yuan, Y.; Zheng, G.; Zhou, D. Assays and Technologies for Developing Proteolysis Targeting Chimera Degraders. Future Med. Chem. 2020, 12, 1155–1179. [Google Scholar] [CrossRef]
- Zou, Y.; Ma, D.; Wang, Y. The PROTAC Technology in Drug Development. Cell Biochem. Funct. 2019, 37, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Smith, B.E.; Wang, S.L.; Jaime-Figueroa, S.; Harbin, A.; Wang, J.; Hamman, B.D.; Crews, C.M. Differential PROTAC Substrate Specificity Dictated by Orientation of Recruited E3 Ligase. Nat. Commun. 2019, 10, 131. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Gao, H.; Yang, Y.; He, M.; Wu, Y.; Song, Y.; Tong, Y.; Rao, Y. PROTACs: Great Opportunities for Academia and Industry. Signal Transduct. Target Ther. 2019, 4, 64. [Google Scholar] [CrossRef] [Green Version]
- Remillard, D.; Buckley, D.L.; Paulk, J.; Brien, G.L.; Sonnett, M.; Seo, H.-S.; Dastjerdi, S.; Wühr, M.; Dhe-Paganon, S.; Armstrong, S.A.; et al. Degradation of the BAF Complex Factor BRD9 by Heterobifunctional Ligands. Angew. Chem. Int. Ed. 2017, 56, 5738–5743. [Google Scholar] [CrossRef] [Green Version]
- Hines, J.; Lartigue, S.; Dong, H.; Qian, Y.; Crews, C.M. MDM2-Recruiting PROTAC Offers Superior, Synergistic Antiproliferative Activity via Simultaneous Degradation of BRD4 and Stabilization of P53. Cancer Res. 2019, 79, 251–262. [Google Scholar] [CrossRef]
- Zoppi, V.; Hughes, S.J.; Maniaci, C.; Testa, A.; Gmaschitz, T.; Wieshofer, C.; Koegl, M.; Riching, K.M.; Daniels, D.L.; Spallarossa, A.; et al. Iterative Design and Optimization of Initially Inactive Proteolysis Targeting Chimeras (PROTACs) Identify VZ185 as a Potent, Fast, and Selective von Hippel–Lindau (VHL) Based Dual Degrader Probe of BRD9 and BRD7. J. Med. Chem. 2019, 62, 699–726. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Wang, W.-L.; Yang, Y.-Y.; Hu, X.-T.; Wang, Q.-W.; Zuo, W.-Q.; Xu, Y.; Feng, Q.; Wang, N.-Y. Identification of a Selective BRD4 PROTAC with Potent Antiproliferative Effects in AR-Positive Prostate Cancer Based on a Dual BET/PLK1 Inhibitor. Eur. J. Med. Chem. 2022, 227, 113922. [Google Scholar] [CrossRef] [PubMed]
- Gechijian, L.N.; Buckley, D.L.; Lawlor, M.A.; Reyes, J.M.; Paulk, J.; Ott, C.J.; Winter, G.E.; Erb, M.A.; Scott, T.G.; Xu, M.; et al. Functional TRIM24 Degrader via Conjugation of Ineffectual Bromodomain and VHL Ligands. Nat. Chem. Biol. 2018, 14, 405–412. [Google Scholar] [CrossRef]
- Wang, X.; Feng, S.; Fan, J.; Li, X.; Wen, Q.; Luo, N. New Strategy for Renal Fibrosis: Targeting Smad3 Proteins for Ubiquitination and Degradation. Biochem. Pharmacol. 2016, 116, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.C.; Nandi, G.; Donovan, K.A.; Cai, Q.; Berry, B.C.; Nowak, R.P.; Fischer, E.S.; Gray, N.S.; Ferguson, F.M.; Haggarty, S.J. Discovery and Optimization of Tau Targeted Protein Degraders Enabled by Patient Induced Pluripotent Stem Cells-Derived Neuronal Models of Tauopathy. Front. Cell. Neurosci. 2022, 16, 801179. [Google Scholar] [CrossRef] [PubMed]
- Tomoshige, S.; Nomura, S.; Ohgane, K.; Hashimoto, Y.; Ishikawa, M. Discovery of Small Molecules That Induce the Degradation of Huntingtin. Angew. Chem. Int. Ed. 2017, 56, 11530–11533. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Jin, W.Y.; Lu, J.; Wang, J.; Wang, Y.T. Rapid and Reversible Knockdown of Endogenous Proteins by Peptide-Directed Lysosomal Degradation. Nat. Neurosci. 2014, 17, 471–480. [Google Scholar] [CrossRef] [Green Version]
- Kargbo, R.B. PROTAC Compounds Targeting α-Synuclein Protein for Treating Neurogenerative Disorders: Alzheimer’s and Parkinson’s Diseases. ACS Med. Chem. Lett. 2020, 11, 1086–1087. [Google Scholar] [CrossRef]
- Bondeson, D.P.; Mares, A.; Smith, I.E.D.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L.; et al. Catalytic in Vivo Protein Knockdown by Small-Molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. [Google Scholar] [CrossRef] [Green Version]
- Itoh, Y.; Kitaguchi, R.; Ishikawa, M.; Naito, M.; Hashimoto, Y. Design, Synthesis and Biological Evaluation of Nuclear Receptor-Degradation Inducers. Bioorg. Med. Chem. 2011, 19, 6768–6778. [Google Scholar] [CrossRef]
- Zhao, Q.; Lan, T.; Su, S.; Rao, Y. Induction of Apoptosis in MDA-MB-231 Breast Cancer Cells by a PARP1-Targeting PROTAC Small Molecule. Chem. Commun. 2019, 55, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Han, L.; Mao, S.; Xu, P.; Xu, X.; Zhao, R.; Wu, Z.; Zhong, K.; Yu, G.; Wang, X. Discovery of A031 as Effective Proteolysis Targeting Chimera (PROTAC) Androgen Receptor (AR) Degrader for the Treatment of Prostate Cancer. Eur. J. Med. Chem. 2021, 216, 113307. [Google Scholar] [CrossRef]
- Papatzimas, J.W.; Gorobets, E.; Maity, R.; Muniyat, M.I.; MacCallum, J.L.; Neri, P.; Bahlis, N.J.; Derksen, D.J. From Inhibition to Degradation: Targeting the Antiapoptotic Protein Myeloid Cell Leukemia 1 (MCL1). J. Med. Chem. 2019, 62, 5522–5540. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Thummuri, D.; He, Y.; Liu, X.; Zhang, P.; Zhou, D.; Zheng, G. Utilizing PROTAC Technology to Address the On-Target Platelet Toxicity Associated with Inhibition of BCL-XL. Chem. Commun. 2019, 55, 14765–14768. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; He, N.; Guo, Z.; Niu, C.; Song, T.; Guo, Y.; Cao, K.; Wang, A.; Zhu, J.; Zhang, X.; et al. Proteolysis Targeting Chimeras for the Selective Degradation of Mcl-1/Bcl-2 Derived from Nonselective Target Binding Ligands. J. Med. Chem. 2019, 62, 8152–8163. [Google Scholar] [CrossRef] [PubMed]
- Zengerle, M.; Chan, K.-H.; Ciulli, A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Phthalimide Conjugation as a Strategy for in Vivo Target Protein Degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef] [Green Version]
- Schneekloth, A.R.; Pucheault, M.; Tae, H.S.; Crews, C.M. Targeted Intracellular Protein Degradation Induced by a Small Molecule: En Route to Chemical Proteomics. Bioorg. Med. Chem. Lett. 2008, 18, 5904–5908. [Google Scholar] [CrossRef] [Green Version]
- Itoh, Y.; Ishikawa, M.; Naito, M.; Hashimoto, Y. Protein Knockdown Using Methyl Bestatin—Ligand Hybrid Molecules: Design and Synthesis of Inducers of Ubiquitination-Mediated Degradation of Cellular Retinoic Acid-Binding Proteins. J. Am. Chem. Soc. 2010, 132, 5820–5826. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, Y.; Shi, L.; Yang, S.; Chang, J.; Zhong, Y.; Li, Q.; Xing, D. Recent Advances in IAP-Based PROTACs (SNIPERs) as Potential Therapeutic Agents. J. Enzym. Inhib. Med. Chem. 2022, 37, 1437–1453. [Google Scholar] [CrossRef]
- Humphreys, L.M.; Smith, P.; Chen, Z.; Fouad, S.; D’Angiolella, V. The Role of E3 Ubiquitin Ligases in the Development and Progression of Glioblastoma. Cell Death Differ. 2021, 28, 522–537. [Google Scholar] [CrossRef] [PubMed]
- Maniaci, C.; Hughes, S.J.; Testa, A.; Chen, W.; Lamont, D.J.; Rocha, S.; Alessi, D.R.; Romeo, R.; Ciulli, A. Homo-PROTACs: Bivalent Small-Molecule Dimerizers of the VHL E3 Ubiquitin Ligase to Induce Self-Degradation. Nat. Commun. 2017, 8, 830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyers, M.; Rottapel, R. VHL: A Very Hip Ligase. Proc. Natl. Acad. Sci. USA 1999, 96, 12230–12232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Li, T.; Miao, Z.; Wang, P.; Sheng, C.; Zhuang, C. Homobivalent, Trivalent, and Covalent PROTACs: Emerging Strategies for Protein Degradation. J. Med. Chem. 2022, 65, 8798–8827. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Liu, T.; Jiao, Q.; Ji, J.; Tao, M.; Liu, Y.; You, Q.; Jiang, Z. Discovery of a Keap1-Dependent Peptide PROTAC to Knockdown Tau by Ubiquitination-Proteasome Degradation Pathway. Eur. J. Med. Chem. 2018, 146, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wu, Z.; Chen, P.; Zhang, J.; Wang, T.; Zhou, J.; Zhang, H. Discovery of a New Class of PROTAC BRD4 Degraders Based on a Dihydroquinazolinone Derivative and Lenalidomide/Pomalidomide. Bioorg. Med. Chem. 2020, 28, 115228. [Google Scholar] [CrossRef] [PubMed]
- Kannt, A.; Đikić, I. Expanding the Arsenal of E3 Ubiquitin Ligases for Proximity-Induced Protein Degradation. Cell Chem. Biol. 2021, 28, 1014–1031. [Google Scholar] [CrossRef]
- Cyrus, K.; Wehenkel, M.; Choi, E.-Y.; Han, H.-J.; Lee, H.; Swanson, H.; Kim, K.-B. Impact of Linker Length on the Activity of PROTACs. Mol. BioSyst. 2011, 7, 359–364. [Google Scholar] [CrossRef] [Green Version]
- Troup, R.I.; Fallan, C.; Baud, M.G.J. Current Strategies for the Design of PROTAC Linkers: A Critical Review. Explor. Target. Anti Tumor Ther. 2020, 1, 273–312. [Google Scholar] [CrossRef]
- Bemis, T.A.; La Clair, J.J.; Burkart, M.D. Unraveling the Role of Linker Design in Proteolysis Targeting Chimeras. J. Med. Chem. 2021, 64, 8042–8052. [Google Scholar] [CrossRef]
- Klein, V.G.; Bond, A.G.; Craigon, C.; Lokey, R.S.; Ciulli, A. Amide-to-Ester Substitution as a Strategy for Optimizing PROTAC Permeability and Cellular Activity. J. Med. Chem. 2021, 64, 18082–18101. [Google Scholar] [CrossRef] [PubMed]
- Goracci, L.; Desantis, J.; Valeri, A.; Castellani, B.; Eleuteri, M.; Cruciani, G. Understanding the Metabolism of Proteolysis Targeting Chimeras (PROTACs): The Next Step toward Pharmaceutical Applications. J. Med. Chem. 2020, 63, 11615–11638. [Google Scholar] [CrossRef] [PubMed]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a Novel MET and VEGFR2 Inhibitor, Simultaneously Suppresses Metastasis, Angiogenesis, and Tumor Growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef] [Green Version]
- Cochin, V.; Gross-Goupil, M.; Ravaud, A.; Godbert, Y.; Le Moulec, S. Cabozantinib: Modalités d’action, Efficacité et Indications. Bull. Cancer 2017, 104, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Bouattour, M.; Raymond, E.; Qin, S.; Cheng, A.-L.; Stammberger, U.; Locatelli, G.; Faivre, S. Recent Developments of c-Met as a Therapeutic Target in Hepatocellular Carcinoma. Hepatology 2018, 67, 1132–1149. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y. Prognostic Value of c-Met in Colorectal Cancer: A Meta-Analysis. World J. Gastroenterol. 2015, 21, 3706. [Google Scholar] [CrossRef] [PubMed]
- Pothula, S.P.; Xu, Z.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Targeting HGF/c-Met Axis in Pancreatic Cancer. Int. J. Mol. Sci. 2020, 21, 9170. [Google Scholar] [CrossRef]
- Scagliotti, G.V.; Novello, S.; von Pawel, J. The Emerging Role of MET/HGF Inhibitors in Oncology. Cancer Treat. Rev. 2013, 39, 793–801. [Google Scholar] [CrossRef]
- Lacy, S.A.; Miles, D.R.; Nguyen, L.T. Clinical Pharmacokinetics and Pharmacodynamics of Cabozantinib. Clin. Pharmacokinet. 2017, 56, 477–491. [Google Scholar] [CrossRef]
- Markowitz, J.N.; Fancher, K.M. Cabozantinib: A Multitargeted Oral Tyrosine Kinase Inhibitor. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2018, 38, 357–369. [Google Scholar] [CrossRef]
- Schmidinger, M.; Danesi, R. Management of Adverse Events Associated with Cabozantinib Therapy in Renal Cell Carcinoma. Oncologist 2018, 23, 306–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lien, V.T.; Pettersen, S.; Haugen, M.H.; Olberg, D.E.; Mælandsmo, G.M.; Klaveness, J. Design, Synthesis and Biological Evaluation of 6-substituted Quinolines Derived from Cabozantinib as c-Met Inhibitors. Arch. Pharm. 2019, 352, 1900101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinebach, C.; Kehm, H.; Lindner, S.; Vu, L.P.; Köpff, S.; López Mármol, Á.; Weiler, C.; Wagner, K.G.; Reichenzeller, M.; Krönke, J.; et al. PROTAC-Mediated Crosstalk between E3 Ligases. Chem. Commun. 2019, 55, 1821–1824. [Google Scholar] [CrossRef]
- Zheng, G.; Zhou, D.; Zhang, X.; Wang, Y.; Chang, J. Compounds That Induce Degradation of Anti-Apoptotic bcl-2 Family Proteins and the Uses Thereof. Patent WO2017184995, 26 October 2017. [Google Scholar]
- Iselt, M.; Holtei, W.; Hilgard, P. The Tetrazolium Dye Assay for Rapid in Vitro Assessment of Cytotoxicity. Arzneimittelforschung 1989, 39, 747–749. [Google Scholar]
- Volkova, Y.A.; Antonov, Y.S.; Komkov, A.V.; Scherbakov, A.M.; Shashkov, A.S.; Menchikov, L.G.; Chernoburova, E.I.; Zavarzin, I.V. Access to Steroidal Pyridazines via Modified Thiohydrazides. RSC Adv. 2016, 6, 42863–42868. [Google Scholar] [CrossRef]
- Scherbakov, A.M.; Lobanova, Y.S.; Shatskaya, V.A.; Onopchenko, O.V.; Gershtein, E.S.; Krasil’nikov, M.A. Activation of Mitogenic Pathways and Sensitization to Estrogen-Induced Apoptosis: Two Independent Characteristics of Tamoxifen-Resistant Breast Cancer Cells? Breast Cancer Res. Treat. 2006, 100, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Mruk, D.D.; Cheng, C.Y. Enhanced Chemiluminescence (ECL) for Routine Immunoblotting. Spermatogenesis 2011, 1, 121–122. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Zhuo, L.-S.; Liu, P.-F.; Fang, W.-R.; Li, Y.-M.; Huang, W. Discovery of 1,6-Naphthyridinone-Based MET Kinase Inhibitor Bearing Quinoline Moiety as Promising Antitumor Drug Candidate. Eur. J. Med. Chem. 2020, 192, 112174. [Google Scholar] [CrossRef]
- Forsyth, T.P.; Mac, M.B.; Leahy, J.W.; Nuss, J.M.; Xu, W. c-Met Modulators and Methods of Use. U.S. Patent 2007054928-A1, 26 September 2003. [Google Scholar]
- Gore, V.G.; Shukla, V.K.; Bhandari, S.S.; Hasbe, S. Process for the Preparation of Lenalidomide. Patent US8946265B2, 3 February 2015. [Google Scholar]
- Sawada, K.; Radjabi, A.R.; Shinomiya, N.; Kistner, E.; Kenny, H.; Becker, A.R.; Turkyilmaz, M.A.; Salgia, R.; Yamada, S.D.; Vande Woude, G.F.; et al. c-Met Overexpression Is a Prognostic Factor in Ovarian Cancer and an Effective Target for Inhibition of Peritoneal Dissemination and Invasion. Cancer Res. 2007, 67, 1670–1679. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Du, R.; Jiang, S.; Wu, C.; Barkauskas, D.S.; Richey, J.; Molter, J.; Lam, M.; Flask, C.; Gerson, S.; et al. Dual MET–EGFR Combinatorial Inhibition against T790M-EGFR-Mediated Erlotinib-Resistant Lung Cancer. Br. J. Cancer 2008, 99, 911–922. [Google Scholar] [CrossRef]
- Mueller, K.L.; Yang, Z.-Q.; Haddad, R.; Ethier, S.P.; Boerner, J.L. EGFR/Met Association Regulates EGFR TKI Resistance in Breast Cancer. J. Mol. Signal. 2010, 5, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, T.R.; Fridlyand, J.; Yan, Y.; Penuel, E.; Burton, L.; Chan, E.; Peng, J.; Lin, E.; Wang, Y.; Sosman, J.; et al. Widespread Potential for Growth-Factor-Driven Resistance to Anticancer Kinase Inhibitors. Nature 2012, 487, 505–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.-J.; Wu, Y.; Hou, W.-H.; Wang, Y.-X.; Yuan, Q.-Y.; Wang, H.-J.; Yu, M. A Novel Bispecific c-Met/PD-1 Antibody with Therapeutic Potential in Solid Cancer. Oncotarget 2017, 8, 29067–29079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Guo, Q.; Zhang, C.; Huang, Z.; Wang, T.; Wang, X.; Wang, X.; Xu, G.; Liu, Y.; Yang, S.; et al. Discovery of a Highly Potent, Selective and Novel CDK9 Inhibitor as an Anticancer Drug Candidate. Bioorg. Med. Chem. Lett. 2017, 27, 3231–3237. [Google Scholar] [CrossRef]
- Taylor, S.C.; Berkelman, T.; Yadav, G.; Hammond, M. A Defined Methodology for Reliable Quantification of Western Blot Data. Mol. Biotechnol. 2013, 55, 217–226. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Li, Y.; Bhattacharya, A.; Zhang, Y. A Recombinant Human Protein Targeting HER2 Overcomes Drug Resistance in HER2-Positive Breast Cancer. Sci. Transl. Med. 2019, 11, eaav1620. [Google Scholar] [CrossRef]
- Organ, S.L.; Tsao, M.-S. An Overview of the c-Met Signaling Pathway. Ther. Adv. Med. Oncol. 2011, 3, S7–S19. [Google Scholar] [CrossRef] [Green Version]
- Yao, H.-P.; Tong, X.-M.; Wang, M.-H. Oncogenic Mechanism-Based Pharmaceutical Validation of Therapeutics Targeting MET Receptor Tyrosine Kinase. Ther. Adv. Med. Oncol. 2021, 13, 175883592110069. [Google Scholar] [CrossRef]
- Peschard, P.; Fournier, T.M.; Lamorte, L.; Naujokas, M.A.; Band, H.; Langdon, W.Y.; Park, M. Mutation of the C-Cbl TKB Domain Binding Site on the Met Receptor Tyrosine Kinase Converts It into a Transforming Protein. Mol. Cell 2001, 8, 995–1004. [Google Scholar] [CrossRef]
- Petrelli, A.; Gilestro, G.F.; Lanzardo, S.; Comoglio, P.M.; Migone, N.; Giordano, S. The Endophilin–CIN85–Cbl Complex Mediates Ligand-Dependent Downregulation of c-Met. Nature 2002, 416, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Bannen, L.C.; Chan, D.S.-M.; Forsyth, T.P.; Khoury, R.G.; Leahy, J.W.; Mac, M.B.; Mann, L.W.; Nuss, J.M.; Parks, J.J.; Wang, Y.; et al. c-Met Modulators and Methods of Use. Patent US7579473B2, 25 August 2009. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | |||||
|---|---|---|---|---|---|
| Compound | A-431 | T47D | MCF7 | HCC1954 | SKBR3 |
| c-Met Status | ++ | - | - | +++ | - |
| 15a | >25 | 6.1 | 28.1 | 5.7 | >25 |
| 15b | >25 | 14.2 | >25 | 9.1 | >25 |
| 15c | >25 | >25 | >50 | 6.7 | 10.8 |
| 15d | >25 | >25 | >50 | 20.3 | >25 |
| 16a | 12.3 | 8.2 | 19.4 | >25 | >25 |
| 16b | >25 | 3.8 | >25 | >25 | >25 |
| 17a | >25 | >25 | >25 | >25 | >25 |
| 17b | >25 | >25 | >25 | >25 | >25 |
| 17c | >25 | >25 | >25 | >25 | >25 |
| 17d | >25 | >25 | >25 | >25 | >25 |
| 3 | 20.6 | 7.2 | 14.8 | 9.8 | 22.6 |
| cabozantinib | 11.2 | 4.4 | 9.0 | 8.7 | 18.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sachkova, A.A.; Andreeva, D.V.; Tikhomirov, A.S.; Scherbakov, A.M.; Salnikova, D.I.; Sorokin, D.V.; Bogdanov, F.B.; Rysina, Y.D.; Shchekotikhin, A.E.; Shchegravina, E.S.; et al. Design, Synthesis and In Vitro Investigation of Cabozantinib-Based PROTACs to Target c-Met Kinase. Pharmaceutics 2022, 14, 2829. https://doi.org/10.3390/pharmaceutics14122829
Sachkova AA, Andreeva DV, Tikhomirov AS, Scherbakov AM, Salnikova DI, Sorokin DV, Bogdanov FB, Rysina YD, Shchekotikhin AE, Shchegravina ES, et al. Design, Synthesis and In Vitro Investigation of Cabozantinib-Based PROTACs to Target c-Met Kinase. Pharmaceutics. 2022; 14(12):2829. https://doi.org/10.3390/pharmaceutics14122829
Chicago/Turabian StyleSachkova, Anastasia A., Daria V. Andreeva, Alexander S. Tikhomirov, Alexander M. Scherbakov, Diana I. Salnikova, Danila V. Sorokin, Fedor B. Bogdanov, Yulia D. Rysina, Andrey E. Shchekotikhin, Ekaterina S. Shchegravina, and et al. 2022. "Design, Synthesis and In Vitro Investigation of Cabozantinib-Based PROTACs to Target c-Met Kinase" Pharmaceutics 14, no. 12: 2829. https://doi.org/10.3390/pharmaceutics14122829