
ABCB1 and ABCG2 Control Brain Accumulation and Intestinal Disposition of the Novel ROS1/TRK/ALK Inhibitor Repotrectinib, While OATP1A/1B, ABCG2, and CYP3A Limit Its Oral Availability

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Lines and Transport Assays
2.3. Animals
2.4. Drug Stock and Working Solution
2.5. Plasma Pharmacokinetics and Tissue Distribution of Repotrectinib in Mice
2.6. LC-MS/MS Analysis
2.7. Data and Statistical Analysis
3. Results
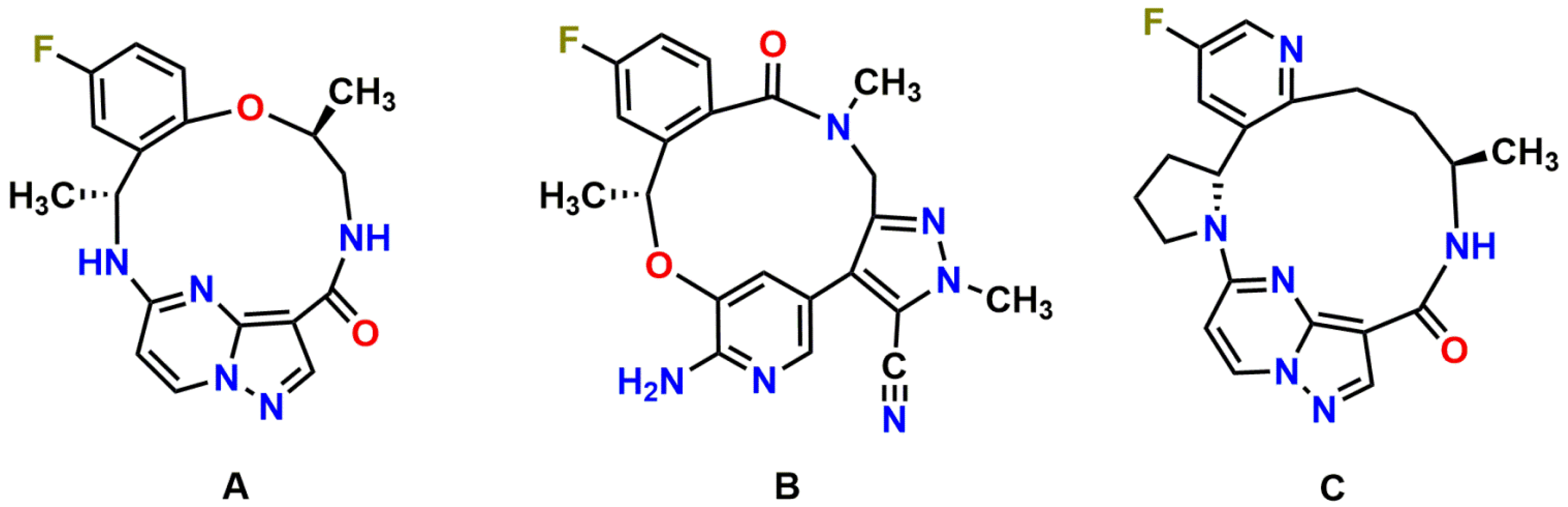
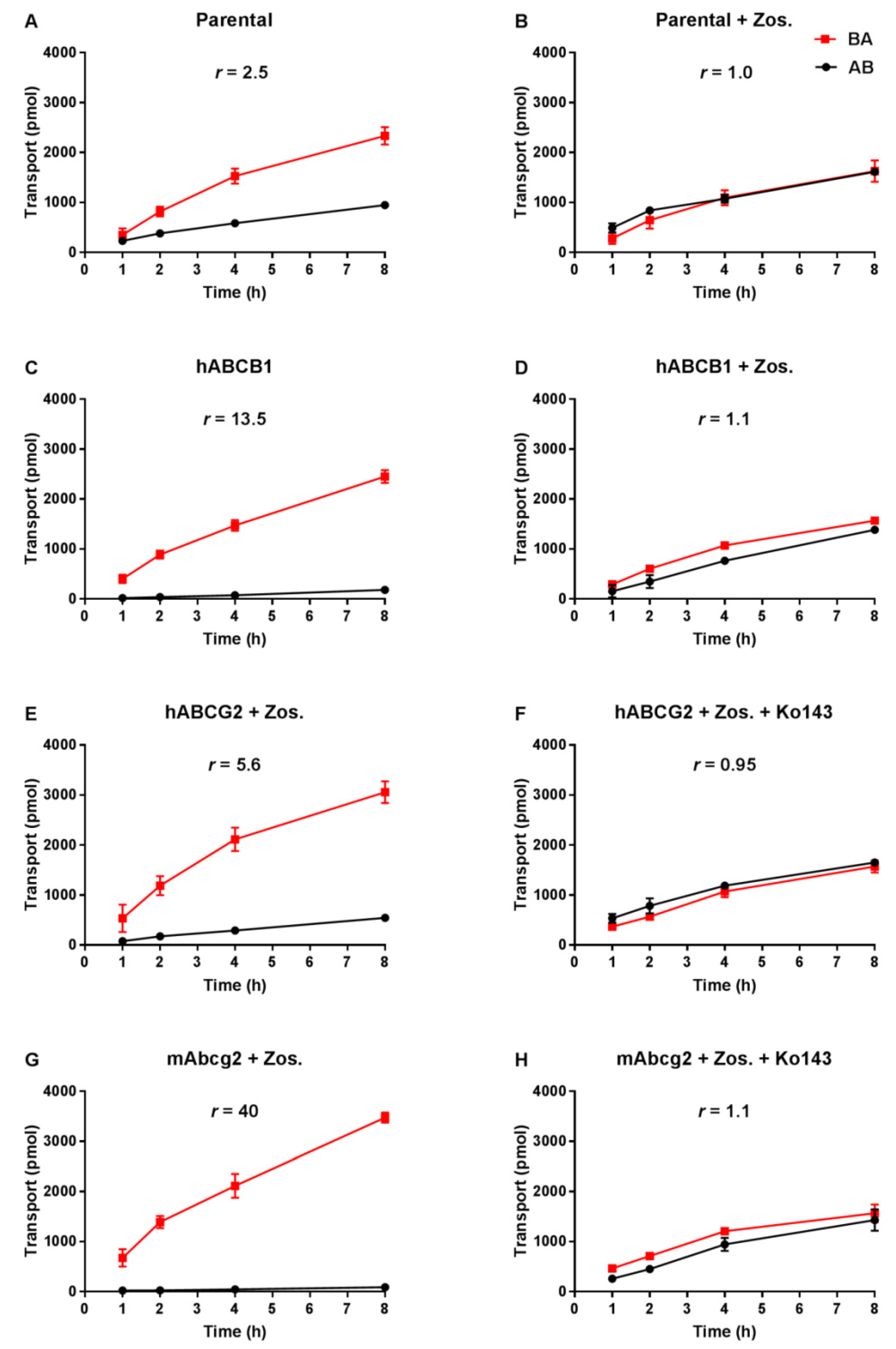
3.1. Repotrectinib Is Efficiently Transported by Human ABCB1 and ABCG2, and by Mouse Abcg2 In Vitro
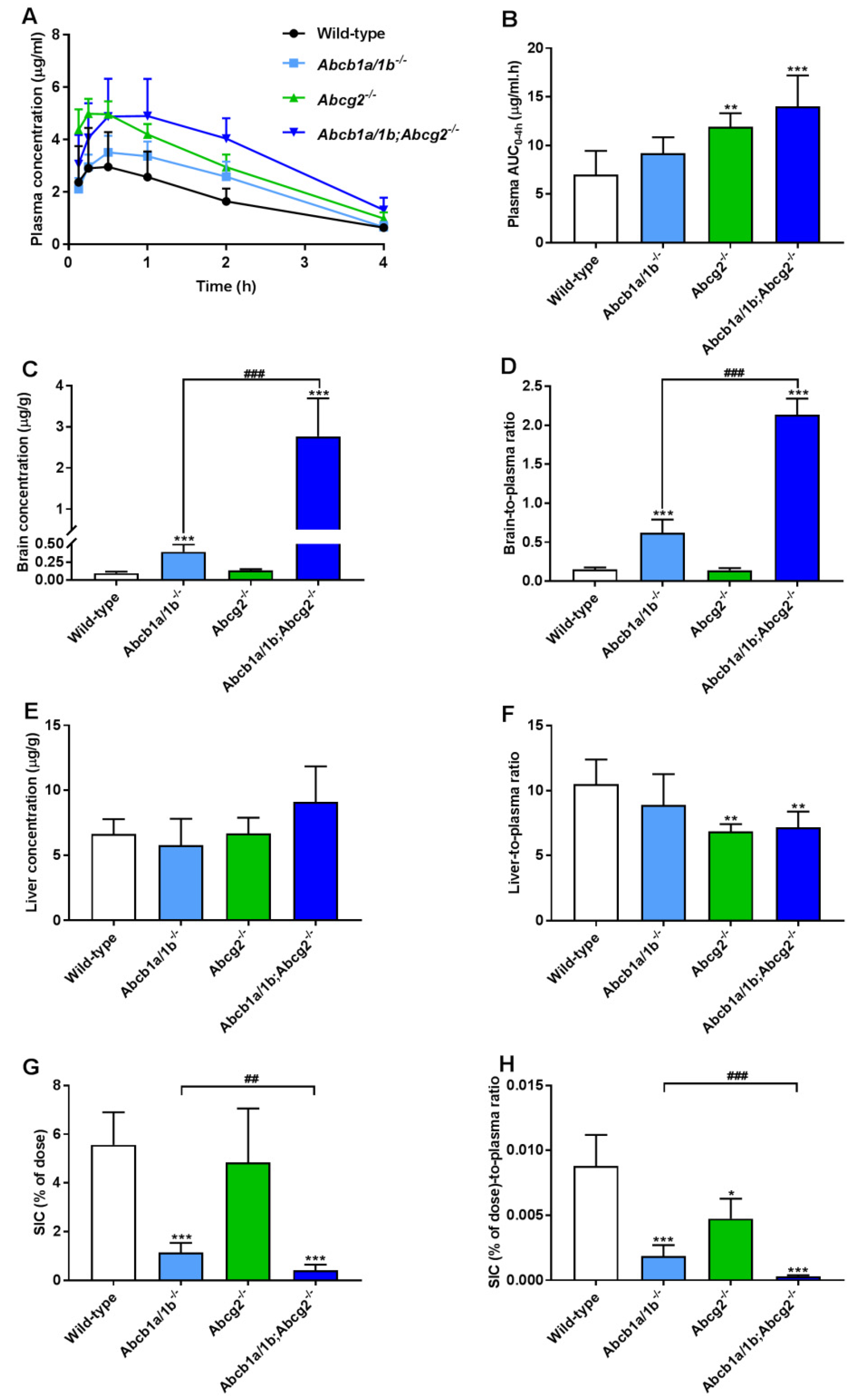
3.2. Both ABCB1 and ABCG2 Control Repotrectinib Plasma Exposure and Tissue Distribution
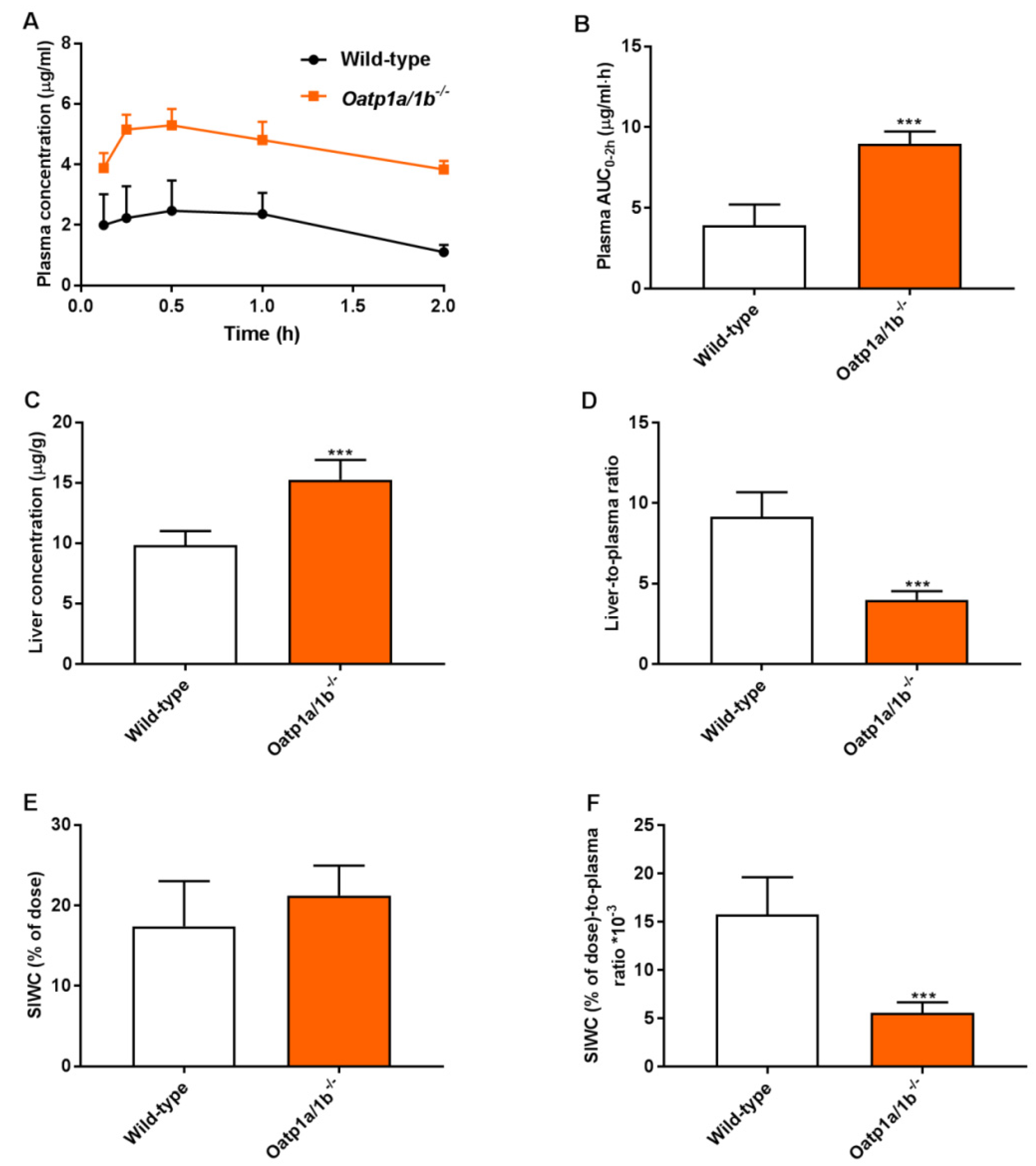
3.3. Mouse Oatp1a/1b Controls Oral Availability, Liver, and Small Intestine Distribution of Repotrectinib
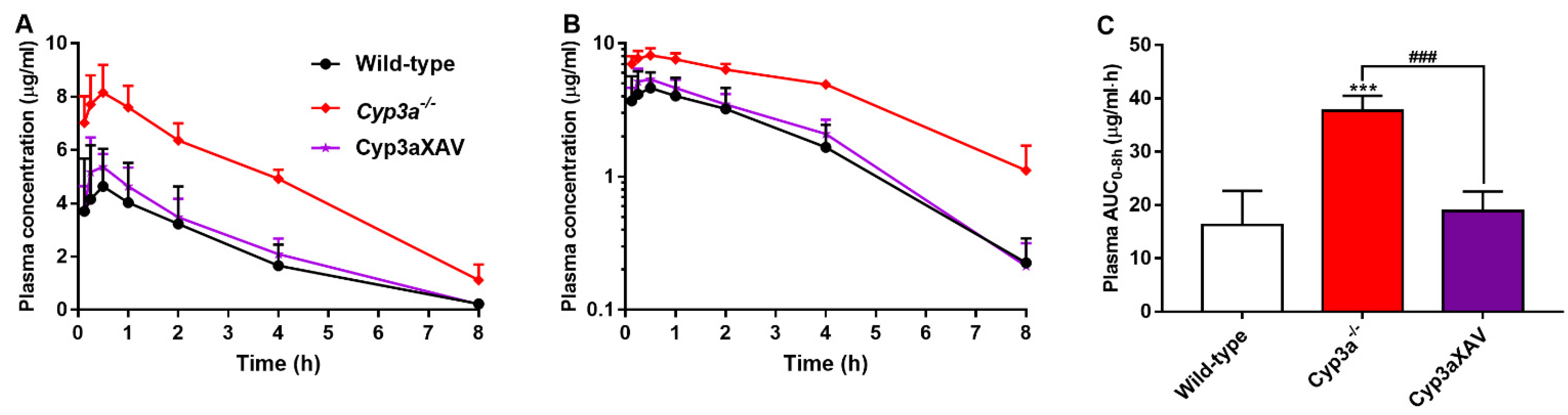
3.4. CYP3A Markedly Limits Repotrectinib Systemic Exposure
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Lin, J.J.; Shaw, A.T. Recent Advances in Targeting ROS1 in Lung Cancer. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer. 2017, 12, 1611–1625. [Google Scholar] [CrossRef] [Green Version]
- Grande, E.; Bolos, M.V.; Arriola, E. Targeting oncogenic ALK: A promising strategy for cancer treatment. Mol. Cancer Ther. 2011, 10, 569–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, S.H.; Tan, J.; Yen, Y.; Soo, R.A. ROS1 as a ‘druggable’ receptor tyrosine kinase: Lessons learned from inhibiting the ALK pathway. Expert Rev. Anticancer Ther. 2012, 12, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.I.; Zhu, V.W. CNS metastasis in ROS1+ NSCLC: An urgent call to action, to understand, and to overcome. Lung Cancer 2019, 130, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Drilon, A.; Siena, S.; Ou, S.I.; Patel, M.; Ahn, M.J.; Lee, J.; Bauer, T.M.; Farago, A.F.; Wheler, J.J.; Liu, S.V.; et al. Safety and Antitumor Activity of the Multitargeted Pan-TRK, ROS1, and ALK Inhibitor Entrectinib: Combined Results from Two Phase I Trials (ALKA-372-001 and STARTRK-1). Cancer Discov. 2017, 7, 400–409. [Google Scholar] [CrossRef] [Green Version]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schiöth, H.B. Trends in kinase drug discovery: Targets, indications and inhibitor design. Nat. Rev. Drug Discov. 2021, 1–23. [Google Scholar]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2021 update. Pharmacol. Res. 2021, 165, 105463. [Google Scholar] [CrossRef]
- Drilon, A.; Ou, S.I.; Cho, B.C.; Kim, D.W.; Lee, J.; Lin, J.J.; Zhu, V.W.; Ahn, M.-J.; Camidge, D.R.; Nguyen, J.; et al. Repotrectinib (TPX-0005) Is a Next-Generation ROS1/TRK/ALK Inhibitor That Potently Inhibits ROS1/TRK/ALK Solvent—Front Mutations. Cancer Discov. 2018, 8, 1227–1236. [Google Scholar] [CrossRef] [Green Version]
- Cervantes-Madrid, D.; Szydzik, J.; Lind, D.E.; Borenäs, M.; Bemark, M.; Cui, J.; Palmer, R.H.; Hallberg, B. Repotrectinib (TPX-0005), effectively reduces growth of ALK driven neuroblastoma cells. Sci. Rep. 2019, 9, 19353. [Google Scholar] [CrossRef] [Green Version]
- Yun, M.R.; Kim, D.H.; Kim, S.Y.; Joo, H.S.; Lee, Y.W.; Choi, H.M.; Park, C.W.; Heo, S.G.; Kang, H.N.; Lee, S.S.; et al. Repotrectinib Exhibits Potent Antitumor Activity in Treatment-Naive and Solvent-Front-Mutant ROS1-Rearranged Non-Small Cell Lung Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 3287–3295. [Google Scholar] [CrossRef] [Green Version]
- Cho, B.C.; Drilon, A.E.; Doebele, R.C.; Kim, D.-W.; Lin, J.J.; Lee, J.; Ahn, M.-J.; Zhu, V.W.; Ejadi, S.; Camidge, D.R.; et al. Safety and preliminary clinical activity of repotrectinib in patients with advanced ROS1 fusion-positive non-small cell lung cancer (TRIDENT-1 study). Ann. Oncol. 2019, 37 Suppl. S15, 9011. [Google Scholar] [CrossRef]
- Drilon, A.; Cho, B.; Kim, D.-W.; Lee, J.; Lin, J.; Zhu, V.; Camidge, R.; Stopatschinskaja, S.; Cui, J.; Hyman, D.; et al. Safety and preliminary clinical activity of repotrectinib in patients with advanced ROS1/TRK fusion-positive solid tumors (TRIDENT-1 study). Ann. Oncol. 2019, 30, v162. [Google Scholar] [CrossRef]
- Nigam, S.K. What do drug transporters really do? Nat. Rev. Drug Discov. 2015, 14, 29–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borst, P.; Elferink, R.O. Mammalian ABC transporters in health and disease. Annu. Rev. Biochem. 2002, 71, 537–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schinkel, A.H.; Jonker, J.W. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: An overview. Adv. Drug Deliv. Rev. 2003, 55, 3–29. [Google Scholar] [CrossRef]
- van de Steeg, E.; Wagenaar, E.; van der Kruijssen, C.M.; Burggraaff, J.E.; de Waart, D.R.; Elferink, R.P.; Kenworthy, K.E.; Schinkel, A.H. Organic anion transporting polypeptide 1a/1b-knockout mice provide insights into hepatic handling of bilirubin, bile acids, and drugs. J. Clin. Investig. 2010, 120, 2942–2952. [Google Scholar] [CrossRef] [PubMed]
- Szakacs, G.; Annereau, J.-P.; Lababidi, S.; Shankavaram, U.; Arciello, A.; Bussey, K.; Reinhold, W.; Guo, Y.; Kruh, G.D.; Reimers, M.; et al. Predicting drug sensitivity and resistance: Profiling ABC transporter genes in cancer cells. Cancer Cell 2004, 6, 129–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buxhofer-Ausch, V.; Secky, L.; Wlcek, K.; Svoboda, M.; Kounnis, V.; Briasoulis, E.; Tzakos, A.G.; Jaeger, W.; Thalhammer, T. Tumor-specific expression of organic anion-transporting polypeptides: Transporters as novel targets for cancer therapy. J. Drug Deliv. 2013, 2013, 863539. [Google Scholar] [CrossRef]
- Urquhart, B.L.; Kim, R.B. Blood-brain barrier transporters and response to CNS-active drugs. Eur. J. Clin. Pharmacol. 2009, 65, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sparidans, R.W.; Wang, Y.; Lebre, M.C.; Beijnen, J.H.; Schinkel, A.H. P-glycoprotein and breast cancer resistance protein restrict brigatinib brain accumulation and toxicity, and, alongside CYP3A, limit its oral availability. Pharmacol. Res. 2018, 137, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Thelen, K.; Dressman, J.B. Cytochrome P450-mediated metabolism in the human gut wall. J. Pharm. Pharmacol. 2009, 61, 541–558. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Perpinioti, N.; Schinkel, A.H.; Beijnen, J.H.; Sparidans, R.W. Bioanalytical assay for the new-generation ROS1/TRK/ALK inhibitor repotrectinib in mouse plasma and tissue homogenate using liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2020, 1144, 122098. [Google Scholar] [CrossRef] [PubMed]
- Jonker, J.; Buitelaar, M.; Wagenaar, E.; van der Valk, M.A.; Scheffer, G.L.; Scheper, R.J.; Plösch, T.; Kuipers, F.; Elferink, R.P.J.O.; Rosing, H.; et al. The breast cancer resistance protein protects against a major chlorophyll-derived dietary phototoxin and protoporphyria. Proc. Natl. Acad. Sci. USA 2002, 99, 15649–15654. [Google Scholar] [CrossRef] [Green Version]
- Van Herwaarden, A.E.; Wagenaar, E.; van der Kruijssen, C.M.; van Waterschoot, R.A.; Smit, J.W.; Song, J.Y.; van der Valk, M.A.; van Tellingen, O.; van der Hoorn, J.W.A.; Rosing, H.; et al. Knockout of cytochrome P450 3A yields new mouse models for understanding xenobiotic metabolism. J. Clin. Investig. 2007, 117, 3583–3592. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Huo, M.; Zhou, J.; Xie, S. PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput. Methods Programs Biomed. 2010, 99, 306–314. [Google Scholar] [CrossRef]
- Tang, S.C.; Nguyen, L.N.; Sparidans, R.W.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. Increased oral availability and brain accumulation of the ALK inhibitor crizotinib by coadministration of the P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) inhibitor elacridar. Int. J. Cancer 2014, 134, 1484–1494. [Google Scholar] [CrossRef]
- Kodaira, H.; Kusuhara, H.; Ushiki, J.; Fuse, E.; Sugiyama, Y. Kinetic analysis of the cooperation of P-glycoprotein (P-gp/Abcb1) and breast cancer resistance protein (Bcrp/Abcg2) in limiting the brain and testis penetration of erlotinib, flavopiridol, and mitoxantrone. J. Pharmacol. Exp. Ther. 2010, 333, 788–796. [Google Scholar] [CrossRef] [Green Version]
- Gainor, J.F.; Tseng, D.; Yoda, S.; Dagogo-Jack, I.; Friboulet, L.; Lin, J.J.; Hubbeling, H.G.; Dardaei, L.; Farago, A.F.; Schultz, K.R.; et al. Patterns of Metastatic Spread and Mechanisms of Resistance to Crizotinib in ROS1-Positive Non-Small-Cell Lung Cancer. JCO Precis. Oncol. 2017, 1, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Smeyne, R.J.; Wurst, W.; Long, L.K.; Auerbach, B.A.; Joyner, A.L.; Barbacid, M. Targeted disruption of the trkB neurotrophin receptor gene results in nervous system lesions and neonatal death. Cell 1993, 75, 113–122. [Google Scholar] [CrossRef]
- Klein, R.; Silos-Santiago, I.; Smeyne, R.; Lira, S.A.; Brambilla, R.; Bryant, S.; Zhang, L.; Snider, W.D.; Barbacid, M. Disruption of the neurotrophin-3 receptor gene trkC eliminates la muscle afferents and results in abnormal movements. Nature 1994, 368, 249–251. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sparidans, R.W.; Wang, Y.; Lebre, M.C.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. P-glycoprotein (MDR1/ABCB1) restricts brain accumulation and cytochrome P450-3A (CYP3A) limits oral availability of the novel ALK/ROS1 inhibitor lorlatinib. Int. J. Cancer 2018, 143, 2029–2038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Sparidans, R.W.; Martins, M.L.F.; El-Lari, M.; Lebre, M.C.; van Tellingen, O.; Beijnen, J.H.; Schinkel, A.H. ABCB1 and ABCG2 restrict brain and testis accumulation and, alongside CYP3A, limit oral availability of the novel TRK inhibitor selitrectinib. Mol. Cancer Ther. 2021, 20, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sparidans, R.W.; Wang, Y.; Lebre, M.C.; Beijnen, J.H.; Schinkel, A.H. Oral coadministration of elacridar and ritonavir enhances brain accumulation and oral availability of the novel ALK/ROS1 inhibitor lorlatinib. Eur. J. Pharm. Biopharm. 2019, 136, 120–130. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Genotype | |||
|---|---|---|---|---|
| Wild-Type | Abcb1a/1b−/− | Abcg2−/− | Abcb1a/1b;Abcg2−/− | |
| AUC0–4h, µg/mL·h | 6.99 ± 2.46 | 9.20 ± 1.64 | 11.90 ± 1.42 ** | 14.01 ± 3.18 *** |
| Fold increase AUC0–4h | 1.00 | 1.32 | 1.70 | 2.00 |
| Cmax, µg/mL | 3.08 ± 1.44 | 3.63 ± 0.62 | 5.08 ± 0.49 ** | 5.23 ± 1.24 ** |
| Tmax, h | 0.5 (0.25–1) | 0.5 (0.5–1) | 0.25 (0.125–0.5) | 1 (0.5–2) |
| Cbrain, ng/g | 95.1 ± 23.5 | 394 ± 102 *** | 134 ± 18 | 2763 ± 928 *** (###) |
| Fold increase Cbrain | 1.00 | 4.14 | 1.41 | 29.1 |
| Brain-to-plasma ratio | 0.15 ± 0.03 | 0.62 ± 0.17 *** | 0.14 ± 0.03 | 2.13 ± 0.21 *** (###) |
| Fold change ratio | 1.00 | 4.13 | 0.93 | 14.2 |
| Ctestis, ng/g | 155 ± 55 | 380 ± 122 *** | 166 ± 27 | 1672 ± 386 *** (###) |
| Fold increase Ctestis | 1.00 | 2.45 | 1.07 | 10.8 |
| Testis-to-plasma ratio | 0.24 ± 0.07 | 0.58 ± 0.14 *** | 0.17 ± 0.02 | 1.35 ± 0.31 *** (###) |
| Fold change ratio | 1.00 | 2.42 | 0.71 | 5.6 |
| Cliver µg/g | 6.63 ± 1.16 | 5.58 ± 2.03 | 6.69 ± 1.21 | 9.09 ± 2.75 |
| Fold change Cliver | 1.00 | 0.84 | 1.01 | 1.37 |
| Liver-to-plasma ratio | 10.5 ± 1.9 | 8.9 ± 2.4 | 6.8 ± 0.6 ** | 7.2 ± 1.2 ** |
| Fold change ratio | 1.00 | 0.85 | 0.65 | 0.69 |
| SIC (% of dose) | 5.56 ± 1.34 | 1.13 ± 0.40 *** | 4.84 ± 2.21 | 0.41 ± 0.23 *** (##) |
| Fold change SIC (% of dose) | 1.00 | 0.20 | 0.87 | 0.07 |
| SIC (%)-to-plasma ratio *10−3 | 8.79 ± 2.41 | 1.86 ± 0.84 *** | 4.75 ± 1.54 * | 0.30 ± 0.09 *** (###) |
| Fold change ratio | 1.00 | 0.21 | 0.54 | 0.03 |
| Parameter | Genotype | |
|---|---|---|
| Wild-Type | Oatp1a/1b−/− | |
| AUC0–2h, µg/mL·h | 3.92 ± 1.29 | 8.96 ± 0.77 *** |
| Fold increase AUC0–2h | 1.00 | 2.29 |
| Cmax, µg/mL | 2.64 ± 0.99 | 5.38 ± 0.52 *** |
| Tmax, h | 0.5 (0.25–1) | 0.5 (0.5–1) |
| Cliver, µg/g | 9.84 ± 1.16 | 15.3 ± 1.6 *** |
| Fold increase Cliver | 1.00 | 1.55 |
| Liver-to-plasma ratio | 9.16 ± 1.53 | 4.00 ± 0.54 *** |
| Fold change ratio | 1.00 | 0.44 |
| CSIWC, µg/g | 84.74 ± 24.13 | 121.1 ± 29.2 * |
| Fold increase CSIWC | 1.00 | 1.43 |
| SIWC (% of dose) | 17.4 ± 5.6 | 21.3 ± 3.7 |
| Fold change | 1.00 | 1.22 |
| SIWC (%)-to-plasma ratio·10−3 | 15.8 ± 3.9 | 5.58 ± 1.09 *** |
| Fold change ratio | 1.00 | 0.35 |
| SIWC (%)-to-AUC0–2h ratio·10−3 | 4.59 ± 1.04 | 2.40 ± 0.54 *** |
| Fold change ratio | 1.00 | 0.52 |
| Parameter | Genotype | ||
|---|---|---|---|
| Wild-Type | Cyp3a−/− | Cyp3aXAV | |
| AUC0–8h, µg/mL·h | 16.28 ± 6.35 | 37.64 ± 2.83 *** | 18.86 ± 3.65 ### |
| Fold change AUC0–8h | 1.00 | 2.31 | 1.16 |
| Cmax, µg/mL | 4.80 ± 1.78 | 8.28 ± 0.93 *** | 5.56 ± 0.61 ## |
| Tmax, h | 0.5 (0.25–1) | 0.5 (0.125–0.5) | 0.5 (0.25–0.5) |
| T1/2, h | 1.6 ± 0.4 | 2.5 ± 0.9 * | 1.6 ± 0.2 # |
| Cbrain, ng/g | 51.2 ± 17.8 | 212.8 ± 53.7 *** | 80.2 ± 35.3 ### |
| Fold increase Cbrain | 1.00 | 4.16 | 1.57 |
| Brain-to-plasma ratio | 0.26 ± 0.12 | 0.22 ± 0.08 | 0.40 ± 0.09 |
| Fold increase ratio | 1.00 | 0.85 | 1.54 |
| Cliver, µg/mL | 1.22 ± 0.65 | 5.02 ± 1.92 *** | 1.38 ± 0.44 ### |
| Fold increase Cliver | 1.00 | 4.11 | 1.13 |
| Liver-to-plasma ratio | 5.36 ± 0.54 | 4.81 ± 0.76 | 7.10 ± 2.11 |
| Fold increase ratio | 1.00 | 0.90 | 1.32 |
| SIC (% of dose) | 2.08 ± 1.35 | 5.11 ± 2.83 * | 1.68 ± 0.63 # |
| Fold change | 1.00 | 2.46 | 0.81 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.; Sparidans, R.W.; Lebre, M.C.; Beijnen, J.H.; Schinkel, A.H. ABCB1 and ABCG2 Control Brain Accumulation and Intestinal Disposition of the Novel ROS1/TRK/ALK Inhibitor Repotrectinib, While OATP1A/1B, ABCG2, and CYP3A Limit Its Oral Availability. Pharmaceutics 2021, 13, 1761. https://doi.org/10.3390/pharmaceutics13111761
Li W, Sparidans RW, Lebre MC, Beijnen JH, Schinkel AH. ABCB1 and ABCG2 Control Brain Accumulation and Intestinal Disposition of the Novel ROS1/TRK/ALK Inhibitor Repotrectinib, While OATP1A/1B, ABCG2, and CYP3A Limit Its Oral Availability. Pharmaceutics. 2021; 13(11):1761. https://doi.org/10.3390/pharmaceutics13111761
Chicago/Turabian StyleLi, Wenlong, Rolf W. Sparidans, Maria C. Lebre, Jos H. Beijnen, and Alfred H. Schinkel. 2021. "ABCB1 and ABCG2 Control Brain Accumulation and Intestinal Disposition of the Novel ROS1/TRK/ALK Inhibitor Repotrectinib, While OATP1A/1B, ABCG2, and CYP3A Limit Its Oral Availability" Pharmaceutics 13, no. 11: 1761. https://doi.org/10.3390/pharmaceutics13111761