
In Vitro and In Vivo Inhibition of MATE1 by Tyrosine Kinase Inhibitors

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cellular Models and Cell Culture Conditions
2.2. Uptake Assays
2.3. Gene Expression Analysis
2.4. Computational Modeling
2.5. Murine Pharmacokinetic Studies
2.6. Immunoblotting Assays
2.7. Human Pharmacokinetic Studies
2.8. Quantification of N-Methylnicotinamide
2.9. Statistical Analyses
3. Results and Discussion
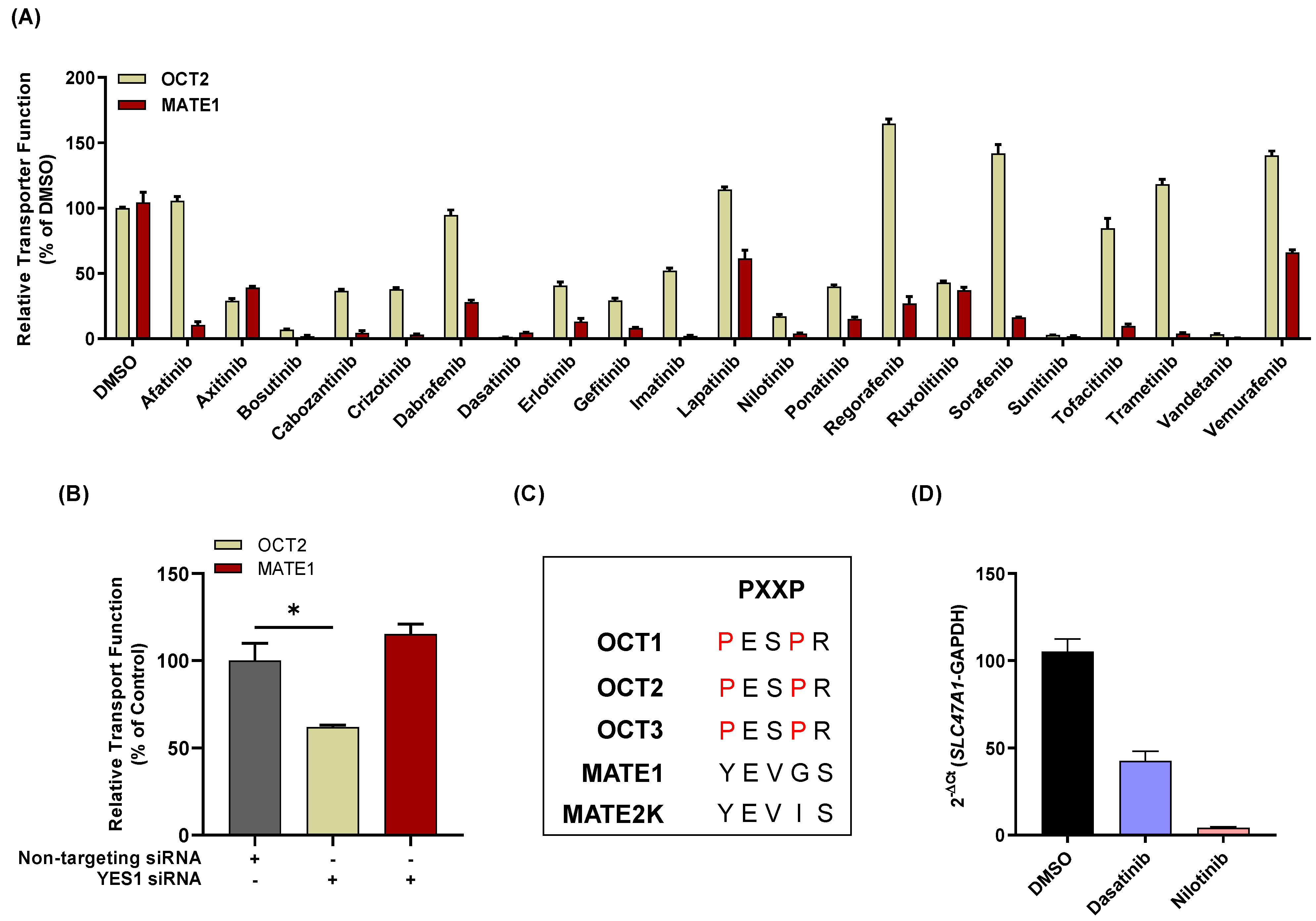
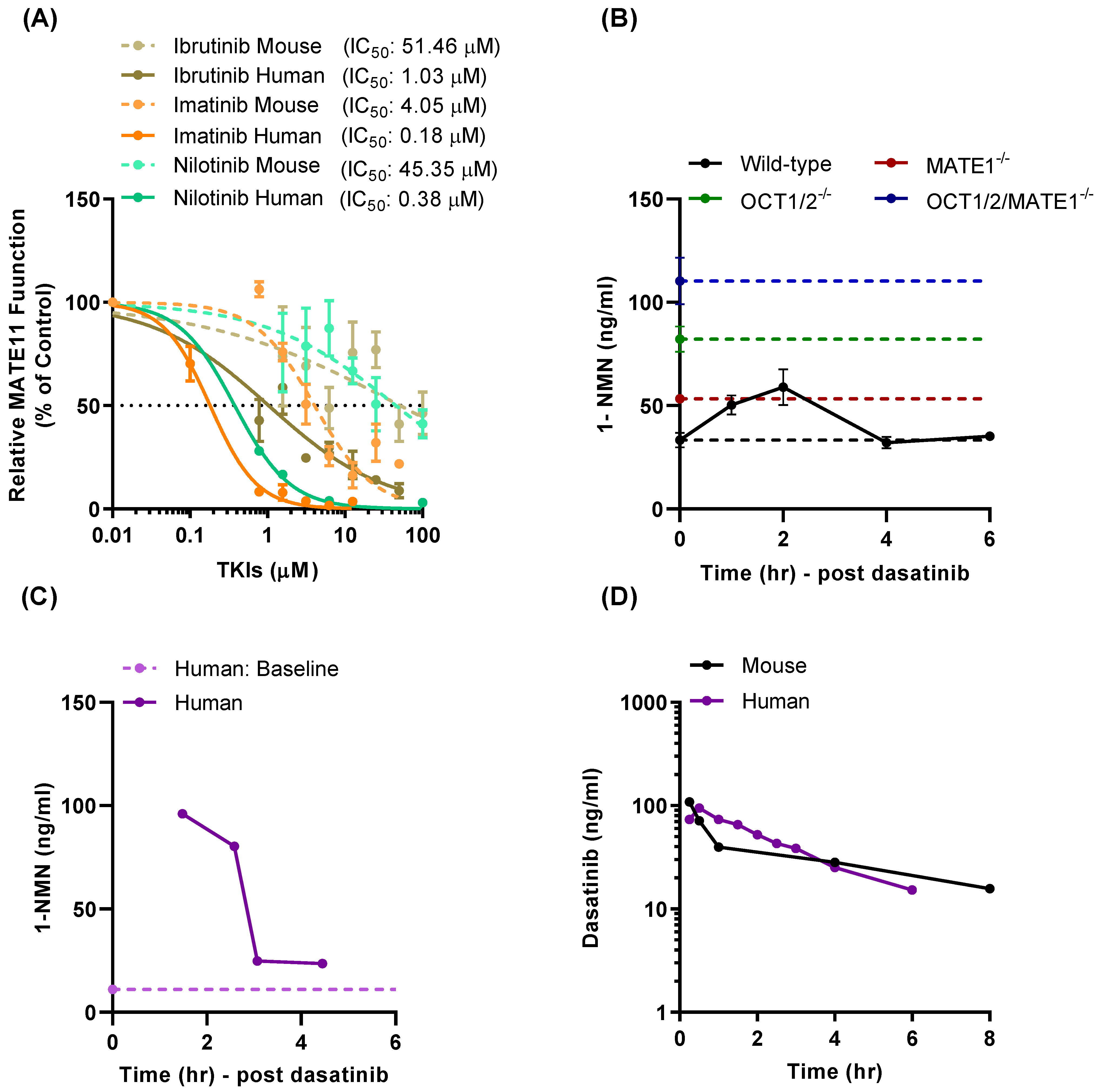
3.1. TKI-Mediated Inhibition of MATE1 In Vitro
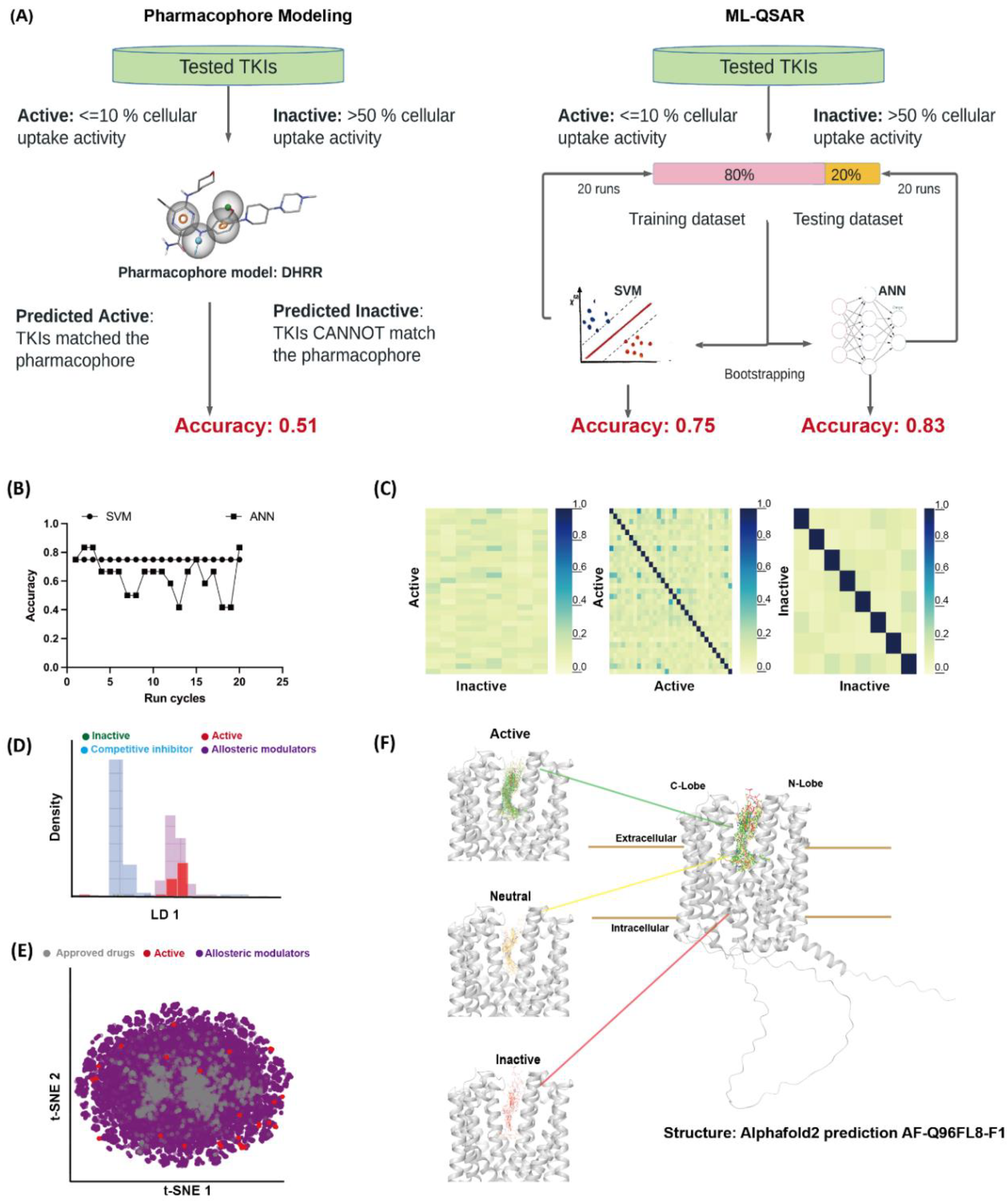
3.2. Computational Evaluation of MATE1-TKIs Interaction
3.3. Influence of TKIs on MATE1 Regulation In Vitro
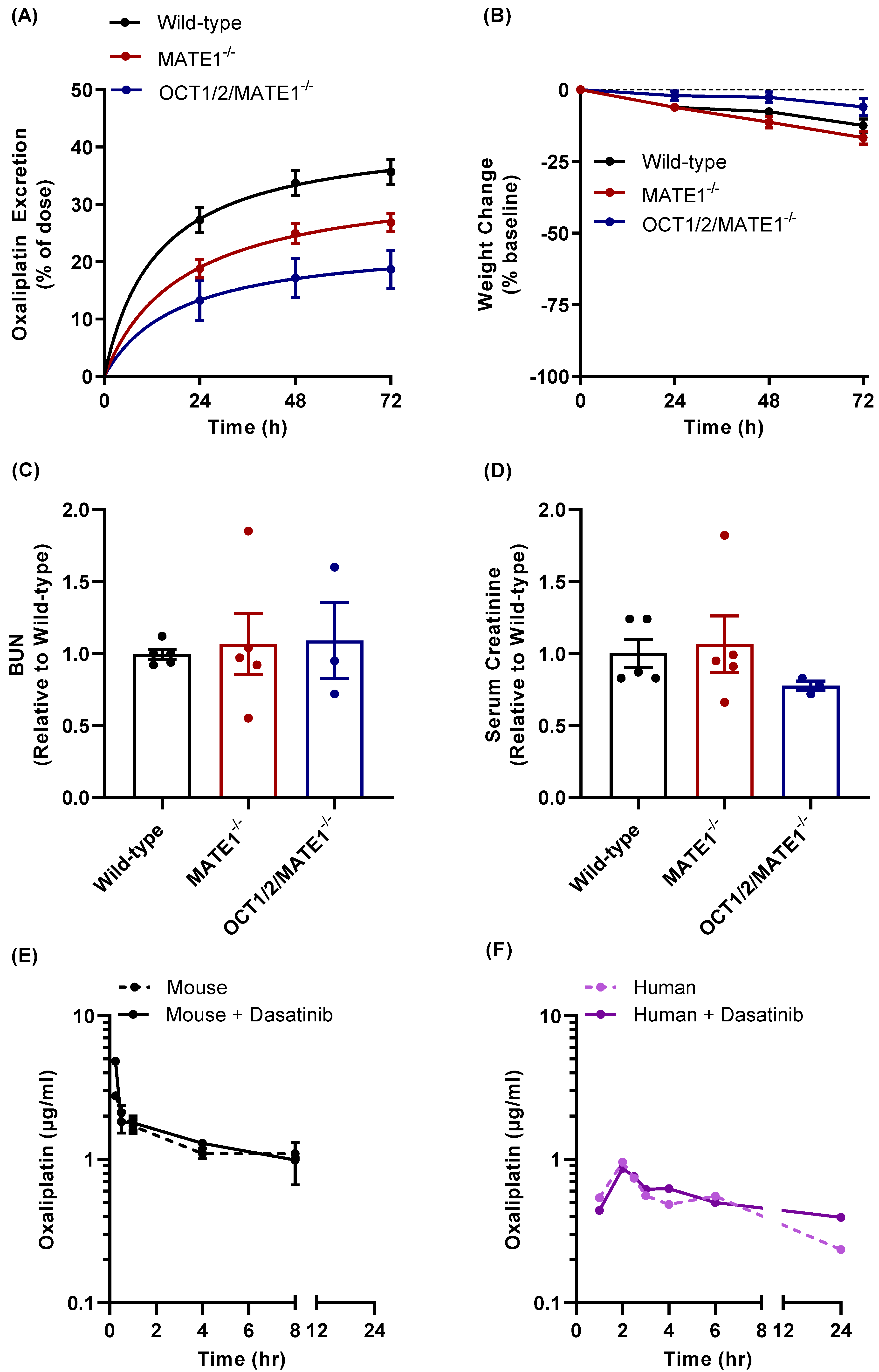
3.4. Effect of TKI Treatment on MATE1 Function In Vivo
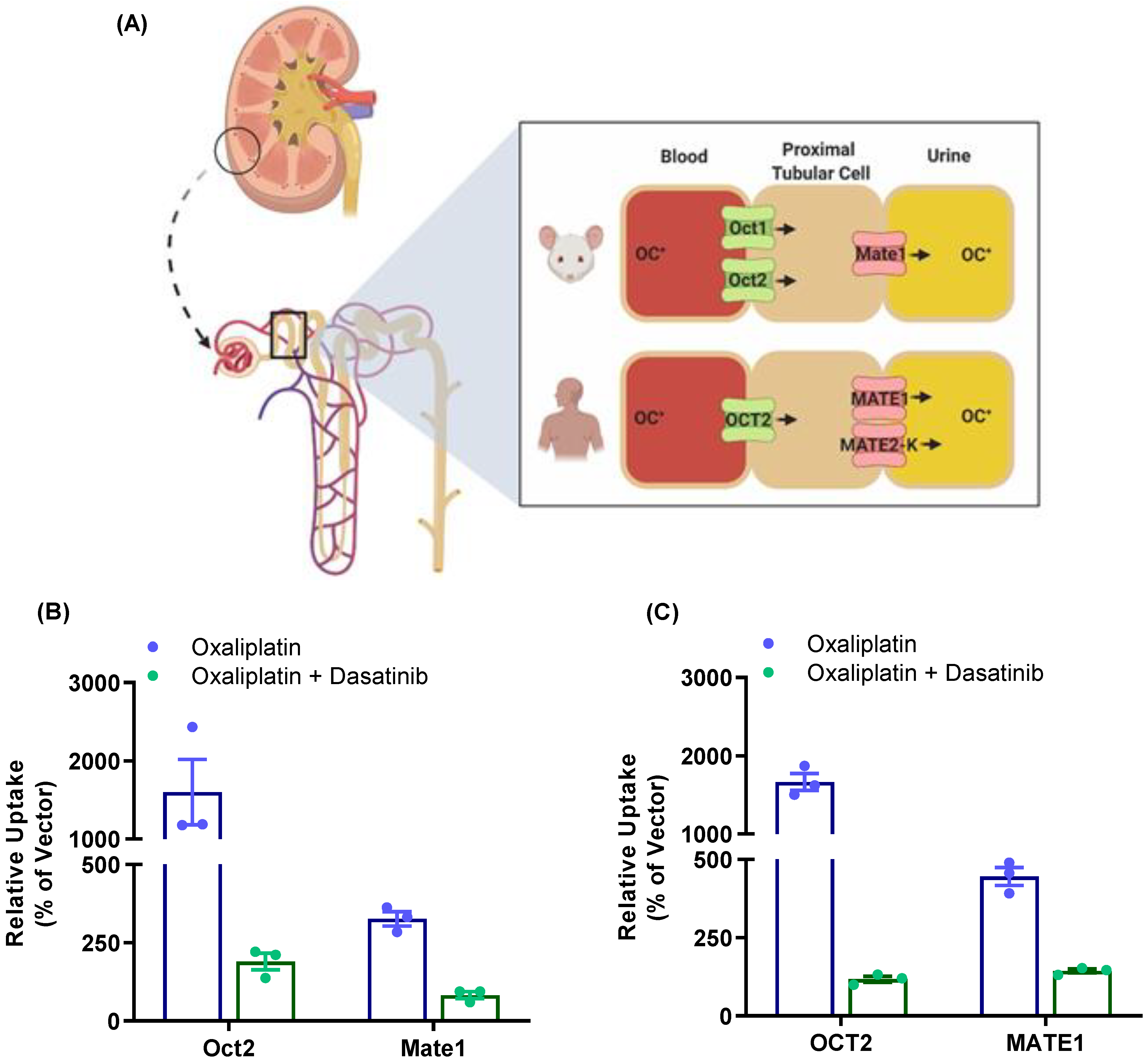
3.5. Modulation of Oxaliplatin Pharmacokinetics by TKI Treatment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kaback, H.R.; Sahin-Tóth, M.; Weinglass, A.B. The kamikaze approach to membrane transport. Nat. Rev. Mol. Cell Biol. 2001, 2, 610–620. [Google Scholar] [CrossRef]
- Borst, P.; Elferink, R.O. Mammalian ABC Transporters in Health and Disease. Annu. Rev. Biochem. 2002, 71, 537–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borst, P.; Evers, R.; Kool, M.; Wijnholds, J. The multidrug resistance protein family. Biochim. Biophys. Acta 1999, 1461, 347–357. [Google Scholar] [CrossRef] [Green Version]
- Sprowl, J.A.; Sparreboom, A. Uptake carriers and oncology drug safety. Drug Metab. Dispos. 2014, 42, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yonezawa, A.; Inui, K. Importance of the multidrug and toxin extrusion MATE/SLC47A family to pharmacokinetics, pharmacodynamics/toxicodynamics and pharmacogenomics. Br. J. Pharmacol. 2011, 164, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- Koepsell, H.; Lips, K.; Volk, C. Polyspecific organic cation transporters: Structure, function, physiological roles, and biopharmaceutical implications. Pharm. Res. 2007, 24, 1227–1251. [Google Scholar] [CrossRef] [PubMed]
- Tanihara, Y.; Masuda, S.; Katsura, T.; Inui, K. Protective effect of concomitant administration of imatinib on cisplatin-induced nephrotoxicity focusing on renal organic cation transporter OCT2. Biochem. Pharm. 2009, 78, 1263–1271. [Google Scholar] [CrossRef]
- Giacomini, K.M.; Yee, S.W.; Ratain, M.J.; Weinshilboum, R.M.; Kamatani, N.; Nakamura, Y. Pharmacogenomics and patient care: One size does not fit all. Sci. Transl. Med. 2012, 4, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koepsell, H. Update on drug-drug interaction at organic cation transporters: Mechanisms, clinical impact, and proposal for advanced in vitro testing. Expert Opin. Drug Metab. Toxicol. 2021, 17, 635–653. [Google Scholar] [CrossRef] [PubMed]
- Sprowl, J.A.; Ong, S.S.; Gibson, A.A.; Hu, S.; Du, G.; Lin, W.; Li, L.; Bharill, S.; Ness, R.A.; Stecula, A.; et al. A phosphotyrosine switch regulates organic cation transporters. Nat. Commun. 2016, 7, 10880. [Google Scholar] [CrossRef]
- Uddin, M.E.; Garrison, D.A.; Kim, K.; Jin, Y.; Eisenmann, E.D.; Huang, K.M.; Gibson, A.A.; Hu, Z.; Sparreboom, A.; Hu, S.; et al. Influence of YES1 Kinase and Tyrosine Phosphorylation on the Activity of OCT1. Front. Pharmacol. 2021, 12, 644342. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.M.; Thomas, M.Z.; Magdy, T.; Eisenmann, E.D.; Uddin, M.E.; DiGiacomo, D.F.; Pan, A.; Keiser, M.; Jin, Y.; Fu, Q.; et al. Targeting OCT3 attenuates doxorubicin-induced cardiac injury. Proc. Natl. Acad. Sci. USA 2021, 118, e2020168118. [Google Scholar] [CrossRef] [PubMed]
- Pabla, N.; Gibson, A.A.; Buege, M.; Ong, S.S.; Li, L.; Hu, S.; Du, G.; Sprowl, J.A.; Vasilyeva, A.; Janke, L.J.; et al. Mitigation of acute kidney injury by cell-cycle inhibitors that suppress both CDK4/6 and OCT2 functions. Proc. Natl. Acad. Sci. USA 2015, 112, 5231–5236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprowl, J.A.; Ciarimboli, G.; Lancaster, C.S.; Giovinazzo, H.; Gibson, A.A.; Du, G.; Janke, L.J.; Cavaletti, G.; Shields, A.F.; Sparreboom, A. Oxaliplatin-induced neurotoxicity is dependent on the organic cation transporter OCT2. Proc. Natl. Acad. Sci. USA 2013, 110, 11199–11204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minematsu, T.; Giacomini, K.M. Interactions of tyrosine kinase inhibitors with organic cation transporters and multidrug and toxic compound extrusion proteins. Mol. Cancer Ther. 2011, 10, 531–539. [Google Scholar] [CrossRef] [Green Version]
- Tátrai, P.; Schweigler, P.; Poller, B.; Domange, N.; de Wilde, R.; Hanna, I.; Gáborik, Z.; Huth, F. A Systematic In Vitro Investigation of the Inhibitor Preincubation Effect on Multiple Classes of Clinically Relevant Transporters. Drug Metab. Dispos. 2019, 47, 768–778. [Google Scholar] [CrossRef]
- Kajiwara, M.; Terada, T.; Ogasawara, K.; Iwano, J.; Katsura, T.; Fukatsu, A.; Doi, T.; Inui, K.-I. Identification of multidrug and toxin extrusion (MATE1 and MATE2-K) variants with complete loss of transport activity. J. Hum. Genet. 2009, 54, 40–46. [Google Scholar] [CrossRef]
- Omote, S.; Matsuoka, N.; Arakawa, H.; Nakanishi, T.; Tamai, I. Effect of tyrosine kinase inhibitors on renal handling of creatinine by MATE1. Sci. Rep. 2018, 8, 9237. [Google Scholar] [CrossRef] [Green Version]
- George, B.; Wen, X.; Jaimes, E.A.; Joy, M.S.; Aleksunes, L.M. In Vitro Inhibition of Renal OCT2 and MATE1 Secretion by Antiemetic Drugs. Int. J. Mol. Sci. 2021, 22, 6439. [Google Scholar] [CrossRef]
- Hiasa, M.; Matsumoto, T.; Komatsu, T.; Moriyama, Y. Wide variety of locations for rodent MATE1, a transporter protein that mediates the final excretion step for toxic organic cations. Am. J. Physiol. Physiol. 2006, 291, C678–C686. [Google Scholar] [CrossRef]
- Terada, T.; Masuda, S.; Asaka, J.I.; Tsuda, M.; Katsura, T.; Inui, K.I. Molecular cloning, functional characterization and tissue distribution of rat H+/organic cation antiporter MATE1. Pharm. Res. 2006, 23, 1696–1701. [Google Scholar] [CrossRef]
- Dangprapai, Y.; Wright, S.H. Interaction of H+ with the extracellular and intracellular aspects of hMATE1. Am. J. Physiol. Renal Physiol. 2011, 301, F520-8. [Google Scholar] [CrossRef] [Green Version]
- Filipski, K.K.; Loos, W.J.; Verweij, J.; Sparreboom, A. Interaction of cisplatin with the human organic cation transporter 2. Clin. Cancer Res. 2008, 14, 3875–3880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided. Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farhat, N.H. Photonit neural networks and learning mathines the role of electron-trapping materials. IEEE Expert Syst. Appl. 1992, 7, 63–72. [Google Scholar] [CrossRef]
- Rumelhart, D.E.; Hinton, G.E.; Williams, R.J. Learning representations by back-propagating errors. Nature 1986, 323, 533–536. [Google Scholar] [CrossRef]
- Leblanc, A.F.; Huang, K.M.; Uddin, M.E.; Anderson, J.T.; Chen, M.; Hu, S. Murine Pharmacokinetic Studies. Bio-Protocol 2018, 8, e3056. [Google Scholar] [CrossRef]
- Filipski, K.K.; Mathijssen, R.H.; Mikkelsen, T.S.; Schinkel, A.H.; Sparreboom, A. Contribution of organic cation transporter 2 (OCT2) to cisplatin-induced nephrotoxicity. Clin. Pharmacol. Ther. 2009, 86, 396–402. [Google Scholar] [CrossRef]
- Furmanski, B.D.; Hu, S.; Fujita, K.I.; Li, L.; Gibson, A.A.; Janke, L.J.; Williams, R.T.; Schuetz, J.D.; Sparreboom, A.; Baker, S.D. Contribution of ABCC4-mediated gastric transport to the absorption and efficacy of dasatinib. Clin. Cancer Res. 2013, 19, 4359–4370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprowl, J.A.; van Doorn, L.; Hu, S.; van Gerven, L.; de Bruijn, P.; Li, L.; Gibson, A.A.; Mathijssen, R.H.; Sparreboom, A. Conjunctive therapy of cisplatin with the OCT2 inhibitor cimetidine: Influence on antitumor efficacy and systemic clearance. Clin. Pharmacol. Ther. 2013, 94, 585–592. [Google Scholar] [CrossRef] [Green Version]
- Belzer, M.; Morales, M.; Jagadish, B.; Mash, E.A.; Wright, S.H. Substrate-dependent ligand inhibition of the human organic cation transporter OCT2. J. Pharmacol. Exp. Ther. 2013, 346, 300–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, R.D.; Lu, J.; Carlson, H.A. Are there physicochemical differences between allosteric and competitive ligands? PLoS Comput Biol. 2017, 13, e1005813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Koepsell, H. Organic cation transporters in health and disease. Pharmacol. Rev. 2020, 72, 253–319. [Google Scholar] [CrossRef]
- Freitas-Lima, L.C.; Budu, A.; Arruda, A.C.; Perilhão, M.S.; Barrera-Chimal, J.; Araujo, R.C.; Estrela, G.R. PPAR-α Deletion Attenuates Cisplatin Nephrotoxicity by Modulating Renal Organic Transporters MATE-1 and OCT-2. Int. J. Mol. Sci. 2020, 21, 7416. [Google Scholar] [CrossRef]
- Arruda, A.C.; Perilhão, M.S.; Santos, W.A.; Gregnani, M.F.; Budu, A.; Neto, J.C.R.; Estrela, G.R.; Araujo, R.C. PPARα-Dependent Modulation by Metformin of the Expression of OCT-2 and MATE-1 in the Kidney of Mice. Molecules 2020, 25, 392. [Google Scholar] [CrossRef] [Green Version]
- Wongwan, T.; Chatsudthipong, V.; Soodvilai, S. Farnesoid X Receptor Activation Stimulates Organic Cations Transport in Human Renal Proximal Tubular Cells. Int. J. Mol. Sci. 2020, 21, 6078. [Google Scholar] [CrossRef]
- Kajiwara, M.; Terada, T.; Asaka, J.I.; Ogasawara, K.; Katsura, T.; Ogawa, O.; Fukatsu, A.; Doi, T.; Inui, K.-I. Critical roles of Sp1 in gene expression of human and rat H+/organic cation antiporter MATE1. Am. J. Physiol. Physiol. 2007, 293, F1564–F1570. [Google Scholar] [CrossRef]
- Ha Choi, J.; Yee, S.W.; Kim, M.J.; Nguyen, L.; Lee, J.H.; Kang, J.O.; Hesselson, S.; Stryke, D.; Johns, S.J.; Kwok, P.; et al. Identification and characterization of novel polymorphisms in the basal promoter of the human transporter, MATE1. Pharm. Genom. 2009, 19, 770–780. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.H.; Kim, K.H.; Park, H.J.; Kim, S.; Choi, J.H. Identification and functional characterization of novel MATE1 genetic variations in Koreans. Biochem. Biophys. Res. Commun. 2013, 434, 334–340. [Google Scholar] [CrossRef]
- Shapira, S.; Granot, G.; Mor-Tzuntz, R.; Raanani, P.; Uziel, O.; Lahav, M.; Shpilberg, O. Second-generation tyrosine kinase inhibitors reduce telomerase activity in K562 cells. Cancer Lett. 2012, 323, 223–231. [Google Scholar] [CrossRef]
- Miyake, T.; Mizuno, T.; Takehara, I.; Mochizuki, T.; Kimura, M.; Matsuki, S.; Irie, S.; Watanabe, N.; Kato, Y.; Ieiri, I.; et al. Elucidation of N (1)-methyladenosine as a Potential Surrogate Biomarker for Drug Interaction Studies Involving Renal Organic Cation Transporters. Drug Metab. Dispos. 2019, 47, 1270–1280. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Peng, X.; Yang, H.; Wang, H.; Shu, Y. Deficiency of multidrug and toxin extrusion 1 enhances renal accumulation of paraquat and deteriorates kidney injury in mice. Mol. Pharm. 2011, 8, 2476–2483. [Google Scholar] [CrossRef]
- Jonker, J.W.; Wagenaar, E.; van Eijl, S.; Schinkel, A.H. Deficiency in the Organic Cation Transporters 1 and 2 (Oct1/Oct2 [Slc22a1/Slc22a2]) in Mice Abolishes Renal Secretion of Organic Cations. Mol. Cell Biol. 2003, 23, 7902–7908. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.M.; Leblanc, A.F.; Uddin, M.E.; Kim, J.Y.; Chen, M.; Eisenmann, E.D.; Gibson, A.A.; Li, Y.; Hong, K.W.; DiGiacomo, D.; et al. Neuronal uptake transporters contribute to oxaliplatin neurotoxicity in mice. J. Clin. Investig. 2020, 130, 4601–4606. [Google Scholar] [CrossRef]
- Miyake, T.; Kimoto, E.; Luo, L.; Mathialagan, S.; Horlbogen, L.M.; Ramanathan, R.; Wood, L.S.; Johnson, J.G.; Le, V.H.; Vourvahis, M.; et al. Identification of Appropriate Endogenous Biomarker for Risk Assessment of Multidrug and Toxin Extrusion Protein-Mediated Drug-Drug Interactions in Healthy Volunteers. Clin. Pharmacol. Ther 2021, 109, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Levêque, D.; Becker, G.; Bilger, K.; Natarajan-Amé, S. Clinical Pharmacokinetics and Pharmacodynamics of Dasatinib. Clin. Pharm. 2020, 59, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Lipka, D.B.; Wagner, M.C.; Dziadosz, M.; Schnöder, T.; Heidel, F.; Schemionek, M.; Fischer, T.; Melo, J.V.; Kindler, T.; Müller-Tidow, C.; et al. Intracellular retention of ABL kinase inhibitors determines commitment to apoptosis in CML cells. PLoS ONE 2012, 7, e40853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, K.; Lago, M.W.; Iyer, R.A.; Shyu, W.-C.; Humphreys, W.G.; Christopher, L.J. Lacteal secretion, fetal and maternal tissue distribution of dasatinib in rats. Drug Metab. Dispos. 2008, 36, 2564–2570. [Google Scholar] [CrossRef] [PubMed]
- Graham, M.A.; Lockwood, G.F.; Greenslade, D.; Brienza, S.; Bayssas, M.; Gamelin, E. Clinical pharmacokinetics of oxaliplatin: A critical review. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2000, 6, 1205–1218. [Google Scholar]
- Zhang, S.; Lovejoy, K.S.; Shima, J.E.; Lagpacan, L.L.; Shu, Y.; Lapuk, A.; Chen, Y.; Komori, T.; Gray, J.W.; Chen, X.; et al. Organic cation transporters are determinants of oxaliplatin cytotoxicity. Cancer Res. 2006, 66, 8847–8857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoo, S.; Yonezawa, A.; Masuda, S.; Fukatsu, A.; Katsura, T.; Inui, K.-I. Differential contribution of organic cation transporters, OCT2 and MATE1, in platinum agent-induced nephrotoxicity. Biochem. Pharm. 2007, 74, 477–487. [Google Scholar] [CrossRef]
- Nakamura, T.; Yonezawa, A.; Hashimoto, S.; Katsura, T.; Inui, K.I. Disruption of multidrug and toxin extrusion MATE1 potentiates cisplatin-induced nephrotoxicity. Biochem. Pharm. 2010, 80, 1762–1767. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Guo, D.; Dong, Z.; Zhang, W.; Zhang, L.; Huang, S.M.; Pollia, J.E.; Shu, Y. Ondansetron can enhance cisplatin-induced nephrotoxicity via inhibition of multiple toxin and extrusion proteins (MATEs). Toxicol. Appl. Pharm. 2013, 273, 100–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, T.; Sato, W.; Ishikawa, K.; Terao, Y.; Takahashi, K.; Noda, Y.; Yuzawa, Y.; Nagamatsu, T. Significance of downregulation of renal organic cation transporter (SLC47A1) in cisplatin-induced proximal tubular injury. OncoTargets Ther. 2015, 8, 1701–1706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauzay, C.; White-Koning, M.; Hennebelle, I.; Deluche, T.; Delmas, C.; Imbs, D.C.; Chatelut, E.; Thomas, F. Inhibition of OCT2, MATE1 and MATE2-K as a possible mechanism of drug interaction between pazopanib and cisplatin. Pharmacol. Res. 2016, 110, 89–95. [Google Scholar] [CrossRef]
- Shen, H.; Yang, Z.; Zhao, W.; Zhang, Y.; Rodrigues, A.D. Assessment of vandetanib as an inhibitor of various human renal transporters: Inhibition of multidrug and toxin extrusion as a possible mechanism leading to decreased cisplatin and creatinine clearance. Drug Metab. Dispos. 2013, 41, 2095–2103. [Google Scholar] [CrossRef] [Green Version]
- Iwata, K.; Aizawa, K.; Kamitsu, S.; Jingami, S.; Fukunaga, E.; Yoshida, M.; Yoshimura, M.; Hamada, A.; Saito, H. Effects of genetic variants in SLC22A2 organic cation transporter 2 and SLC47A1 multidrug and toxin extrusion 1 transporter on cisplatin-induced adverse events. Clin. Exp. Nephrol. 2012, 16, 843–851. [Google Scholar] [CrossRef]
- Qian, C.-Y.; Zheng, Y.; Wang, Y.; Chen, J.; Liu, J.Y.; Zhou, H.H.; Yin, J.-Y.; Liu, Z.-Q. Associations of genetic polymorphisms of the transporters organic cation transporter 2 (OCT2), multidrug and toxin extrusion 1 (MATE1), and ATP-binding cassette subfamily C member 2 (ABCC2) with platinum-based chemotherapy response and toxicity in non-sm. Chin. J. Cancer 2016, 35, 85. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.; Hu, Y.; Hogan, S.L.; Mercke, N.; Gomez, M.; O’Bryant, C.; Bowles, D.W.; George, B.; Wen, X.; Aleksunes, L.M.; et al. Pharmacogenomic Variants May Influence the Urinary Excretion of Novel Kidney Injury Biomarkers in Patients Receiving Cisplatin. Int. J. Mol. Sci. 2017, 18, 1333. [Google Scholar] [CrossRef] [Green Version]
- El-Arabey, A.A. Dual function of OCT2 and MATE1 in cisplatin induced nephrotoxicity. Pharmacol. Res. 2017, 119, 493. [Google Scholar] [CrossRef] [PubMed]
- Vlaming, M.L.H.; van Esch, A.; Pala, Z.; Wagenaar, E.; van de Wetering, K.; van Tellingen, O.; Schinkel, A.H. Abcc2 (Mrp2), Abcc3 (Mrp3), and Abcg2 (Bcrp1) are the main determinants for rapid elimination of methotrexate and its toxic metabolite 7-hydroxymethotrexate in vivo. Mol. Cancer Ther. 2009, 8, 3350–3359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancaster, C.S.; Sprowl, J.A.; Walker, A.L.; Hu, S.; Gibson, A.A.; Sparreboom, A. Modulation of OATP1B-type transporter function alters cellular uptake and disposition of platinum chemotherapeutics. Mol. Cancer Ther. 2013, 12, 1537–1544. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.M.; Uddin, M.E.; DiGiacomo, D.; Lustberg, M.B.; Hu, S.; Sparreboom, A. Role of SLC transporters in toxicity induced by anticancer drugs. Expert Opin. Drug Metab. Toxicol. 2020, 16, 493–506. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uddin, M.E.; Talebi, Z.; Chen, S.; Jin, Y.; Gibson, A.A.; Noonan, A.M.; Cheng, X.; Hu, S.; Sparreboom, A. In Vitro and In Vivo Inhibition of MATE1 by Tyrosine Kinase Inhibitors. Pharmaceutics 2021, 13, 2004. https://doi.org/10.3390/pharmaceutics13122004
Uddin ME, Talebi Z, Chen S, Jin Y, Gibson AA, Noonan AM, Cheng X, Hu S, Sparreboom A. In Vitro and In Vivo Inhibition of MATE1 by Tyrosine Kinase Inhibitors. Pharmaceutics. 2021; 13(12):2004. https://doi.org/10.3390/pharmaceutics13122004
Chicago/Turabian StyleUddin, Muhammad Erfan, Zahra Talebi, Sijie Chen, Yan Jin, Alice A. Gibson, Anne M. Noonan, Xiaolin Cheng, Shuiying Hu, and Alex Sparreboom. 2021. "In Vitro and In Vivo Inhibition of MATE1 by Tyrosine Kinase Inhibitors" Pharmaceutics 13, no. 12: 2004. https://doi.org/10.3390/pharmaceutics13122004