

Micro-Scale Vacuum Compression Molding as a Predictive Screening Tool of Protein Integrity for Potential Hot-Melt Extrusion Processes


Abstract
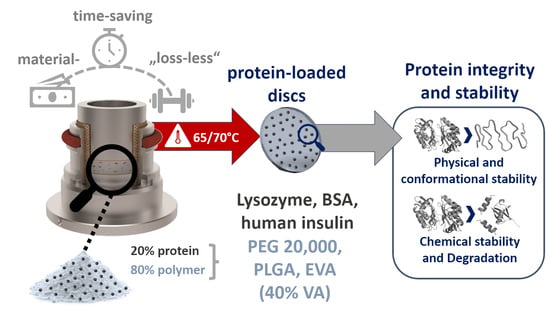
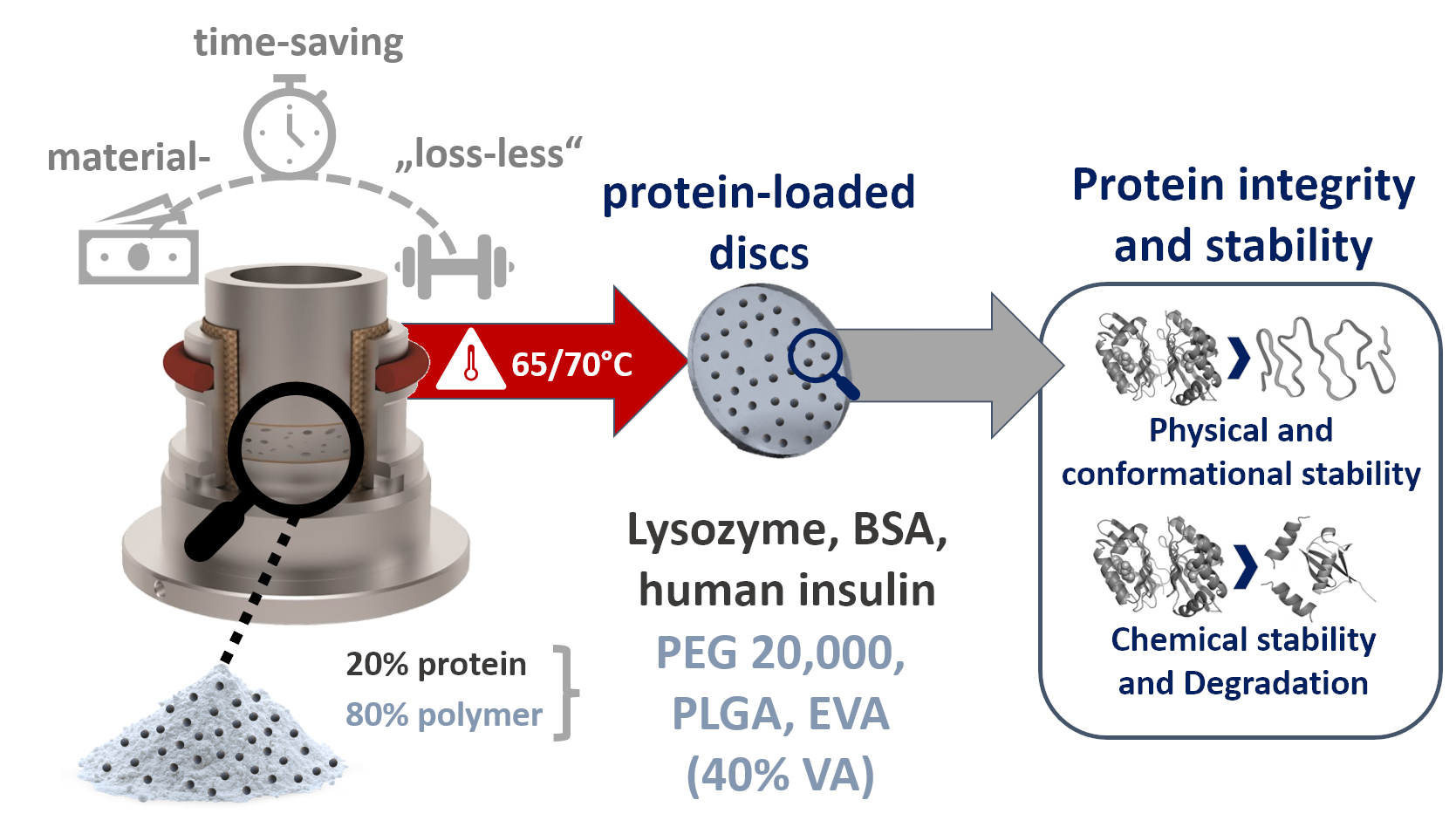
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Physical Mixtures and Sample Discs by VCM
2.3. Sample Preparation for SEC Analysis (Protein Extraction Procedures)
2.4. Size-Exclusion Chromatography (SEC)
2.5. Differential Scanning Calorimetry (DSC)
2.6. Fourier-Transform Infrared Spectroscopy (FT-IR)
2.7. Statistical Analysis
3. Results and Discussion
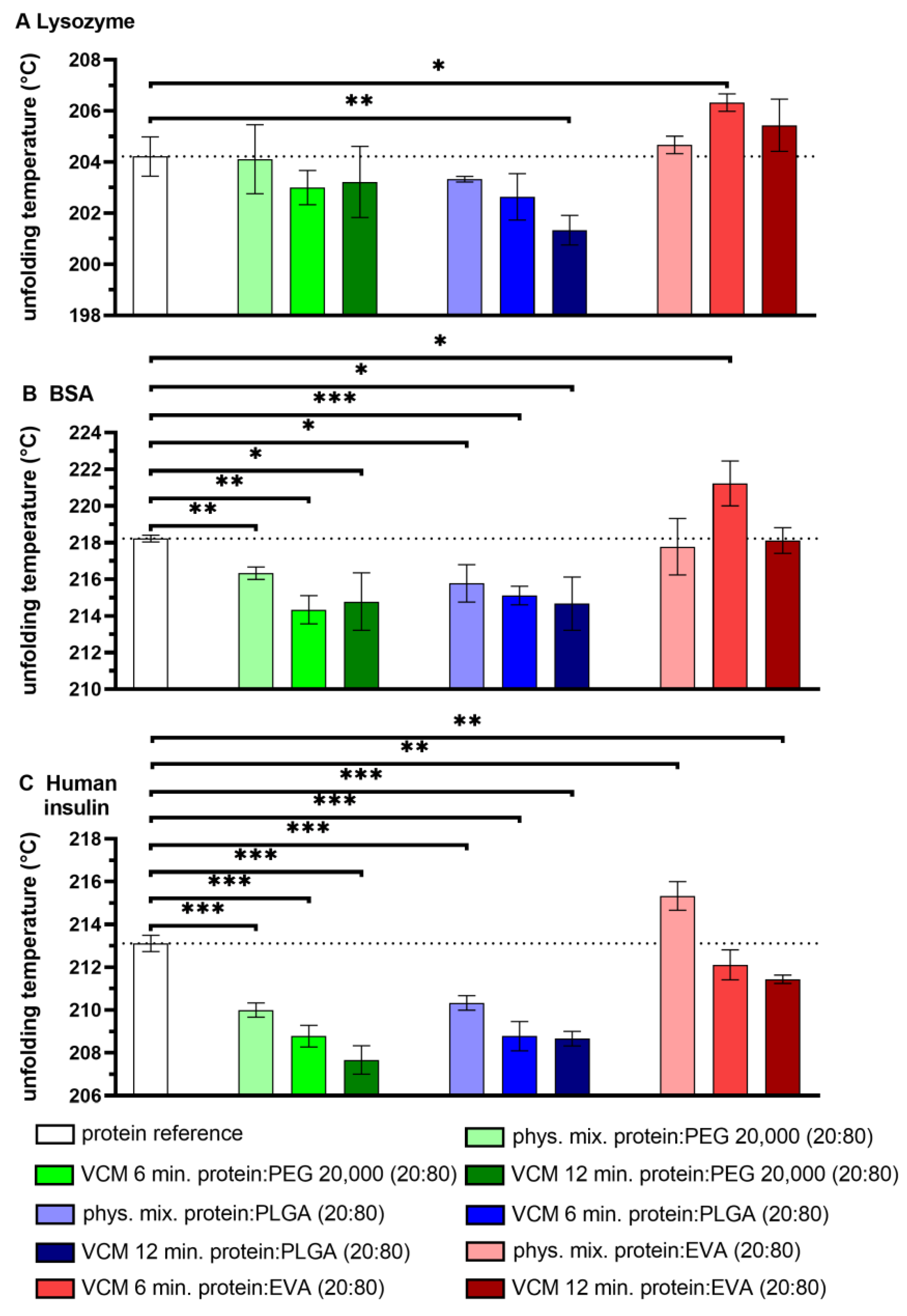
3.1. Unfolding Temperature of Proteins by DSC
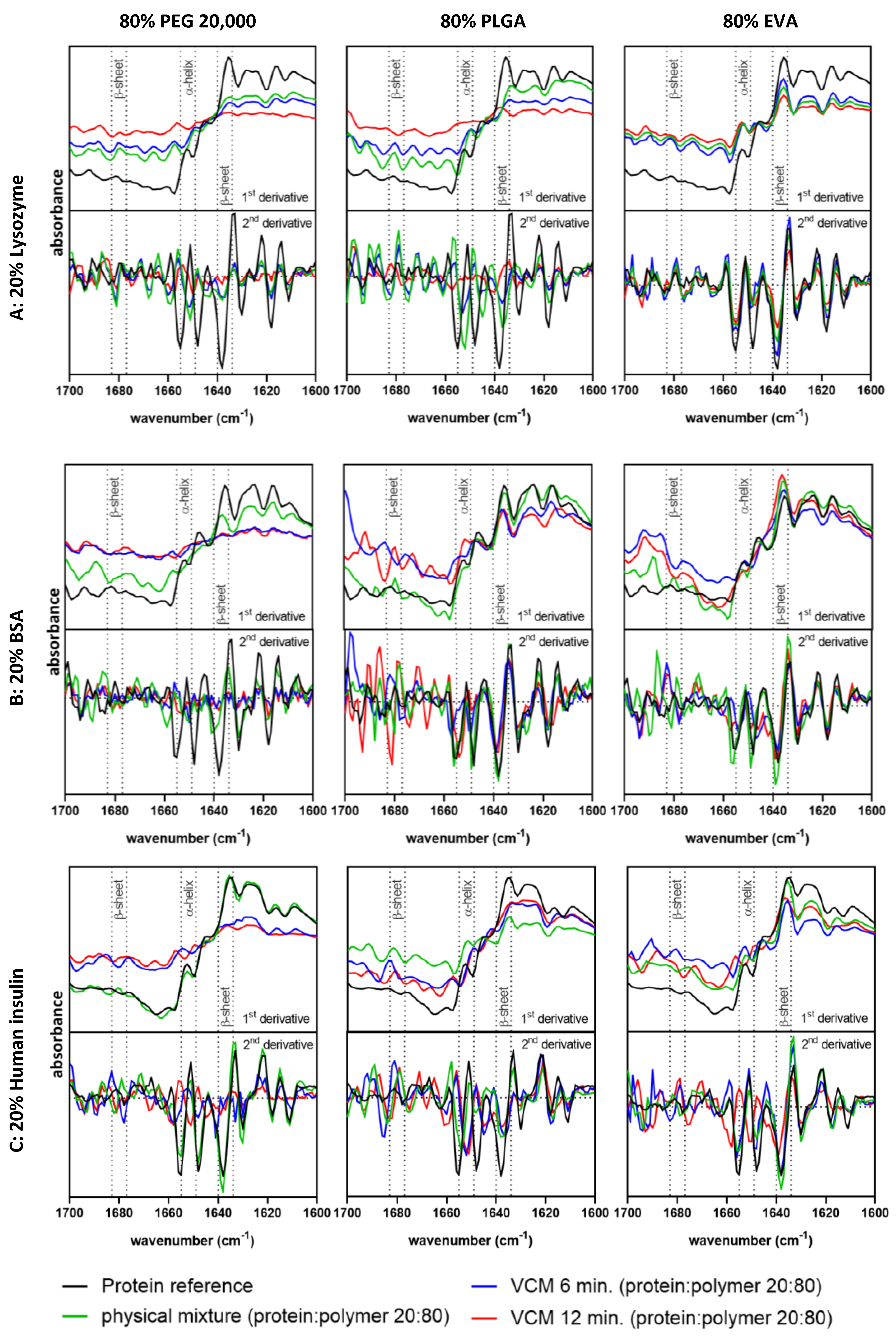
3.2. Secondary Structure Analysis and Conformational Protein Stability by FT-IR Spectroscopy
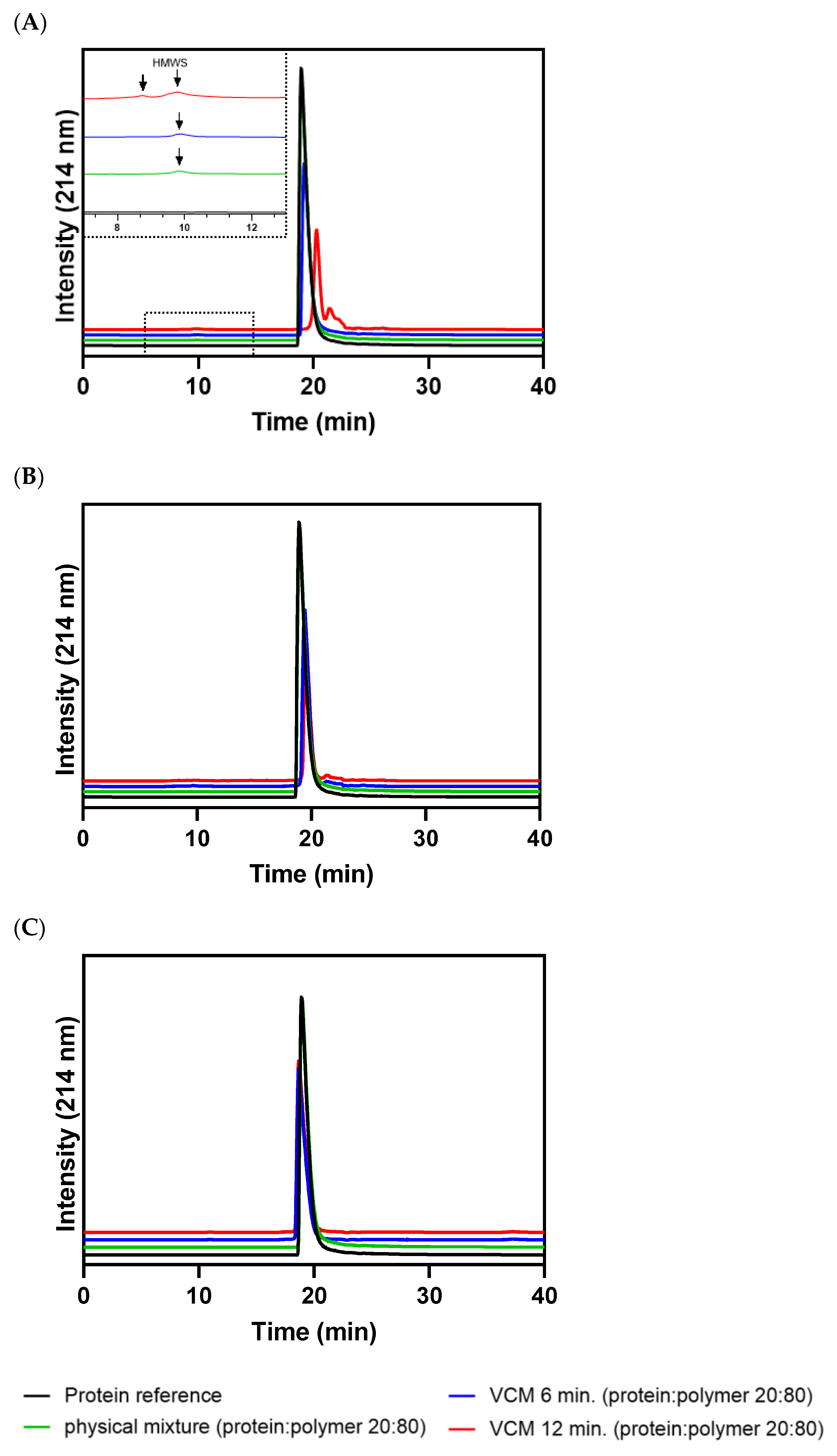
3.3. Process-Induced Formation of Protein Aggregates or Fragments by SEC
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lyu, X. The global landscape of approved antibody therapies. Antib. Ther. 2022, 5, 233–257. [Google Scholar] [CrossRef] [PubMed]
- Gervasi, V.; Agnol, R.D.; Cullen, S.; McCoy, T.; Vucen, S.; Crean, A. Parenteral protein formulations: An overview of approved products within the European Union. Eur. J. Pharm. Biopharm. 2018, 131, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Dauer, K.; Pfeiffer-Marek, S.; Kamm, W.; Wagner, K. Microwell Plate-Based Dynamic Light Scattering as a High-Throughput Characterization Tool in Biopharmaceutical Development. Pharmaceutics 2021, 13, 172. [Google Scholar] [CrossRef]
- Privalova, A.M.; Gulyaeva, N.V.; Bukreeva, T.V. Intranasal administration: A prospective drug delivery route to the brain. Neurochem. J. 2012, 6, 77–88. [Google Scholar] [CrossRef]
- Shaji, J.; Patole, V. Protein and Peptide Drug Delivery: Oral Approaches. Indian J. Pharm. Sci. 2008, 70, 269–277. [Google Scholar] [CrossRef] [Green Version]
- Türker, S.; Onur, E.; Ozer, Y. Nasal route and drug delivery systems. Pharm. World Sci. PWS 2004, 26, 137–142. [Google Scholar] [CrossRef]
- Preston, K.B.; Randolph, T.W. Stability of lyophilized and spray dried vaccine formulations. Adv. Drug Deliv. Rev. 2021, 171, 50–61. [Google Scholar] [CrossRef]
- Cossé, A.; König, C.; Lamprecht, A.; Wagner, K.G. Hot Melt Extrusion for Sustained Protein Release: Matrix Erosion and In Vitro Release of PLGA-Based Implants. AAPS PharmSciTech 2017, 18, 15–26. [Google Scholar] [CrossRef]
- Adali, M.B.; Barresi, A.A.; Boccardo, G.; Pisano, R. Spray Freeze-Drying as a Solution to Continuous Manufacturing of Pharmaceutical Products in Bulk. Processes 2020, 8, 709. [Google Scholar] [CrossRef]
- Dauer, K.; Werner, C.; Lindenblatt, D.; Wagner, K.G. Impact of process stress on protein stability in highly-loaded solid protein/PEG formulations from small-scale melt extrusion. Int. J. Pharm. X 2023, 5, 100154. [Google Scholar] [CrossRef]
- Stanković, M.; Frijlink, H.W.; Hinrichs, W.L.J. Polymeric formulations for drug release prepared by hot melt extrusion: Application and characterization. Drug Discov. Today 2015, 20, 812–823. [Google Scholar] [CrossRef]
- Stanković, M.; de Waard, H.; Steendam, R.; Hiemstra, C.; Zuidema, J.; Frijlink, H.W.; Hinrichs, W.L. Low temperature extruded implants based on novel hydrophilic multiblock copolymer for long-term protein delivery. Eur. J. Pharm. Sci. 2013, 49, 578–587. [Google Scholar] [CrossRef]
- Madan, S.M.S. Hot melt extrusion and its pharmaceutical applications. Asian J. Pharm. Sci. 2012, 7, 123–133. [Google Scholar]
- Gajda, M.; Nartowski, K.P.; Pluta, J.; Karolewicz, B. The role of the polymer matrix in solvent-free hot melt extrusion continuous process for mechanochemical synthesis of pharmaceutical cocrystal. Eur. J. Pharm. Biopharm. 2018, 131, 48–59. [Google Scholar] [CrossRef]
- Ghalanbor, Z.; Körber, M.; Bodmeier, R. Improved Lysozyme Stability and Release Properties of Poly(lactide-co-glycolide) Implants Prepared by Hot-Melt Extrusion. Pharm. Res. 2010, 27, 371–379. [Google Scholar] [CrossRef]
- Lee, S.L.; Hafeman, A.E.; Debenedetti, P.G.; Pethica, B.A.; Moore, D.J. Solid-State Stabilization of α-Chymotrypsin and Catalase with Carbohydrates. Ind. Eng. Chem. Res. 2006, 45, 5134–5147. [Google Scholar] [CrossRef]
- Treffer, D.; Troiss, A.; Khinast, J. A novel tool to standardize rheology testing of molten polymers for pharmaceutical applications. Int. J. Pharm. 2015, 495, 474–481. [Google Scholar] [CrossRef]
- Rupenthal, P.A.I.D. Injectable implants for the sustained release of protein and peptide drugs. Drug Discov. Today 2013, 18, 337–349. [Google Scholar] [CrossRef]
- Gundry, R.L.; White, M.Y.; Murray, C.I.; Kane, L.A.; Fu, Q.; Stanley, B.A.; Van Eyk, J.E. Preparation of Proteins and Peptides for Mass Spectrometry Analysis in a Bottom-Up Proteomics Workflow. Curr. Protoc. Mol. Biol. 2009, 90, 10–25. [Google Scholar] [CrossRef] [Green Version]
- Langer, R.; Brown, L.; Edelman, E. Controlled release and magnetically modulated release systems for macromolecules. Methods Enzymol. 1985, 112, 399–422. [Google Scholar] [CrossRef]
- Klibanov, K.G.A.M. Lyophilization-induced reversible changes in the secondary structure of proteins. Proc. Natl. Acad. Sci. USA 1995, 92, 10969–10976. [Google Scholar] [CrossRef] [Green Version]
- Chao, S.-H.; Matthews, S.S.; Paxman, R.; Aksimentiev, A.; Gruebele, M.; Price, J.L. Two Structural Scenarios for Protein Stabilization by PEG. J. Phys. Chem. B 2014, 118, 8388–8395. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Components (%) | Preparation Method | Process Parameters | |
|---|---|---|---|---|
| Protein | Polymer | |||
| LYS reference | 100% Lysozyme | |||
| LYS PEG PM | 20% Lysozyme | 80% PEG 20,000 | Phys. mix. | |
| LYS PEG VCM 6 | 20% Lysozyme | 80% PEG 20,000 | VCM | 6 min at 65 °C |
| LYS PEG VCM 12 | 20% Lysozyme | 80% PEG 20,000 | VCM | 12 min at 65 °C |
| LYS PLGA PM | 20% Lysozyme | 80% PLGA | Phys. mix. | |
| LYS PLGA VCM 6 | 20% Lysozyme | 80% PLGA | VCM | 6 min at 70 °C |
| LYS PLGA VCM 12 | 20% Lysozyme | 80% PLGA | VCM | 12 min at 70 °C |
| LYS EVA PM | 20% Lysozyme | 80% EVA | Phys. mix. | |
| LYS EVA VCM 6 | 20% Lysozyme | 80% EVA | VCM | 6 min at 70 °C |
| LYS EVA VCM 12 | 20% Lysozyme | 80% EVA | VCM | 12 min at 70 °C |
| BSA reference | 100% BSA | |||
| BSA PEG PM | 20% BSA | 80% PEG 20,000 | Phys. mix. | |
| BSA PEG VCM 6 | 20% BSA | 80% PEG 20,000 | VCM | 6 min at 65 °C |
| BSA PEG VCM 12 | 20% BSA | 80% PEG 20,000 | VCM | 12 min at 65 °C |
| BSA PLGA PM | 20% BSA | 80% PLGA | Phys. mix. | |
| BSA PLGA VCM 6 | 20% BSA | 80% PLGA | VCM | 6 min at 70 °C |
| BSA PLGA VCM 12 | 20% BSA | 80% PLGA | VCM | 12 min at 70 °C |
| BSA EVA PM | 20% BSA | 80% EVA | Phys. mix. | |
| BSA EVA VCM 6 | 20% BSA | 80% EVA | VCM | 6 min at 70 °C |
| BSA EVA VCM 12 | 20% BSA | 80% EVA | VCM | 12 min at 70 °C |
| IHU reference | 100% Human Insulin | |||
| IHU PEG PM | 20% Human Insulin | 80% PEG 20,000 | Phys. mix. | |
| IHU PEG VCM 6 | 20% Human Insulin | 80% PEG 20,000 | VCM | 6 min at 65 °C |
| IHU PEG VCM 12 | 20% Human Insulin | 80% PEG 20,000 | VCM | 12 min at 65 °C |
| IHU PLGA PM | 20% Human Insulin | 80% PLGA | Phys. mix. | |
| IHU PLGA VCM 6 | 20% Human Insulin | 80% PLGA | VCM | 6 min at 70 °C |
| IHU PLGA VCM 12 | 20% Human Insulin | 80% PLGA | VCM | 12 min at 70 °C |
| IHU EVA PM | 20% Human Insulin | 80% EVA | Phys. mix. | |
| IHU EVA VCM 6 | 20% Human Insulin | 80% EVA | VCM | 6 min at 70 °C |
| IHU EVA VCM 12 | 20% Human Insulin | 80% EVA | VCM | 12 min at 70 °C |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dauer, K.; Wagner, K.G. Micro-Scale Vacuum Compression Molding as a Predictive Screening Tool of Protein Integrity for Potential Hot-Melt Extrusion Processes. Pharmaceutics 2023, 15, 723. https://doi.org/10.3390/pharmaceutics15030723
Dauer K, Wagner KG. Micro-Scale Vacuum Compression Molding as a Predictive Screening Tool of Protein Integrity for Potential Hot-Melt Extrusion Processes. Pharmaceutics. 2023; 15(3):723. https://doi.org/10.3390/pharmaceutics15030723
Chicago/Turabian StyleDauer, Katharina, and Karl G. Wagner. 2023. "Micro-Scale Vacuum Compression Molding as a Predictive Screening Tool of Protein Integrity for Potential Hot-Melt Extrusion Processes" Pharmaceutics 15, no. 3: 723. https://doi.org/10.3390/pharmaceutics15030723