
Supersaturation and Solubilization upon In Vitro Digestion of Fenofibrate Type I Lipid Formulations: Effect of Droplet Size, Surfactant Concentration and Lipid Type




Abstract
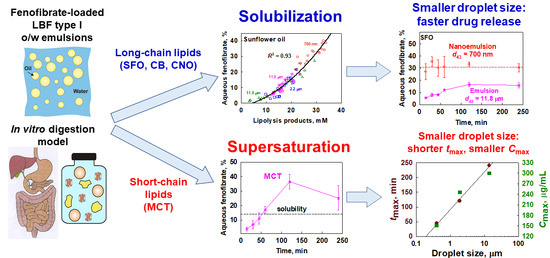
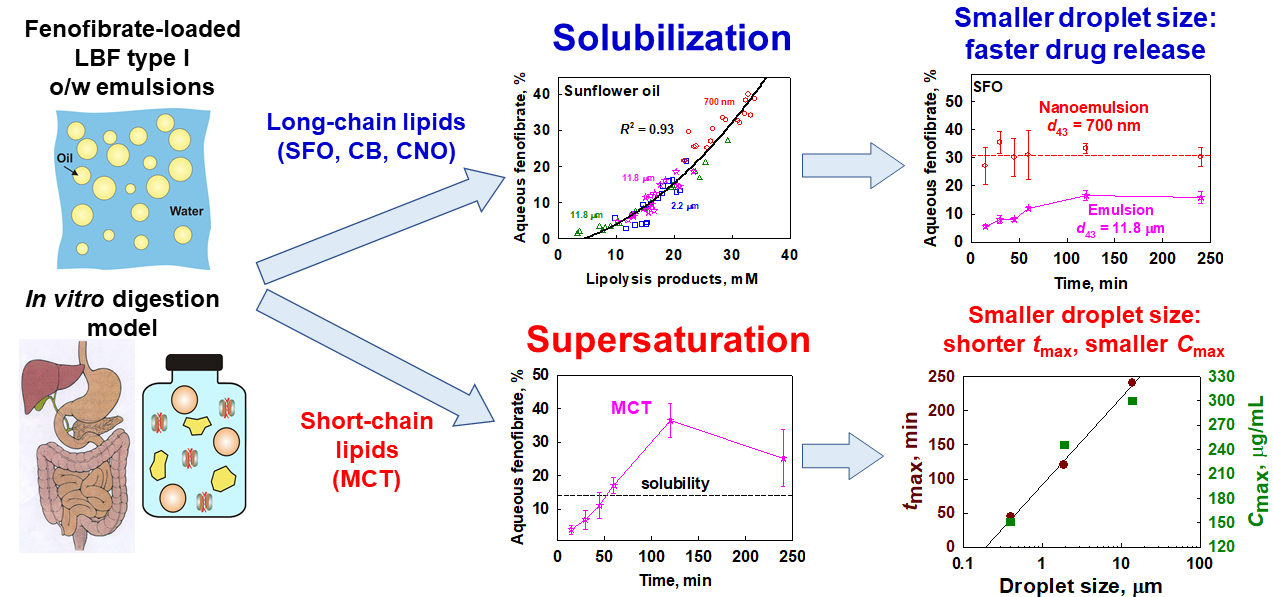
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Emulsion Preparation and Characterization
2.2.1. Aqueous and Oily Phase Preparation
2.2.2. Emulsification Protocols
2.2.3. Emulsion Droplet Size Determination by Laser Diffraction
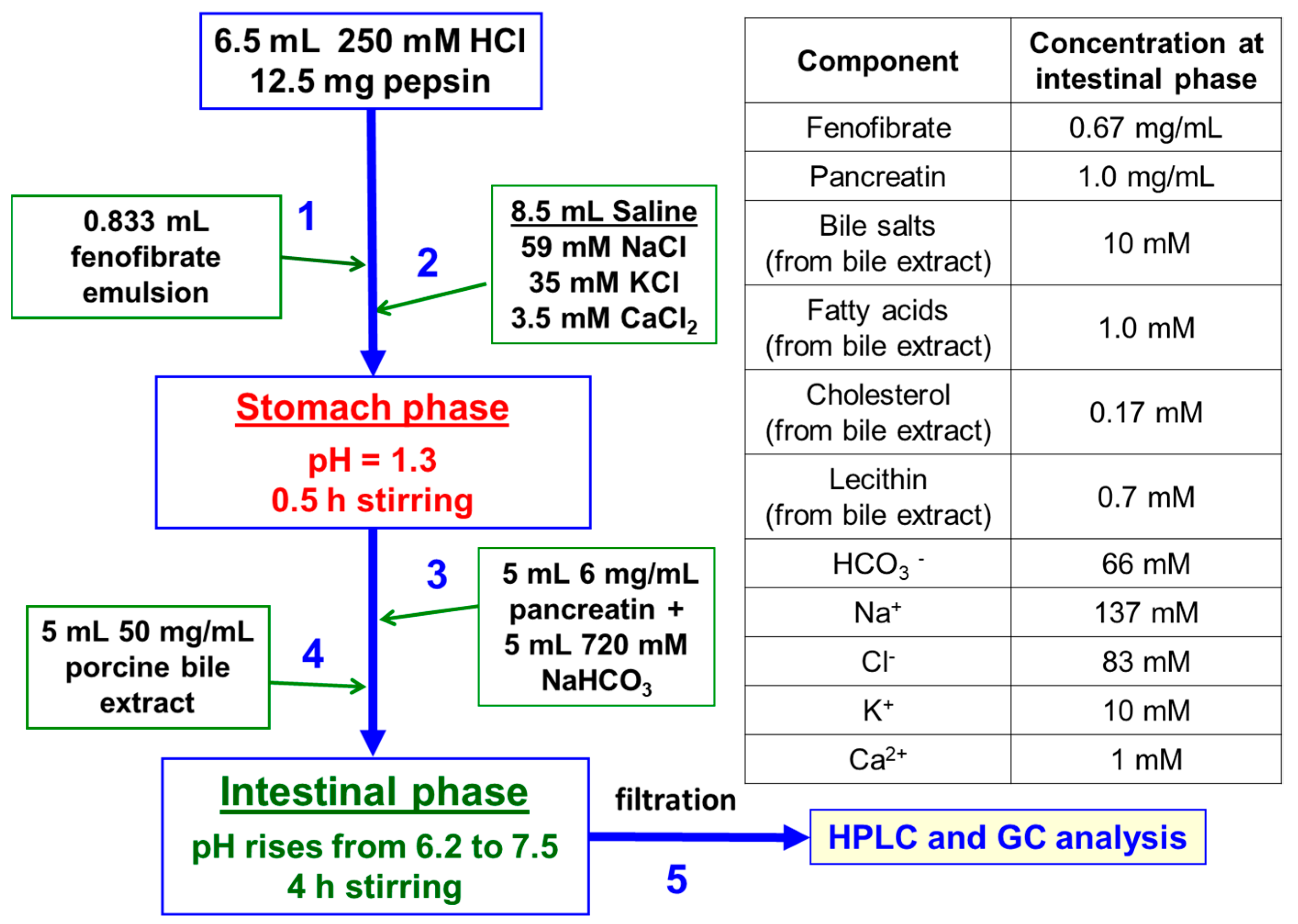
2.3. In Vitro Digestion Model
2.4. Analytical Protocols
2.4.1. Fenofibrate Determination by HPLC
2.4.2. Lipid Extraction and Analysis by GC
2.5. Extent of Lipid Digestion
2.6. Supersaturation Assessment
3. Experimental Results
3.1. Emulsion Droplet Size
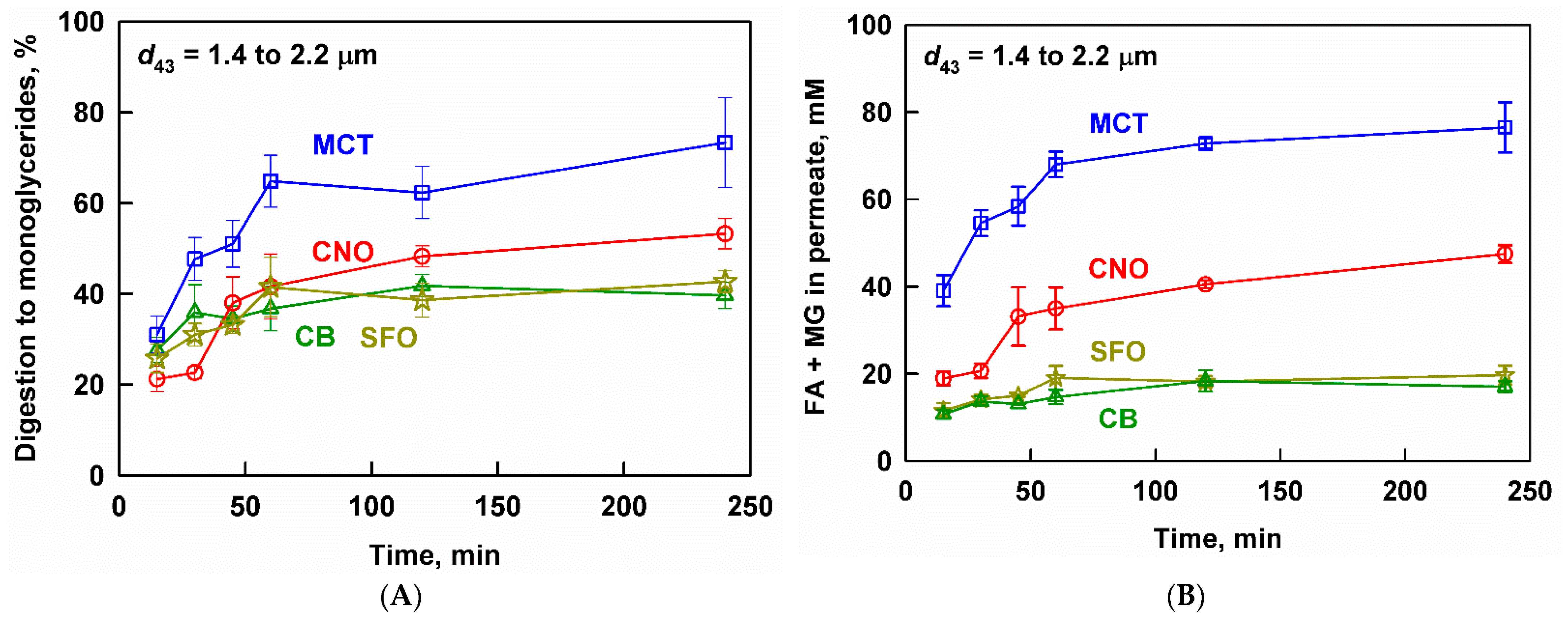
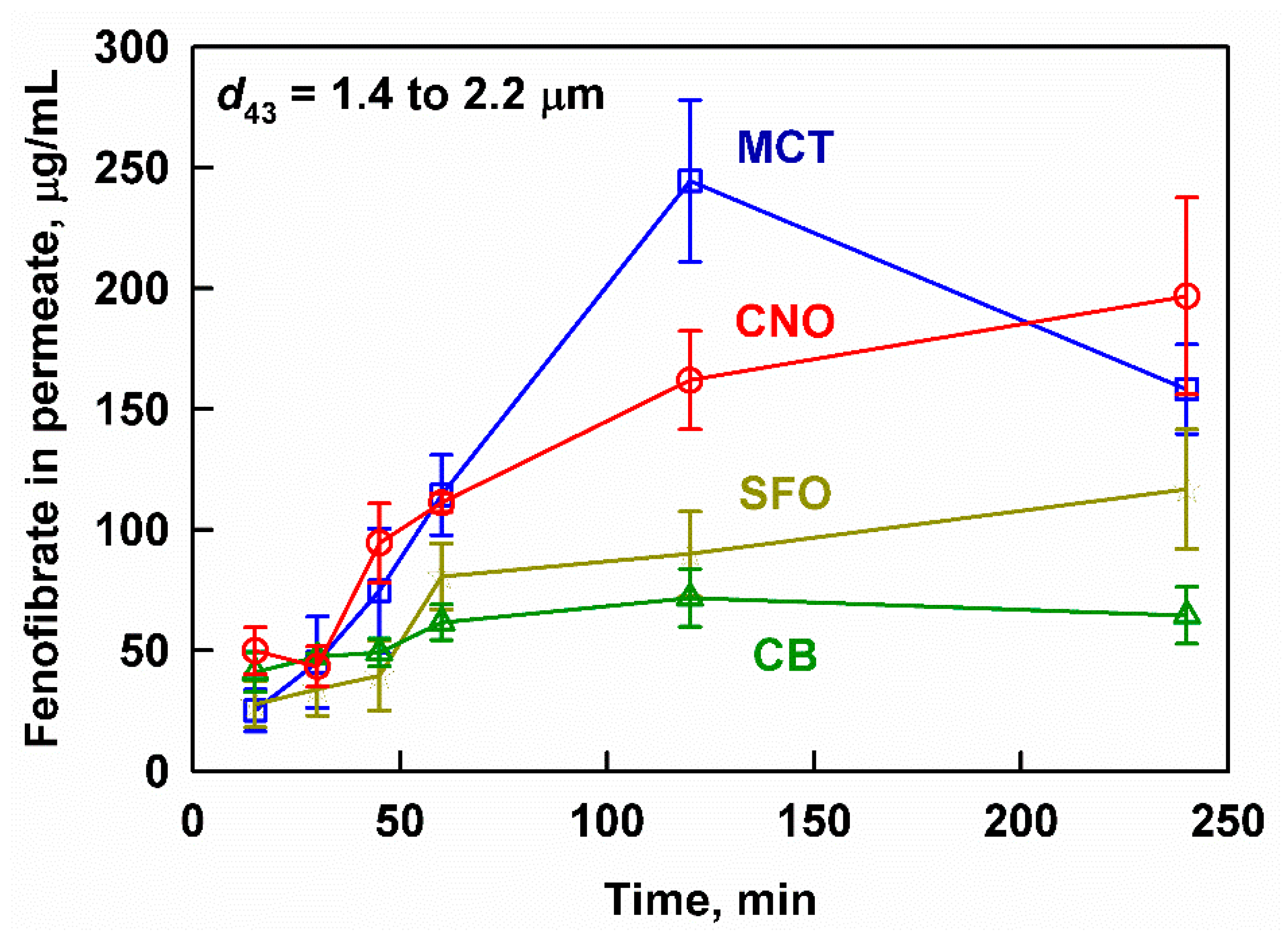
3.2. Effect of Lipid Type on Lipolysis and Fenofibrate Release
3.3. Effect of Drop Size and Surfactant Concentration on Lipolysis and Fenofibrate Release
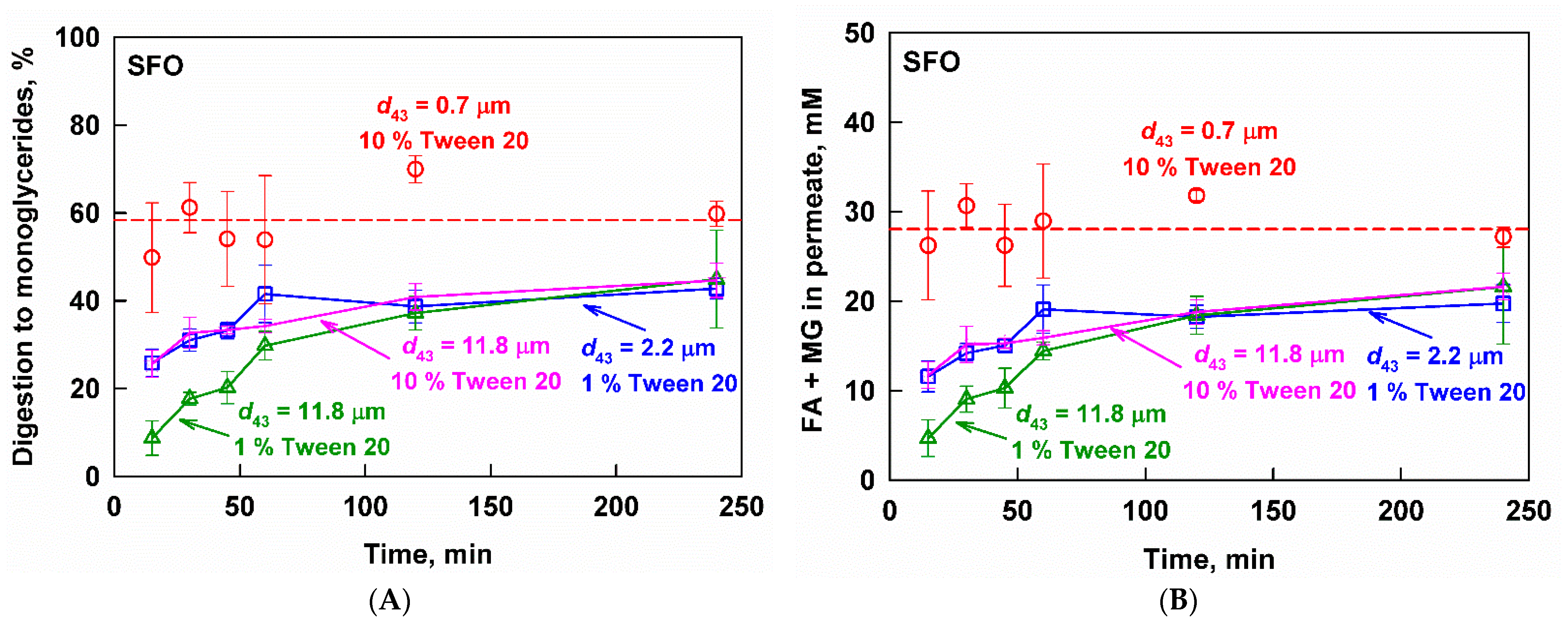
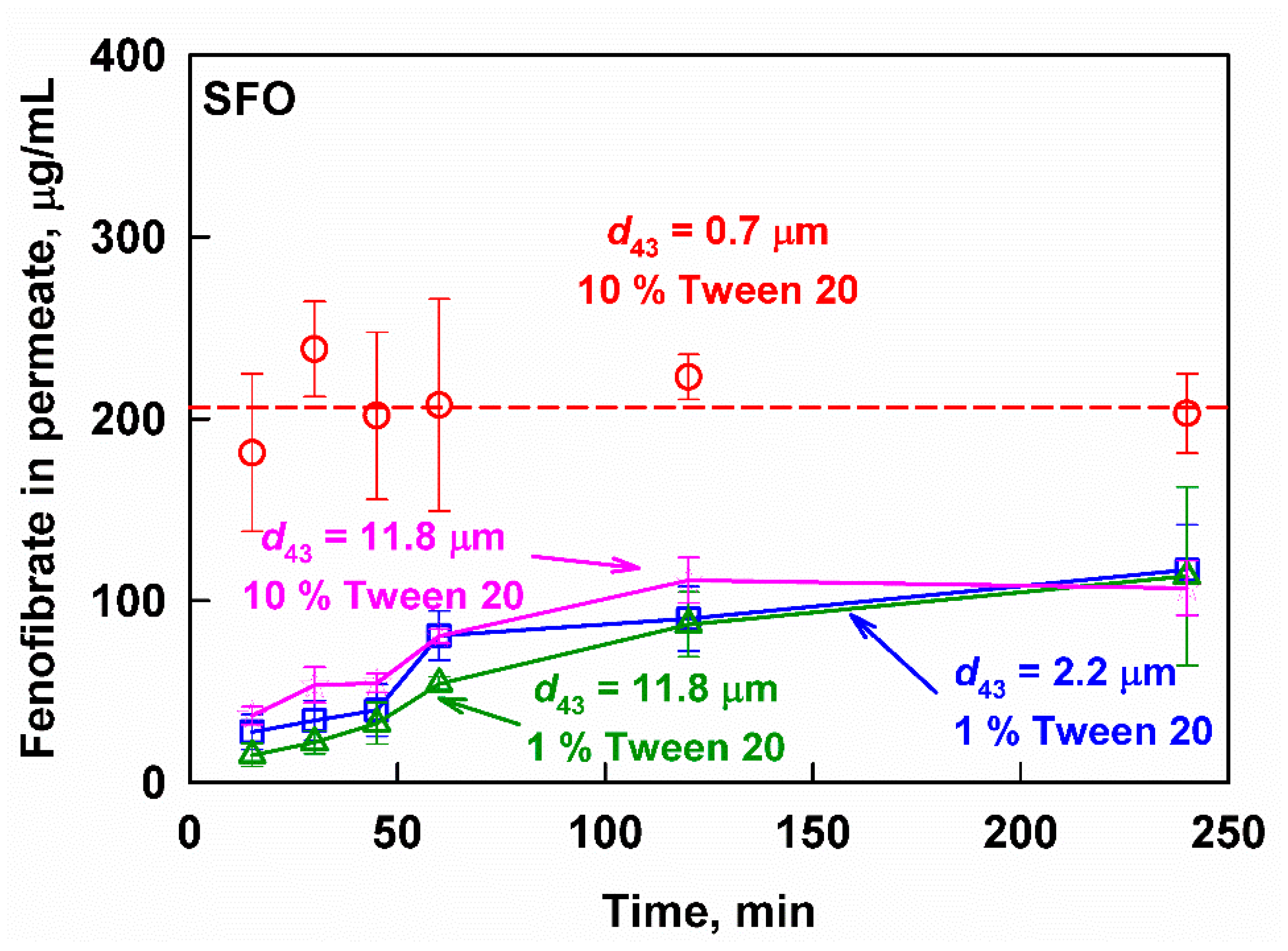
3.3.1. Sunflower Oil Emulsions
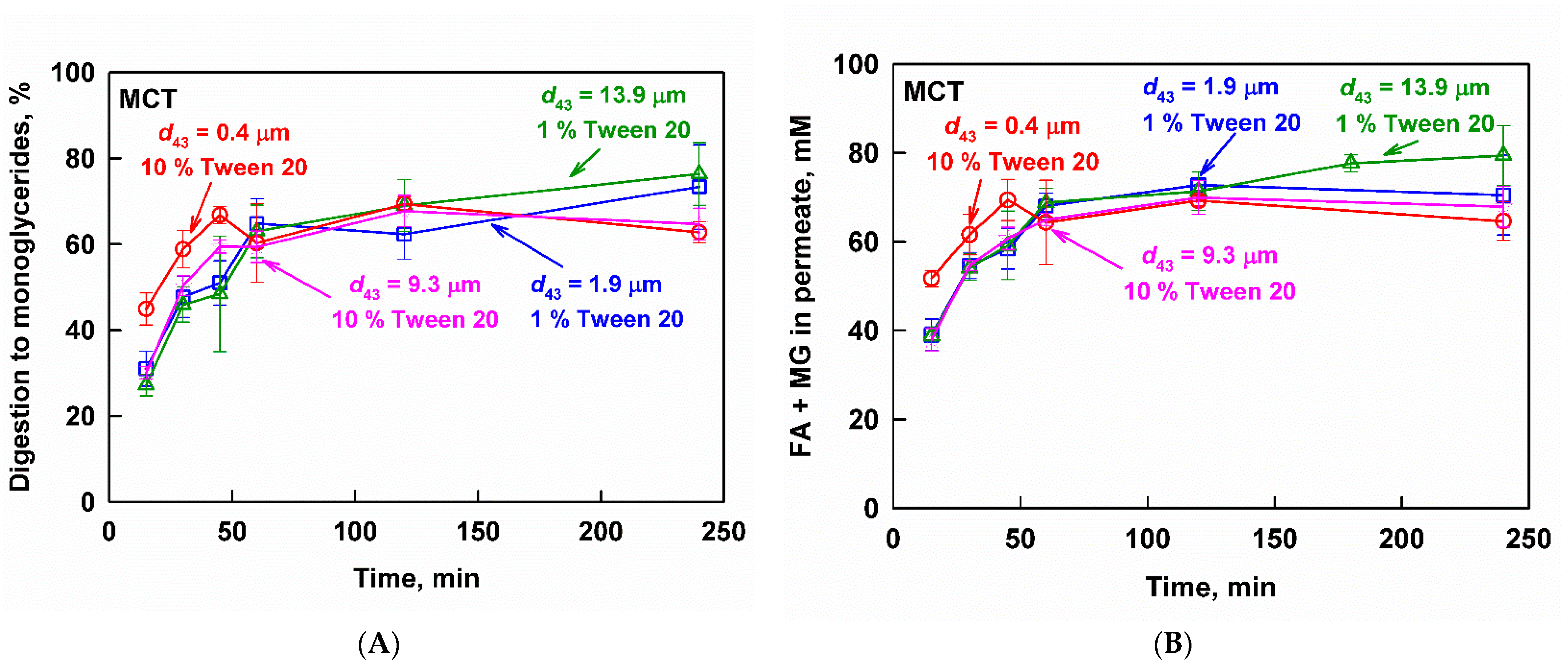
3.3.2. Medium-Chain Triglyceride Emulsions
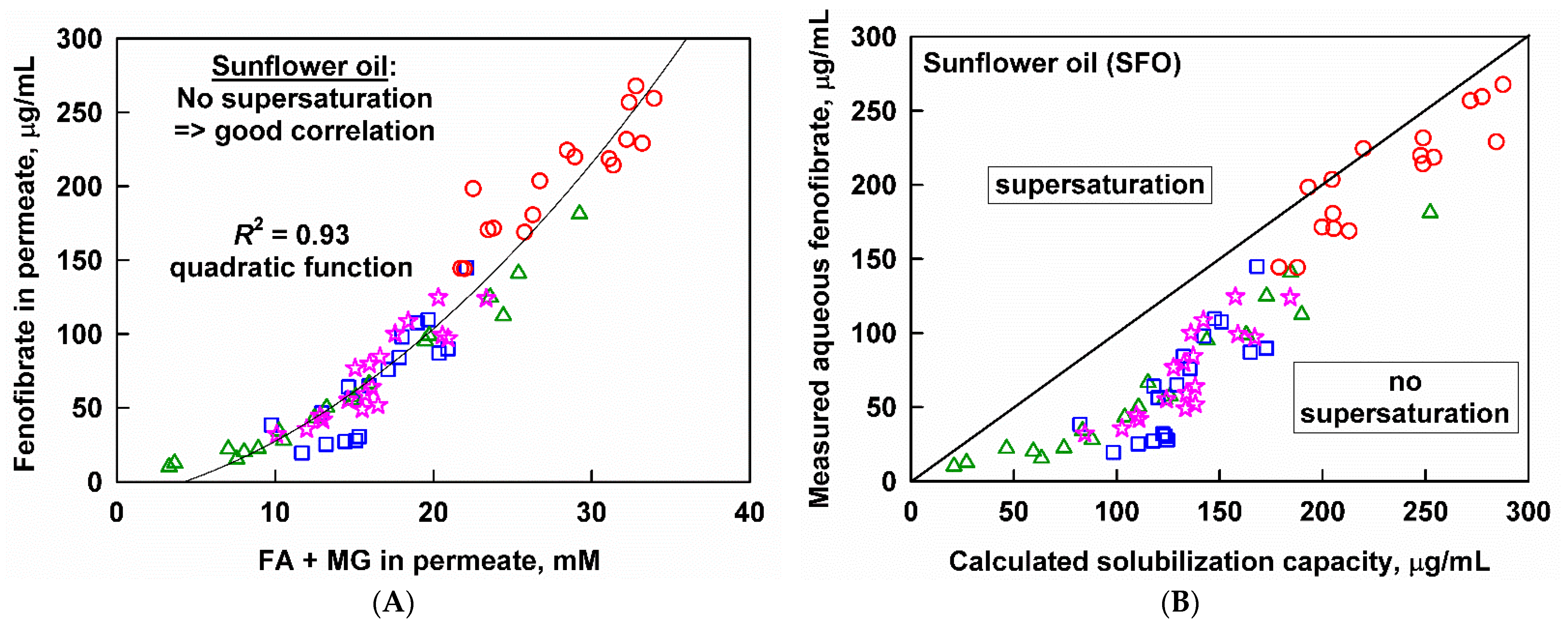
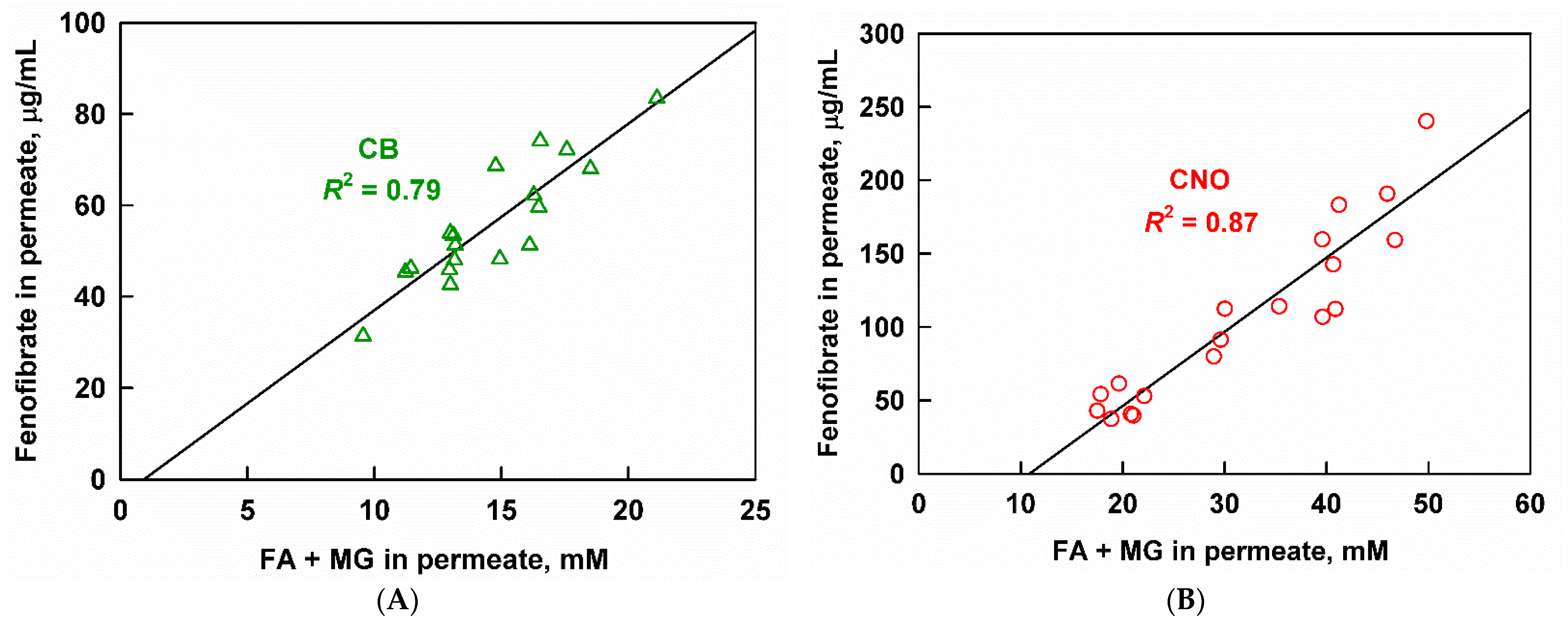
3.4. Interplay between Solubilization, Supersaturation and Drug Release
3.4.1. Effect of Emulsion Drop Size
3.4.2. Effect of Lipid Carrier Type
4. Discussion
4.1. Drug Solubilization and the Role of Lipid Digestion Products
4.2. Supersaturation and Precipitation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Doak, B.; Over, B.; Giordanetto, F.; Kihlberg, J. Oral Druggable Space beyond the Rule of 5: Insights from Drugs and Clinical Candidates. Chem. Biol. 2014, 21, 1115–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keserü, G.M.; Makara, G.M. The influence of lead discovery strategies on the properties of drug candidates. Nat. Rev. Drug Discov. 2009, 8, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Benet, L.Z.; Broccatelli, F.; Oprea, T. BDDCS Applied to Over 900 Drugs. AAPS J. 2011, 13, 519–547. [Google Scholar] [CrossRef] [Green Version]
- Augustijns, P.; Wuyts, B.; Hens, B.; Annaert, P.; Butler, J.; Brouwers, J. A review of drug solubility in human intestinal fluids: Implications for the prediction of oral absorption. Eur. J. Pharm. Sci. 2014, 57, 322–332. [Google Scholar] [CrossRef] [PubMed]
- De la Cruz-Moreno, M.P.; Montejo, C.; Aguilar-Ros, A.; Dewe, W.; Beck, B.; Stappaerts, J.; Tack, J.; Augustijns, P. Exploring drug solubility in fasted human intestinal fluid aspirates: Impact of inter-individual variability, sampling site and dilution. Int. J. Pharm. 2017, 528, 471–484. [Google Scholar] [CrossRef]
- Boyd, B.J.; Bergström, C.A.; Vinarov, Z.; Kuentz, M.; Brouwers, J.; Augustijns, P.; Brandl, M.; Bernkop-Schnürch, A.; Shrestha, N.; Préat, V.; et al. Successful oral delivery of poorly water-soluble drugs both depends on the intraluminal behavior of drugs and of appropriate advanced drug delivery systems. Eur. J. Pharm. Sci. 2019, 137, 104967. [Google Scholar] [CrossRef]
- Porter, C.; Trevaskis, N.; Charman, W. Lipids and lipid-based formulations: Optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug Discov. 2007, 6, 231–248. [Google Scholar] [CrossRef] [PubMed]
- Mu, H.; Holm, R.; Müllertz, A. Lipid-based formulations for oral administration of poorly water-soluble drugs. Int. J. Pharm. 2013, 453, 215–224. [Google Scholar] [CrossRef]
- Pouton, C.W.; Porter, C.J.H. Formulation of lipid-based delivery systems for oral administration: Materials, methods and strategies. Adv. Drug Deliv. Rev. 2008, 60, 625–637. [Google Scholar] [CrossRef]
- Hauss, D.J. Oral Lipid-Based Formulations: Enhancing the Bioavailability of Poorly Water-Soluble Drugs; CRC Press: Boca Raton, FL, USA, 2007; Volume 170. [Google Scholar]
- Müllertz, A.; Ogbonna, A.; Ren, S.; Rades, T. New perspectives on lipid and surfactant based drug delivery systems for oral delivery of poorly soluble drugs. J. Pharm. Pharmacol. 2010, 62, 1622–1636. [Google Scholar] [CrossRef]
- Pouton, C.W. Formulation of poorly water-soluble drugs for oral administration: Physicochemical and physiological issues and the lipid formulation classification system. Eur. J. Pharm. Sci. 2006, 29, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Trevaskis, N.; Charman, W.; Porter, C. Lipid-based delivery systems and intestinal lymphatic drug transport: A mechanistic update. Adv. Drug Deliv. Rev. 2008, 60, 702–716. [Google Scholar] [CrossRef] [PubMed]
- Trevaskis, N.L.; Lee, G.; Escott, A.; Phang, K.L.; Hong, J.; Cao, E.; Katneni, K.; Charman, S.A.; Han, S.; Charman, W.N.; et al. Intestinal Lymph Flow, and Lipid and Drug Transport Scale Allometrically From Pre-clinical Species to Humans. Front. Physiol. 2020, 11, 458. [Google Scholar] [CrossRef] [PubMed]
- Fatouros, D.G.; Deen, G.R.; Arleth, L.; Bergenståhl, B.; Nielsen, F.S.; Pedersen, J.S.; Müllertz, A. Structural Development of Self Nano Emulsifying Drug Delivery Systems (SNEDDS) During In Vitro Lipid Digestion Monitored by Small-angle X-ray Scattering. Pharm. Res. 2007, 24, 1844–1853. [Google Scholar] [CrossRef]
- Thomas, N.; Müllertz, A.; Graf, A.; Rades, T. Influence of Lipid Composition and Drug Load on the In Vitro Performance of Self-Nanoemulsifying Drug Delivery Systems. J. Pharm. Sci. 2012, 101, 1721–1731. [Google Scholar] [CrossRef]
- Kontogiannidou, E.; Meikopoulos, T.; Gika, H.; Panteris, E.; Vizirianakis, I.S.; Müllertz, A.; Fatouros, D.G. In Vitro Evaluation of Self-Nano-Emulsifying Drug Delivery Systems (SNEDDS) Containing Room Temperature Ionic Liquids (RTILs) for the Oral Delivery of Amphotericin B. Pharmaceutics 2020, 12, 699. [Google Scholar] [CrossRef]
- Tran, T.; Bønløkke, P.; Rodriguez-Rodriguez, C.; Nosrati, Z.; Esquinas, P.L.; Borkar, N.; Plum, J.; Strindberg, S.; Karagiozov, S.; Rades, T.; et al. Using in vitro lipolysis and SPECT/CT in vivo imaging to understand oral absorption of fenofibrate from lipid-based drug delivery systems. J. Control. Release 2020, 317, 375–384. [Google Scholar] [CrossRef]
- Crum, M.F.; Trevaskis, N.; Pouton, C.; Porter, C. Transient Supersaturation Supports Drug Absorption from Lipid-Based Formulations for Short Periods of Time, but Ongoing Solubilization Is Required for Longer Absorption Periods. Mol. Pharm. 2017, 14, 394–405. [Google Scholar] [CrossRef]
- Ainousah, B.E.; Perrier, J.; Dunn, C.; Khadra, I.; Wilson, C.G.; Halbert, G. Dual Level Statistical Investigation of Equilibrium Solubility in Simulated Fasted and Fed Intestinal Fluid. Mol. Pharm. 2017, 14, 4170–4180. [Google Scholar] [CrossRef]
- Dunn, C.; Perrier, J.; Khadra, I.; Wilson, C.G.; Halbert, G.W. Topography of Simulated Intestinal Equilibrium Solubility. Mol. Pharm. 2019, 16, 1890–1905. [Google Scholar] [CrossRef]
- Katev, V.; Vinarov, Z.; Tcholakova, S. Mechanisms of drug solubilization by polar lipids in biorelevant media. Eur. J. Pharm. Sci. 2021, 159, 105733. [Google Scholar] [CrossRef]
- Kleberg, K.; Jacobsen, F.; Fatouros, D.G.; Müllertz, A. Biorelevant Media Simulating Fed State Intestinal Fluids: Colloid Phase Characterization and Impact on Solubilization Capacity. J. Pharm. Sci. 2010, 99, 3522–3532. [Google Scholar] [CrossRef]
- McPherson, S.; Perrier, J.; Dunn, C.; Khadra, I.; Davidson, S.; Ainousah, B.; Wilson, C.G.; Halbert, G. Small scale design of experiment investigation of equilibrium solubility in simulated fasted and fed intestinal fluid. Eur. J. Pharm. Biopharm. 2020, 150, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Perrier, J.; Zhou, Z.; Dunn, C.; Khadra, I.; Wilson, C.G.; Halbert, G. Statistical investigation of the full concentration range of fasted and fed simulated intestinal fluid on the equilibrium solubility of oral drugs. Eur. J. Pharm. Sci. 2017, 111, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Dunn, C.; Khadra, I.; Wilson, C.G.; Halbert, G.W. Influence of Physiological Gastrointestinal Surfactant Ratio on the Equilibrium Solubility of BCS Class II Drugs Investigated Using a Four Component Mixture Design. Mol. Pharm. 2017, 14, 4132–4144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Dunn, C.; Khadra, I.; Wilson, C.G.; Halbert, G.W. Statistical investigation of simulated fed intestinal media composition on the equilibrium solubility of oral drugs. Eur. J. Pharm. Sci. 2017, 99, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Jantratid, E.; Janssen, N.; Reppas, C.; Dressman, J.B. Dissolution Media Simulating Conditions in the Proximal Human Gastrointestinal Tract: An Update. Pharm. Res. 2008, 25, 1663–1676. [Google Scholar] [CrossRef] [PubMed]
- Dening, T.J.; Joyce, P.; Prestidge, C.A. Improving Correlations Between Drug Solubilization and In Vitro Lipolysis by Monitoring the Phase Partitioning of Lipolytic Species for Lipid-Based Formulations. J. Pharm. Sci. 2019, 108, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Zangenberg, N.H.; Müllertz, A.; Kristensen, H.G.; Hovgaard, L. A dynamic in vitro lipolysis model: II: Evaluation of the model. Eur. J. Pharm. Sci. 2001, 14, 237–244. [Google Scholar] [CrossRef]
- Borkar, N.; Xia, D.; Holm, R.; Gan, Y.; Müllertz, A.; Yang, M.; Mu, H. Investigating the correlation between in vivo absorption and in vitro release of fenofibrate from lipid matrix particles in biorelevant medium. Eur. J. Pharm. Sci. 2014, 51, 204–210. [Google Scholar] [CrossRef]
- Diril, M.; Türkyılmaz, G.Y.; Karasulu, H.Y.; Karasulua, H.Y. Formulation and In Vitro Evaluation of Self Microemulsifying Drug Delivery System Containing Atorvastatin Calcium. Curr. Drug Deliv. 2019, 16, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Kamoun, J.; Krichen, F.; Koubaa, I.; Zouari, N.; Bougatef, A.; Abousalham, A.; Aloulou, A. In vitro lipolysis and physicochemical characterization of unconventional star anise oil towards the development of new lipid-based drug delivery systems. Heliyon 2021, 7, e06717. [Google Scholar] [CrossRef] [PubMed]
- Kazi, M.; Al-Qarni, H.; Alanazi, F.K. Development of oral solid self-emulsifying lipid formulations of risperidone with improved in vitro dissolution and digestion. Eur. J. Pharm. Biopharm. 2017, 114, 239–249. [Google Scholar] [CrossRef]
- Koehl, N.J.; Holm, R.; Kuentz, M.; Griffin, B.T. New Insights into Using Lipid Based Suspensions for ‘Brick Dust’ Molecules: Case Study of Nilotinib. Pharm. Res. 2019, 36, 56. [Google Scholar] [CrossRef]
- O’Dwyer, P.J.; Box, K.J.; Koehl, N.J.; Bennett-Lenane, H.; Reppas, C.; Holm, R.; Kuentz, M.; Griffin, B.T. Novel Biphasic Lipolysis Method To Predict in Vivo Performance of Lipid-Based Formulations. Mol. Pharm. 2020, 17, 3342–3352. [Google Scholar] [CrossRef] [PubMed]
- Salim, M.; Ramirez, G.; Clulow, A.J.; Zhang, Y.; Ristroph, K.; Feng, J.; McManus, S.A.; Hawley, A.; Prud’Homme, R.K.; Boyd, B.J. Solid-State Behavior and Solubilization of Flash Nanoprecipitated Clofazimine Particles during the Dispersion and Digestion of Milk-Based Formulations. Mol. Pharm. 2019, 16, 2755–2765. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Lv, Y.; Zhao, S.; Wang, B.; Tan, M.; Xie, H.; Lv, G.; Ma, X. Effect of Lipolysis on Drug Release from Self-microemulsifying Drug Delivery Systems (SMEDDS) with Different Core/Shell Drug Location. AAPS PharmSciTech 2014, 15, 731–740. [Google Scholar] [CrossRef] [Green Version]
- Williams, H.D.; Anby, M.U.; Sassene, P.; Kleberg, K.; Bakala-N’Goma, J.-C.; Calderone, M.; Jannin, V.; Igonin, A.; Partheil, A.; Marchaud, D.; et al. Toward the Establishment of Standardized in Vitro Tests for Lipid-Based Formulations. 2. The Effect of Bile Salt Concentration and Drug Loading on the Performance of Type I, II, IIIA, IIIB, and IV Formulations during in Vitro Digestion. Mol. Pharm. 2012, 9, 3286–3300. [Google Scholar] [CrossRef]
- Williams, H.D.; Sassene, P.; Kleberg, K.; Bakala-N’Goma, J.-C.; Calderone, M.; Jannin, V.; Igonin, A.; Partheil, A.; Marchaud, D.; Jule, E.; et al. Toward the Establishment of Standardized In Vitro Tests for Lipid-Based Formulations, Part 1: Method Parameterization and Comparison of In Vitro Digestion Profiles Across a Range of Representative Formulations. J. Pharm. Sci. 2012, 101, 3360–3380. [Google Scholar] [CrossRef]
- Williams, H.D.; Sassene, P.; Kleberg, K.; Calderone, M.; Igonin, A.; Jule, E.; Vertommen, J.; Blundell, R.; Benameur, H.; Müllertz, A.; et al. Toward the Establishment of Standardized In Vitro Tests for Lipid-Based Formulations, Part 4: Proposing a New Lipid Formulation Performance Classification System. J. Pharm. Sci. 2014, 103, 2441–2455. [Google Scholar] [CrossRef]
- Williams, H.D.; Sassene, P.; Kleberg, K.; Calderone, M.; Igonin, A.; Jule, E.; Vertommen, J.; Blundell, R.; Benameur, H.; Mullertz, A.; et al. Toward the Establishment of Standardized In Vitro Tests for Lipid-Based Formulations, Part 3: Understanding Supersaturation Versus Precipitation Potential During the In Vitro Digestion of Type I, II, IIIA, IIIB and IV Lipid-Based Formulations. Pharm. Res. 2013, 30, 3059–3076. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Morozowich, W. Development of supersaturatable self-emulsifying drug delivery system formulations for improving the oral absorption of poorly soluble drugs. Expert Opin. Drug Deliv. 2005, 3, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Tay, E.; Nguyen, T.-H.; Porter, C. Quantifying In Vivo Luminal Drug Solubilization -Supersaturation-Precipitation Profiles to Explain the Performance of Lipid Based Formulations. Pharm. Res. 2020, 37, 47. [Google Scholar] [CrossRef] [PubMed]
- Bannow, J.; Yorulmaz, Y.; Löbmann, K.; Müllertz, A.; Rades, T. Improving the drug load and in vitro performance of supersaturated self-nanoemulsifying drug delivery systems (super-SNEDDS) using polymeric precipitation inhibitors. Int. J. Pharm. 2020, 575, 118960. [Google Scholar] [CrossRef] [PubMed]
- Palmelund, H.; Madsen, C.M.; Plum, J.; Müllertz, A.; Rades, T. Studying the Propensity of Compounds to Supersaturate: A Practical and Broadly Applicable Approach. J. Pharm. Sci. 2016, 105, 3021–3029. [Google Scholar] [CrossRef] [PubMed]
- Strindberg, S.; Plum, J.; Stie, M.B.; Christiansen, M.L.; Nielsen, L.H.; Rades, T.; Müllertz, A. Effect of supersaturation on absorption of indomethacin and tadalafil in a single pass intestinal perfusion rat model, in the absence and presence of a precipitation inhibitor. Eur. J. Pharm. Biopharm. 2020, 151, 108–115. [Google Scholar] [CrossRef]
- Thomas, N.; Holm, R.; Garmer, M.; Karlsson, J.J.; Müllertz, A.; Rades, T. Supersaturated Self-Nanoemulsifying Drug Delivery Systems (Super-SNEDDS) Enhance the Bioavailability of the Poorly Water-Soluble Drug Simvastatin in Dogs. AAPS J. 2012, 15, 219–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, N.; Holm, R.; Müllertz, A.; Rades, T. In vitro and in vivo performance of novel supersaturated self-nanoemulsifying drug delivery systems (super-SNEDDS). J. Control. Release 2012, 160, 25–32. [Google Scholar] [CrossRef]
- Frank, D.; Matzger, A.J. Effect of Polymer Hydrophobicity on the Stability of Amorphous Solid Dispersions and Supersaturated Solutions of a Hydrophobic Pharmaceutical. Mol. Pharm. 2019, 16, 682–688. [Google Scholar] [CrossRef]
- Kawakami, K. Supersaturation and crystallization: Non-equilibrium dynamics of amorphous solid dispersions for oral drug delivery. Expert Opin. Drug Deliv. 2016, 14, 735–743. [Google Scholar] [CrossRef]
- Schver, G.C.R.M.; Lee, P.I. Combined Effects of Supersaturation Rates and Doses on the Kinetic-Solubility Profiles of Amorphous Solid Dispersions Based on Water-Insoluble Poly(2-hydroxyethyl methacrylate) Hydrogels. Mol. Pharm. 2018, 15, 2017–2026. [Google Scholar] [CrossRef]
- Sun, D.D.; Lee, P.I. Evolution of Supersaturation of Amorphous Pharmaceuticals: The Effect of Rate of Supersaturation Generation. Mol. Pharm. 2013, 10, 4330–4346. [Google Scholar] [CrossRef]
- Sun, D.D.; Lee, P.I. Evolution of Supersaturation of Amorphous Pharmaceuticals: Nonlinear Rate of Supersaturation Generation Regulated by Matrix Diffusion. Mol. Pharm. 2015, 12, 1203–1215. [Google Scholar] [CrossRef]
- Bernardo, F.P.; Saraiva, P.M. A Theoretical Model for Transdermal Drug Delivery from Emulsions and its Dependence upon Formulation. J. Pharm. Sci. 2008, 97, 3781–3809. [Google Scholar] [CrossRef] [PubMed]
- Bakala-N’Goma, J.-C.; Williams, H.D.; Sassene, P.J.; Kleberg, K.; Calderone, M.; Jannin, V.; Igonin, A.; Partheil, A.; Marchaud, D.; Jule, E.; et al. Toward the Establishment of Standardized In Vitro Tests for Lipid-Based Formulations. 5. Lipolysis of Representative Formulations by Gastric Lipase. Pharm. Res. 2015, 32, 1279–1287. [Google Scholar] [CrossRef]
- Sassene, P.; Kleberg, K.; Williams, H.D.; Bakala-N’Goma, J.-C.; Carriere, F.; Calderone, M.; Jannin, V.; Igonin, A.; Partheil, A.; Marchaud, D.; et al. Toward the Establishment of Standardized In Vitro Tests for Lipid-Based Formulations, Part 6: Effects of Varying Pancreatin and Calcium Levels. AAPS J. 2014, 16, 1344–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pestieau, A.; Krier, F.; Brouwers, A.; Streel, B.; Evrard, B. Selection of a discriminant and biorelevant in vitro dissolution test for the development of fenofibrate self-emulsifying lipid-based formulations. Eur. J. Pharm. Sci. 2016, 92, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Mohsin, K. Design of Lipid-Based Formulations for Oral Administration of Poorly Water-Soluble Drug Fenofibrate: Effects of Digestion. AAPS PharmSciTech 2012, 13, 637–646. [Google Scholar] [CrossRef] [Green Version]
- Grove, M.; Müllertz, A.; Nielsen, J.L.; Pedersen, G.P. Bioavailability of seocalcitol: II: Development and characterisation of self-microemulsifying drug delivery systems (SMEDDS) for oral administration containing medium and long chain triglycerides. Eur. J. Pharm. Sci. 2006, 28, 233–242. [Google Scholar] [CrossRef]
- Gershanik, T.; Benita, S. Self-dispersing lipid formulations for improving oral absorption of lipophilic drugs. Eur. J. Pharm. Biopharm. 2000, 50, 179–188. [Google Scholar] [CrossRef]
- Griffin, B.T.; Kuentz, M.; Vertzoni, M.; Kostewicz, E.S.; Fei, Y.; Faisal, W.; Stillhart, C.; O’Driscoll, C.M.; Reppas, C.; Dressman, J.B. Comparison of in vitro tests at various levels of complexity for the prediction of in vivo performance of lipid-based formulations: Case studies with fenofibrate. Eur. J. Pharm. Biopharm. 2014, 86, 427–437. [Google Scholar] [CrossRef]
- Porter, C.; Kaukonen, A.M.; Boyd, B.J.; Edwards, G.A.; Charman, W.N. Susceptibility to lipase-mediated digestion reduces the oral bioavailability of danazol after administration as a medium-chain lipid-based microemulsion formulation. Pharm. Res. 2004, 21, 1405–1412. [Google Scholar] [CrossRef]
- Larsen, A.T.; Åkesson, P.; Juréus, A.; Saaby, L.; Abu-Rmaileh, R.; Abrahamsson, B.; Østergaard, J.; Müllertz, A. Bioavailability of Cinnarizine in Dogs: Effect of SNEDDS Loading Level and Correlation with Cinnarizine Solubilization During In Vitro Lipolysis. Pharm. Res. 2013, 30, 3101–3113. [Google Scholar] [CrossRef]
- Larsen, A.T.; Ohlsson, A.G.; Polentarutti, B.; Barker, R.A.; Phillips, A.R.; Abu-Rmaileh, R.; Dickinson, P.A.; Abrahamsson, B.; Østergaard, J.; Müllertz, A. Oral bioavailability of cinnarizine in dogs: Relation to SNEDDS droplet size, drug solubility and in vitro precipitation. Eur. J. Pharm. Sci. 2013, 48, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Mueller, E.A.; Kovarik, J.M.; Van Bree, J.B.; Grevel, J.; Lücker, P.W.; Kutz, K. Influence of a Fat-Rich Meal on the Pharmacokinetics of a New Oral Formulation of Cyclosporine in a Crossover Comparison with the Market Formulation. Pharm. Res. 1994, 11, 151–155. [Google Scholar] [CrossRef]
- De Smidt, P.; Campanero, M.A.; Troconiz, I. Intestinal absorption of penclomedine from lipid vehicles in the conscious rat: Contribution of emulsification versus digestibility. Int. J. Pharm. 2004, 270, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Wen, X.; Meng, Q.; Wu, W.; Everaert, N.; Xie, J.; Zhang, H. Alteration in bile acids profile in Large White pigs during chronic heat exposure. J. Therm. Biol. 2019, 84, 375–383. [Google Scholar] [CrossRef]
- Zangenberg, N.H.; Müllertz, A.; Kristensen, H.G.; Hovgaard, L. A dynamic in vitro lipolysis model: I. Controlling the rate of lipolysis by continuous addition of calcium. Eur. J. Pharm. Sci. 2001, 14, 115–122. [Google Scholar] [CrossRef]
- Vinarov, Z.; Petrova, L.; Tcholakova, S.; Denkov, N.; Stoyanov, S.; Lips, A. In vitro study of triglyceride lipolysis and phase distribution of the reaction products and cholesterol: Effects of calcium and bicarbonate. Food Funct. 2012, 3, 1206–1220. [Google Scholar] [CrossRef] [PubMed]
- Vinarova, L.; Vinarov, Z.; Atanasov, V.; Pantcheva, I.; Tcholakova, S.; Denkov, N.; Stoyanov, S. Lowering of cholesterol bioaccessibility and serum concentrations by saponins: In vitro and in vivo studies. Food Funct. 2014, 6, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Vinarova, L.; Vinarov, Z.; Tcholakova, S.; Denkov, N.; Stoyanov, S.; Lips, A. The mechanism of lowering cholesterol absorption by calcium studied by using an in vitro digestion model. Food Funct. 2015, 7, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Riethorst, D.; Mols, R.; Duchateau, G.S.M.J.E.; Tack, J.; Brouwers, J.; Augustijns, P. Characterization of Human Duodenal Fluids in Fasted and Fed State Conditions. J. Pharm. Sci. 2016, 105, 673–681. [Google Scholar] [CrossRef] [Green Version]
- Devraj, R.; Williams, H.D.; Warren, D.B.; Müllertz, A.; Porter, C.; Pouton, C. In vitro digestion testing of lipid-based delivery systems: Calcium ions combine with fatty acids liberated from triglyceride rich lipid solutions to form soaps and reduce the solubilization capacity of colloidal digestion products. Int. J. Pharm. 2013, 441, 323–333. [Google Scholar] [CrossRef]
- Hens, B.; Abeele, J.V.D.; Rubbens, J.; Keirsebilck, M.; Roelens, J.; Schreurs, C.; Verheyen, K.; Casteels, M.; Laekeman, G.; Augustijns, P. Evaluation of real-life dosing of oral medicines with respect to fluid and food intake in a Dutch-speaking population. J. Clin. Pharm. Ther. 2017, 42, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Vinarov, Z.; Tcholakova, S.; Damyanova, B.; Atanasov, Y.; Denkov, N.D.; Stoyanov, S.D.; Pelan, E.; Lips, A. Effects of Emulsifier Charge and Concentration on Pancreatic Lipolysis: 2. Interplay of Emulsifiers and Biles. Langmuir 2012, 28, 12140–12150. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; McClements, D.J. Inhibition of lipase-catalyzed hydrolysis of emulsified triglyceride oils by low-molecular weight surfactants under simulated gastrointestinal conditions. Eur. J. Pharm. Biopharm. 2011, 79, 423–431. [Google Scholar] [CrossRef]
- Maurer, J.M.; Schellekens, R.C.A.; Van Rieke, H.M.; Wanke, C.; Iordanov, V.; Stellaard, F.; Wutzke, K.D.; Dijkstra, G.; Van Der Zee, M.; Woerdenbag, H.; et al. Gastrointestinal pH and Transit Time Profiling in Healthy Volunteers Using the IntelliCap System Confirms Ileo-Colonic Release of ColoPulse Tablets. PLoS ONE 2015, 10, e0129076. [Google Scholar] [CrossRef] [PubMed]
- Alskär, L.C.; Keemink, J.; Johannesson, J.; Porter, C.; Bergström, C.A.S. Impact of Drug Physicochemical Properties on Lipolysis-Triggered Drug Supersaturation and Precipitation from Lipid-Based Formulations. Mol. Pharm. 2018, 15, 4733–4744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suys, E.J.; Brundel, D.H.; Chalmers, D.K.; Pouton, C.W.; Porter, C.J. Interaction with biliary and pancreatic fluids drives supersaturation and drug absorption from lipid-based formulations of low (saquinavir) and high (fenofibrate) permeability poorly soluble drugs. J. Control. Release 2021, 331, 45–61. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FA Profile of the Studied Lipid Carriers (%) | ||||
|---|---|---|---|---|
| FA | MCT | CNO | CB | SFO |
| C8:0 | 71.0 | 6.6 | n.d. | n.d. |
| C10:0 | 29.0 | 5.6 | n.d. | n.d. |
| C12:0 | n.d. | 48.0 | n.d. | n.d. |
| C14:0 | n.d. | 18.8 | n.d. | n.d. |
| C16:0 | n.d. | 9.5 | 26.6 | 7.1 |
| C18:0 | n.d. | 3.4 | 38.6 | 5.7 |
| C18:1,2 | n.d. | 8.0 | 34.8 | 87.2 |
| average molecular weight, g/mol | 495 | 682 | 866 | 876 |
| Emulsification Equipment | Lipid Type | Tween 20, wt% | d43, µm | ||
|---|---|---|---|---|---|
| AVG | SD | n | |||
| High-pressure homogenizer | SFO | 10 | 0.7 | 1 | |
| Sonicator | SFO | 1 | 2.2 | 0.1 | 2 |
| Rotor–stator homogenizer | SFO | 1 | 11.8 | 0.2 | 2 |
| Rotor–stator homogenizer | SFO | 10 | 11.8 | 0.8 | 2 |
| High-pressure homogenizer | MCT | 10 | 0.4 | 0.1 | 2 |
| Sonicator | MCT | 1 | 1.9 | 0.2 | 3 |
| Rotor–stator homogenizer | MCT | 1 | 13.9 | 1.0 | 2 |
| Rotor–stator homogenizer | MCT | 10 | 9.3 | 0.4 | 2 |
| Sonicator | CNO | 1 | 2.0 | 1 | |
| Sonicator | CB | 1 | 1.4 | 1 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katev, V.; Tsibranska-Gyoreva, S.; Vinarov, Z.; Tcholakova, S. Supersaturation and Solubilization upon In Vitro Digestion of Fenofibrate Type I Lipid Formulations: Effect of Droplet Size, Surfactant Concentration and Lipid Type. Pharmaceutics 2021, 13, 1287. https://doi.org/10.3390/pharmaceutics13081287
Katev V, Tsibranska-Gyoreva S, Vinarov Z, Tcholakova S. Supersaturation and Solubilization upon In Vitro Digestion of Fenofibrate Type I Lipid Formulations: Effect of Droplet Size, Surfactant Concentration and Lipid Type. Pharmaceutics. 2021; 13(8):1287. https://doi.org/10.3390/pharmaceutics13081287
Chicago/Turabian StyleKatev, Vladimir, Sonya Tsibranska-Gyoreva, Zahari Vinarov, and Slavka Tcholakova. 2021. "Supersaturation and Solubilization upon In Vitro Digestion of Fenofibrate Type I Lipid Formulations: Effect of Droplet Size, Surfactant Concentration and Lipid Type" Pharmaceutics 13, no. 8: 1287. https://doi.org/10.3390/pharmaceutics13081287