
Computational and Experimental Evaluation of the Stability of a GLP-1-like Peptide in Ethanol–Water Mixtures

 , ,
, ,
Abstract
:1. Introduction
- (i)
- Provide insight into the potential stabilisation effect of solvents and, where the organic solvent is a known aggregator but inescapable;
- (ii)
- Narrow the choice to the least aggregating candidate.
2. Materials and Methods
2.1. Docking Assay for Transmembrane Component (DAFT) Setup
2.2. Molecular Dynamics Simulations
2.3. Computational Analyses
2.3.1. Interaction Energies
2.3.2. Orientation Analysis
2.3.3. Interaction Pattern
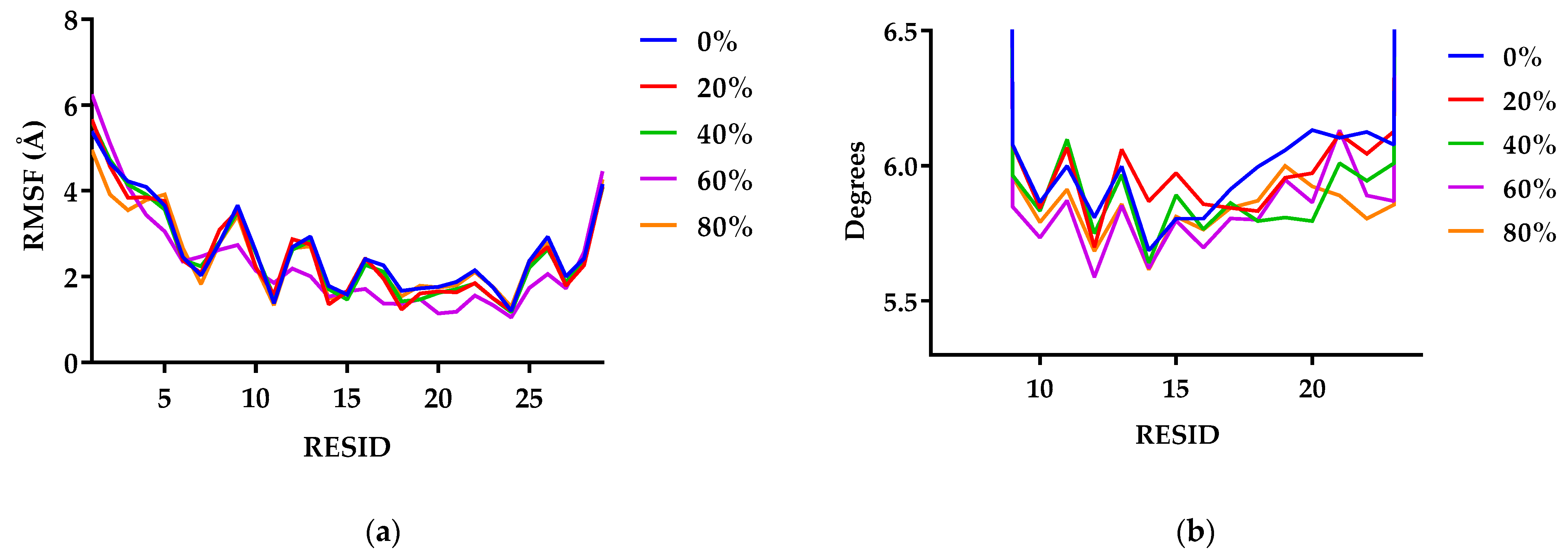
2.3.4. Fluctuation in the Structure
2.4. Experimental Stability Studies
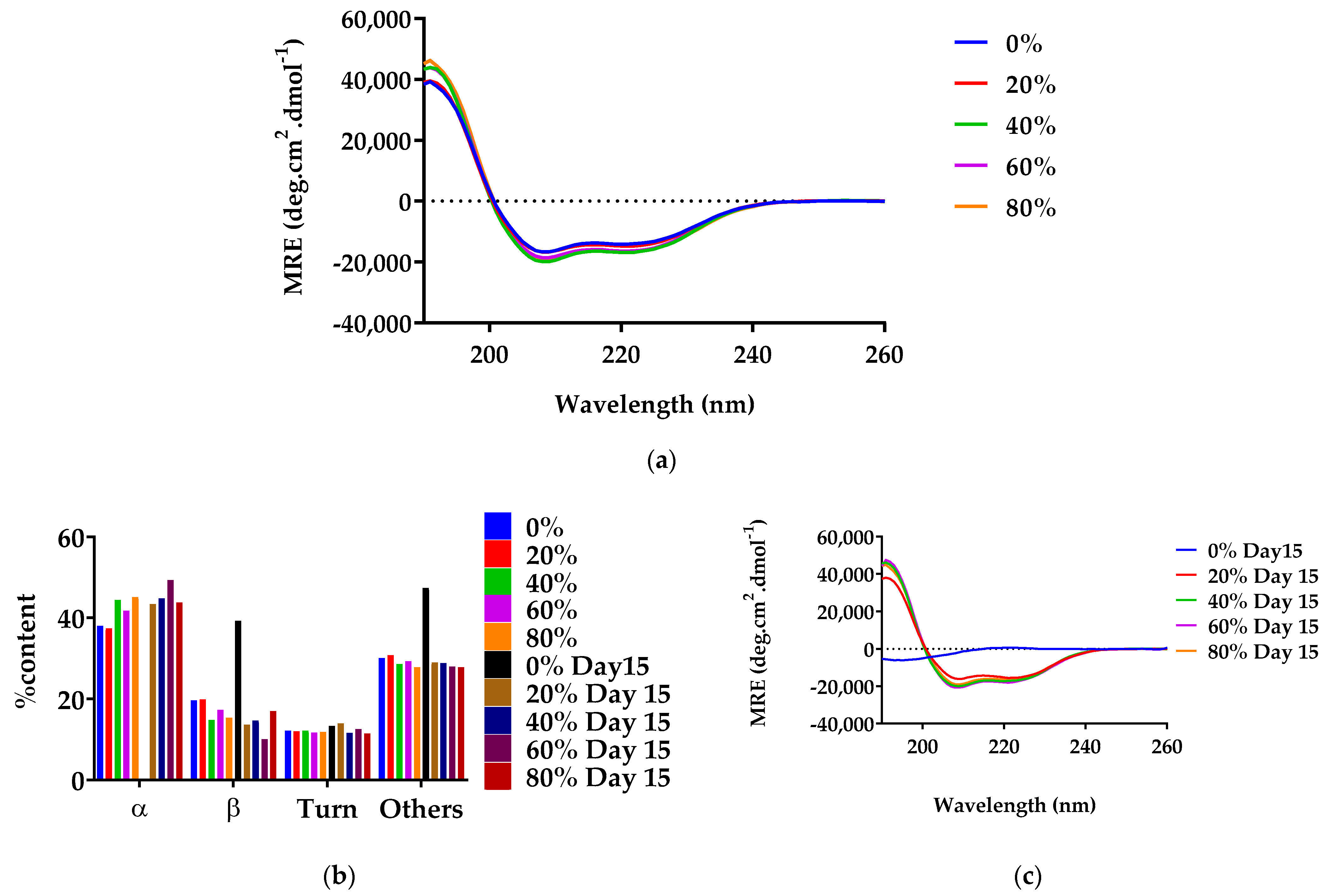
2.4.1. Circular Dichroism
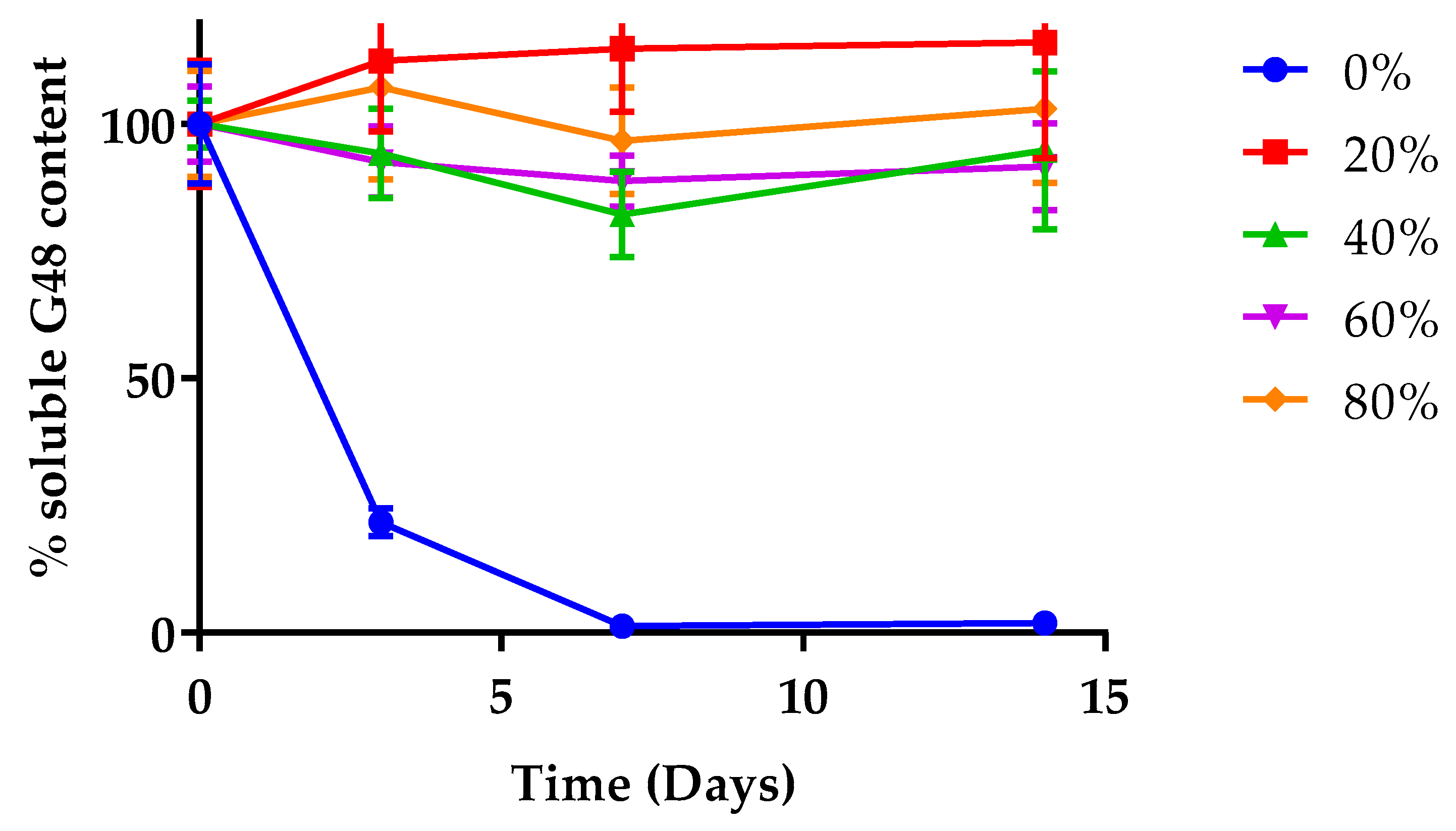
2.4.2. Quantification of Soluble Peptide
3. Results
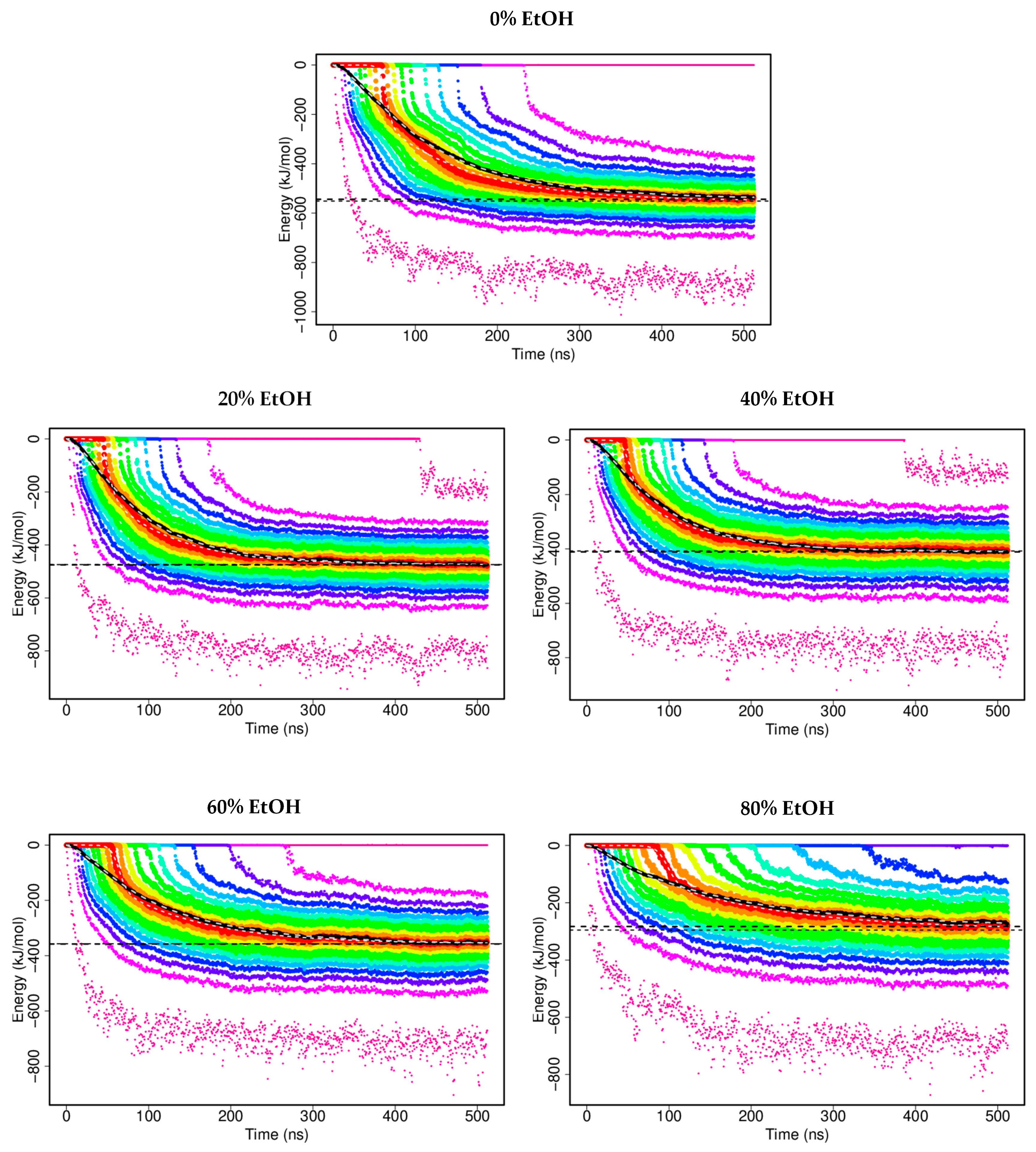
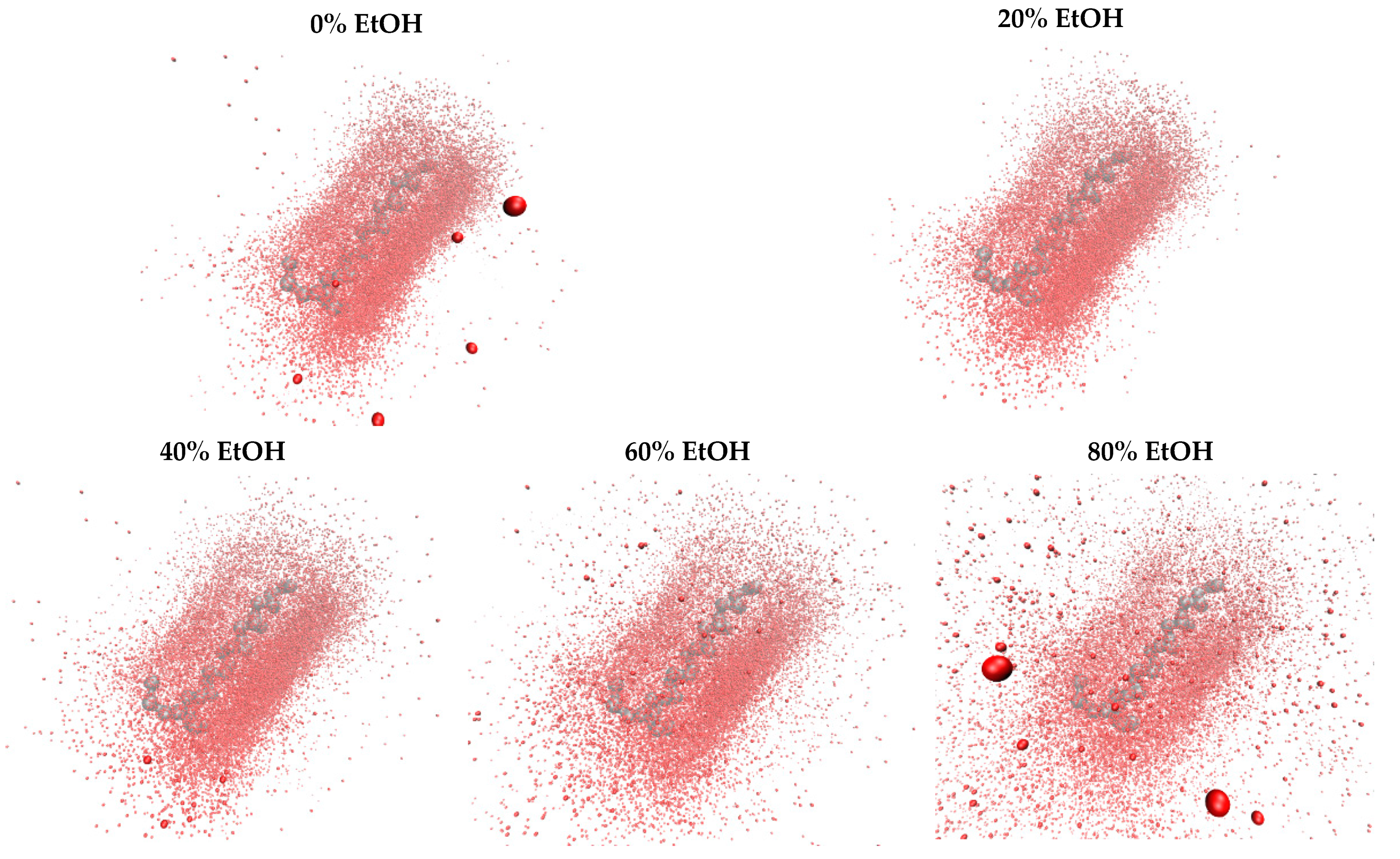
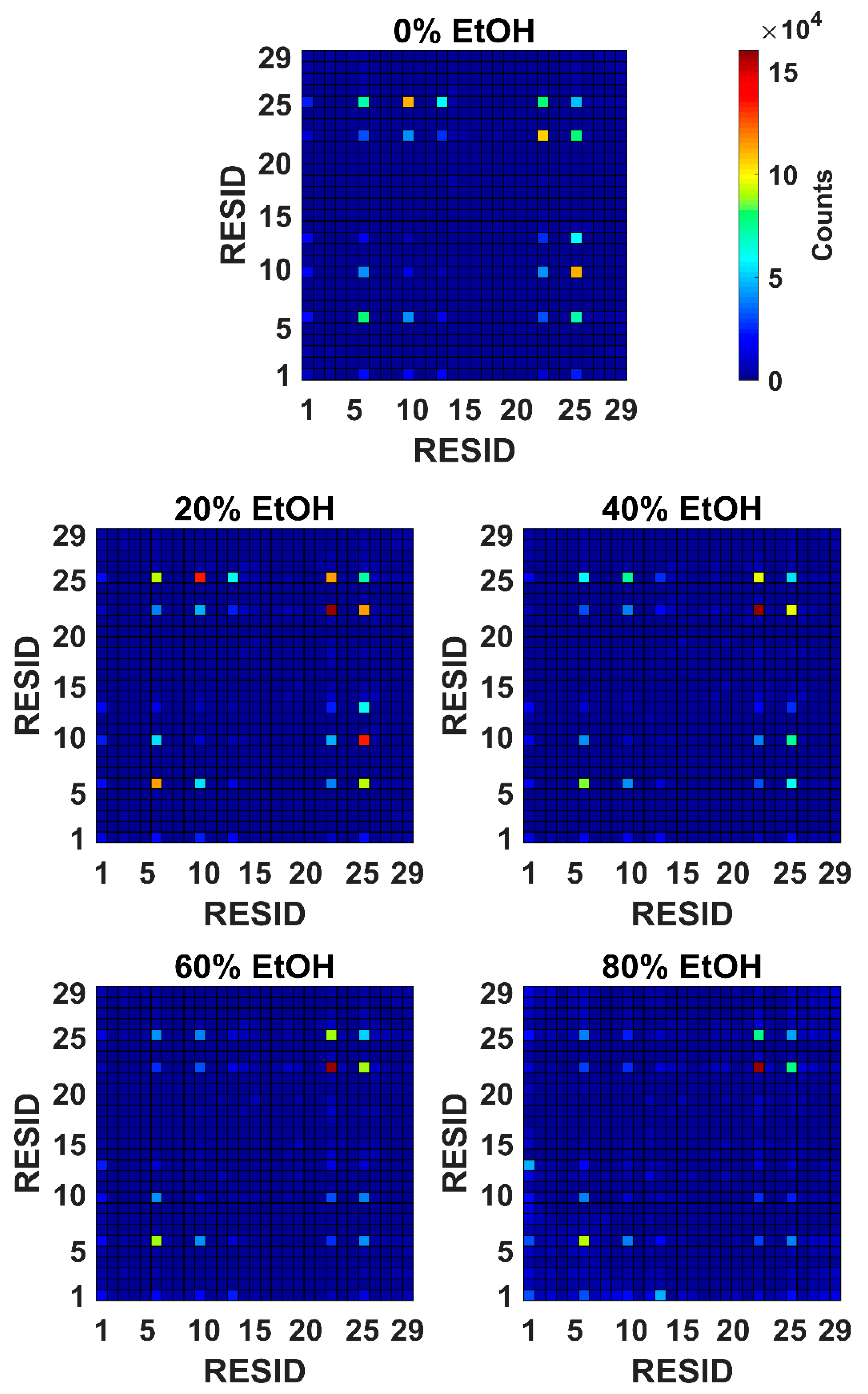
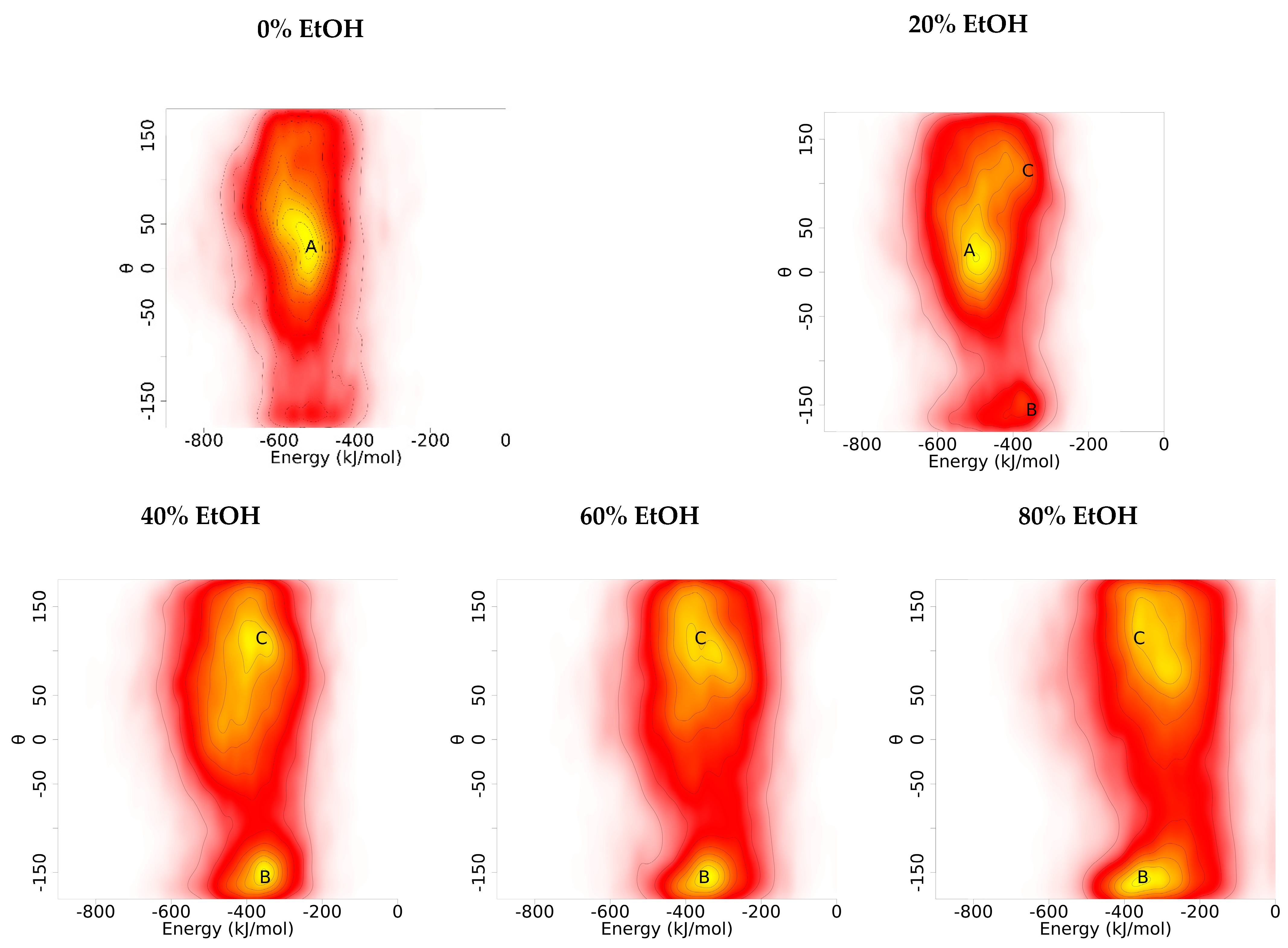
3.1. In Silico G48 Dimerisation
3.1.1. Interaction Energies
3.1.2. Interacting Points
3.1.3. Orientation Analysis
3.1.4. G48 Stability
3.2. Experimental Results
3.2.1. Secondary Structure of G48
3.2.2. Soluble Peptide Quantification
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef]
- Ahren, B. GLP-1 for type 2 diabetes. Exp. Cell Res. 2011, 317, 1239–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, L.; Khurana, R.; Coats, A.; Frokjaer, S.; Brange, J.; Vyas, S.; Uversky, V.N.; Fink, A.L. Effect of Environmental Factors on the Kinetics of Insulin Fibril Formation: Elucidation of the Molecular Mechanism. Biochemistry 2001, 40, 6036–6046. [Google Scholar] [CrossRef]
- Demeule, B.; Gurny, R.; Arvinte, T. Where disease pathogenesis meets protein formulation: Renal deposition of immunoglobulin aggregates. Eur. J. Pharm. Biopharm. 2006, 62, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.R. Insulin amyloid at injection sites of patients with diabetes. Amyloid 2016, 23, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Frokjaer, S.; Otzen, D.E. Protein drug stability: A formulation challenge. Nat. Rev. Drug Discov. 2005, 4, 298–306. [Google Scholar] [CrossRef]
- Bakhtiani, P.A.; Caputo, N.; Castle, J.R.; El Youssef, J.; Carroll, J.M.; David, L.L.; Roberts, C.T.; Ward, W.K. A novel, stable, aqueous glucagon formulation using ferulic acid as an excipient. J. Diabetes Sci. Technol. 2015, 9, 17–23. [Google Scholar] [CrossRef] [Green Version]
- Dong, N.; Zhu, C.; Jiang, J.; Huang, D.; Li, X.; Quan, G.; Liu, Y.; Tan, W.; Pan, X.; Wu, C. Development of composite PLGA microspheres containing exenatide-encapsulated lecithin nanoparticles for sustained drug release. Asian J. Pharm. 2019, 15, 347–355. [Google Scholar] [CrossRef]
- Fleming, K.G.; Engelman, D.M. Specificity in transmembrane helix–helix interactions can define a hierarchy of stability for sequence variants. Proc. Natl. Acad. Sci. USA 2001, 98, 14340–14344. [Google Scholar] [CrossRef] [Green Version]
- Liang, R.; Zhang, R.; Li, X.; Wang, A.; Chen, D.; Sun, K.; Liu, W.; Li, Y. Stability of exenatide in poly(D,L-lactide-co-glycolide) solutions: A simplified investigation on the peptide degradation by the polymer. Eur. J. Pharm. Sci. 2013, 50, 502–510. [Google Scholar] [CrossRef]
- Srisailam, S.; Kumar, T.K.; Srimathi, T.; Yu, C. Influence of backbone conformation on protein aggregation. J. Am. Chem. Soc. 2002, 124, 1884–1888. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Webster, P.; Taddei, N.; Clark, A.; Stefani, M.; Ramponi, G.; Dobson, C.M. Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc. Natl. Acad. Sci. USA 1999, 96, 3590–3594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taluja, A.; Bae, Y.H. Role of a novel excipient poly(ethylene glycol)-b-poly(L-histidine) in retention of physical stability of insulin at aqueous/organic interface. Mol. Pharm. 2007, 4, 561–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grudzielanek, S.; Smirnovas, V.; Winter, R. Solvation-assisted pressure tuning of insulin fibrillation: From novel aggregation pathways to biotechnological applications. J. Mol. Biol. 2006, 356, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.P.; Hoshino, M.; Kuboi, R.; Goto, Y. Clustering of Fluorine-Substituted Alcohols as a Factor Responsible for Their Marked Effects on Proteins and Peptides. J. Am. Chem. Soc. 1999, 121, 8427–8433. [Google Scholar] [CrossRef]
- Shao, O.; Fan, Y.; Yang, L.; Gao, Y.Q. From protein denaturant to protectant: Comparative molecular dynamics study of alcohol/protein interactions. J. Chem. Phys. 2012, 136, 115101. [Google Scholar] [CrossRef]
- Reiersen, H.; Rees, A.R. Trifluoroethanol may form a solvent matrix for assisted hydrophobic interactions between peptide side chains. Protein Eng. Des. Sel. 2000, 13, 739–743. [Google Scholar] [CrossRef] [Green Version]
- Jalili, S.; Akhavan, M. Molecular dynamics simulation study of association in trifluoroethanol/water mixtures. J. Comput. Chem. 2009, 31, 286–294. [Google Scholar] [CrossRef]
- Chang, X.; Keller, D.; O’Donoghue, S.I.; Led, J.J. NMR studies of the aggregation of glucagon-like peptide-1: Formation of a symmetric helical dimer. FEBS Lett. 2002, 515, 165–170. [Google Scholar] [CrossRef] [Green Version]
- European Medicines Agency. ICH Q3C (R8) Residual Solvents. Available online: https://www.ema.europa.eu/en/ich-q3c-r8-residual-solvents (accessed on 31 March 2022).
- Klimov, D.K.; Thirumalai, D. Dissecting the Assembly of Aβ16−22 Amyloid Peptides into Antiparallel β Sheets. Structure 2003, 11, 295–307. [Google Scholar] [CrossRef] [Green Version]
- Riccardi, L.; Nguyen, P.H.; Stock, G. Construction of the Free Energy Landscape of Peptide Aggregation from Molecular Dynamics Simulations. J. Chem. Theory Comput. 2012, 8, 1471–1479. [Google Scholar] [CrossRef] [PubMed]
- Barz, B.; Wales, D.J.; Strodel, B. A Kinetic Approach to the Sequence-Aggregation Relationship in Disease-Related Protein Assembly. J. Phys. Chem. B 2014, 118, 1003–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, A.; Balaji, P.V. Molecular Events During the Early Stages of Aggregation of GNNQQNY: An all Atom MD Simulation Study of Randomly Dispersed Peptides. J. Struct. Biol. 2015, 192, 376–391. [Google Scholar] [CrossRef] [PubMed]
- Wassenaar, T.A.; Ingólfsson, H.I.; Prieß, M.; Marrink, S.J.; Schäfer, L.V. Mixing MARTINI: Electrostatic Coupling in Hybrid Atomistic–Coarse-Grained Biomolecular Simulations. J. Phys. Chem. B 2013, 117, 3516–3530. [Google Scholar] [CrossRef]
- Marrink, S.J.; de Vries, A.H.; Mark, A.E. Coarse Grained Model for Semiquantitative Lipid Simulations. J. Phys. Chem. B 2004, 108, 750–760. [Google Scholar] [CrossRef] [Green Version]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The MARTINI Force Field: Coarse Grained Model for Biomolecular Simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef] [Green Version]
- Monticelli, L.; Kandasamy, S.K.; Periole, X.; Larson, R.G.; Tieleman, D.P.; Marrink, S.J. The MARTINI Coarse-Grained Force Field: Extension to Proteins. J. Chem. Theory Comput. 2008, 4, 819–834. [Google Scholar] [CrossRef]
- Wassenaar, T.A.; Pluhackova, K.; Moussatova, A.; Sengupta, D.; Marrink, S.J.; Tieleman, D.P.; Böckmann, R.A. High-Throughput Simulations of Dimer and Trimer Assembly of Membrane Proteins. The DAFT Approach. J. Chem. Theory Comput. 2015, 11, 2278–2291. [Google Scholar] [CrossRef]
- Thota, N.; Luo, Z.; Hu, Z.; Jiang, J. Self-Assembly of Amphiphilic Peptide (AF)6H5K15: Coarse-Grained Molecular Dynamics Simulation. J. Phys. Chem. B 2013, 117, 9690–9698. [Google Scholar] [CrossRef]
- Guo, C.; Luo, Y.; Zhou, R.; Wei, G. Probing the Self-Assembly Mechanism of Diphenylalanine-Based Peptide Nanovesicles and Nanotubes. ACS Nano 2012, 6, 3907–3918. [Google Scholar] [CrossRef]
- Mondal, J.; Yethiraj, A. Effect of secondary structure on the self-assembly of amphiphilic molecules: A multiscale simulation study. J. Chem. Phys. 2012, 136, 084902. [Google Scholar] [CrossRef]
- Frederix, P.W.J.M.; Ulijn, R.V.; Hunt, N.T.; Tuttle, T. Virtual Screening for Dipeptide Aggregation: Toward Predictive Tools for Peptide Self-Assembly. J. Phys. Chem. Lett. 2011, 2, 2380–2384. [Google Scholar] [CrossRef] [PubMed]
- Lui, L.H.; van der Walle, C.F.; Brocchini, S.; Velayudhan, A. Discovering novel small molecule compound for prevention of monoclonal antibody self-association. Antibodies 2022, 11, 40. [Google Scholar] [CrossRef] [PubMed]
- de Jong, D.H.; Singh, G.; Bennett, W.F.D.; Arnarez, C.; Wassenaar, T.A.; Schäfer, L.V.; Periole, X.; Tieleman, D.P.; Marrink, S.J. Improved Parameters for the Martini Coarse-Grained Protein Force Field. J. Chem. Theory Comput. 2013, 9, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Cheng, X.; Islam, S.M.; Huang, L.; Rui, H.; Zhu, A.; Lee, H.S.; Qi, Y.; Han, W.; Vanommeslaeghe, K.; et al. Chapter Eight—CHARMM-GUI PDB Manipulator for Advanced Modeling and Simulations of Proteins Containing Nonstandard Residues. Adv. Protein Chem. Struct. Biol. 2014, 6, 235–265. [Google Scholar]
- Neidigh, J.W.; Fesinmeyer, R.M.; Prickett, K.S.; Andersen, N.H. Exendin-4 and Glucagon-like-peptide-1: NMR Structural Comparisons in the Solution and Micelle-Associated States. Biochemistry 2001, 40, 13188–13200. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Javanainen, M.; Martinez-Seara, H.; Vattulainen, I. Excessive aggregation of membrane proteins in the Martini model. PLoS ONE 2017, 12, e0187936. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Bansal, M.; Kumar, S.; Velavan, R. HELANAL: A program to characterize helix geometry in proteins. J. Biomol. Struct. Dyn. 2000, 17, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.H.; Goto, Y.; Refregiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E3095–E3103. [Google Scholar] [CrossRef] [Green Version]
- Teska, B.M.; Alarcon, J.; Pettis, R.J.; Randolph, T.W.; Carpenter, J.F. Effects of phenol and meta-cresol depletion on insulin analog stability at physiological temperature. J. Pharm. Sci. 2014, 103, 2255–2267. [Google Scholar] [CrossRef]
- Kelly, S.M.; Jess, T.J.; Price, N.C. How to study proteins by circular dichroism. Biochim. Biophys. Acta 2005, 1751, 119–139. [Google Scholar] [CrossRef]
- Nemzer, L.R.; Flanders, B.N.; Schmit, J.D.; Chakrabarti, A.; Sorensen, C.M. Ethanol shock and lysozyme aggregation. Soft Matter 2013, 9, 2187–2196. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent Environment | Plateau Value (kJ/mol) | Standard Error of the Mean |
|---|---|---|
| 0% EtOH | −544.0 | 0.3 |
| 20% EtOH | −474.9 | 0.2 |
| 40% EtOH | −410.9 | 0.2 |
| 60% EtOH | −357.5 | 0.2 |
| 80% EtOH | −283.6 | 0.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lui, L.H.; Egbu, R.; Graver, T.; Williams, G.R.; Brocchini, S.; Velayudhan, A. Computational and Experimental Evaluation of the Stability of a GLP-1-like Peptide in Ethanol–Water Mixtures. Pharmaceutics 2022, 14, 1462. https://doi.org/10.3390/pharmaceutics14071462
Lui LH, Egbu R, Graver T, Williams GR, Brocchini S, Velayudhan A. Computational and Experimental Evaluation of the Stability of a GLP-1-like Peptide in Ethanol–Water Mixtures. Pharmaceutics. 2022; 14(7):1462. https://doi.org/10.3390/pharmaceutics14071462
Chicago/Turabian StyleLui, Lok Hin, Raphael Egbu, Thomas Graver, Gareth R. Williams, Steve Brocchini, and Ajoy Velayudhan. 2022. "Computational and Experimental Evaluation of the Stability of a GLP-1-like Peptide in Ethanol–Water Mixtures" Pharmaceutics 14, no. 7: 1462. https://doi.org/10.3390/pharmaceutics14071462