

Design, Formulation, and Characterization of Valsartan Nanoethosomes for Improving Their Bioavailability



Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
Chemicals
2.2. Preparation of VLT-Loaded Ethosomes (VLT-ETHs)
2.3. Preparation of VLT-Loaded Ethogel
2.4. Screening Formulations for Build Information
2.5. Optimization and Selection of Most Significant Factors
2.6. Particle Size (PS) and Zeta Potential (ZP)
2.7. Entrapment Efficiency (EE%)
2.8. In Vitro Drug Release
2.9. Ex Vivo Permeation Study through Rat Abdominal Skin
2.10. Differential Scanning Calorimetry (DSC)
2.11. Fourier Transform Infrared Spectroscopy (FTIR)
2.12. Transmission Electron Microscopy (TEM)
2.13. In Vivo Study
2.13.1. Experimental Animals
2.13.2. Pharmacokinetic Study
Chromatographic Conditions
Preparation of Standard Stock Solution of VLT in Methanol
Sample Preparation
Preparation of VLT Oral Solution
3. Results and Discussion
3.1. Screening Formulations for Build Information
3.1.1. Effect of the Independent Factors on Particle Size (PS)
Effect of Lipid Concentration (A) on Particle Size (PS)
Effect of Type of Lipid (D) on Particle Size (PS)
3.1.2. Effect of the Independent Factors on the Zeta Potential (ZP)
Effect of the Ethanol Concentration on the Zeta Potential (ZP)
Effect of the Lipid Type on the Zeta Potential (ZP)
Effect of the Penetration Enhancer Type on the Zeta Potential (ZP)
3.1.3. Effect of the Independent Factors on the Entrapment Efficiency (EE%)
Effect of the Concentration of Lipid on the Entrapment Efficiency (EE%)
Effect of the Type of Lipid on the Entrapment Efficiency (EE%)
3.1.4. Effect of the Independent Factors on the Polydispersity Index (PDI)
Effect of the Lipid Concentration on Polydispersity Index (PDI)
Effect of Ethanol Concentration on Polydispersity Index (PDI)
3.1.5. Summary of the Screening Results
3.2. Optimization Study Selection of the Most Significant Factors
3.2.1. Effect of the Independent Factors on Particle Size (PS)
3.2.2. Effect of the Independent Factors on Polydispersity Index (PDI)
3.2.3. Effect of the Independent Factors on Zeta Potential (ZP)
3.2.4. Effect of the Independent Factors on Entrapment Efficiency (EE%)
3.3. Selection of the Optimized VLT-ETHs Formulation
3.4. Transmission Electron Microscopy (TEM)
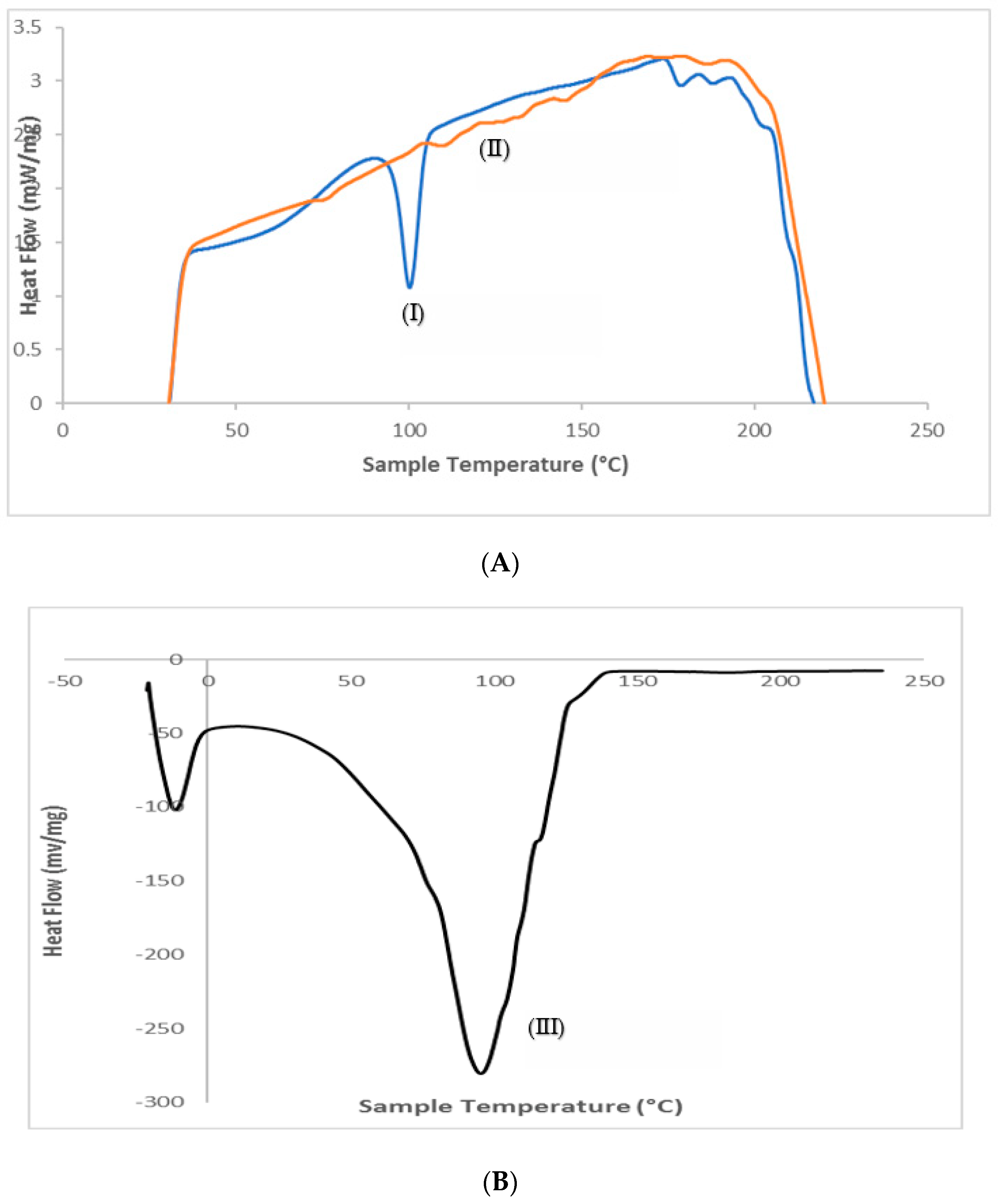
3.5. Differential Scanning Calorimetry (DSC)
3.6. Fourier Transform Infrared Spectroscopy (FTIR)
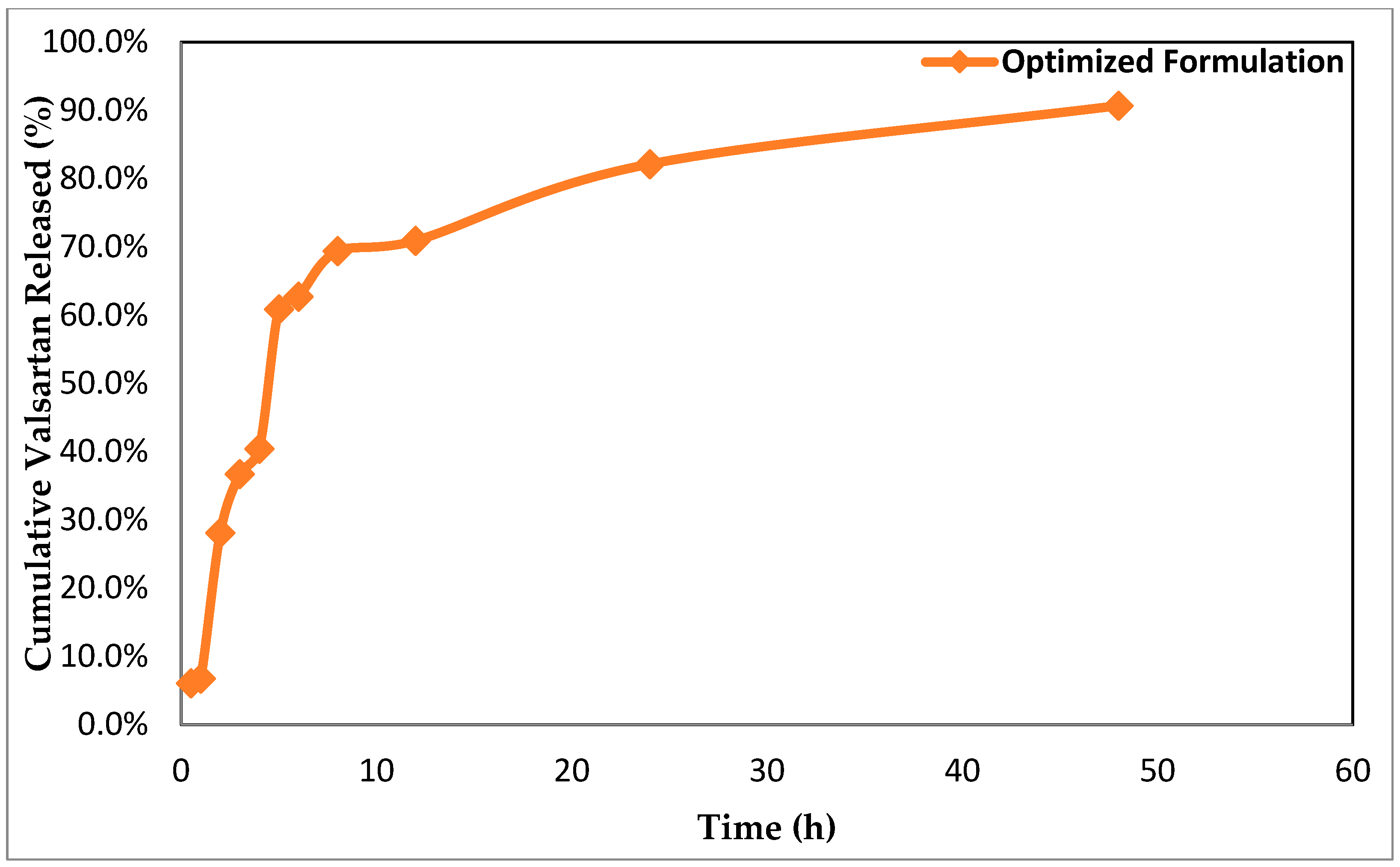
3.7. In Vitro Drug Release Studies
3.8. Ex Vivo Nature of the Valsartan Ethosomal Gel (VLT-Ethogel)
3.9. Pharmacokinetics Study of VLT in the Prepared Gel
Calculating the Pharmacokinetic Parameters
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Godin, B.; Touitou, E. Ethosomes: New Prospects in Transdermal Delivery. Crit. Rev. Ther. Drug Carr. Syst. 2003, 20, 63–102. [Google Scholar] [CrossRef] [PubMed]
- Formulation and Delivery Strategies of Ibuprofen: Challenges and Opportunities: Drug Development and Industrial Pharmacy: Vol 44, No 2. Available online: https://www.tandfonline.com/doi/abs/10.1080/03639045.2017.1391838 (accessed on 17 November 2021).
- El Maghraby, G.M.M.; Williams, A.C.; Barry, B.W. Skin Delivery of Oestradiol from Lipid Vesicles: Importance of Liposome Structure. Int. J. Pharm. 2000, 204, 159–169. [Google Scholar] [CrossRef]
- Manconi, M.; Sinico, C.; Valenti, D.; Loy, G.; Fadda, A.M. Niosomes as Carriers for Tretinoin. I. Preparation and Properties. Int. J. Pharm. 2002, 234, 237–248. [Google Scholar] [CrossRef]
- Touitou, E.; Godin, B. Ethosomes for Skin Delivery. J. Drug Deliv. Sci. Technol. 2007, 17, 303–308. [Google Scholar] [CrossRef]
- Elsayed, M.M.A.; Abdallah, O.Y.; Naggar, V.F.; Khalafallah, N.M. Lipid Vesicles for Skin Delivery of Drugs: Reviewing Three Decades of Research. Int. J. Pharm. 2007, 332, 1–16. [Google Scholar] [CrossRef]
- Opatha, S.A.T.; Titapiwatanakun, V.; Chutoprapat, R. Transfersomes: A Promising Nanoencapsulation Technique for Transdermal Drug Delivery. Pharmaceutics 2020, 12, 855. [Google Scholar] [CrossRef]
- Shumilov, M.; Touitou, E. Buspirone Transdermal Administration for Menopausal Syndromes, In Vitro and in Animal Model Studies. Int. J. Pharm. 2010, 387, 26–33. [Google Scholar] [CrossRef]
- El-Menshawe, S.F.; Ali, A.A.; Halawa, A.A.; Srag El-Din, A.S. A Novel Transdermal Nanoethosomal Gel of Betahistine Dihydrochloride for Weight Gain Control: In-Vitro and in-Vivo Characterization. Drug Des. Dev. Ther. 2017, 11, 3377–3388. [Google Scholar] [CrossRef] [Green Version]
- An, K.; Sun, Y.; Wu, Y.; Yuan, H.; Cui, Z.; Xu, L. Preparation and In Vitro Percutaneous Penetration of Simvastatin Ethosome Gel. Artif. Cells Nanomed. Biotechnol. 2013, 41, 315–318. [Google Scholar] [CrossRef]
- Bhosale, S.S.; Avachat, A.M. Design and Development of Ethosomal Transdermal Drug Delivery System of Valsartan with Preclinical Assessment in Wistar Albino Rats. J. Liposome Res. 2013, 23, 119–125. [Google Scholar] [CrossRef]
- Akhtar, N.; Verma, A.; Pathak, K. Feasibility of Binary Composition in Development of Nanoethosomal Glycolic Vesicles of Triamcinolone Acetonide Using Box-Behnken Design: In Vitro and Ex Vivo Characterization. Artif. Cells Nanomed. Biotechnol. 2017, 45, 1123–1131. [Google Scholar] [CrossRef] [Green Version]
- Peppas, N.A.; Bures, P.; Leobandung, W.; Ichikawa, H. Hydrogels in Pharmaceutical Formulations. Eur. J. Pharm. Biopharm. 2000, 50, 27–46. [Google Scholar] [CrossRef]
- Abdulbaqi, I.M.; Darwis, Y.; Khan, N.A.K.; Assi, R.A.; Khan, A.A. Ethosomal Nanocarriers: The Impact of Constituents and Formulation Techniques on Ethosomal Properties, In Vivo Studies, and Clinical Trials. Int. J. Nanomed. 2016, 11, 2279–2304. [Google Scholar] [CrossRef] [Green Version]
- Saydam, M.; Takka, S. Bioavailability file: Valsartan. FABAD J. Pharm. Sci. 2007, 4, 185–196. [Google Scholar]
- Nishida, N.; Taniyama, K.; Sawabe, T.; Manome, Y. Development and Evaluation of a Monolithic Drug-in-Adhesive Patch for Valsartan. Int. J. Pharm. 2010, 402, 103–109. [Google Scholar] [CrossRef]
- Touitou, E. Composition for Applying Active Substances to or through the Skin. U.S. Patent 5,716,638, 10 February 1998. [Google Scholar]
- Pandey, V.; Golhani, D.; Shukla, R. Ethosomes: Versatile Vesicular Carriers for Efficient Transdermal Delivery of Therapeutic Agents. Drug Deliv. 2015, 22, 988–1002. [Google Scholar] [CrossRef] [Green Version]
- Jain, S.; Umamaheshwari, R.B.; Bhadra, D.; Jain, N.K. Ethosomes: A Novel Vesicular Carrier for Enhanced Transdermal Delivery of an AntiHIV Agent. Indian J. Pharm. Sci. 2004, 66, 72. [Google Scholar]
- Ramakrishna, G.A.; Manohar, S.D.; Bhanudas, S.R. Ethosomes: Carrier for Enhanced Transdermal Drug Delivery System. J. Adv. Pharm. Educ. Res. 2014, 4, 380–387. [Google Scholar]
- Gupta, A.; Aggarwal, G.; Singla, S.; Arora, R. Transfersomes: A Novel Vesicular Carrier for Enhanced Transdermal Delivery of Sertraline: Development, Characterization, and Performance Evaluation. Sci. Pharm. 2012, 80, 1061–1080. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, A.; Ghorab, M.; Gad, S.; Qushawy, M. Design, Formulation, and Evaluation of Piroxicam Niosomal Gel. Int. J. PharmTech Res. 2014, 6, 185–195. [Google Scholar]
- Garg, B.J.; Garg, N.K.; Beg, S.; Singh, B.; Katare, O.P. Nanosized Ethosomes-Based Hydrogel Formulations of Methoxsalen for Enhanced Topical Delivery against Vitiligo: Formulation Optimization, In Vitro Evaluation and Preclinical Assessment. J. Drug Target. 2016, 24, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Rao, Y.; Zheng, F.; Zhang, X.; Gao, J.; Liang, W. In Vitro Percutaneous Permeation and Skin Accumulation of Finasteride Using Vesicular Ethosomal Carriers. AAPS PharmSciTech 2008, 9, 860–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhalaria, M.K.; Naik, S.; Misra, A.N. Ethosomes: A Novel Delivery System for Antifungal Drugs in the Treatment of Topical Fungal Diseases. IJEB 2009, 47, 368–375. [Google Scholar]
- Dayan, N.; Touitou, E. Carriers for Skin Delivery of Trihexyphenidyl HCl: Ethosomes vs. Liposomes. Biomaterials 2000, 21, 1879–1885. [Google Scholar] [CrossRef]
- Mura, S.; Manconi, M.; Sinico, C.; Valenti, D.; Fadda, A.M. Penetration Enhancer-Containing Vesicles (PEVs) as Carriers for Cutaneous Delivery of Minoxidil. Int. J. Pharm. 2009, 380, 72–79. [Google Scholar] [CrossRef]
- Ghanbarzadeh, S.; Hariri, R.; Kouhsoltani, M.; Shokri, J.; Javadzadeh, Y.; Hamishehkar, H. Enhanced Stability and Dermal Delivery of Hydroquinone Using Solid Lipid Nanoparticles. Colloids Surf. B Biointerfaces 2015, 136, 1004–1010. [Google Scholar] [CrossRef]
- Mbah, C.C.; Builders, P.F.; Agubata, C.O.; Attama, A.A. Development of Ethosomal Vesicular Carrier for Topical Application of Griseofulvin: Effect of Ethanol Concentration. J. Pharm. Investig. 2019, 49, 27–36. [Google Scholar] [CrossRef]
- Nava, G.; Piñón, E.; Mendoza, L.; Mendoza, N.; Quintanar, D.; Ganem, A. Formulation and In Vitro, Ex Vivo and In Vivo Evaluation of Elastic Liposomes for Transdermal Delivery of Ketorolac Tromethamine. Pharmaceutics 2011, 3, 954–970. [Google Scholar] [CrossRef] [Green Version]
- Pradhan, M.; Yadav, K.; Singh, D.; Singh, M.R. Topical Delivery of Fluocinolone Acetonide Integrated NLCs and Salicylic Acid Enriched Gel: A Potential and Synergistic Approach in the Management of Psoriasis. J. Drug Deliv. Sci. Technol. 2021, 61, 102282. [Google Scholar] [CrossRef]
- Jain, S.; Tiwary, A.K.; Sapra, B.; Jain, N.K. Formulation and Evaluation of Ethosomes for Transdermal Delivery of Lamivudine. AAPS PharmSciTech 2007, 8, 249. [Google Scholar] [CrossRef] [Green Version]
- Verma, P.; Pathak, K. Nanosized Ethanolic Vesicles Loaded with Econazole Nitrate for the Treatment of Deep Fungal Infections through Topical Gel Formulation. Nanomed. Nanotechnol. Biol. Med. 2012, 8, 489–496. [Google Scholar] [CrossRef]
- Gilani, S.; Mir, S.; Masood, M.; Khan, A.K.; Rashid, R.; Azhar, S.; Rasul, A.; Ashraf, M.N.; Waqas, M.K.; Murtaza, G. Triple-Component Nanocomposite Films Prepared Using a Casting Method: Its Potential in Drug Delivery. J. Food Drug Anal. 2018, 26, 887–902. [Google Scholar] [CrossRef] [Green Version]
- Fathalla, D.; Youssef, E.M.K.; Soliman, G.M. Liposomal and Ethosomal Gels for the Topical Delivery of Anthralin: Preparation, Comparative Evaluation and Clinical Assessment in Psoriatic Patients. Pharmaceutics 2020, 12, 446. [Google Scholar] [CrossRef]
- Ahmed, A.; Ghorab, M.; Gad, S.; Qushawy, M. The Application of Plackett-Burman Design and Response Surface Methodology for Optimization of Formulation Variables to Produce Piroxicam Niosomes. Int. J. Drug Dev. Res. 2013, 5, 121–130. [Google Scholar]
- Ahmed, T.A. Preparation of Transfersomes Encapsulating Sildenafil Aimed for Transdermal Drug Delivery: Plackett–Burman Design and Characterization. J. Liposome Res. 2015, 25, 1–10. [Google Scholar] [CrossRef]
- Garg, V.; Singh, H.; Bhatia, A.; Raza, K.; Singh, S.K.; Singh, B.; Beg, S. Systematic Development of Transethosomal Gel System of Piroxicam: Formulation Optimization, In Vitro Evaluation, and Ex Vivo Assessment. AAPS PharmSciTech 2017, 18, 58–71. [Google Scholar] [CrossRef]
- Guo, J.; Ping, Q.; Sun, G.; Jiao, C. Lecithin Vesicular Carriers for Transdermal Delivery of Cyclosporin A. Int. J. Pharm. 2000, 194, 201–207. [Google Scholar] [CrossRef]
- Paget, G.E.; Barnes, J.M. Inter Species Dosages Conversion Scheme in Evaluation of Results and Quantitative Application in Different Species Toxicity Test. Eval. Drug Act. Pharmacomet. 1964, 1, 160–162. [Google Scholar]
- Ghosh, M.N. Fundamentals of Experimental Pharmacology; Scientific Book Agency: Calcutta, India, 1984. [Google Scholar]
- Piao, Z.-Z.; Lee, E.-S.; Tran, H.T.T.; Lee, B.-J. Improved Analytical Validation and Pharmacokinetics of Valsartan Using HPLC with UV Detection. Arch. Pharm. Res. 2008, 31, 1055–1059. [Google Scholar] [CrossRef]
- Verma, D.D.; Fahr, A. Synergistic Penetration Enhancement Effect of Ethanol and Phospholipids on the Topical Delivery of Cyclosporin A. J. Control. Release 2004, 97, 55–66. [Google Scholar] [CrossRef]
- Limsuwan, T.; Amnuaikit, T. Development of Ethosomes Containing Mycophenolic Acid. Procedia Chem. 2012, 4, 328–335. [Google Scholar] [CrossRef] [Green Version]
- Jin-guang, C.; Wei, L.a.I.; Yu, J. Preparation of Curcumin Ethosomes. AJPP 2013, 7, 2246–2251. [Google Scholar] [CrossRef]
- Verma, D.D.; Verma, S.; Blume, G.; Fahr, A. Particle Size of Liposomes Influences Dermal Delivery of Substances into Skin. Int. J. Pharm. 2003, 258, 141–151. [Google Scholar] [CrossRef]
- Du Plessis, J.; Ramachandran, C.; Weiner, N.; Müller, D.G. The Influence of Particle Size of Liposomes on the Deposition of Drug into Skin. Int. J. Pharm. 1994, 103, 277–282. [Google Scholar] [CrossRef] [Green Version]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Hasanzadeh Davarani, F.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M.R. Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef] [Green Version]
- Hua, S. Lipid-Based Nano-Delivery Systems for Skin Delivery of Drugs and Bioactives. Front. Pharmacol. 2015, 6, 219. [Google Scholar] [CrossRef]
- Anita, C.; Munira, M.; Mural, Q.; Shaily, L. Topical Nanocarriers for Management of Rheumatoid Arthritis: A Review. Biomed. Pharmacother. 2021, 141, 111880. [Google Scholar] [CrossRef]
- Pathan, I.B. Transdermal Delivery of Ethosomes as a Novel Vesicular Carrier for Paroxetine Hydrochloride: In Vitro Evaluation and In Vivo Study. Marmara Pharm. J. 2015, 20, 1. [Google Scholar] [CrossRef] [Green Version]
- Limsuwan, T.; Boonme, P.; Khongkow, P.; Amnuaikit, T. Ethosomes of Phenylethyl Resorcinol as Vesicular Delivery System for Skin Lightening Applications. BioMed Res. Int. 2017, 2017, e8310979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Wu, L.; Wu, D.; Shi, D.; Wang, T.; Zhu, X. Mechanism of Transdermal Permeation Promotion of Lipophilic Drugs by Ethosomes. Int. J. Nanomed. 2017, 12, 3357–3364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trivedi, R.V.; Taksande, J.B.; Awandekar, N.B.; Umekar, M.J. Effect of lipid composition on liposomal formulation of pramipexole. World J. Pharm. Res. 2017, 6, 17. [Google Scholar]
- Iizhar, S.A.; Syed, I.A.; Satar, R.; Ansari, S.A. In Vitro Assessment of Pharmaceutical Potential of Ethosomes Entrapped with Terbinafine Hydrochloride. J. Adv. Res. 2016, 7, 453–461. [Google Scholar] [CrossRef]
- Youssef, A.; Dudhipala, N.; Majumdar, S. Ciprofloxacin Loaded Nanostructured Lipid Carriers Incorporated into In-Situ Gels to Improve Management of Bacterial Endophthalmitis. Pharmaceutics 2020, 12, 572. [Google Scholar] [CrossRef]
- Finnin, B.C.; Morgan, T.M. Transdermal Penetration Enhancers: Applications, Limitations, and Potential. J. Pharm. Sci. 1999, 88, 955–958. [Google Scholar] [CrossRef]
- Puri, M.; Sharma, D.; Barrow, C.J. Enzyme-Assisted Extraction of Bioactives from Plants. Trends Biotechnol. 2012, 30, 37–44. [Google Scholar] [CrossRef]
- Dubey, V.; Mishra, D.; Dutta, T.; Nahar, M.; Saraf, D.K.; Jain, N.K. Dermal and Transdermal Delivery of an Anti-Psoriatic Agent via Ethanolic Liposomes. J. Control. Release 2007, 123, 148–154. [Google Scholar] [CrossRef]
- Ogiso, T.; Yamaguchi, T.; Iwaki, M.; Tanino, T.; Miyake, Y. Effect of Positively and Negatively Charged Liposomes on Skin Permeation of Drugs. J. Drug Target. 2001, 9, 49–59. [Google Scholar] [CrossRef]
- Abdulbaqi, I.M.; Darwis, Y.; Assi, R.A.; Khan, N.A.K. Transethosomal Gels as Carriers for the Transdermal Delivery of Colchicine: Statistical Optimization, Characterization, and Ex Vivo Evaluation. Drug Des. Dev. Ther. 2018, 12, 795–813. [Google Scholar] [CrossRef]
- Agrawal, U.; Mehra, N.K.; Gupta, U.; Jain, N.K. Hyperbranched Dendritic Nano-Carriers for Topical Delivery of Dithranol. J. Drug Target. 2013, 21, 497–506. [Google Scholar] [CrossRef]
- Kateh Shamshiri, M.; Momtazi-Borojeni, A.A.; Khodabandeh Shahraky, M.; Rahimi, F. Lecithin Soybean Phospholipid Nano-Transfersomes as Potential Carriers for Transdermal Delivery of the Human Growth Hormone. J. Cell. Biochem. 2019, 120, 9023–9033. [Google Scholar] [CrossRef]
- Akhtar, N.; Arkvanshi, S.; Bhattacharya, S.S.; Verma, A.; Pathak, K. Preparation and Evaluation of a Buflomedil Hydrochloride Niosomal Patch for Transdermal Delivery. J. Liposome Res. 2015, 25, 191–201. [Google Scholar] [CrossRef]
- Mura, P.; Bragagni, M.; Mennini, N.; Cirri, M.; Maestrelli, F. Development of Liposomal and Microemulsion Formulations for Transdermal Delivery of Clonazepam: Effect of Randomly Methylated β-Cyclodextrin. Int. J. Pharm. 2014, 475, 306–314. [Google Scholar] [CrossRef]
- Tefas, L.; Muntean, D.; Vlase, L.; Porfire, A.; Achim, M.; Tomuţă, I. Quercetin-Loaded Liposomes: Formulation Optimization through a D-Optimal Experimental Design. Farmacia 2015, 63, 26–33. [Google Scholar]
- Taymouri, S.; Varshosaz, J. Effect of Different Types of Surfactants on the Physical Properties and Stability of Carvedilol Nano-Niosomes. Adv. Biomed. Res. 2016, 5, 48. [Google Scholar] [CrossRef]
- Ahad, A.; Al-Saleh, A.A.; Al-Mohizea, A.M.; Al-Jenoobi, F.I.; Raish, M.; Yassin, A.E.B.; Alam, M.A. Pharmacodynamic Study of Eprosartan Mesylate-Loaded Transfersomes Carbopol® Gel under Dermaroller® on Rats with Methyl Prednisolone Acetate-Induced Hypertension. Biomed. Pharmacother. 2017, 89, 177–184. [Google Scholar] [CrossRef]
- Brittain, S.; Ibbett, H.; de Lange, E.; Dorward, L.; Hoyte, S.; Marino, A.; Milner-Gulland, E.J.; Newth, J.; Rakotonarivo, S.; Veríssimo, D.; et al. Ethical Considerations When Conservation Research Involves People. Conserv. Biol. 2020, 34, 925–933. [Google Scholar] [CrossRef]
- Shaji, J.; Lal, M. Preparation, optimization and evaluation of transferosomal formulation for enhanced transdermal delivery of a cox-2 inhibitor. Int. J. Pharm. Pharm. Sci. 2014, 6, 11. [Google Scholar]
- Prieto, C.; Evtoski, Z.; Pardo-Figuerez, M.; Hrakovsky, J.; Lagaron, J.M. Nanostructured Valsartan Microparticles with Enhanced Bioavailability Produced by High-Throughput Electrohydrodynamic Room-Temperature Atomization. Mol. Pharm. 2021, 18, 2947–2958. [Google Scholar] [CrossRef]
- Hathout, R.M.; Mansour, S.; Mortada, N.D.; Guinedi, A.S. Liposomes as an Ocular Delivery System for Acetazolamide: In Vitro and In Vivo Studies. AAPS PharmSciTech 2007, 8, E1–E12. [Google Scholar] [CrossRef]
- Mohammed, M.I.; Makky, A.M.A.; Teaima, M.H.M.; Abdellatif, M.M.; Hamzawy, M.A.; Khalil, M.A.F. Transdermal Delivery of Vancomycin Hydrochloride Using Combination of Nano-Ethosomes and Iontophoresis: In Vitro and In Vivo Study. Drug Deliv. 2016, 23, 1558–1564. [Google Scholar] [CrossRef]
- Mahapatra, A.K.; Pn, M.; Biswal, S.; Mahapatra, A.; Pradhan, S. Dissolution Enhancement and Physicochemical Characterization of Valsartan in Solid Dispersions with β-CD, HP β-CD, and PVP K-30. Dissolution Technol. 2011, 18, 39–45. [Google Scholar] [CrossRef]
- Bodade, S.S.; Shaikh, K.S.; Kamble, M.S.; Chaudhari, P.D. A Study on Ethosomes as Mode for Transdermal Delivery of an Antidiabetic Drug. Drug Deliv. 2013, 20, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Soliman, G.; Fathalla, D.; Fouad, E. Development and In Vitro/In Vivo Evaluation of Liposomal Gels for the Sustained Ocular Delivery of Latanoprost. J. Clin. Exp. Ophthalmol. 2015, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Chourasia, M.K.; Kang, L.; Chan, S.Y. Nanosized Ethosomes Bearing Ketoprofen for Improved Transdermal Delivery. Results Pharma Sci. 2011, 1, 60–67. [Google Scholar] [CrossRef] [Green Version]
- Caddeo, C.; Sales, O.D.; Valenti, D.; Saurí, A.R.; Fadda, A.M.; Manconi, M. Inhibition of Skin Inflammation in Mice by Diclofenac in Vesicular Carriers: Liposomes, Ethosomes and PEVs. Int. J. Pharm. 2013, 443, 128–136. [Google Scholar] [CrossRef]
- Zhao, Y.-Z.; Lu, C.-T.; Zhang, Y.; Xiao, J.; Zhao, Y.-P.; Tian, J.-L.; Xu, Y.-Y.; Feng, Z.-G.; Xu, C.-Y. Selection of High Efficient Transdermal Lipid Vesicle for Curcumin Skin Delivery. Int. J. Pharm. 2013, 454, 302–309. [Google Scholar] [CrossRef]
- Mahmood, S.; Mandal, U.K.; Chatterjee, B. Transdermal Delivery of Raloxifene HCl via Ethosomal System: Formulation, Advanced Characterizations and Pharmacokinetic Evaluation. Int. J. Pharm. 2018, 542, 36–46. [Google Scholar] [CrossRef]
- Deshpande, P.B.; Gurram, A.K.; Deshpande, A.; Shavi, G.V.; Musmade, P.; Arumugam, K.; Averineni, R.K.; Mutalik, S.; Reddy, M.S.; Udupa, N. A Novel Nanoproliposomes of Lercanidipine: Development, In Vitro and Preclinical Studies to Support Its Effectiveness in Hypertension Therapy. Life Sci. 2016, 162, 125–137. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factor 1 | Factor 2 | Factor 3 | Factor 4 | Factor 5 | |
|---|---|---|---|---|---|
| Formulation | A: Lipid Concentration | B: Ethanol Concentration | C: Sonication Time | D: Lipid Type | E: Penetration Enhancer Type (10%) |
| % w/v | % w/v | min | (%) | ||
| 1 | 4 | 10 | 5 | Phospholipone | Transcutol |
| 2 | 4 | 40 | 5 | Lecithin | Transcutol |
| 3 | 4 | 10 | 5 | Phospholipone | Transcutol |
| 5 | 1 | 10 | 5 | Lecithin | Propylene glycol |
| 6 | 4 | 40 | 10 | Lecithin | Propylene glycol |
| 7 | 2.5 | 25 | 7.5 | Lecithin | Transcutol |
| 8 | 1 | 10 | 10 | Lecithin | Transcutol |
| 9 | 1 | 40 | 5 | Lecithin | Transcutol |
| 10 | 1.75 | 32.5 | 6.25 | Phospholipone | Propylene glycol |
| 11 | 4 | 10 | 10 | Lecithin | Transcutol |
| 11 | 4 | 10 | 10 | Lecithin | Transcutol |
| 12 | 1 | 40 | 10 | Phospholipone | Propylene glycol |
| 13 | 4 | 40 | 10 | Lecithin | Propylene glycol |
| 14 | 1 | 40 | 10 | Phospholipone | Propylene glycol |
| 15 | 1 | 10 | 10 | Lecithin | Transcutol |
| 16 | 1 | 10 | 5 | Lecithin | Propylene glycol |
| Drug (mg) | 80 in all formulations | ||||
| Water (%v/v) | To 100 in all formulations | ||||
| F | A | B | C | D | E | R1 | R2 | R3 | R4 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 4 | 10 | 5 | PL 90G | Trans P | 177.00 ± 3.20 | −3.42 ± 0.30 | 91.44 ± 0.01 | 0.62 ± 0.00 |
| 2 | 4 | 40 | 5 | Lecithin | Trans P | 1723.00 ± 18.00 | −4.15 ± 0.10 | 90.01 ± 0.01 | 0.24 ± 0.20 |
| 3 | 4 | 10 | 5 | PL 90G | Trans P | 389.57 ± 4.50 | −2.58 ± 0.30 | 94.77 ± 0.02 | 0.68 ± 0.20 |
| 4 | 1 | 10 | 5 | Lecithin | Prop. glycol | 390.30 ± 3.10 | −20.03 ± 0.30 | 96.38 ± 0.01 | 1.00 ± 0.10 |
| 5 | 4 | 40 | 10 | Lecithin | Prop. glycol | 133.80 ± 4.10 | −1.10 ± 1.40 | 83.51 ± 0.02 | 0.38 ± 0.00 |
| 6 | 2.5 | 25 | 7.5 | Lecithin | Trans P | 369.73 ± 0.80 | −11.13 ± 0.10 | 88.69 ± 0.02 | 0.47 ± 0.00 |
| 7 | 1 | 10 | 10 | Lecithin | Trans P | 313.83 ± 1.50 | −25.73 ± 0.40 | 95.03 ± 0.01 | 1.00 ± 0.00 |
| 8 | 1 | 40 | 5 | Lecithin | Trans P | 635.83 ± 3.00 | −7.43 ± 0.30 | 95.19 ± 0.01 | 0.41 ± 0.00 |
| 9 | 1.75 | 32.5 | 6.25 | PL 90G | Prop. glycol | 671.20 ± 2.80 | 0.44 ± 0.10 | 74.27 ± 0.02 | 0.98 ± 0.10 |
| 10 | 4 | 10 | 10 | Lecithin | Trans P | 1855.33 ± 4.00 | −19.03 ± 0.00 | 84.48 ± 0.02 | 0.60 ± 0.00 |
| 11 | 4 | 10 | 10 | Lecithin | Trans P | 1855.33 ± 72.60 | −19.03 ± 0.40 | 84.48 ± 0.01 | 0.60 ± 0.10 |
| 12 | 1 | 40 | 10 | PL 90G | Prop. glycol | 873.87 ± 10.80 | 0.18 ± 0.20 | 91.53 ± 0.02 | 1.00 ± 0.00 |
| 13 | 4 | 40 | 10 | Lecithin | Prop. glycol | 223.17 ± 5.20 | −6.15 ± 0.20 | 95.90 ± 0.01 | 0.45 ± 0.00 |
| 14 | 1 | 40 | 10 | PL 90G | Prop. glycol | 1079.67 ± 46.30 | 0.68 ± 0.00 | 92.18 ± 0.01 | 0.64 ± 0.00 |
| 15 | 1 | 10 | 10 | Lecithin | Trans P | 435.03 ± 3.50 | −11.80 ± 0.50 | 86.93 ± 0.01 | 0.43 ± 0.00 |
| 16 | 1 | 10 | 5 | Lecithin | Prop. glycol | 187.37 ± 6.30 | −25.70 ± 0.30 | 92.97 ± 0.01 | 1.00 ± 0.00 |
| Intercept | A | B | C | D | E | |
|---|---|---|---|---|---|---|
| R1:PS | 16.9112 | 11.5183 | −12.436 | |||
| p-values | 0.0004 | 0.0003 | ||||
| R2:ZP | 9.89743 | −5.2385 | −6.4237 | 3.50959 | ||
| p-values | 0.0094 | 0.0033 | 0.0482 | |||
| R3:EE | 94.3599 | −4.3202 | 3.9952 | |||
| p-values | 0.0010 | 0.0022 | ||||
| R4:PDI | 0.59507 | −0.1425 | −0.2017 | |||
| p-values | 0.0029 | 0.0005 |
| F | A | B | C | R1 | R2 | R3 | R4 |
|---|---|---|---|---|---|---|---|
| 1 | 22.50 | 2.50 | 5.50 | 120.97 ± 17.90 | 0.39 ± 0.00 | −41.33 ± 1.30 | 93.40 ± 0.00 |
| 2 | 22.50 | 1.00 | 7.00 | 253.27 ± 9.40 | 0.38 ± 0.00 | −48.00 ± 2.90 | 91.56 ± 0.5 |
| 3 | 27.50 | 1.00 | 5.50 | 248.20 ± 9.10 | 0.36 ± 0.00 | −47.30 ± 2.40 | 92.27 ± 0.9 |
| 4 | 27.50 | 2.50 | 5.50 | 101.60 ± 13.40 | 0.48 ± 0.10 | −62.13 ± 4.10 | 91.44 ± 0.2 |
| 5 | 25.00 | 1.75 | 7.51 | 139.77 ± 15.80 | 0.48 ± 0.10 | −49.27 ± 3.70 | 87.30 ± 2.1 |
| 6 | 25.00 | 0.49 | 6.25 | 120.87 ± 2.3 | 0.32 ± 0.00 | −40.70 ± 0.80 | 90.10 ± 0.5 |
| 7 | 25.00 | 1.75 | 6.25 | 126.33 ± 0.50 | 0.28 ± 0.00 | −52.73 ± 1.80 | 95.75 ± 0.5 |
| 8 | 29.20 | 1.75 | 6.25 | 214.70 ± 16.90 | 0.29 ± 0.00 | −51.47 ± 3.00 | 93.89 ± 2.9 |
| 9 | 25.00 | 1.75 | 6.25 | 51.33 ± 0.60 | 0.24 ± 0.00 | −49.53 ± 1.10 | 92.27 ± 0.4 |
| 10 | 22.50 | 2.50 | 7.00 | 51.68 ± 0.90 | 0.42 ± 0.10 | −54.67 ± 2.00 | 93.65 ± 0.2 |
| 11 | 27.50 | 2.50 | 7.00 | 156.50 ± 3.20 | 0.27 ± 0.00 | −58.07 ± 1.00 | 91.18 ± 0.2 |
| 12 | 20.80 | 1.75 | 6.25 | 202.90 ± 2.50 | 0.28 ± 0.00 | −57.87 ± 1.20 | 94.14 ± 2.2 |
| 13 | 27.50 | 1.00 | 7.00 | 170.17 ± 6.20 | 0.34 ± 0.00 | −58.20 ± 0.60 | 89.97 ± 0.3 |
| 14 | 25.00 | 1.75 | 6.25 | 123.27 ± 0.50 | 0.23 ± 0.00 | −50.10 ± 1.30 | 92.69 ± 1.3 |
| 15 | 25.00 | 1.75 | 4.99 | 160.47 ± 2.80 | 0.26 ± 0.00 | −51.40 ± 3.00 | 95.25 ± 1.4 |
| 16 | 22.50 | 1.00 | 5.50 | 59.39 ± 1.30 | 0.48 ± 0.10 | −45.10 ± 5.30 | 96.90 ± 0.3 |
| 17 | 25.00 | 3.01 | 6.25 | 45.90 ± 0.40 | 0.32 ± 0.00 | −58.70 ± 2.70 | 93.54 ± 2.1 |
| 18 | 25.00 | 1.75 | 6.25 | 81.87 ± 5.40 | 0.37 ± 0.00 | −62.30 ± 3.20 | 91.09 ± 1.7 |
| Intercept | A | B | C | AÂ2 | |
|---|---|---|---|---|---|
| PS | 107.15 | 15.45 | −31.22 | 36.65 | |
| p-values | 0.2878 | 0.0424 | 0.0202 | ||
| ZP | −52.16 | −3.51 | |||
| p-values | 0.0417 | ||||
| EE% | 92.58 | −1.54 | |||
| p-values | 0.0087 |
| Number | Independent Variables | Dependent Variables Observed Values | |||||
|---|---|---|---|---|---|---|---|
| F | A | B | C | R1 | R2 | R3 | R4 |
| %v/v | %w/v | minute | nm | mV | % | ||
| 1 | 24.47 | 2.50 | 5.50 | 45.8 ± 0.5 | −51.4 ± 6.3 | 94.24 ± 0.2 | 0.32 ± 0.02 |
| Tmax (h) | Cmax (µg/mL) | T½ (h) | AUC (0–24) (µg.h/mL) | AUC (0–∞) (µg.h/mL) | |
|---|---|---|---|---|---|
| Optimized VAL-nanoethosomal gel | 16 | 4.94 ± 8.02 | 12.02 ± 0.45 | 85.8 ± 37.33 | 137.2 ± 49.88 |
| Oral VAL suspension | 2 | 0.86 ± 2.14 | 2.038 ± 0.50 | 1.3 ± 12.62 | 7.0 ± 2.94 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nasr, A.M.; Moftah, F.; Abourehab, M.A.S.; Gad, S. Design, Formulation, and Characterization of Valsartan Nanoethosomes for Improving Their Bioavailability. Pharmaceutics 2022, 14, 2268. https://doi.org/10.3390/pharmaceutics14112268
Nasr AM, Moftah F, Abourehab MAS, Gad S. Design, Formulation, and Characterization of Valsartan Nanoethosomes for Improving Their Bioavailability. Pharmaceutics. 2022; 14(11):2268. https://doi.org/10.3390/pharmaceutics14112268
Chicago/Turabian StyleNasr, Ali M., Fayrouz Moftah, Mohammed A. S. Abourehab, and Shadeed Gad. 2022. "Design, Formulation, and Characterization of Valsartan Nanoethosomes for Improving Their Bioavailability" Pharmaceutics 14, no. 11: 2268. https://doi.org/10.3390/pharmaceutics14112268