

Alternative Excipients for Protein Stabilization in Protein Therapeutics: Overcoming the Limitations of Polysorbates



Abstract
:1. Introduction
- The excipient’s effect on the overall quality, stability, and effectiveness of the drug product;
- Physical, chemical, and biological compatibility of excipient with the drug as well as the packaging system [54];
- Compatibility of the excipient with the manufacturing process;
- Amount of excipients that can be added to drug product, both from the formulation, safety and toxicological perspectives.
2. The Benefits and Drawbacks of Polysorbates in Protein Stabilization
3. Potential Polysorbate Alternatives for Protein Stabilization in Injectable Formulations
3.1. Surfactants
3.1.1. Surfactants Comprising Ester Bonds
- Sucrose fatty acid esters and sugar monoesters
- Polyethylene glycol (PEG) stearates and PEG fatty esters
3.1.2. Non-Ester Surfactants
- Block polyethylene-propylene glycol
- Polyoxyethylene fatty ethers
- Non-ester sugar-based surfactants
- N-alkyl amino acid polyether amides
3.2. Carbohydrates and Their Derivatives
- Disaccharides
- Sugar alcohols
- Cyclodextrins
- Hydroxypropyl methylcellulose
- Dextrans
3.3. Amino Acid-Based Stabilizers
- Amino acids
- Natural polyamines
- Albumin
3.4. Synthetic Amphiphilic Polymers
- Polyether polyols
- Polyampholytes
3.5. Ionic Liquids
4. Discussion
Supplementary Materials
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vulto, A.G.; Jaquez, O.A. The Process Defines the Product: What Really Matters in Biosimilar Design and Production? Rheumatology 2017, 56, iv14–iv29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Center for Biologics Evaluation and Research (CBER). What Are “Biologics” Questions and Answers; FDA, Center for Biologics Evaluation and Research: Silver Spring, MA, USA, 2019. [Google Scholar]
- Dimitrov, D.S. Therapeutic Proteins. Methods Mol. Biol. 2012, 899, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Kroon, D.J.; Baldwin-Ferro, A.; Lalan, P. Identification of Sites of Degradation in a Therapeutic Monoclonal Antibody by Peptide Mapping. Pharm Res. 1992, 9, 1386–1393. [Google Scholar] [CrossRef] [PubMed]
- Tous, G.I.; Wei, Z.; Feng, J.; Bilbulian, S.; Bowen, S.; Smith, J.; Strouse, R.; McGeehan, P.; Casas-Finet, J.; Schenerman, M.A. Characterization of a Novel Modification to Monoclonal Antibodies: Thioether Cross-Link of Heavy and Light Chains. Anal. Chem. 2005, 77, 2675–2682. [Google Scholar] [CrossRef]
- Harris, R.J.; Kabakoff, B.; Macchi, F.D.; Shen, F.J.; Kwong, M.; Andya, J.D.; Shire, S.J.; Bjork, N.; Totpal, K.; Chen, A.B. Identification of Multiple Sources of Charge Heterogeneity in a Recombinant Antibody. J. Chromatogr. B Biomed. Sci. Appl. 2001, 752, 233–245. [Google Scholar] [CrossRef]
- Wang, W. Instability, Stabilization, and Formulation of Liquid Protein Pharmaceuticals. Int. J. Pharm. 1999, 185, 129–188. [Google Scholar] [CrossRef]
- Cleland, J.L.; Powell, M.F.; Shire, S.J. The Development of Stable Protein Formulations: A Close Look at Protein Aggregation, Deamidation, and Oxidation. Crit. Rev. Ther. Drug Carr. Syst. 1993, 10, 307–377. [Google Scholar]
- Xie, M.; Schowen, R.L. Secondary Structure and Protein Deamidation. J. Pharm. Sci. 1999, 88, 8–13. [Google Scholar] [CrossRef]
- Usami, A.; Ohtsu, A.; Takahama, S.; Fujii, T. The Effect of PH, Hydrogen Peroxide and Temperature on the Stability of Human Monoclonal Antibody. J. Pharm. Biomed. Anal. 1996, 14, 1133–1140. [Google Scholar] [CrossRef]
- Lam, X.M.; Yang, J.Y.; Cleland, J.L. Antioxidants for Prevention of Methionine Oxidation in Recombinant Monoclonal Antibody HER2. J. Pharm. Sci. 1997, 86, 1250–1255. [Google Scholar] [CrossRef]
- Kennedy, D.M.; Skillen, A.W.; Self, C.H. Glycation of Monoclonal Antibodies Impairs Their Ability to Bind Antigen. Clin. Exp. Immunol. 1994, 98, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Cacia, J.; Keck, R.; Presta, L.G.; Frenz, J. Isomerization of an Aspartic Acid Residue in the Complementarity-Determining Regions of a Recombinant Antibody to Human IgE: Identification and Effect on Binding Affinity. Biochemistry 1996, 35, 1897–1903. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.Y.; Janis, L.J. Influence of PH, Buffer Species, and Storage Temperature on Physicochemical Stability of a Humanized Monoclonal Antibody LA298. Int. J. Pharm. 2006, 308, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Singh, S.; Zeng, D.L.; King, K.; Nema, S. Antibody Structure, Instability, and Formulation. J. Pharm. Sci. 2007, 96, 1–26. [Google Scholar] [CrossRef]
- Liu, D.; Ren, D.; Huang, H.; Dankberg, J.; Rosenfeld, R.; Cocco, M.J.; Li, L.; Brems, D.N.; Remmele, R.L. Structure and Stability Changes of Human IgG1 Fc as a Consequence of Methionine Oxidation. Biochemistry 2008, 47, 5088–5100. [Google Scholar] [CrossRef]
- Kozlowski, S.; Swann, P. Current and Future Issues in the Manufacturing and Development of Monoclonal Antibodies. Adv. Drug Deliv. Rev. 2006, 58, 707–722. [Google Scholar] [CrossRef]
- Geiger, T.; Clarke, S. Deamidation, Isomerization, and Racemization at Asparaginyl and Aspartyl Residues in Peptides. Succinimide-Linked Reactions That Contribute to Protein Degradation. J. Biol. Chem. 1987, 262, 785–794. [Google Scholar] [CrossRef]
- Chen, B.; Bautista, R.; Yu, K.; Zapata, G.A.; Mulkerrin, M.G.; Chamow, S.M. Influence of Histidine on the Stability and Physical Properties of a Fully Human Antibody in Aqueous and Solid Forms. Pharm. Res. 2003, 20, 1952–1960. [Google Scholar] [CrossRef]
- Li, S.-Q.; Bomser, J.A.; Zhang, Q.H. Effects of Pulsed Electric Fields and Heat Treatment on Stability and Secondary Structure of Bovine Immunoglobulin G. J. Agric. Food Chem. 2005, 53, 663–670. [Google Scholar] [CrossRef]
- Hermeling, S.; Crommelin, D.J.A.; Huub, S.; Huub, W. Structure-Immunogenicity Relationships of Therapeutic Proteins. Pharm. Res. 2004, 21, 897–903. [Google Scholar] [CrossRef]
- Shire, S.J.; Shahrokh, Z.; Liu, J. Challenges in the Development of High Protein Concentration Formulations. J. Pharm. Sci. 2004, 93, 1390–1402. [Google Scholar] [CrossRef] [PubMed]
- Braun, A.; Kwee, L.; Labow, M.A.; Alsenz, J. Protein Aggregates Seem to Play a Key Role among the Parameters Influencing the Antigenicity of Interferon Alpha (IFN-Alpha) in Normal and Transgenic Mice. Pharm. Res. 1997, 14, 1472–1478. [Google Scholar] [CrossRef]
- Sukumar, M.; Doyle, B.L.; Combs, J.L.; Pekar, A.H. Opalescent Appearance of an IgG1 Antibody at High Concentrations and Its Relationship to Noncovalent Association. Pharm. Res. 2004, 21, 1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Andya, J.D.; Hsu, C.C.; Shire, S.J. Mechanisms of Aggregate Formation and Carbohydrate Excipient Stabilization of Lyophilized Humanized Monoclonal Antibody Formulations. AAPS PharmSci. 2003, 5, E10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doran, P.M. Loss of Secreted Antibody from Transgenic Plant Tissue Cultures Due to Surface Adsorption. J. Biotechnol. 2006, 122, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Kumru, O.S.; Yi, L.; Wang, Y.J.; Zhang, J.; Kim, J.H.; Joshi, S.B.; Middaugh, C.R.; Volkin, D.B. Effect of Ionic Strength and PH on the Physical and Chemical Stability of a Monoclonal Antibody Antigen-Binding Fragment. J. Pharm. Sci. 2013, 102, 2520–2537. [Google Scholar] [CrossRef]
- Wang, W. Protein Aggregation and Its Inhibition in Biopharmaceutics. Int. J. Pharm. 2005, 289, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Garidel, P.; Blume, A.; Wagner, M. Prediction of Colloidal Stability of High Concentration Protein Formulations. Pharm. Dev. Technol. 2015, 20, 367–374. [Google Scholar] [CrossRef]
- Garidel, P.; Pevestorf, B.; Bahrenburg, S. Stability of Buffer-Free Freeze-Dried Formulations: A Feasibility Study of a Monoclonal Antibody at High Protein Concentrations. Eur. J. Pharm. Biopharm. 2015, 97, 125–139. [Google Scholar] [CrossRef]
- Kalonia, C.; Toprani, V.; Toth, R.; Wahome, N.; Gabel, I.; Middaugh, C.R.; Volkin, D.B. Effects of Protein Conformation, Apparent Solubility, and Protein-Protein Interactions on the Rates and Mechanisms of Aggregation for an IgG1 Monoclonal Antibody. J. Phys. Chem. B 2016, 120, 7062–7075. [Google Scholar] [CrossRef]
- Saluja, A.; Badkar, A.V.; Zeng, D.L.; Kalonia, D.S. Ultrasonic Rheology of a Monoclonal Antibody (IgG2) Solution: Implications for Physical Stability of Proteins in High Concentration Formulations. J. Pharm. Sci. 2007, 96, 3181–3195. [Google Scholar] [CrossRef] [PubMed]
- Chi, E.E.; Krishnan, S.; Kendrick, B.S.; Change, B.S.; Carpenter, J.F.; Chi, T.w. Roles of Conformational Stability and Colloidal Stability in the Aggregation of Recombinant Human Granulocyte Colony-Stimulating Factor. Protein Sci. A Publ. Protein Soc. 2003, 12, 903–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, S.; Hasegawa, J.; Kobayashi, N.; Tomitsuka, T.; Uchiyama, S.; Fukui, K. Effects of Ionic Strength and Sugars on the Aggregation Propensity of Monoclonal Antibodies: Influence of Colloidal and Conformational Stabilities. Pharm. Res. 2013, 30, 1263–1280. [Google Scholar] [CrossRef]
- Reiche, K.; Harti, J.; Blume, A.; Garidel, P. Liquid-Liquid Phase Separation of a Monoclonal Antibody at Low Ionic Strength: Influence of Anion Charge and Concentration. Biophys. Chem. 2017, 220, 7–19. [Google Scholar] [CrossRef]
- Jiskoot, W.; Randolph, T.W.; Volkin, D.B.; Middaugh, C.R.; Schöneich, C.; Winter, G.; Friess, W.; Crommelin, D.J.A.; Carpenter, J.F. Protein Instability and Immunogenicity: Roadblocks to Clinical Application of Injectable Protein Delivery Systems for Sustained Release. J. Pharm. Sci. 2012, 101, 946–954. [Google Scholar] [CrossRef]
- Schiefelbein, L.K.J. Sugar-Based Surfactants for Pharmaceutical Protein Formulations; Ludwig-Maximilians-Universität München: Munich, Germany, 2011. [Google Scholar]
- Gidalevitz, D.; Huang, Z.; Rice, S.A. Protein Folding at the Air-Water Interface Studied with X-ray Reflectivity. Proc. Natl. Acad. Sci. USA 1999, 96, 2608–2611. [Google Scholar] [CrossRef] [Green Version]
- Jones, L.S.; Kaufmann, A.; Middaugh, R. Silicone Oil Induced Aggregation of Proteins. J. Pharm. Sci. 2005, 94, 918–927. [Google Scholar] [CrossRef]
- Liu, L.; Ammar, D.A.; Ross, L.A.; Mandava, N.; Kahook, M.Y.; Carpenter, J.F. Silicone Oil Microdroplets and Protein Aggregates in Repackaged Bevacizumab and Ranibizumab: Effects of Long-Term Storage and Product Mishandling. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1023–1034. [Google Scholar] [CrossRef]
- Li, J.; Pinnamaneni, S.; Quan, Y.; Jaiswal, A.; Andersson, F.I.; Zhang, X. Mechanistic Understanding of Protein-Silicone Oil Interactions. Pharm. Res. 2012, 29, 1689–1697. [Google Scholar] [CrossRef]
- Krayukhina, E.; Tsumoto, K.; Uchiyama, S.; Fukui, K. Effects of Syringe Material and Silicone Oil Lubrication on the Stability of Pharmaceutical Proteins. J. Pharm. Sci. 2015, 104, 527–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerhardt, A.; McUmber, A.C.; Nguyen, B.H.; Lewus, R.; Schwartz, D.K.; Carpenter, J.F.; Randolph, T.W. Surfactant Effects on Particle Generation in Antibody Formulations in Pre-Filled Syringes. J. Pharm. Sci. 2015, 104, 4056–4064. [Google Scholar] [CrossRef] [PubMed]
- Jordan, S.; Katz, J. Alternative Surfactants for Biologics Stabilization. 2020. Available online: https://www.pharma.dupont.com/content/dam/dupont/amer/us/en/nutrition-health/general/pharmaceuticals/documents/Biologics%20stabilization%20white%20paper.pdf (accessed on 29 September 2022).
- van Beers, M.M.C.; Sauerborn, M.; Gilli, F.; Brinks, V.; Schellekens, H.; Jiskoot, W. Oxidized and Aggregated Recombinant Human Interferon Beta Is Immunogenic in Human Interferon Beta Transgenic Mice. Pharm. Res. 2011, 28, 2393–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bessa, J.; Boeckle, S.; Beck, H.; Buckel, T.; Schlicht, S.; Ebeling, M.; Kiialainen, A.; Koulov, A.; Boll, B.; Weiser, T.; et al. The Immunogenicity of Antibody Aggregates in a Novel Transgenic Mouse Model. Pharm. Res. 2015, 32, 2344–2359. [Google Scholar] [CrossRef] [PubMed]
- Palazzi, L.; Leri, M.; Cesaro, S.; Stefani, M.; Bucciantini, M.; Polverino de Laureto, P. Insight into the Molecular Mechanism Underlying the Inhibition of α-Synuclein Aggregation by Hydroxytyrosol. Biochem. Pharmacol. 2020, 173, 113722. [Google Scholar] [CrossRef] [PubMed]
- Moussa, E.M.; Panchal, J.P.; Moorthy, B.S.; Blum, J.S.; Joubert, M.K.; Narhi, L.O.; Topp, E.M. Immunogenicity of Therapeutic Protein Aggregates. J. Pharm. Sci. 2016, 105, 417–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, S.; Laue, T.M.; Kalonia, D.S.; Singh, S.N.; Shire, S.J. The Influence of Charge Distribution on Self-Association and Viscosity Behavior of Monoclonal Antibody Solutions. Mol. Pharm. 2012, 9, 791–802. [Google Scholar] [CrossRef]
- Yadav, S.; Liu, J.; Shire, S.J.; Kalonia, D.S. Specific Interactions in High Concentration Antibody Solutions Resulting in High Viscosity. J. Pharm. Sci. 2010, 99, 1152–1168. [Google Scholar] [CrossRef]
- Rao, V.A.; Kim, J.J.; Patel, D.S.; Rains, K.; Estoll, C.R. A Comprehensive Scientific Survey of Excipients Used in Currently Marketed, Therapeutic Biological Drug Products. Pharm. Res. 2020, 37, 200. [Google Scholar] [CrossRef]
- Rayaprolu, B.M.; Strawser, J.J.; Anyarambhatla, G. Excipients in Parenteral Formulations: Selection Considerations and Effective Utilization with Small Molecules and Biologics. Drug Dev. Ind. Pharm. 2018, 44, 1565–1571. [Google Scholar] [CrossRef]
- Jorgensen, L.; Hostrup, S.; Moeller, E.H.; Grohganz, H. Recent Trends in Stabilising Peptides and Proteins in Pharmaceutical Formulation-Considerations in the Choice of Excipients. Expert Opin. Drug Deliv. 2009, 6, 1219–1230. [Google Scholar] [CrossRef]
- Akers, M.J. Excipient-Drug Interactions in Parenteral Formulations. J. Pharm. Sci. 2002, 91, 2283–2300. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wu, G.; Zhang, X.; Tian, Z.; Zhang, N.; Hu, T.; Dai, W.; Qian, F. Stabilizing Two IgG1 Monoclonal Antibodies by Surfactants: Balance between Aggregation Prevention and Structure Perturbation. Eur. J. Pharm. Biopharm. 2017, 114, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Garidel, P.; Blech, M.; Buske, J.; Blume, A. Surface Tension and Self-Association Properties of Aqueous Polysorbate 20 HP and 80 HP Solutions: Insights into Protein Stabilisation Mechanisms. J. Pharm. Innov. 2021, 16, 726–734. [Google Scholar] [CrossRef]
- Garidel, P.; Hoffmann, C.; Blume, A. A Thermodynamic Analysis of the Binding Interaction between Polysorbate 20 and 80 with Human Serum Albumins and Immunoglobulins: A Contribution to Understand Colloidal Protein Stabilisation. Biophys. Chem. 2009, 143, 70–78. [Google Scholar] [CrossRef] [Green Version]
- Otzen, D. Protein-Surfactant Interactions: A Tale of Many States. Biochim. Biophys. Acta 2011, 1814, 562–591. [Google Scholar] [CrossRef]
- Kaplon, H.; Reichert, J.M. Antibodies to Watch in 2019. MAbs 2019, 11, 219–238. [Google Scholar] [CrossRef]
- Kaplon, H.; Muralidharan, M.; Schneider, Z.; Reichert, J.M. Antibodies to Watch in 2020. MAbs 2020, 12, 1703531. [Google Scholar] [CrossRef] [Green Version]
- Kaplon, H.; Reichert, J.M. Antibodies to Watch in 2021. MAbs 2021, 13, 1860476. [Google Scholar] [CrossRef]
- Kreilgaard, L.; Jones, L.S.; Randolph, T.W.; Frokjaer, S.; Flink, J.M.; Manning, M.C.; Carpenter, J.F. Effect of Tween 20 on Freeze-Thawing- and Agitation-Induced Aggregation of Recombinant Human Factor XIII. J. Pharm. Sci. 1998, 87, 1597–1603. [Google Scholar] [CrossRef]
- Hillgren, A.; Lindgren, J.; Aldén, M. Protection Mechanism of Tween 80 during Freeze–Thawing of a Model Protein, LDH. Int. J. Pharm. 2002, 237, 57–69. [Google Scholar] [CrossRef]
- Bam, N.B.; Cleland, J.L.; Yang, J.; Manning, M.C.; Carpenter, J.F.; Kelley, R.F.; Randolph, T.W. Tween Protects Recombinant Human Growth Hormone against Agitation-Induced Damage via Hydrophobic Interactions. J. Pharm. Sci. 1998, 87, 1554–1559. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yadav, S.; Demeule, B.; Wang, Y.J.; Mozziconacci, O.; Schöneich, C. Degradation Mechanisms of Polysorbate 20 Differentiated by 18O-Labeling and Mass Spectrometry. Pharm. Res. 2017, 34, 84–100. [Google Scholar] [CrossRef] [PubMed]
- Edelmann, F.; Ravuri, K. Instability of Polysorbates in Protein Biopharmaceutics. In Proceedings of the 10th Global Drug Delivery and Formulation Summit, Berlin, Germany, 11–13 March 2019. [Google Scholar]
- Santana, H.; González, Y.; Campana, P.T.; Noda, J.; Amarantes, O.; Itri, R.; Beldarraín, A.; Páez, R. Screening for Stability and Compatibility Conditions of Recombinant Human Epidermal Growth Factor for Parenteral Formulation: Effect of PH, Buffers, and Excipients. Int. J. Pharm. 2013, 452, 52–62. [Google Scholar] [CrossRef]
- Ha, E.; Wang, W.; Wang, Y.J. Peroxide Formation in Polysorbate 80 and Protein Stability. J. Pharm. Sci. 2002, 91, 2252–2264. [Google Scholar] [CrossRef] [PubMed]
- Reyes, N.; Ruiz, L.; Aroche, K.; Gerónimo, H.; Brito, O.; Hardy, E. Stability of Ala 125 Recombinant Human Interleukin-2 in Solution. J. Pharm. Pharmacol. 2005, 57, 31–37. [Google Scholar] [CrossRef]
- Kerwin, B.A. Polysorbates 20 and 80 Used in the Formulation of Protein Biotherapeutics: Structure and Degradation Pathways. J. Pharm. Sci. 2008, 97, 2924–2935. [Google Scholar] [CrossRef] [PubMed]
- Kishore, R.S.K.; Kiese, S.; Fischer, S.; Pappenberger, A.; Grauschopf, U.; Mahler, H.-C. The Degradation of Polysorbates 20 and 80 and Its Potential Impact on the Stability of Biotherapeutics. Pharm. Res. 2011, 28, 1194–1210. [Google Scholar] [CrossRef]
- Brange, J.; Langkjaer, L.; Havelund, S.; Vølund, A. Chemical Stability of Insulin. 1. Hydrolytic Degradation during Storage of Pharmaceutical Preparations. Pharm. Res. 1992, 9, 715–726. [Google Scholar] [CrossRef]
- Donbrow, M.; Azaz, E.; Pillersdorf, A. Autoxidation of Polysorbates. J. Pharm. Sci. 1978, 67, 1676–1681. [Google Scholar] [CrossRef]
- Labrenz, R. Ester Hydrolysis of Polysorbate 80 in MAb Drug Product: Evidence in Support of the Hypothesized Risk after the Observation of Visible Particulate in MAb Formulations. J. Pharm. Sci. 2014, 103, 2268–2277. [Google Scholar] [CrossRef]
- Lipiäinen, T.; Peltoniemi, M.; Sarkhel, S.; Yrjönen, T.; Vuorela, H.; Urtti, A.; Juppo, A. Formulation and Stability of Cytokine Therapeutics. J. Pharm. Sci. 2015, 104, 307–326. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, J.; Sorensen, K.; Wolff, S.P. Peroxide Accumulation in Detergents. J. Biochem. Biophys. Methods 1994, 29, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Schöneich, C.; Borchardt, R.T. Chemical Instability of Protein Pharmaceuticals: Mechanisms of Oxidation and Strategies for Stabilization. Biotechnol. Bioeng. 1995, 48, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Currie, F.; Andersson, M.; Holmber, K. Oxidation of Self-Organized Nonionic Surfactants. Langmuir ACS J. Surf. Colloids 2004, 20, 3835–3837. [Google Scholar] [CrossRef]
- Wuchner, K.; Yi, L.; Chery, C.; Nikels, F.; Junge, F.; Crotts, G.; Rinaldi, G.; Starkey, J.A.; Bechtold-Peters, K.; Shuman, M.; et al. Industry Perspective on the Use and Characterization of Polysorbates for Biopharmaceutical Products Part 1: Survey Report on Current State and Common Practices for Handling and Control of Polysorbates. J. Pharm. Sci. 2022, 111, 1280–1291. [Google Scholar] [CrossRef]
- Wuchner, K.; Yi, L.; Chery, C.; Nikels, F.; Junge, F.; Crotts, G.; Rinaldi, G.; Starkey, J.A.; Bechtold-Peters, K.; Shuman, M.; et al. Industry Perspective on the Use and Characterization of Polysorbates for Biopharmaceutical Products Part 2: Survey Report on Control Strategy Preparing for the Future. J. Pharm. Sci. 2022, 111, 2955–2967. [Google Scholar] [CrossRef]
- Ionova, Y.; Wilson, L. Biologic Excipients: Importance of Clinical Awareness of Inactive Ingredients. PLoS ONE 2020, 15, e0235076. [Google Scholar] [CrossRef] [PubMed]
- Nema, S.; Brendel, R.J. Excipients and Their Role in Approved Injectable Products: Current Usage and Future Directions. PDA J. Pharm. Sci. Technol. 2011, 65, 287–332. [Google Scholar] [CrossRef]
- Liu, L.; Qi, W.; Schwartz, D.K.; Randolph, T.W.; Carpenter, J.F. The Effects of Excipients on Protein Aggregation during Agitation: An Interfacial Shear Rheology Study. J. Pharm. Sci. 2013, 102, 2460–2470. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, J.F.; Chang, B.S.; Garzon-Rodriguez, W.; Randolph, T.W. Rational Design of Stable Lyophilized Protein Formulations: Theory and Practice. Pharm. Biotechnol. 2002, 13, 109–133. [Google Scholar] [CrossRef]
- Jayasundera, M.; Adhikari, B.; Adhikari, R.; Aldred, P. The Effect of Protein Types and Low Molecular Weight Surfactants on Spray Drying of Sugar-Rich Foods. Food Hydrocoll. 2011, 25, 459–469. [Google Scholar] [CrossRef]
- Gerhardt, A.; Bonam, K.; Bee, J.S.; Carpenter, J.F.; Randolph, T.W. Ionic Strength Affects Tertiary Structure and Aggregation Propensity of a Monoclonal Antibody Adsorbed to Silicone Oil-Water Interfaces. J. Pharm. Sci. 2013, 102, 429–440. [Google Scholar] [CrossRef]
- Lee, H.J.; McAuley, A.; Schilke, K.F.; McGuire, J. Molecular Origins of Surfactant-Mediated Stabilization of Protein Drugs. Adv. Drug Deliv. Rev. 2011, 63, 1160–1171. [Google Scholar] [CrossRef]
- Bam, N.B.; Cleland, J.L.; Randolph, T.W. Molten Globule Intermediate of Recombinant Human Growth Hormone: Stabilization with Surfactants. Biotechnol. Prog. 1996, 12, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.L.; Lee, J.C. Thermal Stability of Proteins in the Presence of Poly(Ethylene Glycols). Biochemistry 1987, 26, 7813–7819. [Google Scholar] [CrossRef]
- Arakawa, T.; Timasheff, S.N. Mechanism of Poly(Ethylene Glycol) Interaction with Proteins. Biochemistry 1985, 24, 6756–6762. [Google Scholar] [CrossRef] [PubMed]
- Dixit, N.; Maloney, K.M.; Kalonia, D.S. Protein-Silicone Oil Interactions: Comparative Effect of Nonionic Surfactants on the Interfacial Behavior of a Fusion Protein. Pharm. Res. 2013, 30, 1848–1859. [Google Scholar] [CrossRef]
- Ayorinde, F.O.; Gelain, S.V.; Johnson, J.H.; Wan, L.W. Analysis of Some Commercial Polysorbate Formulations Using Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry. Rapid Commun. Mass Spectrom. 2000, 14, 2116–2124. [Google Scholar] [CrossRef]
- Frison-Norrie, S.; Sporns, P. Investigating the Molecular Heterogeneity of Polysorbate Emulsifiers by MALDI-TOF MS. J. Agric. Food Chem. 2001, 49, 3335–3340. [Google Scholar] [CrossRef]
- Dwivedi, M.; Blech, M.; Presser, I.; Garidel, P. Polysorbate Degradation in Biotherapeutic Formulations: Identification and Discussion of Current Root Causes. Int. J. Pharm. 2018, 552, 422–436. [Google Scholar] [CrossRef]
- Hewitt, D.; Alvarez, M.; Robinson, K.; Ji, J.; Wang, Y.J.; Kao, Y.-H.; Zhang, T. Mixed-Mode and Reversed-Phase Liquid Chromatography–Tandem Mass Spectrometry Methodologies to Study Composition and Base Hydrolysis of Polysorbate 20 and 80. J. Chromatogr. A 2011, 1218, 2138–2145. [Google Scholar] [CrossRef]
- Tomlinson, A.; Zarraga, I.E.; Demeule, B. Characterization of Polysorbate Ester Fractions and Implications in Protein Drug Product Stability. Mol. Pharm. 2020, 17, 2345–2353. [Google Scholar] [CrossRef]
- Pan, J.; Ji, Y.; Du, Z.; Zhang, J. Rapid Characterization of Commercial Polysorbate 80 by Ultra-High Performance Supercritical Fluid Chromatography Combined with Quadrupole Time-of-Flight Mass Spectrometry. J. Chromatogr. A 2016, 1465, 190–196. [Google Scholar] [CrossRef]
- European Pharmacopoeia Commission. European Pharmacopoeia PS80. In European Pharmacopoeia; European Pharmacopoeia Commission: Strasbourg, France, 2017; pp. 3591–3593. [Google Scholar]
- European Pharmacopoeia Commission. European Pharmacopoeia PS20. In European Pharmacopoeia; European Pharmacopoeia Commission: Strasbourg, France, 2017; pp. 3589–3590. [Google Scholar]
- Li, Y.; Hewitt, D.; Lentz, Y.K.; Ji, J.A.; Zhang, T.Y.; Zhang, K. Characterization and Stability Study of Polysorbate 20 in Therapeutic Monoclonal Antibody Formulation by Multidimensional Ultrahigh-Performance Liquid Chromatography-Charged Aerosol Detection-Mass Spectrometry. Anal. Chem. 2014, 86, 5150–5157. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Yang, R.; Wang, J.; Yang, X.; Tu, J.; Xie, L.; Li, C.; Lao, Q.; Sun, C. Component-Based Biocompatibility and Safety Evaluation of Polysorbate 80. RSC Adv. 2017, 7, 15127–15138. [Google Scholar] [CrossRef] [Green Version]
- Ravuri, K.S.K. Polysorbate Degradation and Quality. In Challenges in Protein Product Development; Warne, N.W., Mahler, H.-C., Eds.; AAPS Advances in the Pharmaceutical Sciences Series; Springer International Publishing: Cham, Switzerland, 2018; pp. 25–62. ISBN 978-3-319-90603-4. [Google Scholar]
- Kishore, R.S.K.; Pappenberger, A.; Dauphin, I.B.; Ross, A.; Buergi, B.; Staempfli, A.; Mahler, H.-C. Degradation of Polysorbates 20 and 80: Studies on Thermal Autoxidation and Hydrolysis. J. Pharm. Sci. 2011, 100, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Khossravi, M.; Kao, Y.-H.; Mrsny, R.J.; Sweeney, T.D. Analysis Methods of Polysorbate 20: A New Method to Assess the Stability of Polysorbate 20 and Established Methods That May Overlook Degraded Polysorbate 20. Pharm. Res. 2002, 19, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Bates, T.R.; Nightingale, C.H.; Dixon, E. Kinetics of Hydrolysis of Polyoxyethylene (20) Sorbitan Fatty Acid Ester Surfactants. J. Pharm. Pharmacol. 1973, 25, 470–477. [Google Scholar] [CrossRef]
- Donbrow, M.; Hamburger, R.; Azaz, E.; Pillersdorf, A. Development of Acidity in Non-Ionic Surfactants: Formic and Acetic Acid. Analyst 1978, 103, 400–402. [Google Scholar] [CrossRef]
- Donbrow, M.; Hamburger, R.; Azaz, E. Surface Tension and Cloud Point Changes of Polyoxyethylenic Non-Ionic Surfactants during Autoxidation. J. Pharm. Pharmacol. 1975, 27, 160–166. [Google Scholar] [CrossRef]
- Carey, F.A.; Sundberg, R.J. Advanced Organic Chemistry Part A: Structure and Mechanism. In Advanced Organic Chemistry; Springer Science & Business Media: Boston, MA, USA, 2002; pp. 289–472. ISBN 978-0-387-68354-6. [Google Scholar]
- Dixit, N.; Salamat-Miller, N.; Salinas, P.A.; Taylor, K.D.; Basu, S.K. Residual Host Cell Protein Promotes Polysorbate 20 Degradation in a Sulfatase Drug Product Leading to Free Fatty Acid Particles. J. Pharm. Sci. 2016, 105, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Roy, I.; Patel, A.; Kumar, V.; Nanda, T.; Assenberg, R.; Wuchner, K.; Amin, K. Polysorbate Degradation and Particle Formation in a High Concentration MAb: Formulation Strategies to Minimize Effect of Enzymatic Polysorbate Degradation. J. Pharm. Sci. 2021, 110, 3313–3323. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.; Sandefur, S.L.; Frye, C.C.; Tuley, T.L.; Huang, L. Polysorbates 20 and 80 Degradation by Group XV Lysosomal Phospholipase A2 Isomer X1 in Monoclonal Antibody Formulations. J. Pharm. Sci. 2016, 105, 1633–1642. [Google Scholar] [CrossRef]
- McShan, A.C.; Kei, P.; Ji, J.A.; Kim, D.C.; Wang, Y.J. Hydrolysis of Polysorbate 20 and 80 by a Range of Carboxylester Hydrolases. PDA J. Pharm. Sci. Technol. 2016, 70, 332–345. [Google Scholar] [CrossRef]
- Siska, C.C.; Pierini, C.J.; Lau, H.R.; Latypov, R.F.; Fesinmeyer, R.M.; Litowski, J.R. Free Fatty Acid Particles in Protein Formulations, Part 2: Contribution of Polysorbate Raw Material. J. Pharm. Sci. 2015, 104, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Honemann, M.N.; Wendler, J.; Graf, T.; Bathke, A.; Bell, C.H. Monitoring Polysorbate Hydrolysis in Biopharmaceuticals Using a QC-Ready Free Fatty Acid Quantification Method. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2019, 1116, 1–8. [Google Scholar] [CrossRef]
- Tomlinson, A.; Demeule, B.; Lin, B.; Yadav, S. Polysorbate 20 Degradation in Biopharmaceutical Formulations: Quantification of Free Fatty Acids, Characterization of Particulates, and Insights into the Degradation Mechanism. Mol. Pharm. 2015, 12, 3805–3815. [Google Scholar] [CrossRef]
- Larson, N.R.; Wei, Y.; Prajapati, I.; Chakraborty, A.; Peters, B.; Kalonia, C.; Hudak, S.; Choudhary, S.; Esfandiary, R.; Dhar, P.; et al. Comparison of Polysorbate 80 Hydrolysis and Oxidation on the Aggregation of a Monoclonal Antibody. J. Pharm. Sci. 2020, 109, 633–639. [Google Scholar] [CrossRef] [Green Version]
- Chiu, J.; Valente, K.N.; Levy, N.E.; Min, L.; Lenhoff, A.M.; Lee, K.H. Knockout of a Difficult-to-Remove CHO Host Cell Protein, Lipoprotein Lipase, for Improved Polysorbate Stability in Monoclonal Antibody Formulations. Biotechnol. Bioeng. 2017, 114, 1006–1015. [Google Scholar] [CrossRef] [Green Version]
- Mihara, K.; Ito, Y.; Hatano, Y.; Komurasaki, Y.; Sugimura, A.; Jones, M.; Liu, H.; Mai, S.; Lara-Velasco, O.; Bai, L.; et al. Host Cell Proteins: The Hidden Side of Biosimilarity Assessment. J. Pharm. Sci. 2015, 104, 3991–3996. [Google Scholar] [CrossRef]
- Aboulaich, N.; Chung, W.K.; Thompson, J.H.; Larkin, C.; Robbins, D.; Zhu, M. A Novel Approach to Monitor Clearance of Host Cell Proteins Associated with Monoclonal Antibodies. Biotechnol. Prog. 2014, 30, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Graf, T.; Tomlinson, A.; Yuk, I.H.; Kufer, R.; Spensberger, B.; Falkenstein, R.; Shen, A.; Li, H.; Duan, D.; Liu, W.; et al. Identification and Characterization of Polysorbate-Degrading Enzymes in a Monoclonal Antibody Formulation. J. Pharm. Sci. 2021, 110, 3558–3567. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Riccardi, C.; Kamen, D.; Reilly, J.; Mattila, J.; Bak, H.; Xiao, H.; Li, N. Identification of the Specific Causes of Polysorbate 20 Degradation in Monoclonal Antibody Formulations Containing Multiple Lipases. Pharm. Res. 2022, 39, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chandra, D.; Letarte, S.; Adam, G.C.; Welch, J.; Yang, R.-S.; Rivera, S.; Bodea, S.; Dow, A.; Chi, A.; et al. Profiling Active Enzymes for Polysorbate Degradation in Biotherapeutics by Activity-Based Protein Profiling. Anal. Chem. 2021, 93, 8161–8169. [Google Scholar] [CrossRef]
- Zhang, S.; Xiao, H.; Molden, R.; Qiu, H.; Li, N. Rapid Polysorbate 80 Degradation by Liver Carboxylesterase in a Monoclonal Antibody Formulated Drug Substance at Early Stage Development. J. Pharm. Sci. 2020, 109, 3300–3307. [Google Scholar] [CrossRef]
- Zhang, S.; Xiao, H.; Li, N. Degradation of Polysorbate 20 by Sialate O-Acetylesterase in Monoclonal Antibody Formulations. J. Pharm. Sci. 2021, 110, 3866–3873. [Google Scholar] [CrossRef]
- Li, X.; Wang, F.; Li, H.; Richardson, D.D.; Roush, D. The Measurement and Control of High-Risk Host Cell Proteins for Polysorbate Degradation in Biologics Formulation. Antib. Ther. 2022, 5, 42–54. [Google Scholar] [CrossRef]
- Putative Phospholipase B-like 2 Is Not Responsible for Polysorbate Degradation in Monoclonal Antibody Drug Products. J. Pharm. Sci. 2020, 109, 2710–2718. [CrossRef]
- Glücklich, N.; Carle, S.; Buske, J.; Mäder, K.; Garidel, P. Assessing the Polysorbate Degradation Fingerprints and Kinetics of Lipases—How the Activity of Polysorbate Degrading Hydrolases Is Influenced by the Assay and Assay Conditions. Eur. J. Pharm. Sci. 2021, 166, 105980. [Google Scholar] [CrossRef]
- Saito, H.; Tomioka, H.; Watanabe, T.; Yoneyama, T. Mycobacteriocins Produced by Rapidly Growing Mycobacteria Are Tween-Hydrolyzing Esterases. J. Bacteriol. 1983, 153, 1294–1300. [Google Scholar] [CrossRef] [Green Version]
- Tomioka, H. Purification and Characterization of the Tween-Hydrolyzing Esterase of Mycobacterium Smegmatis. J. Bacteriol. 1983, 155, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.T.; Mahler, H.-C.; Yadav, S.; Bindra, D.; Corvari, V.; Fesinmeyer, R.M.; Gupta, K.; Harmon, A.M.; Hinds, K.D.; Koulov, A.; et al. Considerations for the Use of Polysorbates in Biopharmaceuticals. Pharm. Res. 2018, 35, 148. [Google Scholar] [CrossRef] [PubMed]
- Bodin, A.; Linnerborg, M.; Nilsson, J.L.G.; Karlberg, A.-T. Novel Hydroperoxides as Primary Autoxidation Products of a Model Ethoxylated Surfactant. J. Surfact Deterg. 2002, 5, 107–110. [Google Scholar] [CrossRef]
- Bäcktorp, C.; Börje, A.; Nilsson, J.L.G.; Karlberg, A.-T.; Norrby, P.-O.; Nyman, G. Mechanisms of Air Oxidation of Ethoxylated Surfactants--Computational Estimations of Energies and Reaction Behaviors. Chemistry 2008, 14, 9549–9554. [Google Scholar] [CrossRef]
- Singh, S.R.; Zhang, J.; O’Dell, C.; Hsieh, M.-C.; Goldstein, J.; Liu, J.; Srivastava, A. Effect of Polysorbate 80 Quality on Photostability of a Monoclonal Antibody. AAPS PharmSciTech. 2012, 13, 422–430. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Dokuru, D.K.; Noestheden, M.; Park, S.S.; Kerwin, B.A.; Jona, J.; Ostovic, D.; Reid, D.L. A Quantitative Kinetic Study of Polysorbate Autoxidation: The Role of Unsaturated Fatty Acid Ester Substituents. Pharm. Res. 2009, 26, 2303–2313. [Google Scholar] [CrossRef]
- Kerwin, B.A.; Remmele, R.L. Protect from Light: Photodegradation and Protein Biologics. J. Pharm. Sci. 2007, 96, 1468–1479. [Google Scholar] [CrossRef]
- Du, C.; Barnett, G.; Borwankar, A.; Lewandowski, A.; Singh, N.; Ghose, S.; Borys, M.; Li, Z.J. Protection of Therapeutic Antibodies from Visible Light Induced Degradation: Use Safe Light in Manufacturing and Storage. Eur. J. Pharm. Biopharm. 2018, 127, 37–43. [Google Scholar] [CrossRef]
- Nejadnik, M.R.; Randolph, T.W.; Volkin, D.B.; Schöneich, C.; Carpenter, J.F.; Crommelin, D.J.A.; Jiskoot, W. Postproduction Handling and Administration of Protein Pharmaceuticals and Potential Instability Issues. J. Pharm. Sci. 2018, 107, 2013–2019. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-H.; Lee, Y.M.; Suh, J.-K.; Song, N.W. Photodegradation Mechanism and Reaction Kinetics of Recombinant Human Interferon-Alpha2a. Photochem. Photobiol. Sci. 2007, 6, 171–180. [Google Scholar] [CrossRef]
- Sreedhara, A.; Yin, J.; Joyce, M.; Lau, K.; Wecksler, A.T.; Deperalta, G.; Yi, L.; John Wang, Y.; Kabakoff, B.; Kishore, R.S.K. Effect of Ambient Light on IgG1 Monoclonal Antibodies during Drug Product Processing and Development. Eur. J. Pharm. Biopharm. 2016, 100, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Maity, H.; O’Dell, C.; Srivastava, A.; Goldstein, J. Effects of Arginine on Photostability and Thermal Stability of IgG1 Monoclonal Antibodies. Curr. Pharm. Biotechnol. 2009, 10, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Bane, J.; Mozziconacci, O.; Yi, L.; Wang, Y.J.; Sreedhara, A.; Schöneich, C. Photo-Oxidation of IgG1 and Model Peptides: Detection and Analysis of Triply Oxidized His and Trp Side Chain Cleavage Products. Pharm. Res. 2017, 34, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Pattison, D.I.; Rahmanto, A.S.; Davies, M.J. Photo-Oxidation of Proteins. Photochem. Photobiol. Sci. 2012, 11, 38–53. [Google Scholar] [CrossRef]
- Roy, S.; Mason, B.D.; Schöneich, C.S.; Carpenter, J.F.; Boone, T.C.; Kerwin, B.A. Light-Induced Aggregation of Type I Soluble Tumor Necrosis Factor Receptor. J. Pharm. Sci. 2009, 98, 3182–3199. [Google Scholar] [CrossRef]
- Li, Y.; Polozova, A.; Gruia, F.; Feng, J. Characterization of the Degradation Products of a Color-Changed Monoclonal Antibody: Tryptophan-Derived Chromophores. Anal. Chem. 2014, 86, 6850–6857. [Google Scholar] [CrossRef]
- Mason, B.D.; Schöneich, C.; Kerwin, B.A. Effect of PH and Light on Aggregation and Conformation of an IgG1 MAb. Mol. Pharm. 2012, 9, 774–790. [Google Scholar] [CrossRef] [Green Version]
- Fradkin, A.H.; Mozziconacci, O.; Schöneich, C.; Carpenter, J.F.; Randolph, T.W. UV Photodegradation of Murine Growth Hormone: Chemical Analysis and Immunogenicity Consequences. Eur. J. Pharm. Biopharm. 2014, 87, 395–402. [Google Scholar] [CrossRef] [Green Version]
- Torosantucci, R.; Schöneich, C.; Jiskoot, W. Oxidation of Therapeutic Proteins and Peptides: Structural and Biological Consequences. Pharm. Res. 2014, 31, 541–553. [Google Scholar] [CrossRef]
- Pham, N.B.; Meng, W.S. Protein Aggregation and Immunogenicity of Biotherapeutics. Int. J. Pharm. 2020, 585, 119523. [Google Scholar] [CrossRef]
- Coors, E.A.; Seybold, H.; Merk, H.F.; Mahler, V. Polysorbate 80 in Medical Products and Nonimmunologic Anaphylactoid Reactions. Ann. Allergy Asthma Immunol. 2005, 95, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Palacios Castaño, M.I.; Venturini Díaz, M.; Lobera Labairu, T.; González Mahave, I.; Del Pozo Gil, M.D.; Blasco Sarramián, A. Anaphylaxis Due to the Excipient Polysorbate 80. J. Investig. Allergol. Clin. Immunol. 2016, 26, 394–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badiu, I.; Guida, G.; Heffler, E.; Rolla, G. Multiple Drug Allergy Due to Hypersensitivity to Polyethylene Glycols of Various Molecular Weights. J. Investig. Allergol. Clin. Immunol. 2015, 25, 368–369. [Google Scholar] [PubMed]
- Stone, C.A.; Liu, Y.; Relling, M.V.; Krantz, M.S.; Pratt, A.L.; Abreo, A.; Hemler, J.A.; Phillips, E.J. Immediate Hypersensitivity to Polyethylene Glycols and Polysorbates: More Common than We Have Recognized. J. Allergy Clin. Immunol. Pract. 2019, 7, 1533–1540.e8. [Google Scholar] [CrossRef]
- Price, K.S.; Hamilton, R.G. Anaphylactoid Reactions in Two Patients after Omalizumab Administration after Successful Long-Term Therapy. Allergy Asthma Proc. 2007, 28, 313–319. [Google Scholar] [CrossRef]
- Dreyfus, D.H.; Randolph, C.C. Characterization of an Anaphylactoid Reaction to Omalizumab. Ann. Allergy Asthma Immunol. 2006, 96, 624–627. [Google Scholar] [CrossRef]
- Pérez-Pérez, L.; García-Gavín, J.; Piñeiro, B.; Zulaica, A. Biologic-Induced Urticaria Due to Polysorbate 80: Usefulness of Prick Test. Br. J. Dermatol. 2011, 164, 1119–1120. [Google Scholar] [CrossRef]
- Bergmann, K.C.; Maurer, M.; Church, M.K.; Zuberbier, T. Anaphylaxis to Mepolizumab and Omalizumab in a Single Patient: Is Polysorbate the Culprit? J. Investig. Allergol. Clin. Immunol. 2020, 30, 285–287. [Google Scholar] [CrossRef]
- Badiu, I.; Geuna, M.; Heffler, E.; Rolla, G. Hypersensitivity Reaction to Human Papillomavirus Vaccine Due to Polysorbate 80. BMJ Case Rep. 2012, 2012, bcr0220125797. [Google Scholar] [CrossRef] [Green Version]
- Steele, R.H.; Limaye, S.; Cleland, B.; Chow, J.; Suranyi, M.G. Hypersensitivity Reactions to the Polysorbate Contained in Recombinant Erythropoietin and Darbepoietin. Nephrology 2005, 10, 317–320. [Google Scholar] [CrossRef]
- Center for Biologics Evaluation and Research (CBER). Pilot Program for the Review of Innovation and Modernization of Excipients (PRIME); FDA: Silver Spring, MA, USA, 2022. [Google Scholar]
- Bollenbach, L.; Buske, J.; Mäder, K.; Garidel, P. Poloxamer 188 as Surfactant in Biological Formulations—An Alternative for Polysorbate 20/80? Int. J. Pharm. 2022, 620, 121706. [Google Scholar] [CrossRef] [PubMed]
- Inactive Ingredient Search for Approved Drug Products. Available online: https://www.accessdata.fda.gov/scripts/cder/iig/index.cfm (accessed on 12 July 2022).
- Csóka, G.; Marton, S.; Zelko, R.; Otomo, N.; Antal, I. Application of Sucrose Fatty Acid Esters in Transdermal Therapeutic Systems. Eur. J. Pharm. Biopharm. 2007, 65, 233–237. [Google Scholar] [CrossRef] [PubMed]
- CFR-Code of Federal Regulations Title 21. Available online: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm?fr=172.859 (accessed on 2 June 2022).
- Scott, L.N.; Bergfeld, W.F.; Belsito, D.V.; Hill, R.A.; Klaassen, C.D.; Liebler, D.C.; Marks, J.G.; Shank, R.C.; Slaga, T.J.; Snyder, P.W.; et al. Safety Assessment of Saccharide Esters as Used in Cosmetics. Int. J. Toxicol. 2021, 40, 52S–116S. [Google Scholar] [CrossRef]
- Obikili, A.; Deyme, M.; Wouessidjewe, D.; Duchěne, D. Improvement of Aqueous Solubility and Dissolution Kinetics of Canrenone by Solid Dispersion in Sucroester. Drug Dev. Ind. Pharm. 1988, 14, 791–803. [Google Scholar] [CrossRef]
- Hahn, L.; Sucker, H. Solid Surfactant Solutions of Active Ingredients in Sugar Esters. Pharm. Res. 1989, 6, 958–960. [Google Scholar] [CrossRef] [PubMed]
- Abd-Elbary, A.; El-laithy, H.M.; Tadros, M.I. Sucrose Stearate-Based Proniosome-Derived Niosomes for the Nebulisable Delivery of Cromolyn Sodium. Int. J. Pharm. 2008, 357, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Lerk, P.C.; Sucker, H. Application of Sucrose Laurate in Topical Preparations of Cyclosporin A. Int. J. Pharm. 1993, 92, 203–210. [Google Scholar] [CrossRef]
- Okamoto, H.; Sakai, T.; Danjo, K. Effect of Sucrose Fatty Acid Esters on Transdermal Permeation of Lidocaine and Ketoprofen. Biol. Pharm. Bull. 2005, 28, 1689–1694. [Google Scholar] [CrossRef] [Green Version]
- Ahsan, F.; Arnold, J.J.; Meezan, E.; Pillion, D.J. Sucrose Cocoate, a Component of Cosmetic Preparations, Enhances Nasal and Ocular Peptide Absorption. Int. J. Pharm. 2003, 251, 195–203. [Google Scholar] [CrossRef]
- Thevenin, M.A.; Grossiord, J.L.; Poelman, M.C. Sucrose Esters/Cosurfactant Microemulsion Systems for Transdermal Delivery: Assessment of Bicontinuous Structures. Int. J. Pharm. 1996, 137, 177–186. [Google Scholar] [CrossRef]
- Fanun, M. Surfactant Chain Length Effect on the Structural Parameters of Nonionic Microemulsions. J. Dispers. Sci. Technol. 2008, 29, 289–296. [Google Scholar] [CrossRef]
- Mollee, H.; de Vrind, J.; De Vringer, T. Stable Reversed Vesicles in Oil: Characterization Studies and Encapsulation of Model Compounds. J. Pharm. Sci. 2000, 89, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Honeywell-Nguyen, P.L.; Bouwstra, J.A. The in Vitro Transport of Pergolide from Surfactant-Based Elastic Vesicles through Human Skin: A Suggested Mechanism of Action. J. Control. Release 2003, 86, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Klang, V.; Matsko, N.; Raupach, K.; El-Hagin, N.; Valenta, C. Development of Sucrose Stearate-Based Nanoemulsions and Optimisation through γ-Cyclodextrin. Eur. J. Pharm. Biopharm. 2011, 79, 58–67. [Google Scholar] [CrossRef]
- Zheng, Y.; Zheng, M.; Ma, Z.; Xin, B.; Guo, R.; Xu, X. Sugar Fatty Acid Esters. In Polar Lipids; Elsevier Inc.: Amsterdam, The Netherlands, 2015; pp. 215–243. ISBN 978-1-63067-045-0. [Google Scholar]
- Soultani, S.; Ognier, S.; Engasser, J.-M.; Ghoul, M. Comparative Study of Some Surface Active Properties of Fructose Esters and Commercial Sucrose Esters. Colloids Surf. A Physicochem. Eng. Asp. 2003, 227, 35–44. [Google Scholar] [CrossRef]
- Younes, M.; Aggett, P.; Aguilar, F.; Crebelli, R.; Dusemund, B.; Filipič, M.; Frutos, M.J.; Galtier, D.; Gundert-Remy, U.; Kuhnle, G.G. Scientific opinion on the safety of sucrose esters of fatty acids prepared from vinyl esters of fatty acids and on the extension of use of sucrose esters of fatty acids in flavourings|EFSA. EFSA J. 2010, 8, 1512. [Google Scholar] [CrossRef] [Green Version]
- Thomson, A.B.; Hunt, R.H.; Zorich, N.L. Review Article: Olestra and Its Gastrointestinal Safety. Aliment Pharmacol. 1998, 12, 1185–1200. [Google Scholar] [CrossRef]
- Daniel, J.W.; Marshall, C.J.; Jones, H.F.; Snodin, D.J. The Metabolism of Beef Tallow Sucrose Esters in Rat and Man. Food Cosmet. Toxicol. 1979, 17, 19–21. [Google Scholar] [CrossRef]
- Shigeoka, T.; Izawa, O.; Kitazawa, K.; Yamauchi, F.; Murata, T. Studies on the Metabolic Fate of Sucrose Esters in Rats. Food Chem. Toxicol. 1984, 22, 409–414. [Google Scholar] [CrossRef]
- Noker, P.E.; Lin, T.H.; Hill, D.L.; Shigeoka, T. Metabolism of 14C-Labelled Sucrose Esters of Stearic Acid in Rats. Food Chem. Toxicol. 1997, 35, 589–595. [Google Scholar] [CrossRef]
- Sucrose Esters of Fatty Acids, and Sucroglycerides (WHO Food Additives Series 5). Available online: https://inchem.org/documents/jecfa/jecmono/v05je52.htm (accessed on 2 June 2022).
- Drummond, C.J.; Fong, C.; Krodkiewksa, I.; Boyd, B.; Baker, I. Sugar Fatty Acid Esters. In Novel Surfactants: Preparation, Applications and Biodegradability; Marcell Dekker Inc: New York, NY, USA, 2003; pp. 95–128. [Google Scholar]
- Savić, S.; Tamburić, S.; Savić, M.M. From Conventional towards New-Natural Surfactants in Drug Delivery Systems Design: Current Status and Perspectives. Expert Opin. Drug Deliv. 2010, 7, 353–369. [Google Scholar] [CrossRef] [PubMed]
- Szűts, A.; Szabó-Révész, P. Sucrose Esters as Natural Surfactants in Drug Delivery Systems—A Mini-Review. Int. J. Pharm. 2012, 433, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gaudin, T.; Lu, H.; Fayet, G.; Berthauld-Drelich, A.; Rotureau, P.; Pourceau, G.; Wadouachi, A.; Van Hecke, E.; Nesterenko, A.; Pezron, I. Impact of the Chemical Structure on Amphiphilic Properties of Sugar-Based Surfactants: A Literature Overview. Adv. Colloid Interface Sci. 2019, 270, 87–100. [Google Scholar] [CrossRef]
- Lerk, P.C.; Sucker, H.H.; Eicke, H.F. Micellization and Solubilization Behavior of Sucrose Laurate, a New Pharmaceutical Excipient. Pharm. Dev. Technol. 1996, 1, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.L. Parenteral Formulations Comprising Sugar-Based Esters and Ethers. U.S. Patent 20100125051A1, 20 May 2010. [Google Scholar]
- Weber, N.; Benning, H. Metabolism of Orally Administered Alkyl Beta-Glycosides in the Mouse. J. Nutr. 1984, 114, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Garofalakis, G.; Murray, B.S.; Sarney, D.B. Surface Activity and Critical Aggregation Concentration of Pure Sugar Esters with Different Sugar Headgroups. J. Colloid Interface Sci. 2000, 229, 391–398. [Google Scholar] [CrossRef]
- The European Agency for the Evaluation of Medicinal Products. Polyethylene Glycol Searates and Polyethylene Glycol 15 Hydroxystearate; The European Agency for the Evaluation of Medicinal Products: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Elder, E. Final Report on the Safety Assessment of PEG-2,-6, -8, -12, -20, -32, -40, -50, -100, and -150 Stearates. J. Am. Coll. Toxicol. 1983, 2, 17–34. [Google Scholar]
- Fruijtier-Pölloth, C. Safety Assessment on Polyethylene Glycols (PEGs) and Their Derivatives as Used in Cosmetic Products. Toxicology 2005, 214, 1–38. [Google Scholar] [CrossRef]
- Bárány, E.; Lindberg, M.; Lodén, M. Unexpected Skin Barrier Influence from Nonionic Emulsifiers. Int. J. Pharm. 2000, 195, 189–195. [Google Scholar] [CrossRef]
- Johns, C.H.; Pepper, W.P. 5 Final Report on the Safety Assessment of Propylene Glycol Stearate and Propylene Glycol Stearate Self-Emulsifying. J. Am. Coll. Toxicol. 1983, 2, 101–124. [Google Scholar] [CrossRef]
- Johnson, W. Cosmetic Ingredient Review Expert Panel Final Report on the Safety Assessment of PEG-25 Propylene Glycol Stearate, PEG-75 Propylene Glycol Stearate, PEG-120 Propylene Glycol Stearate, PEG-10 Propylene Glycol, PEG-8 Propylene Glycol Cocoate, and PEG-55 Propylene Glycol Oleate. Int. J. Toxicol. 2001, 20 (Suppl. S4), 13–26. [Google Scholar] [CrossRef]
- Ali, S.; Kolter, K. Kolliphor® HS 15-An Enabler for Parenteral and Oral Formulations. Am. Pharm. Rev. 2019, 22, 22–34. [Google Scholar]
- Reintjes, T. Solubility Enhancement with BASF Pharma Polymers-Solubilizer Compendium; BASD SE Pharma Ingredients and Services: Lampretheim, Germany, 2011. [Google Scholar]
- Shubber, S.; Vllasaliu, D.; Rauch, C.; Jordan, F.; Illum, L.; Stolnik, S. Mechanism of Mucosal Permeability Enhancement of CriticalSorb® (Solutol® HS15) Investigated in Vitro in Cell Cultures. Pharm. Res. 2015, 32, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Pilotaz, F.; Mercier, F.; Chibret, H. Preservative-Free Prostaglandin-Based Ophthalmic Solution. Portugal Patent PT2178504E, 31 January 2011. [Google Scholar]
- Dai, W.-G.; Dong, L.C.; Li, S.; Pollock-Dove, C.; Chen, J.; Mansky, P.; Eichenbaum, G. Parallel Screening Approach to Identify Solubility-Enhancing Formulations for Improved Bioavailability of a Poorly Water-Soluble Compound Using Milligram Quantities of Material. Int. J. Pharm. 2007, 336, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Berkó, S.; Regdon, G.; Erös, I. Solutol and Cremophor Products as New Additives in Suppository Formulation. Drug Dev. Ind. Pharm. 2002, 28, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Zakeri, B.; Kroll, S.; Romanski, F.S. Expanding the Toolbox of Surfactants Available for Biologics Formulations with Kolliphor® HS 15 and Kolliphor® ELP; BASF: Ludwigshafen, Germany, 2020. [Google Scholar]
- Ruchatz, F.; Schuch, H. Physicochemical Properties of Solutol HS 15 and Its Solubilizates. BASF ExAct 1988, 1, 6–7. [Google Scholar]
- Zhao, X.; Chen, D.; Gao, P.; Ding, P.; Li, K. Synthesis of Ibuprofen Eugenol Ester and Its Microemulsion Formulation for Parenteral Delivery. Chem. Pharm. Bull. 2005, 53, 1246–1250. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.-H.; Lee, S.-H.; Park, D.-Y.; Ki, K.-H.; Lee, E.-K.; Lee, D.-H.; Noh, G.-J. Physicochemical Properties, Pharmacokinetics, and Pharmacodynamics of a Reformulated Microemulsion Propofol in Rats. Anesthesiology 2008, 109, 436–447. [Google Scholar] [CrossRef]
- Bartels, M.; Vilsendorf, A.; Kassahun, T.; Gerstenbergk, B.; Engelhart, K.; Vilsendorf, E.; Faber, S.; Biesalski, H. Protective Effect of Antioxidative Vitamins against Lipid Peroxidation in Liver Ischemia and Reperfusion–an Animal Experimental Study. EXCLI J. 2007, 6, 152–166. [Google Scholar] [CrossRef]
- Volker, B. Pharmaceutical Technology of BASF Excipients, 3rd ed.; BASF: Ludwigshafen, Germany, 2008. [Google Scholar]
- BASF. Product Regulations Safety Data Sheet- Kolliphor HS 15; Version 4; BASF: Ludwigshafen, Germany, 2016. [Google Scholar]
- Borisov, O.V.; Ji, J.A.; Wang, Y.J. Oxidative Degradation of Polysorbate Surfactants Studied by Liquid Chromatography–Mass Spectrometry. J. Pharm. Sci. 2015, 104, 1005–1018. [Google Scholar] [CrossRef]
- Hvattum, E.; Yip, W.L.; Grace, D.; Dyrstad, K. Characterization of Polysorbate 80 with Liquid Chromatography Mass Spectrometry and Nuclear Magnetic Resonance Spectroscopy: Specific Determination of Oxidation Products of Thermally Oxidized Polysorbate 80. J. Pharm. Biomed. Anal. 2012, 62, 7–16. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Z.; Liu, O. Simultaneous Determination of Kolliphor HS15 and Miglyol 812 in Microemulsion Formulation by Ultra-High Performance Liquid Chromatography Coupled with Nano Quantity Analyte Detector. J. Pharm. Anal. 2016, 6, 11–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coon, J.S.; Knudson, W.; Clodfelter, K.; Lu, B.; Weinstein, R.S. Solutol HS 15, Nontoxic Polyoxyethylene Esters of 12-Hydroxystearic Acid, Reverses Multidrug Resistance. Cancer Res. 1991, 51, 897–902. [Google Scholar]
- Bhaskar, V.; Middha, A.; Tiwari, S.; Shivakumar, S. Identification and Reduction of Matrix Effects Caused by Solutol Hs15 in Bioanalysis Using Liquid Chromatography/Tandem Mass Spectrometry. J. Anal. Bioanal. Tech. 2013, 4, 2. [Google Scholar] [CrossRef]
- Bhaskar, V.V.; Middha, A.; Srivastava, P.; Rajagopal, S. Liquid Chromatography/Tandem Mass Spectrometry Method for Quantitative Estimation of Solutol HS15 and Its Applications. J. Pharm. Anal. 2015, 5, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Martinez, C.V.; Emo, M.; Lebeau, B.; García-Celma, M.-J.; Stébé, M.-J.; Blin, J.-L. Insights of the Kolliphor/Water System for the Design of Mesostructured Silica Materials. Microporous Mesoporous Mater. 2019, 285, 231–240. [Google Scholar] [CrossRef]
- BASF. Product Regulations Technical Information Sheet- Kolliphor EL; BASF: Ludwigshafen, Germany, 2019. [Google Scholar]
- European Medicine Agency. Polyolyl Caster Oil, Polyoxyl Hydrogenated Caster Oil; Summary Report; European Medicine Agency Committee for Veterinary Medicinal Products: Amsterdam, The Netherlands, 1999. [Google Scholar]
- Irizarry, L.; McKoy, J.; Samaras, A.; Fisher, M.; Carias, E.; Raisch, D.; Calhoun, E.; Bennett, C.; Brown, J.; Lurie, R. Cremophor EL-Containing Paclitaxel-Induced Anaphylaxis: A Call to Action. Community Oncol. 2009, 6, 132–134. [Google Scholar] [CrossRef]
- Riegert-Johnson, D.L.; Volcheck, G.W. The Incidence of Anaphylaxis Following Intravenous Phytonadione (Vitamin K1): A 5-Year Retrospective Review. Ann. Allergy Asthma Immunol. 2002, 89, 400–406. [Google Scholar] [CrossRef]
- Baker, M.T.; Naguib, M. Propofol: The Challenges of Formulation. Anesthesiology 2005, 103, 860–876. [Google Scholar] [CrossRef]
- Ebo, D.G.; Piel, G.C.; Conraads, V.; Stevens, W.J. IgE-Mediated Anaphylaxis after First Intravenous Infusion of Cyclosporine. Ann. Allergy Asthma Immunol. 2001, 87, 243–245. [Google Scholar] [CrossRef]
- Jenkins, D.J.; Wolever, T.M.; Taylor, R.H.; Reynolds, D.; Nineham, R.; Hockaday, T.D. Diabetic Glucose Control, Lipids, and Trace Elements on Long-Term Guar. Br. Med. J. 1980, 280, 1353–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volcheck, G.W.; Van Dellen, R.G. Anaphylaxis to Intravenous Cyclosporine and Tolerance to Oral Cyclosporine: Case Report and Review. Ann. Allergy Asthma Immunol. 1998, 80, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Wenande, E.; Garvey, L.H. Immediate-Type Hypersensitivity to Polyethylene Glycols: A Review. Clin. Exp. Allergy 2016, 46, 907–922. [Google Scholar] [CrossRef]
- Rosenberg, A.S. Effects of Protein Aggregates: An Immunologic Perspective. AAPS J. 2006, 8, E501–E507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, W.V.; Leppert, P. Role of Aggregated Human Growth Hormone (HGH) in Development of Antibodies to HGH. J. Clin. Endocrinol. Metab. 1980, 51, 691–697. [Google Scholar] [CrossRef]
- Baert, F.; Noman, M.; Vermeire, S.; Van Assche, G.; D’ Haens, G.; Carbonez, A.; Rutgeerts, P. Influence of Immunogenicity on the Long-Term Efficacy of Infliximab in Crohn’s Disease. N. Engl. J. Med. 2003, 348, 601–608. [Google Scholar] [CrossRef]
- Cox, F.; Khalib, K.; Conlon, N. PEG That Reaction: A Case Series of Allergy to Polyethylene Glycol. J. Clin. Pharmacol. 2021, 61, 832–835. [Google Scholar] [CrossRef]
- Nagarajan, R. Solubilization of Hydrocarbons and Resulting Aggregate Shape Transitions in Aqueous Solutions of Pluronic® (PEO–PPO–PEO) Block Copolymers. Colloids Surf. B Biointerfaces 1999, 16, 55–72. [Google Scholar] [CrossRef]
- Kabanov, A.V.; Batrakova, E.V.; Alakhov, V.Y. Pluronic Block Copolymers as Novel Polymer Therapeutics for Drug and Gene Delivery. J. Control. Release 2002, 82, 189–212. [Google Scholar] [CrossRef]
- Emanuele, M.; Balasubramaniam, B. Differential Effects of Commercial-Grade and Purified Poloxamer 188 on Renal Function. Drugs R D 2014, 14, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Strickley, R.G.; Lambert, W.J. A Review of Formulations of Commercially Available Antibodies. J. Pharm. Sci. 2021, 110, 2590–2608.e56. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, S. Liquid Formulation for Antibody Drugs. Biochim. Biophys. Acta 2014, 1844, 2041–2052. [Google Scholar] [CrossRef]
- Gervasi, V.; Dall Agnol, R.; Cullen, S.; McCoy, T.; Vucen, S.; Crean, A. Parenteral Protein Formulations: An Overview of Approved Products within the European Union. Eur. J. Pharm. Biopharm. 2018, 131, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Highlights of Prescribing Information: Norditropin® (Somatropin) Injection, for Subcutaneous Use. 2016. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/021148s049lbl.pdf (accessed on 5 December 2021).
- Erlandsson, B. Stability-Indicating Changes in Poloxamers: The Degradation of Ethylene Oxide-Propylene Oxide Block Copolymers at 25 and 40 °C. Polym. Degrad. Stab. 2002, 78, 571–575. [Google Scholar] [CrossRef]
- Gallet, G.; Carroccio, S.; Rizzarelli, P.; Karlsson, S. Thermal Degradation of Poly(Ethylene Oxide–Propylene Oxide–Ethylene Oxide) Triblock Copolymer: Comparative Study by SEC/NMR, SEC/MALDI-TOF-MS and SPME/GC-MS. Polymer 2002, 43, 1081–1094. [Google Scholar] [CrossRef]
- Wang, T.; Markham, A.; Thomas, S.J.; Wang, N.; Huang, L.; Clemens, M.; Rajagopalan, N. Solution Stability of Poloxamer 188 under Stress Conditions. J. Pharm. Sci. 2019, 108, 1264–1271. [Google Scholar] [CrossRef]
- Grapentin, C.; Müller, C.; Kishore, R.S.K.; Adler, M.; ElBialy, I.; Friess, W.; Huwyler, J.; Khan, T.A. Protein-Polydimethylsiloxane Particles in Liquid Vial Monoclonal Antibody Formulations Containing Poloxamer 188. J. Pharm. Sci. 2020, 109, 2393–2404. [Google Scholar] [CrossRef]
- Wang, T.; Richard, C.A.; Dong, X.; Shi, G.H. Impact of Surfactants on the Functionality of Prefilled Syringes. J. Pharm. Sci. 2020, 109, 3413–3422. [Google Scholar] [CrossRef]
- Jiao, J. Polyoxyethylated Nonionic Surfactants and Their Applications in Topical Ocular Drug Delivery. Adv. Drug Deliv. Rev. 2008, 60, 1663–1673. [Google Scholar] [CrossRef]
- Sahoo, R.K.; Biswas, N.; Guha, A.; Sahoo, N.; Kuotsu, K. Nonionic Surfactant Vesicles in Ocular Delivery: Innovative Approaches and Perspectives. Biomed. Res. Int. 2014, 2014, 263604. [Google Scholar] [CrossRef] [Green Version]
- Mesiha, M.S.; El-Bitar, H.I. Hypoglycaemic Effect of Oral Insulin Preparations Containing Brij 35, 52, 58 or 92 and Stearic Acid. J. Pharm. Pharmacol. 1981, 33, 733–734. [Google Scholar] [CrossRef] [PubMed]
- Bam, N.B.; Randolph, T.W.; Cleland, J.L. Stability of Protein Formulations: Investigation of Surfactant Effects by a Novel EPR Spectroscopic Technique. Pharm. Res. 1995, 12, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.S.; Bam, N.B.; Randolph, T.W. Surfactant-Stabilized Protein Formulations: A Review of Protein-Surfactant Interactions and Novel Analytical Methodologies. In Therapeutic Protein and Peptide Formulation and Delivery; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1997; Volume 675, pp. 206–222. ISBN 978-0-8412-3528-1. [Google Scholar]
- Krause, M.; Rudolph, R.; Schwarz, E. The Non-Ionic Detergent Brij 58P Mimics Chaperone Effects. FEBS Lett. 2002, 532, 253–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, L.; Yan, Z.; Li, H.; Liu, X.; Sun, P. Brij-58, a Potential Injectable Protein-Stabilizer Used in Therapeutic Protein Formulation. Eur. J. Pharm. Biopharm. 2020, 146, 73–83. [Google Scholar] [CrossRef]
- Agarkhed, M.; O’Dell, C.; Hsieh, M.-C.; Zhang, J.; Goldstein, J.; Srivastava, A. Effect of Surfactants on Mechanical, Thermal, and Photostability of a Monoclonal Antibody. AAPS PharmSciTech. 2018, 19, 79–92. [Google Scholar] [CrossRef]
- Eng, Y.Y.; Sharma, V.K.; Ray, A.K. Photocatalytic Degradation of Nonionic Surfactant, Brij 35 in Aqueous TiO2 Suspensions. Chemosphere 2010, 79, 205–209. [Google Scholar] [CrossRef]
- U.S. Government Accountability Office. Alkyl (C10–C16) Polyglycosides; Exemptions from the Requirement of a Tolerance; U.S. Government Accountability Office: Washington, DC, USA, 2005. [Google Scholar]
- Maggio, E.T. Stabilizing Alkylglycoside Compositions and Methods Thereof. U.S. Patent US12/050,038, 17 March 2008. [Google Scholar]
- Rifkin, R.A.; Maggio, E.T.; Dike, S.; Kerr, D.A.; Levy, M. N-Dodecyl-β-D-Maltoside Inhibits Aggregation of Human Interferon-β-1b and Reduces Its Immunogenicity. J. Neuroimmune Pharm. 2011, 6, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Katz, J.S.; Tan, Y.; Kuppannan, K.; Song, Y.; Brennan, D.J.; Young, T.; Yao, L.; Jordan, S. Amino-Acid-Incorporating Nonionic Surfactants for Stabilization of Protein Pharmaceuticals. ACS Biomater. Sci. Eng. 2016, 2, 1093–1096. [Google Scholar] [CrossRef]
- Katz, J.S.; Nolin, A.; Yezer, B.A.; Jordan, S. Dynamic Properties of Novel Excipient Suggest Mechanism for Improved Performance in Liquid Stabilization of Protein Biologics. Mol. Pharm. 2019, 16, 282–291. [Google Scholar] [CrossRef]
- Katz, J.S.; Brennan, D.J.; Dan, F.; Tan, Y.; Jordan, S.L.; Young, T.J.; Song, Y. Polyalkoxy Fatty Compound. U.S. Patent US20180273683A1, 27 September 2018. [Google Scholar]
- Mensink, M.A.; Frijlink, H.W.; van der Voort Maarschalk, K.; Hinrichs, W.L.J. How Sugars Protect Proteins in the Solid State and during Drying (Review): Mechanisms of Stabilization in Relation to Stress Conditions. Eur. J. Pharm. Biopharm. 2017, 114, 288–295. [Google Scholar] [CrossRef]
- Olsson, C.; Swenson, J. The Role of Disaccharides for Protein–Protein Interactions—A SANS Study. Mol. Phys. 2019, 117, 3408–3416. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, J.F.; Kendrick, B.S.; Chang, B.S.; Manning, M.C.; Randolph, T.W. Inhibition of Stress-Induced Aggregation of Protein Therapeutics. In Methods in Enzymology; Amyloid, Prions, and Other Protein Aggregates; Academic Press: Cambridge, MA, USA, 1999; Volume 309, pp. 236–255. ISBN 978-0-08-049667. [Google Scholar]
- Panzica, M.; Emanuele, A.; Cordone, L. Thermal Aggregation of Bovine Serum Albumin in Trehalose and Sucrose Aqueous Solutions. J. Phys. Chem. B 2012, 116, 11829–11836. [Google Scholar] [CrossRef]
- Sun, W.Q.; Davidson, P. Protein Inactivation in Amorphous Sucrose and Trehalose Matrices: Effects of Phase Separation and Crystallization. Biochim. Et Biophys. Acta (BBA)-Gen. Subj. 1998, 1425, 235–244. [Google Scholar] [CrossRef]
- Uritani, M.; Takai, M.; Yoshinaga, K. Protective Effect of Disaccharides on Restriction Endonucleases during Drying under Vacuum. J. Biochem. 1995, 117, 774–779. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, S.; Wang, Y.J. Trehalose: Current Use and Future Applications. J. Pharm. Sci. 2011, 100, 2020–2053. [Google Scholar] [CrossRef]
- Schiefelbein, L.; Keller, M.; Weissmann, F.; Luber, M.; Bracher, F.; Frieß, W. Synthesis, Characterization and Assessment of Suitability of Trehalose Fatty Acid Esters as Alternatives for Polysorbates in Protein Formulation. Eur. J. Pharm. Biopharm. 2010, 76, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Messina, M.S.; Ko, J.H.; Yang, Z.; Strouse, M.J.; Houk, K.N.; Maynard, H.D. Effect of Trehalose Polymer Regioisomers on Protein Stabilization. Polym. Chem. 2017, 8, 4781–4788. [Google Scholar] [CrossRef]
- Mastrotto, F.; Francini, N.; Allen, S.; van der Walle, C.F.; Stolnik, S.; Mantovani, G. Synthetic Glycopolymers as Modulators of Protein Aggregation: Influences of Chemical Composition, Topology and Concentration. J. Mater. Chem. B 2018, 6, 1044–1054. [Google Scholar] [CrossRef]
- Mancini, R.J.; Lee, J.; Maynard, H.D. Trehalose Glycopolymers for Stabilization of Protein Conjugates to Environmental Stressors. J. Am. Chem. Soc. 2012, 134, 8474–8479. [Google Scholar] [CrossRef]
- Lee, J.; Lin, E.-W.; Lau, U.Y.; Hedrick, J.L.; Bat, E.; Maynard, H.D. Trehalose Glycopolymers as Excipients for Protein Stabilization. Biomacromolecules 2013, 14, 2561–2569. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lee, J.; Mansfield, K.M.; Ko, J.H.; Sallam, S.; Wesdemiotis, C.; Maynard, H.D. Trehalose Glycopolymer Enhances Both Solution Stability and Pharmacokinetics of a Therapeutic Protein. Bioconjugate Chem. 2017, 28, 836–845. [Google Scholar] [CrossRef] [Green Version]
- Schebor, C.; Burin, L.; del PilarBuera, M.; Chirife, J. Stability to Hydrolysis and Browning of Trehalose, Sucrose and Raffinose in Low-Moisture Systems in Relation to Their Use as Protectants of Dry Biomaterials. LWT-Food Sci. Technol. 1999, 32, 481–485. [Google Scholar] [CrossRef]
- Li, J.; Chen, J.; An, L.; Yuan, X.; Yao, L. Polyol and Sugar Osmolytes Can Shorten Protein Hydrogen Bonds to Modulate Function. Commun. Biol. 2020, 3, 528. [Google Scholar] [CrossRef] [PubMed]
- Timasheff, S.N. Control of Protein Stability and Reactions by Weakly Interacting Cosolvents: The Simplicity of the Complicated. Adv. Protein Chem. 1998, 51, 355–432. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, J.F.; Crowe, J.H. An Infrared Spectroscopic Study of the Interactions of Carbohydrates with Dried Proteins. Biochemistry 1989, 28, 3916–3922. [Google Scholar] [CrossRef]
- Vagenende, V.; Yap, M.G.S.; Trout, B.L. Mechanisms of Protein Stabilization and Prevention of Protein Aggregation by Glycerol. Biochemistry 2009, 48, 11084–11096. [Google Scholar] [CrossRef]
- Jena, S.; Suryanarayanan, R.; Aksan, A. Mutual Influence of Mannitol and Trehalose on Crystallization Behavior in Frozen Solutions. Pharm. Res. 2016, 33, 1413–1425. [Google Scholar] [CrossRef]
- Cui, Y.; Cui, P.; Chen, B.; Li, S.; Guan, H. Monoclonal Antibodies: Formulations of Marketed Products and Recent Advances in Novel Delivery System. Drug Dev. Ind. Pharm. 2017, 43, 519–530. [Google Scholar] [CrossRef]
- Andya, J.D.; Maa, Y.-F.; Costantino, H.R.; Nguyen, P.-A.; Dasovich, N.; Sweeney, T.D.; Hsu, C.C.; Shire, S.J. Effect of Formulation Excipients on Protein Stability and Aerosol Performance of Spray-Dried Powders of a Recombinant Humanized Anti-IgE Monoclonal Antibody1. Pharm. Res. 1999, 16, 350–358. [Google Scholar] [CrossRef]
- Al-Hussein, A.; Gieseler, H. The Effect of Mannitol Crystallization in Mannitol–Sucrose Systems on LDH Stability during Freeze-Drying. J. Pharm. Sci. 2012, 101, 2534–2544. [Google Scholar] [CrossRef]
- Izutsu, K.; Yoshioka, S.; Terao, T. Effect of Mannitol Crystallinity on the Stabilization of Enzymes during Freeze-Drying. Chem. Pharm. Bull. 1994, 42, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Izutsu, K.; Kojima, S. Excipient Crystallinity and Its Protein-Structure-Stabilizing Effect during Freeze-Drying. J. Pharm. Pharmacol. 2002, 54, 1033–1039. [Google Scholar] [CrossRef]
- Costantino, H.R.; Andya, J.D.; Nguyen, P.-A.; Dasovich, N.; Sweeney, T.D.; Shire, S.J.; Hsu, C.C.; Maa, Y.-F. Effect of Mannitol Crystallization on the Stability and Aerosol Performance of a Spray-Dried Pharmaceutical Protein, Recombinant Humanized Anti-IgE Monoclonal Antibody. J. Pharm. Sci. 1998, 87, 1406–1411. [Google Scholar] [CrossRef] [PubMed]
- Pyne, A.; Chatterjee, K.; Suryanarayanan, R. Solute Crystallization in Mannitol–Glycine Systems—Implications on Protein Stabilization in Freeze-dried Formulations. J. Pharm. Sci. 2003, 92, 2272–2283. [Google Scholar] [CrossRef]
- Thakral, S.; Sonje, J.; Munjal, B.; Bhatnagar, B.; Suryanarayanan, R. Mannitol as an Excipient for Lyophilized Injectable Formulations. J. Pharm. Sci. 2022. [Google Scholar] [CrossRef] [PubMed]
- Cleland, J.L.; Lam, X.; Kendrick, B.; Yang, J.; Yang, T.; Overcashier, D.; Brooks, D.; Hsu, C.; Carpenter, J.F. A Specific Molar Ratio of Stabilizer to Protein Is Required for Storage Stability of a Lyophilized Monoclonal Antibody. J. Pharm. Sci. 2001, 90, 310–321. [Google Scholar] [CrossRef]
- Wang, S.S.; Yan, Y.S.; Ho, K. US FDA-Approved Therapeutic Antibodies with High-Concentration Formulation: Summaries and Perspectives. Antib. Ther. 2021, 4, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Piedmonte, D.M.; Summers, C.; McAuley, A.; Karamujic, L.; Ratnaswamy, G. Sorbitol Crystallization Can Lead to Protein Aggregation in Frozen Protein Formulations. Pharm. Res. 2007, 24, 136–146. [Google Scholar] [CrossRef]
- Piedmonte, D.M.; Hair, A.; Baker, P.; Brych, L.; Nagapudi, K.; Lin, H.; Cao, W.; Hershenson, S.; Ratnaswamy, G. Sorbitol Crystallization-Induced Aggregation in Frozen MAb Formulations. J. Pharm. Sci. 2015, 104, 686–697. [Google Scholar] [CrossRef]
- Davis, J.M.; Zhang, N.; Payne, R.W.; Murphy, B.M.; Abdul-Fattah, A.M.; Matsuura, J.E.; Herman, A.C.; Manning, M.C. Stability of Lyophilized Sucrose Formulations of an IgG1: Subvisible Particle Formation. Pharm. Dev. Technol. 2013, 18, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.L.; Shepherd, D.; Sun, J.; Tang, X.C.; Pikal, M.J. Effect of Sorbitol and Residual Moisture on the Stability of Lyophilized Antibodies: Implications for the Mechanism of Protein Stabilization in the Solid State. J. Pharm. Sci. 2005, 94, 1445–1455. [Google Scholar] [CrossRef]
- Rowe, R.C.; Quinn, M.E. Handbook of Pharmaceutical Excipients, 6th ed.; Pharmaceutical Press: Washington, DC, USA, 2009. [Google Scholar]
- Kumar, A. Adverse Effects of Pharmaceutical Excipients. Advers. Drug React. Bull. 2003, 222, 851–854. [Google Scholar] [CrossRef]
- Pawar, S.; Kumar, A. Issues in the Formulation of Drugs for Oral Use in Children: Role of Excipients. Paediatr. Drugs 2002, 4, 371–379. [Google Scholar] [CrossRef]
- Sweetman, S.C. Martindale: The Complete Drug Reference, 36th ed.; Pharmaceutical Press: London, UK, 2009. [Google Scholar]
- Hägnevik, K.; Gordon, E.; Lins, L.E.; Wilhelmsson, S.; Forster, D. Glycerol-Induced Haemolysis with Haemoglobinuria and Acute Renal Failure. Report of Three Cases. Lancet 1974, 1, 75–77. [Google Scholar] [CrossRef] [PubMed]
- Welch, K.M.; Meyer, J.S.; Okamoto, S.; Mathew, N.T.; Rivera, V.M.; Bond, J. Letter: Glycerol-Induced Haemolysis. Lancet 1974, 1, 416–417. [Google Scholar] [CrossRef] [PubMed]
- Staples, R.; Misher, A.; Wardell, J., Jr. Gastrointestinal Irritant Effect of Glycerin as Compared with Sorbitol and Propylene Glycol in Rats and Dogs. J. Pharm. Sci. 1967, 56, 398–400. [Google Scholar] [CrossRef]
- Porter, G.A.; Starr, A.; Kimsey, J.; Lenertz, H. Mannitol Hemodilution-Perfusion: The Kinetics of Mannitol Distribution and Excretion during Cardiopulmonary Bypass. J. Surg. Res. 1967, 7, 447–456. [Google Scholar] [CrossRef]
- Safety Assessment of Mannitol, Sorbitol, and Xylitol as Used in Cosmetics 2019. Available online: https://www.cir-safety.org/sites/default/files/Mannitol,%20Sorbitol,%20Xylitol_0.pdf (accessed on 29 September 2022).
- McNeill, I.Y. Hypersensitivity Reaction to Mannitol. Drug Intell. Clin. Pharm. 1985, 19, 552–553. [Google Scholar] [CrossRef] [PubMed]
- Lamb, J.D.; Keogh, J.A. Anaphylactoid Reaction to Mannitol. Can. Anaesth Soc. J. 1979, 26, 435–436. [Google Scholar] [CrossRef] [Green Version]
- Spaeth, G.L.; Spaeth, E.B.; Spaeth, P.G.; Lucier, A.C. Anaphylactic Reaction to Mannitol. Arch. Ophthalmol. 1967, 78, 583–584. [Google Scholar] [CrossRef]
- Serno, T.; Geidobler, R.; Winter, G. Protein Stabilization by Cyclodextrins in the Liquid and Dried State. Adv. Drug Deliv. Rev. 2011, 63, 1086–1106. [Google Scholar] [CrossRef]
- Vyas, A.; Saraf, S.; Saraf, S. Cyclodextrin Based Novel Drug Delivery Systems. J. Incl. Phenom. Macrocycl. Chem. 2008, 62, 23–42. [Google Scholar] [CrossRef]
- Gidwani, B.; Vyas, A. A Comprehensive Review on Cyclodextrin-Based Carriers for Delivery of Chemotherapeutic Cytotoxic Anticancer Drugs. Biomed. Res. Int. 2015, 2015, 198268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, L.; Campos, J.; Veiga, F.; Cardoso, C.; Paiva-Santos, A.C. Cyclodextrin-Based Delivery Systems in Parenteral Formulations: A Critical Update Review. Eur. J. Pharm. Biopharm. 2022, 178, 35–52. [Google Scholar] [CrossRef]
- Charman, S.A.; Mason, K.L.; Charman, W.N. Techniques for Assessing the Effects of Pharmaceutical Excipients on the Aggregation of Porcine Growth Hormone. Pharm. Res. 1993, 10, 954–962. [Google Scholar] [CrossRef]
- Tavornvipas, S.; Tajiri, S.; Hirayama, F.; Arima, H.; Uekama, K. Effects of Hydrophilic Cyclodextrins on Aggregation of Recombinant Human Growth Hormone. Pharm. Res. 2004, 21, 2369–2376. [Google Scholar] [CrossRef]
- Taneri, F.; Güneri, T.; Aigner, Z.; Kata, M. Improvement in the Physicochemical Properties of Ketoconazole through Complexation with Cyclodextrin Derivatives. J. Incl. Phenom. 2002, 44, 257–260. [Google Scholar] [CrossRef]
- Yoshida, A.; Arima, H.; Uekama, K.; Pitha, J. Pharmaceutical Evaluation of Hydroxyalkyl Ethers of β-Cyclodextrins. Int. J. Pharm. 1988, 46, 217–222. [Google Scholar] [CrossRef]
- Samra, H.S.; He, F.; Bhambhani, A.; Pipkin, J.D.; Zimmerer, R.; Joshi, S.B.; Middaugh, C.R. The Effects of Substituted Cyclodextrins on the Colloidal and Conformational Stability of Selected Proteins. J. Pharm. Sci. 2010, 99, 2800–2818. [Google Scholar] [CrossRef]
- Serno, T.; Carpenter, J.F.; Randolph, T.W.; Winter, G. Inhibition of Agitation-Induced Aggregation of an IgG-Antibody by Hydroxypropyl-Beta-Cyclodextrin. J. Pharm. Sci. 2010, 99, 1193–1206. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.H.; Garidel, P.; Michaela, B. HP-β-CD for the Formulation of IgG and Ig-Based Biotherapeutics. Int. J. Pharm. 2021, 601, 120531. [Google Scholar] [CrossRef]
- Aachmann, F.L.; Otzen, D.E.; Larsen, K.L.; Wimmer, R. Structural Background of Cyclodextrin–Protein Interactions. Protein Eng. Des. Sel. 2003, 16, 905–912. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Hong, S.; Tan, S.S.K.; Peng, T.; Goh, L.Y.H.; Lam, K.H.; Chow, K.T.; Gokhale, R. Polysorbates versus Hydroxypropyl Beta-Cyclodextrin (HPβCD): Comparative Study on Excipient Stability and Stabilization Benefits on Monoclonal Antibodies. Molecules 2022, 27, 6497. [Google Scholar] [CrossRef]
- Stam, M.R.; Danchin, E.G.J.; Rancurel, C.; Coutinho, P.M.; Henrissat, B. Dividing the Large Glycoside Hydrolase Family 13 into Subfamilies: Towards Improved Functional Annotations of Alpha-Amylase-Related Proteins. Protein Eng. Des. Sel. 2006, 19, 555–562. [Google Scholar] [CrossRef] [Green Version]
- Karginov, V.A. Cyclodextrin Derivatives as Anti-Infectives. Curr. Opin. Pharmacol. 2013, 13, 717–725. [Google Scholar] [CrossRef]
- Frömming, K.-H.; Szejtli, J. Cyclodextrins in Pharmacy; Springer Science and Business Media: Berlin, Germany, 1994; ISBN 978-94-015-8277-3. [Google Scholar]
- Buedenbender, S.; Schulz, G.E. Structural Base for Enzymatic Cyclodextrin Hydrolysis. J. Mol. Biol. 2009, 385, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Atwood, J.L.; Lehn, J.-M.; Szejtli, J. The Metabolism, Toxicity Abd Biological Effects of Cyclodextrins. In Comprehensive Supramolecular Chemistry; Pergamon: Oxford, UK, 1996; Volume 3, ISBN 978-0-08-042715-7. [Google Scholar]
- Munro, I.C.; Newberne, P.M.; Young, V.R.; Bär, A. Safety Assessment of Gamma-Cyclodextrin. Regul. Toxicol. Pharmacol. 2004, 39 (Suppl. S1), S3–S13. [Google Scholar] [CrossRef]
- Jodál, I.; Kandra, L.; Harangi, J.; Nánási, P.; Szejtli, J. Enzymatic Degradation of Cyclodextrins; Preparation and Application of Their Fragments. J. Incl. Phenom. 1984, 2, 877–884. [Google Scholar] [CrossRef]
- Turner, P.; Labes, A.; Fridjonsson, O.H.; Hreggvidson, G.O.; Schönheit, P.; Kristjansson, J.K.; Holst, O.; Karlsson, E.N. Two Novel Cyclodextrin-Degrading Enzymes Isolated from Thermophilic Bacteria Have Similar Domain Structures but Differ in Oligomeric State and Activity Profile. J. Biosci. Bioeng. 2005, 100, 380–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connolly, B.; HAMBURG, L.; HOLZ, E. Formulations with Reduced Degradation of Polysorbate. French Patent WO2017117311A1, 6 July 2017. [Google Scholar]
- Uchida, K.; Kawakishi, S. Oxidative Degradation of β-Cyclodextrin Induced by an Ascorbic Acid-Copper Ion System. Agric. Biol. Chem. 1986, 50, 367–373. [Google Scholar] [CrossRef]
- Mamani, P.L.; Ruiz-Caro, R.; Veiga, M.D. Matrix Tablets: The Effect of Hydroxypropyl Methylcellulose/Anhydrous Dibasic Calcium Phosphate Ratio on the Release Rate of a Water-Soluble Drug through the Gastrointestinal Tract I. in vitro Tests. AAPS PharmSciTech. 2012, 13, 1073–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, T.; Watanabe, Y.; Takayama, K.; Endo, H.; Matsumoto, M. Effect of Hydroxypropylmethylcellulose (HPMC) on the Release Profiles and Bioavailability of a Poorly Water-Soluble Drug from Tablets Prepared Using Macrogol and HPMC. Int. J. Pharm. 2000, 202, 173–178. [Google Scholar] [CrossRef]
- Mašková, E.; Kubová, K.; Raimi-Abraham, B.T.; Vllasaliu, D.; Vohlídalová, E.; Turánek, J.; Mašek, J. Hypromellose-A Traditional Pharmaceutical Excipient with Modern Applications in Oral and Oromucosal Drug Delivery. J. Control. Release 2020, 324, 695–727. [Google Scholar] [CrossRef]
- CiNii. Final Report on the Safety Assessment of Hydroxyethylcellulose, Hydroxypropylcellulose, Methylcellulose, Hydroxypropyl Methylcellulose, and Cellulose Gum. J. Am. Coll. Toxicol. 1986, 5, 1–59. [Google Scholar] [CrossRef]
- Soane, D.S.; Mahoney, R.P.; Wuthrich, P.; GREENE, D.G. Stabilizing Excipients for Therapeutic Protein Formulations. French Patent WO2019036619, 21 February 2019. [Google Scholar]
- Lynch, S.; Turowski, M.; Yokoyama, W.; Hong, Y.; Conklin, J.; Hung, S.; Young, S. Uses of Water-Soluble Cellulose Derivatives for Preventing or Treating Metabolic Syndrome. French Patent WO2008051794, 2 May 2008. [Google Scholar]
- Sasahara, K.; McPhie, P.; Minton, A.P. Effect of Dextran on Protein Stability and Conformation Attributed to Macromolecular Crowding. J. Mol. Biol. 2003, 326, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Flood, A.; Estrada, M.; McAdams, D.; Ji, Y.; Chen, D. Development of a Freeze-Dried, Heat-Stable Influenza Subunit Vaccine Formulation. PLoS ONE 2016, 11, e0164692. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.Q.; Davidson, P. Effect of Dextran Molecular Weight on Protein Stabilization during Freeze-Drying and Storage. Cryo Lett. 2001, 22, 285–292. [Google Scholar]
- Jones, B.; Mahajan, A.; Aksan, A. Dextranol: A Better Lyoprotectant. bioRxiv 2018, 490441. [Google Scholar] [CrossRef]
- Rudin, C.; Günthard, J.; Halter, C.; Staehelin, J.; Berglund, A. Anaphylactoid Reaction to BCG Vaccine Containing High Molecular Weight Dextran. Eur. J. Pediatr. 1995, 154, 941–942. [Google Scholar] [CrossRef]
- Uchida, S.; Takekawa, D.; Kitayama, M.; Hirota, K. Two Cases of Circulatory Collapse Due to Suspected Remimazolam Anaphylaxis. JA Clin. Rep. 2022, 8, 18. [Google Scholar] [CrossRef]
- Bozorgmehr, M.R.; Monhemi, H. How Can a Free Amino Acid Stabilize a Protein? Insights from Molecular Dynamics Simulation. J. Solut. Chem. 2015, 44, 45–53. [Google Scholar] [CrossRef]
- Bruździak, P.; Panuszko, A.; Kaczkowska, E.; Piotrowski, B.; Daghir, A.; Demkowicz, S.; Stangret, J. Taurine as a Water Structure Breaker and Protein Stabilizer. Amino Acids 2018, 50, 125–140. [Google Scholar] [CrossRef] [Green Version]
- Platts, L.; Falconer, R.J. Controlling Protein Stability: Mechanisms Revealed Using Formulations of Arginine, Glycine and Guanidinium HCl with Three Globular Proteins. Int. J. Pharm. 2015, 486, 131–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiraki, K.; Kudou, M.; Fujiwara, S.; Imanaka, T.; Takagi, M. Biophysical Effect of Amino Acids on the Prevention of Protein Aggregation. J. Biochem. 2002, 132, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Mojtabavi, S.; Samadi, N.; Faramarzi, M.A. Osmolyte-Induced Folding and Stability of Proteins: Concepts and Characterization. Iran. J. Pharm. Res. 2019, 18, 13–30. [Google Scholar] [CrossRef]
- Arakawa, T.; Tsumoto, K. The Effects of Arginine on Refolding of Aggregated Proteins: Not Facilitate Refolding, but Suppress Aggregation. Biochem. Biophys. Res. Commun. 2003, 304, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Inoue, N.; Takai, E.; Arakawa, T.; Shiraki, K. Arginine and Lysine Reduce the High Viscosity of Serum Albumin Solutions for Pharmaceutical Injection. J. Biosci. Bioeng. 2014, 117, 539–543. [Google Scholar] [CrossRef] [Green Version]
- Inoue, N.; Takai, E.; Arakawa, T.; Shiraki, K. Specific Decrease in Solution Viscosity of Antibodies by Arginine for Therapeutic Formulations. Mol. Pharm. 2014, 11, 1889–1896. [Google Scholar] [CrossRef]
- Das, U.; Hariprasad, G.; Ethayathulla, A.S.; Manral, P.; Das, T.K.; Pasha, S.; Mann, A.; Ganguli, M.; Verma, A.K.; Bhat, R.; et al. Inhibition of Protein Aggregation: Supramolecular Assemblies of Arginine Hold the Key. PLoS ONE 2007, 2, e1176. [Google Scholar] [CrossRef] [Green Version]
- Shukla, D.; Trout, B.L. Interaction of Arginine with Proteins and the Mechanism by Which It Inhibits Aggregation. J. Phys. Chem. B 2010, 114, 13426–13438. [Google Scholar] [CrossRef]
- Stärtzel, P. Arginine as an Excipient for Protein Freeze-Drying: A Mini Review. J. Pharm. Sci. 2018, 107, 960–967. [Google Scholar] [CrossRef]
- Falconer, R.J.; Chan, C.; Hughes, K.; Munro, T.P. Stabilization of a Monoclonal Antibody during Purification and Formulation by Addition of Basic Amino Acid Excipients. J. Chem. Technol. Biotechnol. 2011, 86, 942–948. [Google Scholar] [CrossRef]
- Holeček, M. Histidine in Health and Disease: Metabolism, Physiological Importance, and Use as a Supplement. Nutrients 2020, 12, E848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bramham, J.E.; Davies, S.A.; Podmore, A.; Golovanov, A.P. Stability of a High-Concentration Monoclonal Antibody Solution Produced by Liquid–Liquid Phase Separation. MAbs 2021, 13, 1940666. [Google Scholar] [CrossRef]
- Burnett, C.L.; Heldreth, B.; Bergfeld, W.F.; Belsito, D.V.; Hill, R.A.; Klaassen, C.D.; Liebler, D.C.; Marks, J.G.; Shank, R.C.; Slaga, T.J.; et al. Safety Assessment of α-Amino Acids as Used in Cosmetics. Int. J. Toxicol. 2013, 32, 41S–64S. [Google Scholar] [CrossRef] [PubMed]
- Label: R-Gene® 10 Arginine Hydrochloride Injection, USP for Intravenous Use. Available online: https://labeling.pfizer.com/ShowLabeling.aspx?id=642 (accessed on 15 October 2022).
- Tangphao, O.; Grossmann, M.; Chalon, S.; Hoffman, B.B.; Blaschke, T.F. Pharmacokinetics of Intravenous and Oral L-Arginine in Normal Volunteers. Br. J. Clin. Pharmacol. 1999, 47, 261–266. [Google Scholar] [CrossRef]
- Bode-Böger, S.M.; Böger, R.H.; Creutzig, A.; Tsikas, D.; Gutzki, F.M.; Alexander, K.; Frölich, J.C. L-Arginine Infusion Decreases Peripheral Arterial Resistance and Inhibits Platelet Aggregation in Healthy Subjects. Clin. Sci. 1994, 87, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Registration Dossier-ECHA. Available online: https://echa.europa.eu/de/registration-dossier/-/registered-dossier/13725/7/3/1 (accessed on 27 September 2022).
- Tsubuku, S.; Hatayama, K.; Mawatari, K.; Smriga, M.; Kimura, T. Thirteen-Week Oral Toxicity Study of l-Arginine in Rats. Int. J. Toxicol. 2004, 23, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Tsai-Turton, M. Pharmacology/Toxicology NDA/BLA Review and Evaluation; FDA: Silver Spring, MA, USA, 2016. [Google Scholar]
- Minois, N.; Carmona-Gutierrez, D.; Madeo, F. Polyamines in Aging and Disease. Aging 2011, 3, 716–732. [Google Scholar] [CrossRef] [Green Version]
- Kudou, M.; Shiraki, K.; Fujiwara, S.; Imanaka, T.; Takagi, M. Prevention of Thermal Inactivation and Aggregation of Lysozyme by Polyamines. Eur. J. Biochem. 2003, 270, 4547–4554. [Google Scholar] [CrossRef]
- Okanojo, M.; Shiraki, K.; Kudou, M.; Nishikori, S.; Takagi, M. Diamines Prevent Thermal Aggregation and Inactivation of Lysozyme. J. Biosci. Bioeng. 2005, 100, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, C.; Stekovic, S.; Wirth, M.; Benson, G.; Royer, P.; Sigrist, S.J.; Pieber, T.; Dammbrueck, C.; Magnes, C.; Eisenberg, T.; et al. Safety and Tolerability of Spermidine Supplementation in Mice and Older Adults with Subjective Cognitive Decline. Aging 2018, 10, 19–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Til, H.P.; Falke, H.E.; Prinsen, M.K.; Willems, M.I. Acute and Subacute Toxicity of Tyramine, Spermidine, Spermine, Putrescine and Cadaverine in Rats. Food Chem. Toxicol. 1997, 35, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Truong-Le, V.; Lovalenti, P.M.; Abdul-Fattah, A.M. Stabilization Challenges and Formulation Strategies Associated with Oral Biologic Drug Delivery Systems. Adv. Drug Deliv. Rev. 2015, 93, 95–108. [Google Scholar] [CrossRef]
- Sönksen, P.H.; Ellis, J.P.; Lowy, C.; Rutherford, A.; Nabarro, J.D.N. A Quantitative Evaluation of the Relative Efficiency of Gelatine and Albumin in Preventing Insulin Adsorption to Glass. Diabetologia 1966, 1, 208–210. [Google Scholar] [CrossRef] [Green Version]
- Jeyachandran, Y.L.; Mielczarski, E.; Rai, B.; Mielczarski, J.A. Quantitative and Qualitative Evaluation of Adsorption/Desorption of Bovine Serum Albumin on Hydrophilic and Hydrophobic Surfaces. Langmuir 2009, 25, 11614–11620. [Google Scholar] [CrossRef]
- Haeuser, C.; Goldbach, P.; Huwyler, J.; Friess, W.; Allmendinger, A. Excipients for Room Temperature Stable Freeze-Dried Monoclonal Antibody Formulations. J. Pharm. Sci. 2020, 109, 807–817. [Google Scholar] [CrossRef] [Green Version]
- Sønderby, P.; Bukrinski, J.T.; Hebditch, M.; Peters, G.H.J.; Curtis, R.A.; Harris, P. Self-Interaction of Human Serum Albumin: A Formulation Perspective. ACS Omega 2018, 3, 16105–16117. [Google Scholar] [CrossRef] [Green Version]
- Castro, L.S.; Lobo, G.S.; Pereira, P.; Freire, M.G.; Neves, M.C.; Pedro, A.Q. Interferon-Based Biopharmaceuticals: Overview on the Production, Purification, and Formulation. Vaccines 2021, 9, 328. [Google Scholar] [CrossRef]
- Taverna, M.; Marie, A.-L.; Mira, J.-P.; Guidet, B. Specific Antioxidant Properties of Human Serum Albumin. Ann. Intensive Care 2013, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- Chou, D.K.; Krishnamurthy, R.; Manning, M.C.; Randolph, T.W.; Carpenter, J.F. Physical Stability of Albinterferon-α(2b) in Aqueous Solution: Effects of Conformational Stability and Colloidal Stability on Aggregation. J. Pharm. Sci. 2012, 101, 2702–2719. [Google Scholar] [CrossRef]
- Tarelli, E.; Mire-Sluis, A.; Tivnann, H.A.; Bolgiano, B.; Crane, D.T.; Gee, C.; Lemercinier, X.; Athayde, M.L.; Sutcliffe, N.; Corran, P.H.; et al. Recombinant Human Albumin as a Stabilizer for Biological Materials and for the Preparation of International Reference Reagents. Biologicals 1998, 26, 331–346. [Google Scholar] [CrossRef]
- Rajan, R.S.; Li, T.; Aras, M.; Sloey, C.; Sutherland, W.; Arai, H.; Briddell, R.; Kinstler, O.; Lueras, A.M.K.; Zhang, Y.; et al. Modulation of Protein Aggregation by Polyethylene Glycol Conjugation: GCSF as a Case Study. Protein Sci. 2006, 15, 1063–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, S.-H.; Matthews, S.S.; Paxman, R.; Aksimentiev, A.; Gruebele, M.; Price, J.L. Two Structural Scenarios for Protein Stabilization by PEG. J. Phys. Chem. B 2014, 118, 8388–8395. [Google Scholar] [CrossRef]
- Rawat, S.; Raman Suri, C.; Sahoo, D.K. Molecular Mechanism of Polyethylene Glycol Mediated Stabilization of Protein. Biochem. Biophys. Res. Commun. 2010, 392, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Vrkljan, M.; Foster, T.M.; Powers, M.E.; Henkin, J.; Porter, W.R.; Staack, H.; Carpenter, J.F.; Manning, M.C. Thermal Stability of Low Molecular Weight Urokinase during Heat Treatment. II. Effect of Polymeric Additives. Pharm. Res. 1994, 11, 1004–1008. [Google Scholar] [CrossRef]
- El-hoshoudy, A.N. Experimental and Theoretical Investigation of Glycol-Based Hydrogels through Waterflooding Processes in Oil Reservoirs Using Molecular Dynamics and Dissipative Particle Dynamics Simulation. ACS Omega 2021, 6, 30224–30240. [Google Scholar] [CrossRef] [PubMed]
- McCallen, J.; Prybylski, J.; Yang, Q.; Lai, S.K. Cross-Reactivity of Select PEG-Binding Antibodies to Other Polymers Containing a C-C-O Backbone. ACS Biomater. Sci. Eng. 2017, 3, 1605–1615. [Google Scholar] [CrossRef]
- Rajan, R.; Ahmed, S.; Sharma, N.; Kumar, N.; Debas, A.; Matsumura, K. Review of the Current State of Protein Aggregation Inhibition from a Materials Chemistry Perspective: Special Focus on Polymeric Materials. Mater. Adv. 2021, 2, 1139–1176. [Google Scholar] [CrossRef]
- Rajan, R.; Matsumura, K. Inhibition of Protein Aggregation by Zwitterionic Polymer-Based Core-Shell Nanogels. Sci. Rep. 2017, 7, 45777. [Google Scholar] [CrossRef] [Green Version]
- Rajan, R.; Suzuki, Y.; Matsumura, K. Zwitterionic Polymer Design That Inhibits Aggregation and Facilitates Insulin Refolding: Mechanistic Insights and Importance of Hydrophobicity. Macromol. Biosci. 2018, 18, e1800016. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Rajan, R.; Matsumura, K. Dual Thermo- and PH-Responsive Behavior of Double Zwitterionic Graft Copolymers for Suppression of Protein Aggregation and Protein Release. ACS Appl. Mater. Interfaces 2019, 11, 39459–39469. [Google Scholar] [CrossRef] [PubMed]
- Reslan, M.; Kayser, V. Ionic Liquids as Biocompatible Stabilizers of Proteins. Biophys. Rev. 2018, 10, 781–793. [Google Scholar] [CrossRef]
- Shukla, S.K.; Mikkola, J.-P. Use of Ionic Liquids in Protein and DNA Chemistry. Front. Chem. 2020, 8, 598662. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Kumari, M.; Khan, A.B. Recent Advances in the Applications of Ionic Liquids in Protein Stability and Activity: A Review. Appl. Biochem. Biotechnol. 2014, 172, 3701–3720. [Google Scholar] [CrossRef]
- Bui-Le, L.; Clarke, C.J.; Bröhl, A.; Brogan, A.P.S.; Arpino, J.A.J.; Polizzi, K.M.; Hallett, J.P. Revealing the Complexity of Ionic Liquid–Protein Interactions through a Multi-Technique Investigation. Commun. Chem. 2020, 3, 55. [Google Scholar] [CrossRef]
- Kumar, A.; Venkatesu, P. Prevention of Insulin Self-Aggregation by a Protic Ionic Liquid. RSC Adv. 2012, 3, 362–367. [Google Scholar] [CrossRef]
- Summers, C.A.; Flowers II, R.A. Protein Renaturation by the Liquid Organic Salt Ethylammonium Nitrate. Protein Sci. 2000, 9, 2001–2008. [Google Scholar] [CrossRef] [Green Version]
- Bisht, M.; Kumar, A.; Venkatesu, P. Refolding Effects of Partially Immiscible Ammonium-Based Ionic Liquids on the Urea-Induced Unfolded Lysozyme Structure. Phys. Chem. Chem. Phys. 2016, 18, 12419–12422. [Google Scholar] [CrossRef] [Green Version]
- Mann, J.P.; McCluskey, A.; Atkin, R. Activity and Thermal Stability of Lysozyme in Alkylammonium Formate Ionic Liquids—Influence of Cation Modification. Green Chem. 2009, 11, 785–792. [Google Scholar] [CrossRef]
- Satish, L.; Rana, S.; Arakha, M.; Rout, L.; Ekka, B.; Jha, S.; Dash, P.; Sahoo, H. Impact of Imidazolium-Based Ionic Liquids on the Structure and Stability of Lysozyme. Spectrosc. Lett. 2016, 49, 383–390. [Google Scholar] [CrossRef]
- Singh, U.K.; Kumari, M.; Khan, S.H.; Bohidar, H.B.; Patel, R. Mechanism and Dynamics of Long-Term Stability of Cytochrome c Conferred by Long-Chain Imidazolium Ionic Liquids at Low Concentration. ACS Sustain. Chem. Eng. 2018, 6, 803–815. [Google Scholar] [CrossRef]
- Lange, C.; Patil, G.; Rudolph, R. Ionic Liquids as Refolding Additives: N′-Alkyl and N′-(ω-Hydroxyalkyl) N-Methylimidazolium Chlorides. Protein Sci. 2005, 14, 2693–2701. [Google Scholar] [CrossRef]
- Sindhu, A.; Bhakuni, K.; Sankaranarayanan, K.; Venkatesu, P. Implications of Imidazolium-Based Ionic Liquids as Refolding Additives for Urea-Induced Denatured Serum Albumins. ACS Sustain. Chem. Eng. 2020, 8, 604–612. [Google Scholar] [CrossRef]
- Constatinescu, D.; Herrmann, C.; Weingärtner, H. Patterns of Protein Unfolding and Protein Aggregation in Ionic Liquids. Phys. Chem. Chem. Phys. 2010, 12, 1756–1763. [Google Scholar] [CrossRef]
- Zhao, H. Protein Stabilization and Enzyme Activation in Ionic Liquids: Specific Ion Effects. J. Chem. Technol. Biotechnol. 2016, 91, 25–50. [Google Scholar] [CrossRef] [PubMed]
- Bisht, M.; Kumar, A.; Venkatesu, P. Analysis of the Driving Force That Rule the Stability of Lysozyme in Alkylammonium-Based Ionic Liquids. Int. J. Biol. Macromol. 2015, 81, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Baker, G.A.; Heller, W.T. Small-Angle Neutron Scattering Studies of Model Protein Denaturation in Aqueous Solutions of the Ionic Liquid 1-Butyl-3-Methylimidazolium Chloride. Chem. Eng. J. 2009, 147, 6–12. [Google Scholar] [CrossRef]
- Kumar, A.; Venkatesu, P. The Stability of Insulin in the Presence of Short Alkyl Chain Imidazolium-Based Ionic Liquids. RSC Adv. 2014, 4, 4487–4499. [Google Scholar] [CrossRef]
- Kumar, P.K.; Jha, I.; Venkatesu, P.; Bahadur, I.; Ebenso, E.E. A Comparative Study of the Stability of Stem Bromelain Based on the Variation of Anions of Imidazolium-Based Ionic Liquids. J. Mol. Liq. 2017, 246, 178–186. [Google Scholar] [CrossRef]
- Ajloo, D.; Sangian, M.; Ghadamgahi, M.; Evini, M.; Saboury, A.A. Effect of Two Imidazolium Derivatives of Ionic Liquids on the Structure and Activity of Adenosine Deaminase. Int. J. Biol. Macromol. 2013, 55, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Veríssimo, N.V.; Saponi, C.F.; Ryan, T.M.; Greaves, T.L.; Pereira, J.F.B. Imidazolium-Based Ionic Liquids as Additives to Preserve the Enhanced Green Fluorescent Protein Fluorescent Activity. Green Chem. Eng. 2021, 2, 412–422. [Google Scholar] [CrossRef]
- Cheng, K.; Wu, Q.; Jiang, L.; Liu, M.; Li, C. Protein Stability Analysis in Ionic Liquids by 19F NMR. Anal. Bioanal. Chem. 2019, 411, 4929–4935. [Google Scholar] [CrossRef] [PubMed]
- Roethlisberger, D.; Mahler, H.-C.; Altenburger, U.; Pappenberger, A. If Euhydric and Isotonic Do Not Work, What Are Acceptable PH and Osmolality for Parenteral Drug Dosage Forms? J. Pharm. Sci. 2017, 106, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Foureau, D.M.; Vrikkis, R.M.; Jones, C.P.; Weaver, K.D.; Macfarlane, D.R.; Salo, J.C.; McKillop, I.H.; Elliott, G.D. In Vitro Assessment of Choline Dihydrogen Phosphate (CDHP) as a Vehicle for Recombinant Human Interleukin-2 (RhIL-2). Cell Mol. Bioeng. 2012, 5, 390–401. [Google Scholar] [CrossRef]
- Nony, P.; Girard, P.; Chabaud, S.; Hessel, L.; Thébault, C.; Boissel, J.P. Impact of Osmolality on Burning Sensations during and Immediately after Intramuscular Injection of 0.5 Ml of Vaccine Suspensions in Healthy Adults. Vaccine 2001, 19, 3645–3651. [Google Scholar] [CrossRef]
- Kumar, A.; Bisht, M.; Jha, I.; Venkatesu, P. The Role of Ionic Liquids in Protein Folding/ Unfolding Studies. In Progress and Developments in Ionic Liquids; IntechOpen: London, UK, 2017; ISBN 978-953-51-2902-8. [Google Scholar]
- Gonçalves, A.R.P.; Paredes, X.; Cristino, A.F.; Santos, F.J.V.; Queirós, C.S.G.P. Ionic Liquids—A Review of Their Toxicity to Living Organisms. Int. J. Mol. Sci. 2021, 22, 5612. [Google Scholar] [CrossRef] [PubMed]
- Thuy Pham, T.P.; Cho, C.-W.; Yun, Y.-S. Environmental Fate and Toxicity of Ionic Liquids: A Review. Water Res. 2010, 44, 352–372. [Google Scholar] [CrossRef]
- Musiał, M.; Zorębski, E.; Malarz, K.; Kuczak, M.; Mrozek-Wilczkiewicz, A.; Jacquemin, J.; Dzida, M. Cytotoxicity of Ionic Liquids on Normal Human Dermal Fibroblasts in the Context of Their Present and Future Applications. ACS Sustain. Chem. Eng. 2021, 9, 7649–7657. [Google Scholar] [CrossRef]
- Gouveia, W.; Jorge, T.F.; Martins, S.; Meireles, M.; Carolino, M.; Cruz, C.; Almeida, T.V.; Araújo, M.E.M. Toxicity of Ionic Liquids Prepared from Biomaterials. Chemosphere 2014, 104, 51–56. [Google Scholar] [CrossRef]
- Moshikur, R.M.; Chowdhury, M.R.; Moniruzzaman, M.; Goto, M. Biocompatible Ionic Liquids and Their Applications in Pharmaceutics. Green Chem. 2020, 22, 8116–8139. [Google Scholar] [CrossRef]
- Weaver, K.D.; Kim, H.J.; Sun, J.; MacFarlane, D.R.; Elliott, G.D. Cyto-Toxicity and Biocompatibility of a Family of Choline Phosphate Ionic Liquids Designed for Pharmaceutical Applications. Green Chem. 2010, 12, 507–513. [Google Scholar] [CrossRef]
- Raiguel, S.; Dehaen, W.; Binnemans, K. Stability of Ionic Liquids in Brønsted-Basic Media. Green Chem. 2020, 22, 5225–5252. [Google Scholar] [CrossRef]






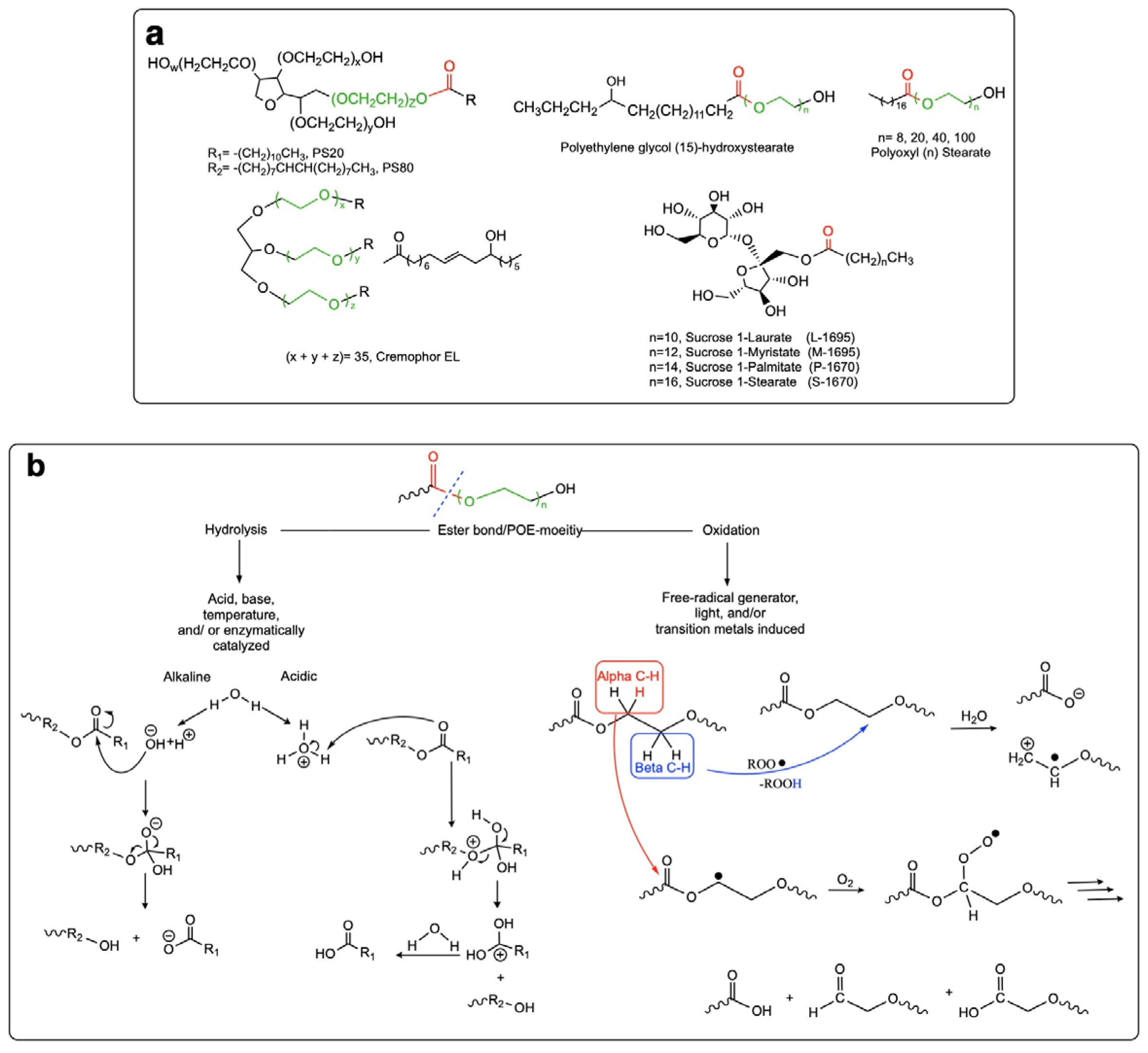{kind=link}
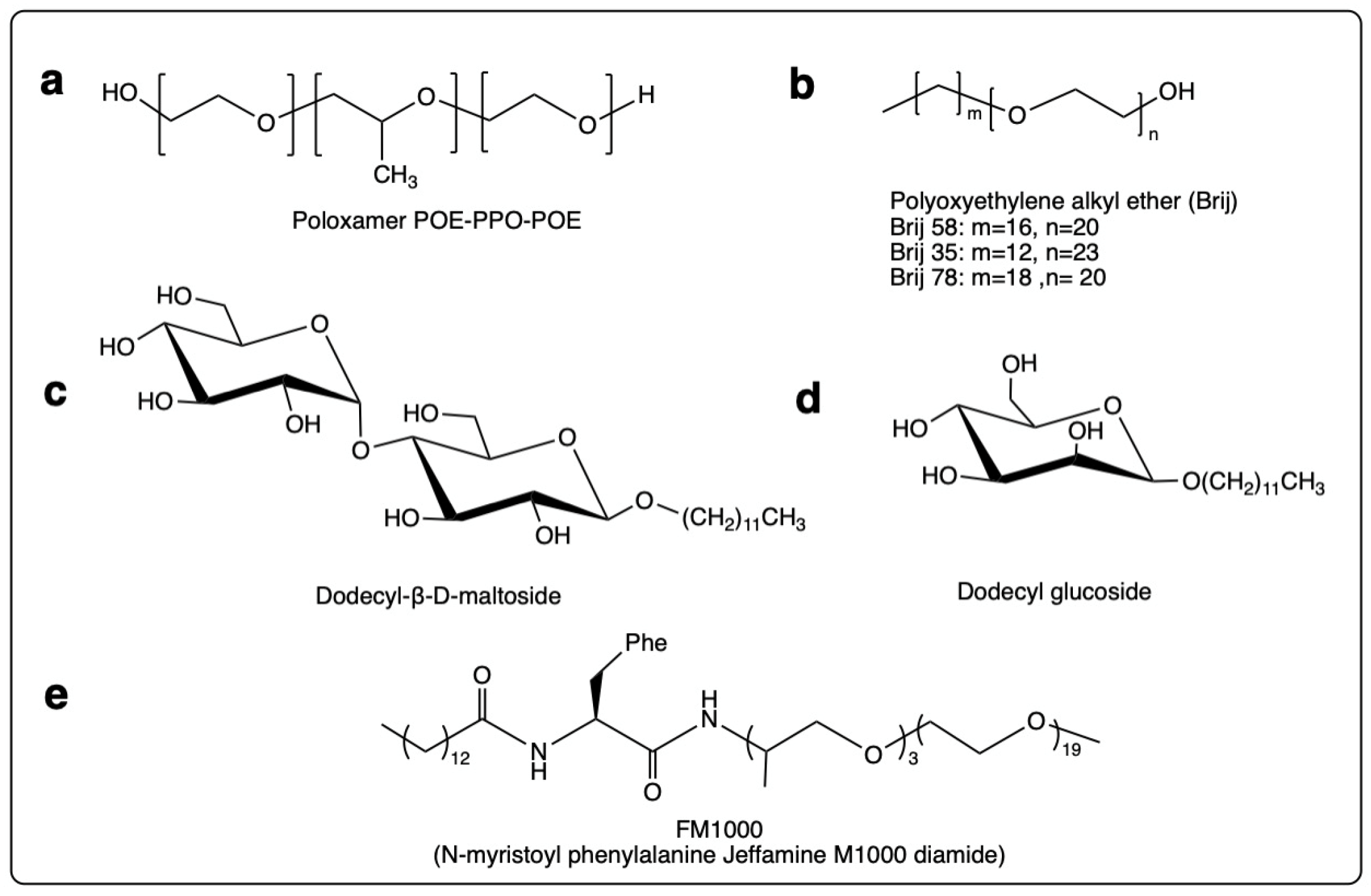{kind=link}
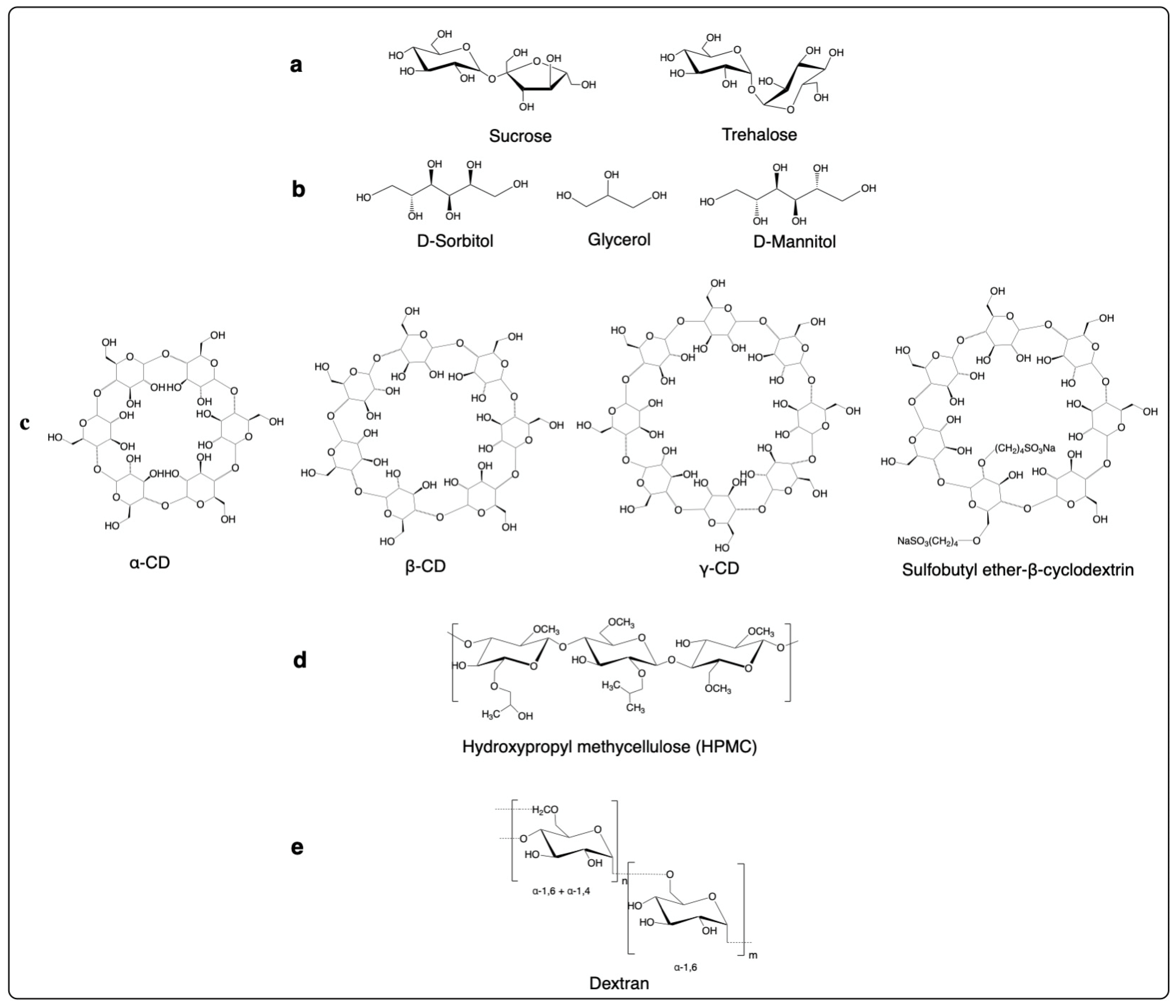{kind=link}
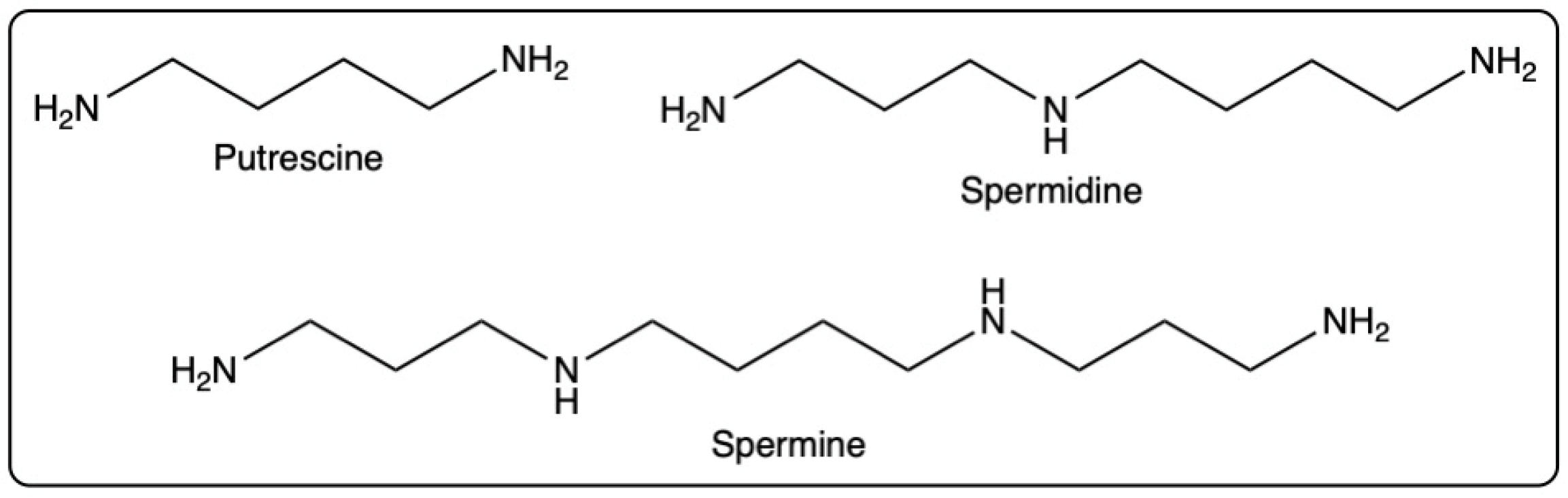{kind=link}
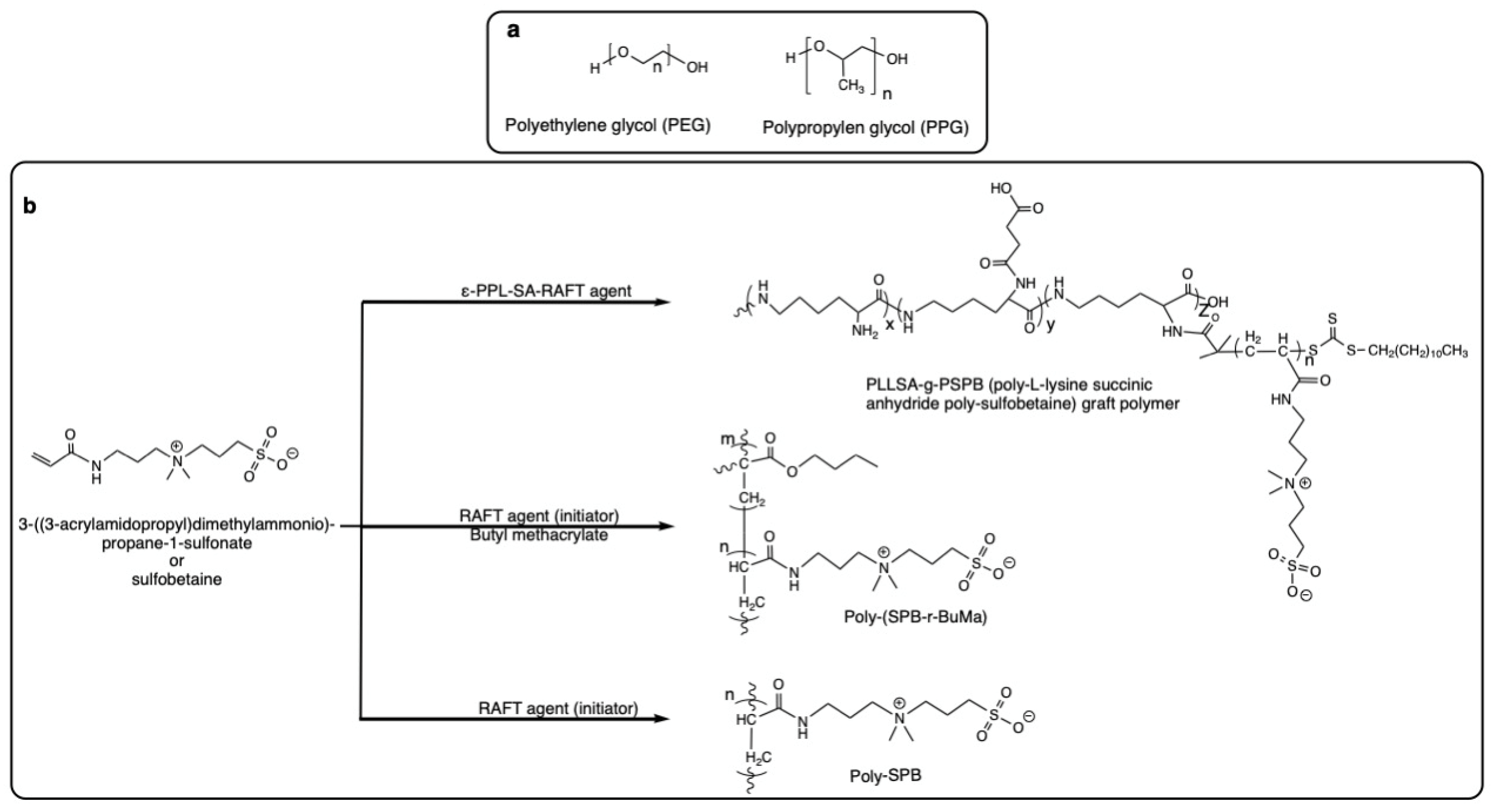{kind=link}
| Fatty Acid | Polysorbate 20 [%] | Polysorbate 80 [%] |
|---|---|---|
| Caproic acid | max. 1.0 | - |
| Caprylic acid | max. 1.0 | - |
| Capric acid | max. 10.0 | - |
| Lauric acid | 40.0–60.0 | - |
| Myristic acid | 14.0–25.0 | max. 5.0 |
| Palmitic acid | 7.0–15.0 | max. 16.0 |
| Palmitoleic acid | - | max. 8.0 |
| Stearic acid | max. 7.0 | max. 6.0 |
| Oleic acid | max. 11.0 | Min. 58.0 |
| Linoleic acid | max. 3.0 | max. 18.0 |
| Linolenic acid | - | max. 4.0 |
| Excipient | Protein Stabilization Efficiency | Physicochemical Stability | Regulatory Status and Safety | Comparative Studies to PS | |
|---|---|---|---|---|---|
| Subcategory | |||||
| Surfactants | Sucrose fatty acid esters | remains to be investigated. As in the case of other surfactants, protection of the protein integrity through direct interaction as well as competitive interface adsorption is expected. | are susceptible to chemical and enzymatic cleavage of ester bond and enzymatic degradation of the sugar moiety | - GRAS - FDA-approved for food and cosmetic use - sucrose stearate and sucrose palmitate: FDA-approved for oral and topical formulations - sucrose monopalmitate (0.5 g/kg): resulted in hemolytic reactions [183,184] | Not available |
| Sugar monoesters | remains to be investigated. As in the case of other surfactants, protection of the protein integrity through direct interaction, as well as competitive interface adsorption being expected. | are susceptible to chemical and enzymatic cleavage of ester bond and enzymatic degradation of the sugar moiety | nontoxic and biodegradable emulsifiers for use in the food industry [190] | Not available | |
| Polyethylene glycol (PEG) stearates and PEG fatty esters | polyoxyl 15 hydroxy stearate and polyoxyl 35 castor oil (Kolliphor® HS 15 and Kolliphor® EL): protect amylase and bovine serum albumin (BSA) from chemical and mechanical stresses [204]. As in the case of other surfactants, protection of the protein integrity through direct interaction therewith as well as competitive interface adsorption is expected. | are susceptible to chemical and enzymatic cleavage of ester bond and oxidation of POE [203] | - PEG 2 stearate: FDA-approved for topical application - PEG 8 and 100 stearates: FDA-approved for oral and topical application - PEG 40 stearate: FDA-approved for ophthalmic, oral, and topical applications - polyoxyl 15 hydroxy stearate and polyoxyl 35 castor oil: FDA approval for oral, parenteral, and ophthalmic application - potential PEG-mediated immunogenicity and hypersensitivity | - polyoxyl 15 hydroxy stearate: superior toxicity profile to PS80 [209] - polyoxyl 15 hydroxy stearate and polyoxyl 35 castor oil: comparable efficiency in protecting BSA against mechanical stress for 7 days [204] - polyoxyl 35 castor oil: superior efficiency to stabilize amylase in the presence of H2O2 over a period of two months [204] | |
| Block polyethylene-propylene glycols (Poloxamers®) | protect the protein through direct interaction as well as competitive interface adsorption | are susceptible to thermal oxidation of PPO block in solid and liquid states | - P188: FDA-approved for intravenous formulations, part of commercial biotherapeutic formulations such as Gazyva®, Orencia®, Norditropin®, and Hemlibra® - potential PEG-mediated immunogenicity and hypersensitivity | P188: Comparable efficiency with PSs in stabilizing mAb formulations, production of PDMS during long-term storage due to interactions with the silicone oil in the stopper in case of P188 [241], increase of glide force in prefilled syringes by PS, but not P188 | |
| Polyoxyethylene fatty ethers (Brij®) | protect the protein through direct interaction as well as competitive interface adsorption [247,248] | might be susceptible to metal- and photo-induced oxidation [78,251] | - FDA-approved for topical administration - Brij® 58: no side effects when administrated in a concentration of 20 mg/mL in animal toxicological studies [249] | - Brij® 92: comparable efficiency to PSs in stabilizing rhGH [247] - Brij® 35: performed inferior to PSs in improving the mechanical and thermal stability of mAb, but superior in protecting the protein against photo-oxidation [250] - Brij® 58: superior inherent stability of the surfactant in protein formulations when compared to PS20 and PS80 [249] | |
| Non-ester sugar-based surfactants | n-dodecyl-β-D-maltoside: serves as cryoprotectant for D-alanine Peptide T-amide [253], reduces the aggregation and immunogenicity of human IFN- β [254], prevents insulin self-association under mechanical stress in liquid state [253] | susceptible to enzymatic degradation of the sugar moiety | - n-dodecyl-β-D-maltoside: classified by EPA as non-mutagenic, non-toxic, and non-irritating [252] - biodegradable and biocompatible | Not available | |
| N-alkyl amino acid polyether amides | N-myristoyl phenylalanine Jeffamine M1000 diamide (FM1000): protects proteins from thermal and mechanical stresses [255,256] | FM1000: potential susceptibility of the amide bond to hydrolysis, through to a significantly lower extent compared with ester bond in ester surfactants | Not available | FM1000: 3-fold more reduction of thermal-induced abatacept aggregation compared to PS20 and PS80 [255], inferior, and superior IgG protection from agitation-mediated stress compared to PS80 and PS20, respectively [256], protein stabilization from agitation-mediated stress in IV bags was superior to both PS20 and PS80 [256] | |
| Carbohydrates | Disaccharides and sugar alcohols | - protect the proteins against thermal and to some extent mechanical stresses in solid and liquid state, act mostly by altering protein–solvent interactions, also serve as molecular crowders [259,272] - mostly improve the thermal stability of the protein - act as cryoprotectants - are associated with inadequate protein protection necessitating the co-application of surfactants - chemical modification, e.g., esterification with fatty acids, etc. improves the protein stabilization efficiency | are susceptible to enzymatic degradation | FDA-approved, GRAS, established excipients in the formulation of intravenously injected formulations | are often used in combination with PSs to confer proteins adequate stability |
| Cyclo- dextrins | protect protein either through direct interaction (in case of certain proteins with substantial solvent exposure to hydrophobic amino-acid residues [311]) or competitive interfacial adsorption (in case of CD with higher surface-active properties [307]), dominant mechanism also depends on protein concentration | are potentially susceptible to enzymatic degradation | - 2-hydroxypropyl-γ-cyclodextrin; FDA-approved for ophthalmic and topical administration - α-cyclodextrin: FDA-approved for intracavitary administration - β-cyclodextrin and sulfobutylether-β-cyclodextrin: FDA-approved for oral, topical, intramuscular, subcutaneous, and intravenous administration (GRAS) | hydroxypropyl-β-cyclodextrin: reduced the interface-induced precipitation of porcine growth hormone through a mechanism similar to PS20 [307], protects PS20 from enzymatic degradation [324] thereby combination of the two might be beneficial for overcoming the drawbacks associated with PS degradation | |
| Hydroxypropyl methyl cellulose (HPMC) | efficiently stabilizes mAb formulations such as cetuximab and abatacept [330] | is potentially susceptible to enzymatic degradation | FDA-approved, GRAS excipient for oral, buccal, vaginal, and nasal administration | stabilization of cetuximab comparable with PS80 [330] | |
| Dextran | stabilize the proteins through a crowding effect [332], reduce the surface adsorption of the proteins in the liquid state [53], maintain protein structural integrity during freezing, drying, and storage in the solid state [333] | potential susceptibility to enzymatic degradation | used in several injectable formulations | Not available | |
| Amino acid-based stabilizers | Amino acids | - certain amino acids promote protein solubilization and refolding mostly by altering protein–solvent interactions, also serve as molecular crowders, stabilizing effect of amino acids is protein dependent - arginine protects protein in solution, and acts as a cryoprotectant [255,256] | are not prone to degradation under formulation and storage conditions | - GRAS - FDA-approved for injectable (intramuscular, intravenous), and oral formulations | Not available |
| Polyamines | protect proteins against thermal aggregation and denaturation | Not available | - oral acute toxicity dose of 2000, 600, and 600 mg/kg body weight for putrescine, spermidine, and spermine, respectively [363] - no-observed adverse effect level (NOAEL) following oral administration was 2000 ppm (180 mg/kg body weight/day) for putrescine, 1000 ppm (83 mg/ kg body weight/day) for spermidine and 200 ppm (19 mg/kg body weight/day) for spermine [363] oral administration of spermidine in human and animal models has been well-tolerated [362] | Not available | |
| Albumin | protects protein through direct interaction as well as competitive interface adsorption [365,366,368] | is potentially prone to forming aggregates with itself or other proteins | - is marketed as an injectable product on its own, and is approved as an excipient for injectable formulations - hypersensitivity and immunological reactions to albumin-containing IFN formulations have been reported [23] - concerns regarding blood-transmitted diseases in case of human and bovine serum albumin | Not available | |
| Synthetic amphiphilic polymers | Polyether polyols (PEG) and Polypropylene glycols (PPG) | - PEGs: increase protein stability through covalent binding, protection of the free protein from thermal and lyophilization stresses [373,374] - PPGs: protect cetuximab from agitation-induced stress, potentially through direct interaction with the protein [330] | are potentially susceptible to oxidation | - PEGs: FDA-approved for intravenous administration, immunogenicity, and hypersensitivity to formulations containing PEGs and their derivatives have been reported [226] - PPGs: FDA-approved for oral and topical formulations, cross-reactivity of anti-PEG antibodies with PPGs is possible [378] | PPGs: comparable efficiency with PS80 in protecting cetuximab from agitation-induced stress [378] |
| Polyampholytes | protect proteins from thermal and agitation-induced stress [380] | Not available | Not available | Not available | |
| Ionic liquids | - can protect the proteins against thermal and storage-mediated unfolding [387,390,391,392] - can induce refolding of denatured proteins [388,389] - stabilizing effects depends on the concentration, the length of the alkyl chain and the nature and size of the anion [385] | are generally recognized as highly stable [414] | Toxicity studies have been limited to bacteria, fungi, plants, small animals, and human cell lines [408,409] | Not available | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castañeda Ruiz, A.J.; Shetab Boushehri, M.A.; Phan, T.; Carle, S.; Garidel, P.; Buske, J.; Lamprecht, A. Alternative Excipients for Protein Stabilization in Protein Therapeutics: Overcoming the Limitations of Polysorbates. Pharmaceutics 2022, 14, 2575. https://doi.org/10.3390/pharmaceutics14122575
Castañeda Ruiz AJ, Shetab Boushehri MA, Phan T, Carle S, Garidel P, Buske J, Lamprecht A. Alternative Excipients for Protein Stabilization in Protein Therapeutics: Overcoming the Limitations of Polysorbates. Pharmaceutics. 2022; 14(12):2575. https://doi.org/10.3390/pharmaceutics14122575
Chicago/Turabian StyleCastañeda Ruiz, Angel J., Maryam A. Shetab Boushehri, Tamara Phan, Stefan Carle, Patrick Garidel, Julia Buske, and Alf Lamprecht. 2022. "Alternative Excipients for Protein Stabilization in Protein Therapeutics: Overcoming the Limitations of Polysorbates" Pharmaceutics 14, no. 12: 2575. https://doi.org/10.3390/pharmaceutics14122575