
Assessing the Functional Redundancy between P-gp and BCRP in Controlling the Brain Distribution and Biliary Excretion of Dual Substrates with PET Imaging in Mice

 , , , and
, , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Radiotracer Synthesis and Formulation
2.2. PET Imaging
2.3. Analysis of PET Data
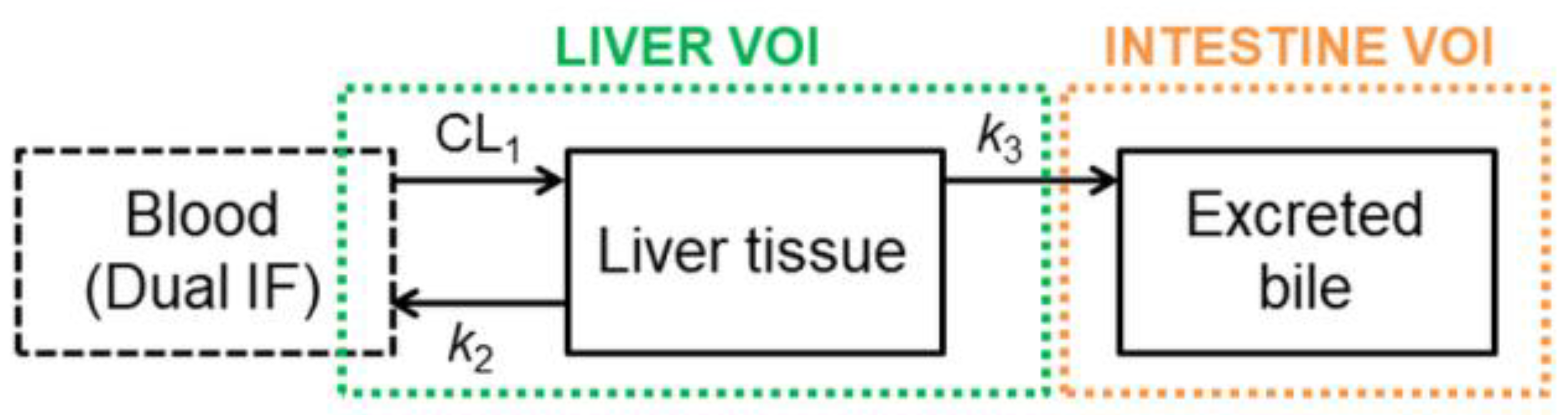
2.4. Pharmacokinetic Modeling
2.5. Statistical Analysis
3. Results
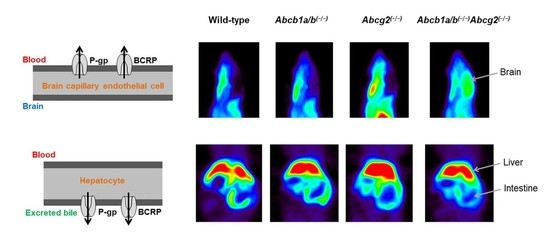
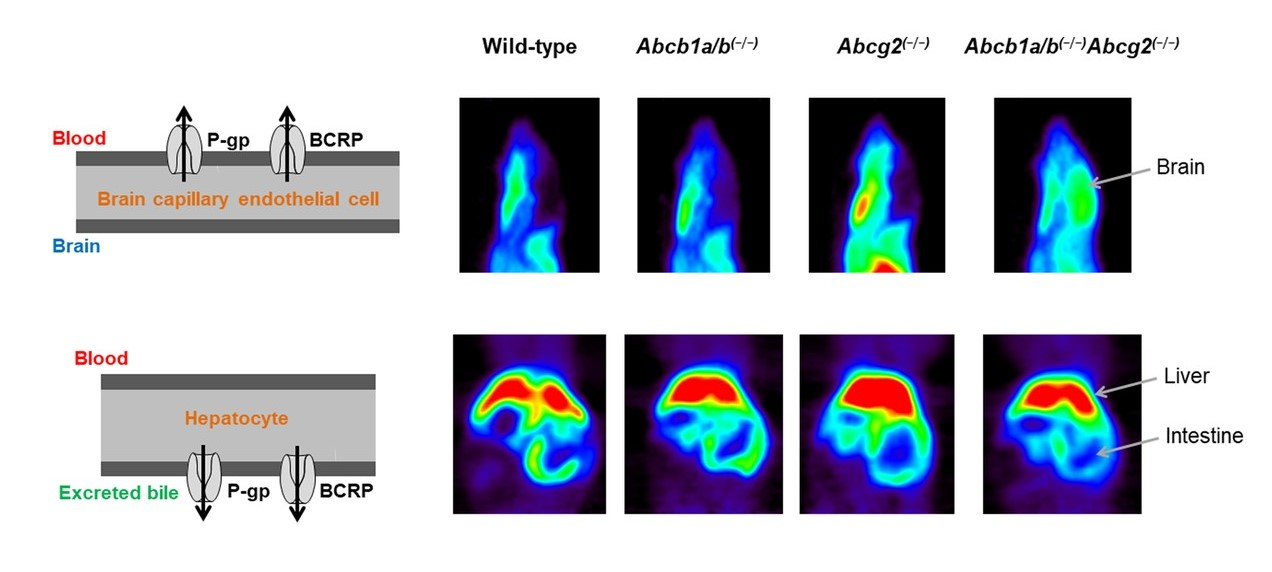
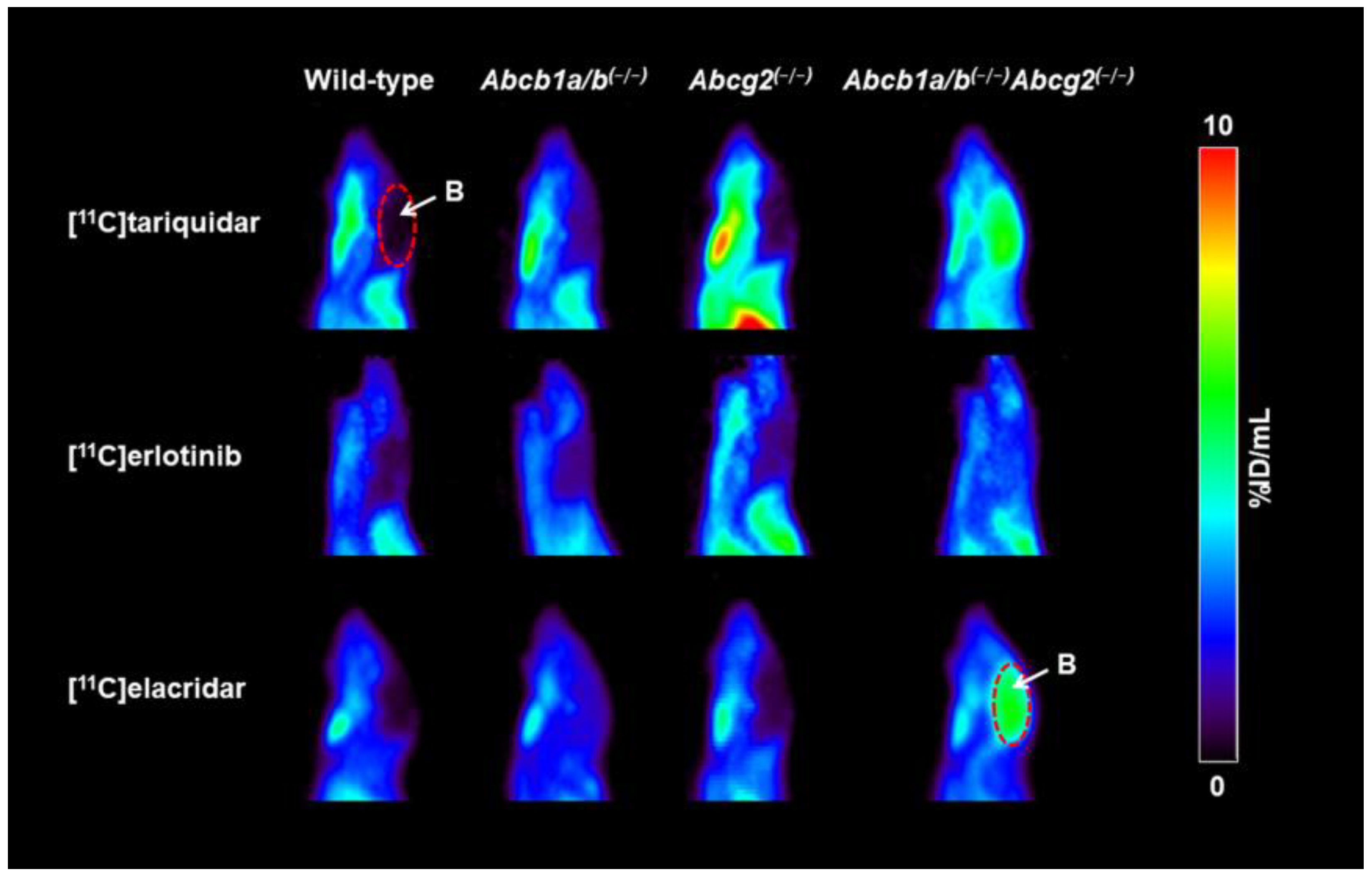
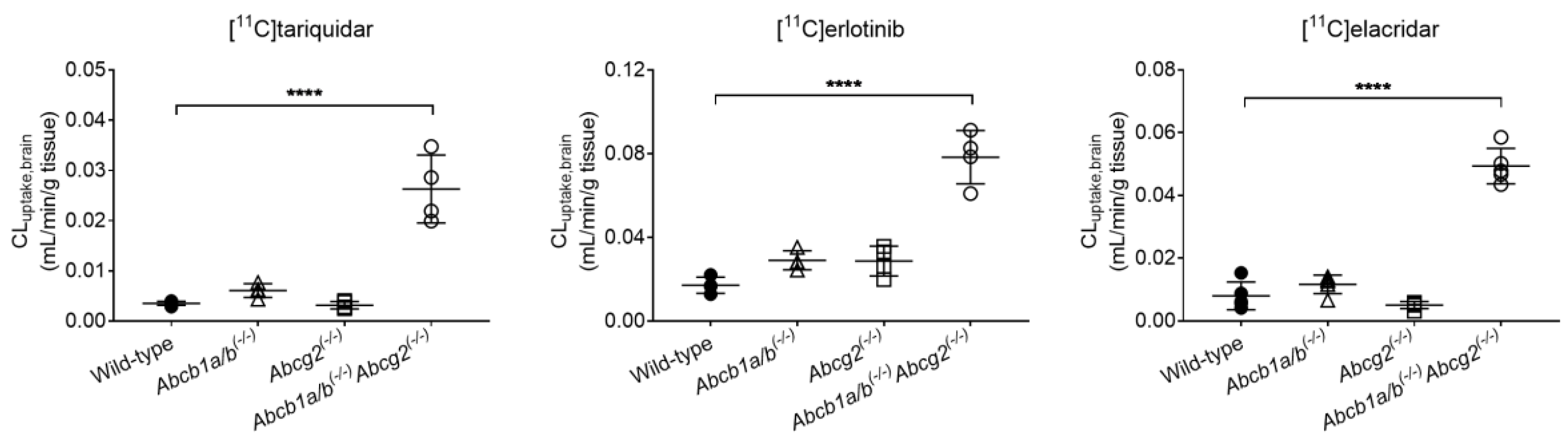
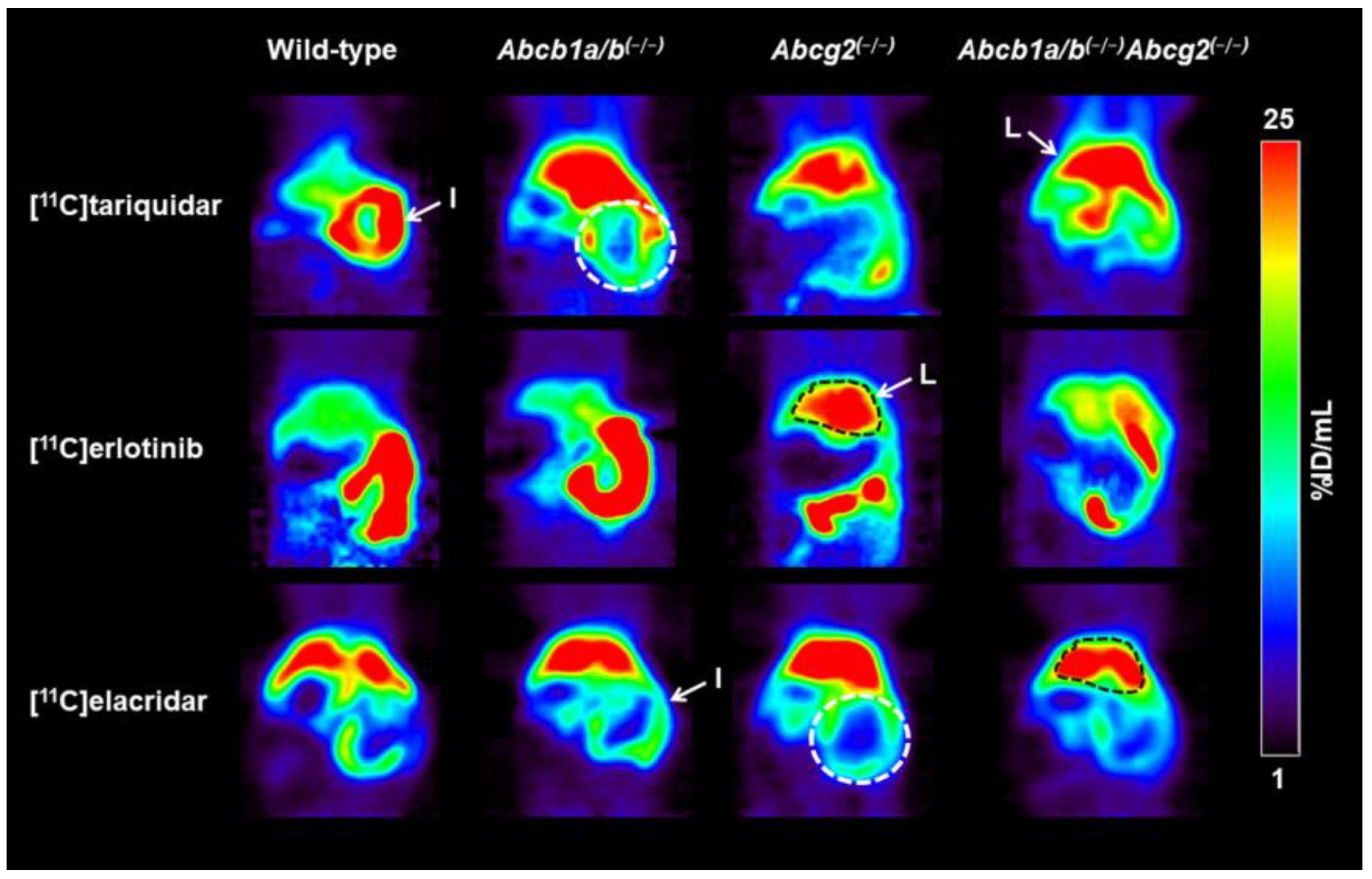
3.1. Functional Redundancy between P-gp and BCRP in Controlling the Brain Distribution of [11C]tariquidar, [11C]erlotinib, and [11C]elacridar
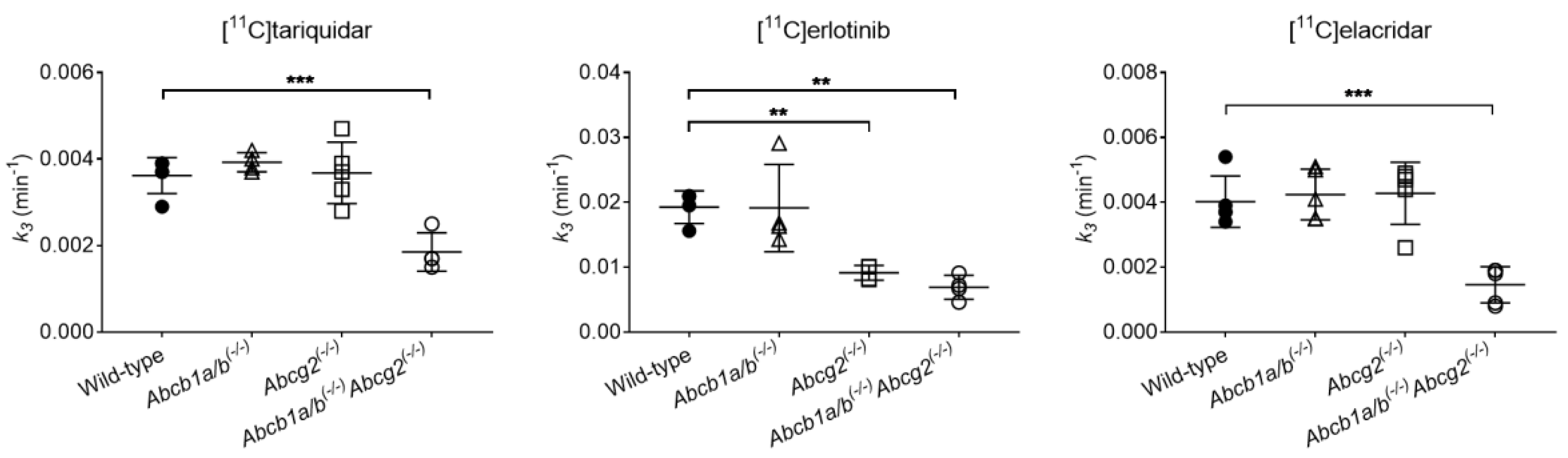
3.2. Functional Redundancy between P-gp and BCRP in Mediating the Biliary Excretion of [11C]tariquidar, [11C]erlotinib, and [11C]elacridar
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Giacomini, K.; Huang, S.; Tweedie, D. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar]
- Agarwal, S.; Hartz, A.M.S.; Elmquist, E.F.; Bauer, B. Breast cancer resistance protein and p-glycoprotein in brain cancer: Two gatekeepers team up. Curr. Pharm. Des. 2011, 17, 2793–2802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durmus, S.; Hendrikx, J.J.; Schinkel, A.H. Apical ABC transporters and cancer chemotherapeutic drug disposition. Adv. Cancer. Res. 2015, 125, 1–41. [Google Scholar] [PubMed]
- Kalvass, J.C.; Polli, J.W.; Bourdet, D.L.; Feng, B.; Huang, S.M.; Liu, X.; Smith, Q.R.; Zhang, L.K.; Zamek-Gliszczynski, M.J.; International Transporter, C. Why clinical modulation of efflux transport at the human blood-brain barrier is unlikely: The ITC evidence-based position. Clin. Pharmacol. Ther. 2013, 94, 80–94. [Google Scholar] [CrossRef] [PubMed]
- Polli, J.W.; Olson, K.L.; Chism, J.P.; John-Williams, L.S.; Yeager, R.L.; Woodard, S.M.; Otto, V.; Castellino, S.; Demby, V.E. An unexpected synergist role of P-glycoprotein and breast cancer resistance protein on the central nervous system penetration of the tyrosine kinase inhibitor lapatinib (N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethyl]amino }methyl)-2-furyl]-4-quinazolinamine; GW572016). Drug Metab. Dispos. 2009, 37, 439–442. [Google Scholar] [PubMed]
- Kodaira, H.; Kusuhara, H.; Ushiki, J.; Fuse, E.; Sugiyama, Y. Kinetic analysis of the cooperation of P-glycoprotein (P-gp/Abcb1) and breast cancer resistance protein (Bcrp/Abcg2) in limiting the brain and testis penetration of erlotinib, flavopiridol, and mitoxantrone. J. Pharmacol. Exp. Ther. 2010, 333, 788–796. [Google Scholar] [CrossRef] [Green Version]
- Bauer, M.; Romermann, K.; Karch, R.; Wulkersdorfer, B.; Stanek, J.; Philippe, C.; Maier-Salamon, A.; Haslacher, H.; Jungbauer, C.; Wadsak, W.; et al. Pilot PET study to assess the functional interplay between ABCB1 and ABCG2 at the human blood-brain barrier. Clin. Pharmacol. Ther. 2016, 100, 131–141. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Tournier, N.; Stieger, B.; Langer, O. Imaging techniques to study drug transporter function in vivo. Pharmacol. Ther. 2018, 189, 104–122. [Google Scholar] [CrossRef] [Green Version]
- Hernández Lozano, I.; Langer, O. Use of imaging to assess the activity of hepatic transporters. Expert Opin. Drug Metab. Toxicol. 2020, 16, 149–164. [Google Scholar] [CrossRef] [Green Version]
- Bankstahl, J.P.; Bankstahl, M.; Römermann, K.; Wanek, T.; Stanek, J.; Windhorst, A.D.; Fedrowitz, M.; Erker, T.; Müller, M.; Löscher, W.; et al. Tariquidar and elacridar are dose-dependently transported by P-glycoprotein and Bcrp at the blood-brain barrier: A small-animal positron emission tomography and in vitro study. Drug Metab. Dispos. 2013, 41, 754–762. [Google Scholar] [CrossRef] [Green Version]
- Wanek, T.; Kuntner, C.; Bankstahl, J.P.; Mairinger, S.; Bankstahl, M.; Stanek, J.; Sauberer, M.; Filip, T.; Erker, T.; Muller, M.; et al. A novel PET protocol for visualization of breast cancer resistance protein function at the blood-brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 2002–2011. [Google Scholar] [CrossRef] [Green Version]
- Bauer, M.; Karch, R.; Zeitlinger, M.; Stanek, J.; Philippe, C.; Wadsak, W.; Mitterhauser, M.; Jager, W.; Haslacher, H.; Muller, M.; et al. Interaction of [11C]tariquidar and [11C]elacridar with P-glycoprotein and breast cancer resistance protein at the human blood-brain barrier. J. Nucl. Med. 2013, 54, 1181–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traxl, A.; Mairinger, S.; Filip, T.; Sauberer, M.; Stanek, J.; Poschner, S.; Jäger, W.; Zoufal, V.; Novarino, G.; Tournier, N.; et al. Inhibition of ABCB1 and ABCG2 at the mouse blood-brain barrier with marketed drugs to improve brain delivery of the model ABCB1/ABCG2 substrate [11C]erlotinib. Mol. Pharm. 2019, 16, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Karch, R.; Wulkersdorfer, B.; Philippe, C.; Nics, L.; Klebermass, E.M.; Weber, M.; Poschner, S.; Haslacher, H.; Jager, W.; et al. A proof-of-concept study to inhibit ABCG2- and ABCB1-mediated efflux transport at the human blood-brain barrier. J. Nucl. Med. 2019, 60, 486–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traxl, A.; Wanek, T.; Mairinger, S.; Stanek, J.; Filip, T.; Sauberer, M.; Müller, M.; Kuntner, C.; Langer, O. Breast cancer resistance protein and P-Glycoprotein influence in vivo disposition of [11C]erlotinib. J. Nucl. Med. 2015, 56, 1930–1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tournier, N.; Goutal, S.; Mairinger, S.; Hernandez-Lozano, I.; Filip, T.; Sauberer, M.; Caille, F.; Breuil, L.; Stanek, J.; Freeman, A.F.; et al. Complete inhibition of ABCB1 and ABCG2 at the blood-brain barrier by co-infusion of erlotinib and tariquidar to improve brain delivery of the model ABCB1/ABCG2 substrate [11C]erlotinib. J. Cereb. Blood Flow Metab. 2021, 41, 1634–1646. [Google Scholar] [CrossRef]
- Ling, J.; Johnson, K.A.; Miao, Z.; Rakhit, A.; Pantze, M.P.; Hamilton, M.; Lum, B.L.; Prakash, C. Metabolism and excretion of erlotinib, a small molecule inhibitor of epidermal growth factor receptor tyrosine kinase, in healthy male volunteers. Drug Metab. Dispos. 2006, 34, 420–426. [Google Scholar] [CrossRef] [Green Version]
- Bauer, M.; Blaickner, M.; Philippe, C.; Wadsak, W.; Hacker, M.; Zeitlinger, M.; Langer, O. Whole-body distribution and radiation dosimetry of [11C]elacridar and [11C]tariquidar in humans. J. Nucl. Med. 2016, 57, 1265–1268. [Google Scholar] [CrossRef] [Green Version]
- Bauer, F.; Kuntner, C.; Bankstahl, J.P.; Wanek, T.; Bankstahl, M.; Stanek, J.; Mairinger, S.; Dörner, B.; Löscher, W.; Müller, M.; et al. Synthesis and in vivo evaluation of [11C]tariquidar, a positron emission tomography radiotracer based on a third-generation P-glycoprotein inhibitor. Bioorg. Med. Chem. 2010, 18, 5489–5497. [Google Scholar] [CrossRef] [Green Version]
- Philippe, C.; Mairinger, S.; Pichler, V.; Stanek, J.; Nics, L.; Mitterhauser, M.; Hacker, M.; Wanek, T.; Langer, O.; Wadsak, W. Comparison of fully-automated radiosyntheses of [11C]erlotinib for preclinical and clinical use starting from in target produced [11C]CO2 or [11C]CH4. EJNMMI Radiopharm. Chem. 2018, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Dörner, B.; Kuntner, C.; Bankstahl, J.P.; Bankstahl, M.; Stanek, J.; Wanek, T.; Stundner, G.; Mairinger, S.; Loscher, W.; Muller, M.; et al. Synthesis and small-animal positron emission tomography evaluation of [11C]-elacridar as a radiotracer to assess the distribution of P-glycoprotein at the blood-brain barrier. J. Med. Chem. 2009, 52, 6073–6082. [Google Scholar] [CrossRef] [Green Version]
- Loening, A.M.; Gambhir, S.S. AMIDE: A free software tool for multimodality medical image analysis. Mol. Imaging 2003, 2, 131–137. [Google Scholar] [CrossRef]
- Takashima, T.; Yokoyama, C.; Mizuma, H.; Yamanaka, H.; Wada, Y.; Onoe, K.; Nagata, H.; Tazawa, S.; Doi, H.; Takahashi, K.; et al. Developmental changes in P-glycoprotein function in the blood-brain barrier of nonhuman primates: PET study with R-11C-verapamil and 11C-oseltamivir. J. Nucl. Med. 2011, 52, 950–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusuhara, H.; Suzuki, H.; Terasaki, T.; Kakee, A.; Lemaire, M.; Sugiyama, Y. P-glycoprotein mediates the efflux of quinidine across the blood-brain barrier. J. Pharmacol. Exp. Ther. 1997, 283, 574–580. [Google Scholar]
- Logan, J.; Fowler, J.S.; Volkow, N.D.; Wolf, A.P.; Dewey, S.L.; Schlyer, D.J.; MacGregor, R.R.; Hitzemann, R.; Bendriem, B.; Gatley, S.J.; et al. Graphical analysis of reversible radioligand binding from time-activity measurements applied to [N-11C-methyl]-(-)-cocaine PET studies in human subjects. J. Cereb. Blood Flow Metab. 1990, 10, 740–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández Lozano, I.; Bauer, M.; Wulkersdorfer, B.; Traxl, A.; Philippe, C.; Weber, M.; Häusler, S.; Stieger, B.; Jäger, W.; Mairinger, S.; et al. Measurement of hepatic ABCB1 and ABCG2 transport activity with [11C]tariquidar and PET in humans and mice. Mol. Pharm. 2020, 17, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Hernández Lozano, I.; Karch, R.; Bauer, M.; Blaickner, M.; Matsuda, A.; Wulkersdorfer, B.; Hacker, M.; Zeitlinger, M.; Langer, O. Towards improved pharmacokinetic models for the analysis of transporter-mediated hepatic disposition of drug molecules with positron emission tomography. AAPS. J. 2019, 21, 61. [Google Scholar] [CrossRef] [PubMed]
- Kamiie, J.; Ohtsuki, S.; Iwase, R.; Ohmine, K.; Katsukura, Y.; Yanai, K.; Sekine, Y.; Uchida, Y.; Ito, S.; Terasaki, T. Quantitative atlas of membrane transporter proteins: Development and application of a highly sensitive simultaneous LC/MS/MS method combined with novel in-silico peptide selection criteria. Pharm. Res. 2008, 25, 1469–1483. [Google Scholar] [CrossRef]
- Zamek-Gliszczynski, M.J.; Kalvass, J.C.; Pollack, G.M.; Brouwer, K.L. Relationship between drug/metabolite exposure and impairment of excretory transport function. Drug Metab. Dispos. 2009, 37, 386–390. [Google Scholar] [CrossRef] [Green Version]
- Munk, O.L.; Keiding, S.; Bass, L. Impulse-response function of splanchnic circulation with model-independent constraints: Theory and experimental validation. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 285, 671–680. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, K.; Yamasaki, T.; Konno, F.; Yui, J.; Hatori, A.; Yanamoto, K.; Wakizaka, H.; Takei, M.; Kimura, Y.; Fukumura, T.; et al. Evaluation of limiting brain penetration related to P-glycoprotein and breast cancer resistance protein using [11C]GF120918 by PET in mice. Mol. Imaging Biol. 2011, 13, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Uchida, Y.; Mittapalli, R.K.; Sane, R.; Terasaki, T.; Elmquist, W.F. Quantitative proteomics of transporter expression in brain capillary endothelial cells isolated from P-glycoprotein (P-gp), breast cancer resistance protein (Bcrp), and P-gp/Bcrp knockout mice. Drug Metab. Dispos. 2012, 40, 1164–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Chen, H.L.; Liu, L.; Sheps, J.A.; Phillips, M.J.; Ling, V. Compensatory role of P-glycoproteins in knockout mice lacking the bile salt export pump. Hepatology 2009, 50, 948–956. [Google Scholar] [CrossRef]
- Mennone, A.; Soroka, C.J.; Harry, K.M.; Boyer, J.L. Role of breast cancer resistance protein in the adaptive response to cholestasis. Drug Metab. Dispos. 2010, 38, 1673–1678. [Google Scholar] [CrossRef] [Green Version]
- Fallon, J.K.; Smith, P.C.; Xia, C.Q.; Kim, M.S. Quantification of four efflux drug transporters in liver and kidney across species using targeted quantitative proteomics by isotope dilution nanoLC-MS/MS. Pharm. Res. 2016, 33, 2280–2288. [Google Scholar] [CrossRef] [PubMed]
- Badawi, R.D.; Shi, H.; Hu, P.; Chen, S.; Xu, T.; Price, P.M.; Ding, Y.; Spencer, B.A.; Nardo, L.; Liu, W.; et al. First human imaging studies with the EXPLORER total-body PET scanner. J. Nucl. Med. 2019, 60, 299–303. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Radiotracer | Parameter | Wild-Type | Abcb1a/b(−/−) | Abcg2(−/−) | Abcb1a/b(−/−)Abcg2(−/−) |
|---|---|---|---|---|---|
| [11C]tariquidar | CLuptake,brain (mL/min/g tissue) | 0.0035 ± 0.0004 | 0.0061 ± 0.0014 | 0.0031 ± 0.0007 | 0.0263 ± 0.0068 * |
| VT,brain | 0.1269 ± 0.0133 | 0.2169 ± 0.0227 | 0.1045 ± 0.0065 | 0.7460 ± 0.1504 * | |
| [11C]erlotinib | CLuptake,brain (mL/min/g tissue) | 0.0171 ± 0.0038 | 0.0290 ± 0.0045 | 0.0287 ± 0.0071 | 0.0784 ± 0.0128 * |
| VT,brain | 0.2016 ± 0.0141 | 0.2622 ± 0.0079 | 0.2335 ± 0.0174 | 0.5015 ± 0.0497 * | |
| [11C]elacridar | CLuptake,brain (mL/min/g tissue) | 0.0080 ± 0.0044 | 0.0116 ± 0.0030 | 0.0051 ± 0.0011 | 0.0493 ± 0.0564 * |
| VT,brain | 0.1547 ± 0.0489 | 0.2460 ± 0.0673 | 0.1097 ± 0.0315 | 0.6048 ± 0.2069 * |
| Radiotracer | Parameter | Wild-Type | Abcb1a/b(−/−) | Abcg2(−/−) | Abcb1a/b(−/−)Abcg2(−/−) |
|---|---|---|---|---|---|
| [11C]tariquidar | CL1 (mL/min) | 2.2798 ± 0.5058 (12.9–32.3) | 1.7207 ± 0.1377 (10.7–23.1) | 1.5836 ± 0.1993 (9.6–22.1) | 1.9316 ± 0.6933 (9.6–34.2) |
| k2 (min−1) | 0.2600 ± 0.0475 (11.0–26.4) | 0.2421 ± 0.0227 (10.5–19.0) | 0.3244 ± 0.0204 (9.0–20.5) | 0.3441 ± 0.1549 (9.0–29.7) | |
| k3 (min−1) | 0.0036 ± 0.0004 (4.1–7.6) | 0.0039 ± 0.0002 (4.2–7.6) | 0.0037 ± 0.0007 (2.8–9.1) | 0.0019 ± 0.0004 * (5.3–8.4) | |
| [11C]erlotinib | CL1 (mL/min) | 5.9437 ± 2.4249 (5.3–51.5) | 5.2978 ± 1.8375 (12.1–45.4) | 6.3856 ± 1.7690 (10.6–33.4) | 7.3708 ± 0.2309 (20.7–36.7) |
| k2 (min−1) | 0.6431 ± 0.2567 (5.1–47.3) | 0.5815 ± 0.1818 (10.8–41.4) | 0.6051 ± 0.0664 (10.7–30.8) | 0.5484 ± 0.1021 (17.6–32.2) | |
| k3 (min−1) | 0.0193 ± 0.0025 (1.2–4.2) | 0.0191 ± 0.0067 (1.7–4.7) | 0.0091 ± 0.0011 * (1.9–3.3) | 0.0069 ± 0.0018 * (2.4–4.1) | |
| [11C]elacridar | CL1 (mL/min) | 0.5338 ± 0.1759 (6.0–14.3) | 0.6887 ± 0.1939 (11.3–22.4) | 0.4304 ± 0.1392 (4.0–21.0) | 0.5777 ± 0.2241 (4.9–24.6) |
| k2 (min−1) | 0.1121 ± 0.0641 (7.7–13.5) | 0.1872 ± 0.1165 (10.5–17.5) | 0.1674 ± 0.1366 (4.8–20.6) | 0.1426 ± 0.0728 (7.1–18.8) | |
| k3 (min−1) | 0.0040 ± 0.0008 (4.0–6.8) | 0.0042 ± 0.0008 (5.2–8.9) | 0.0043 ± 0.0010 (3.2–18.6) | 0.0015 ± 0.0006 * (7.0–23.3) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández-Lozano, I.; Mairinger, S.; Traxl, A.; Sauberer, M.; Filip, T.; Stanek, J.; Kuntner, C.; Wanek, T.; Langer, O. Assessing the Functional Redundancy between P-gp and BCRP in Controlling the Brain Distribution and Biliary Excretion of Dual Substrates with PET Imaging in Mice. Pharmaceutics 2021, 13, 1286. https://doi.org/10.3390/pharmaceutics13081286
Hernández-Lozano I, Mairinger S, Traxl A, Sauberer M, Filip T, Stanek J, Kuntner C, Wanek T, Langer O. Assessing the Functional Redundancy between P-gp and BCRP in Controlling the Brain Distribution and Biliary Excretion of Dual Substrates with PET Imaging in Mice. Pharmaceutics. 2021; 13(8):1286. https://doi.org/10.3390/pharmaceutics13081286
Chicago/Turabian StyleHernández-Lozano, Irene, Severin Mairinger, Alexander Traxl, Michael Sauberer, Thomas Filip, Johann Stanek, Claudia Kuntner, Thomas Wanek, and Oliver Langer. 2021. "Assessing the Functional Redundancy between P-gp and BCRP in Controlling the Brain Distribution and Biliary Excretion of Dual Substrates with PET Imaging in Mice" Pharmaceutics 13, no. 8: 1286. https://doi.org/10.3390/pharmaceutics13081286