
Ultrasound-Mediated Blood-Brain Barrier Opening Improves Whole Brain Gene Delivery in Mice

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Focused Ultrasound Device
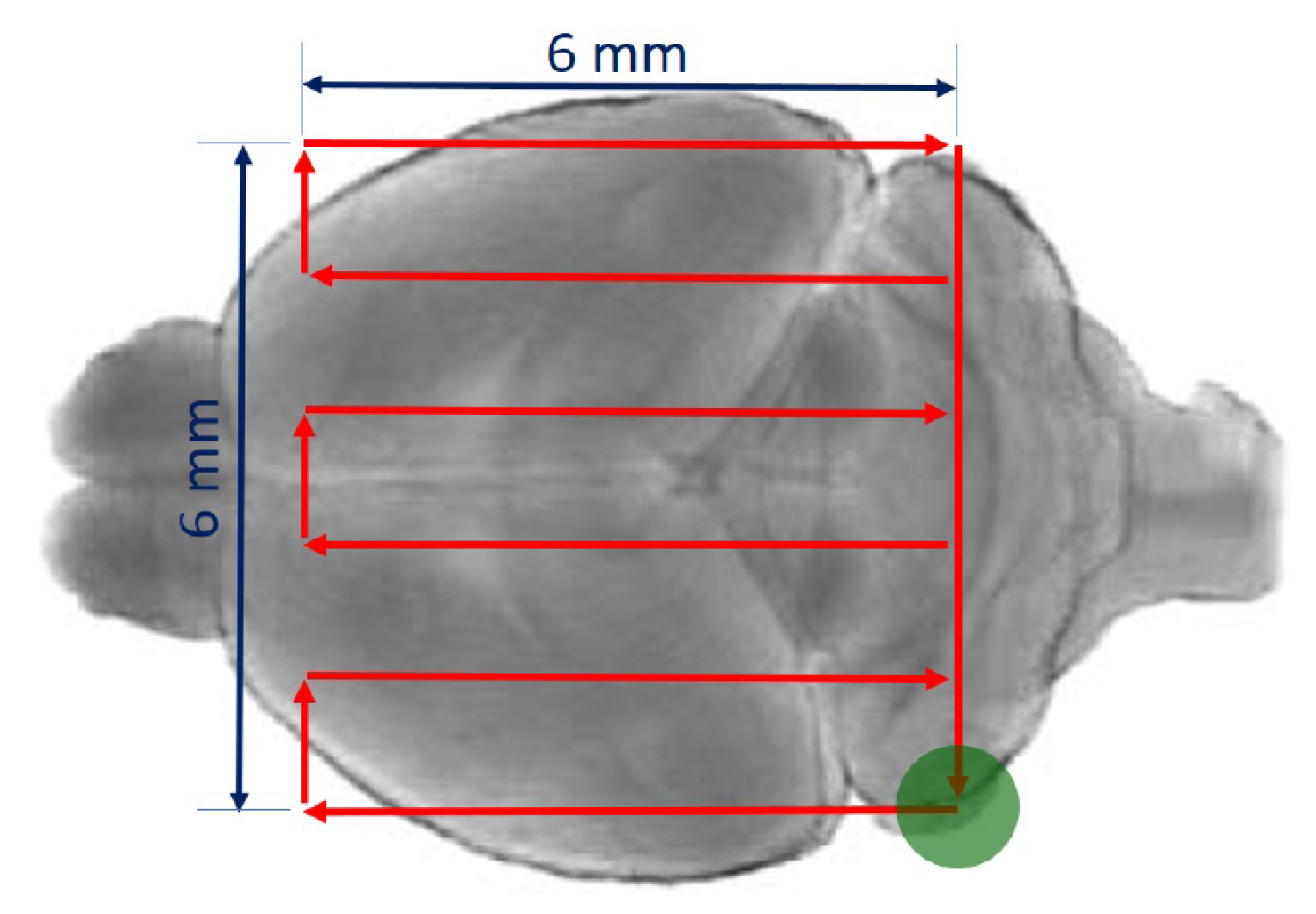
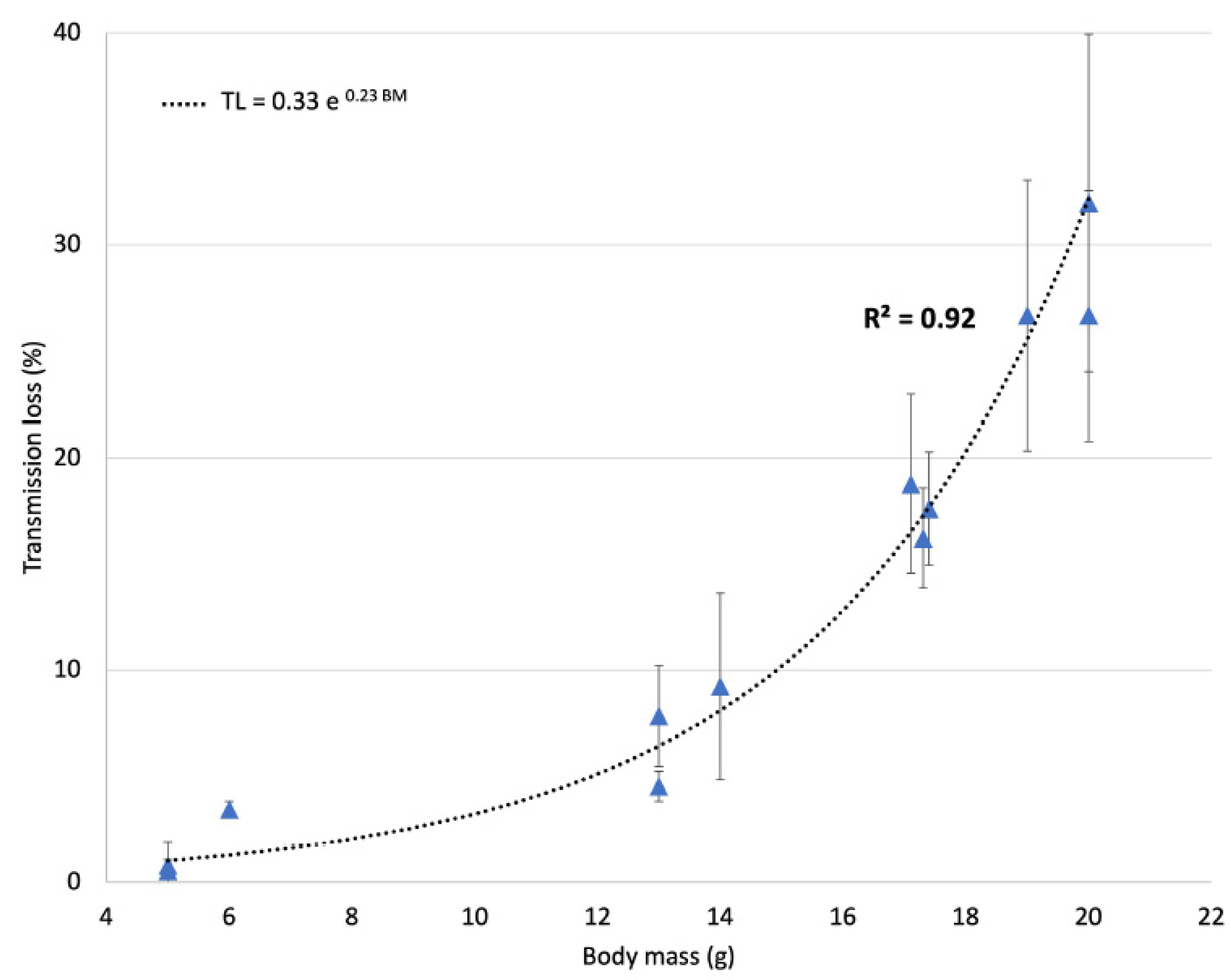
2.3. Ultrasound Transmission through the Skull
2.4. Viral Vector Preparation and In Vivo AAV Injections
2.5. FUS-Induced BBB Disruption
2.6. Tissue Collection
- (1)
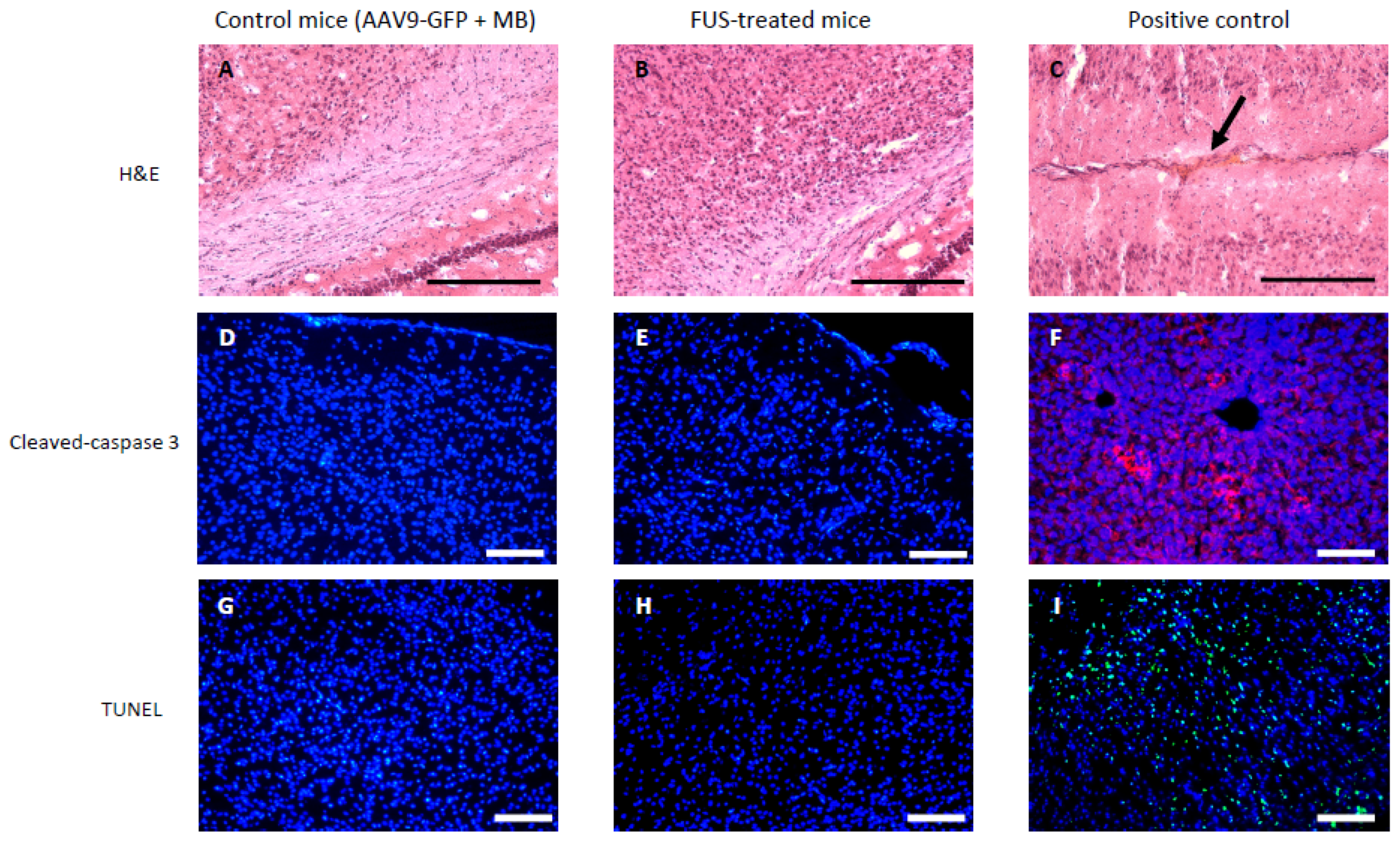
- For H&E staining, TUNEL staining assay, and cleaved caspase-3 immunohistofluorescence, mice were transcardially perfused with 0.01 M PBS followed by 4% paraformaldehyde (PFA) in phosphate buffer, then brains were cryopreserved in 20% sucrose for 2 days and frozen in dry ice. Tissues were embedded in Tissue-Tek CRYO-OCT compound (Fisher Scientific, Illkirch, France), coronally sectioned from bregma 1.34 mm to bregma −3.28 mm into 20 or 40 μm sections using a Leica VT1200s cryostat (Leica Biosystems, Nanterre, France), mounted on SuperFrost Plus slides (Fisher Scientific, Illkirch, France), and stored at −80 °C until assay.
- (2)
- For Iba1, Olig2 and GFAP immunohistofluorescence and GFP immunohistochemistry, mice were transcardially perfused with 0.01 M PBS followed by 4% paraformaldehyde (PFA) in phosphate buffer, then brains were kept at 4 °C in PBS azide (0.02% S2002 Sigma-Aldrich, St Louis, MO, USA), cut in 100 µm sections using a vibratome (Leica VT1200s, Leica, Nanterre, France), and collected serially. Floating sections were kept at 4 °C in PBS azide (0.02% S2002 Sigma-Aldrich, St Louis, MO, USA) until assay.
- (3)
- For GFP quantification (DNA, mRNA, and protein preparations), brains were rapidly dissected and frozen in dry ice.
2.7. H&E Staining
2.8. Cleaved Caspase-3 Immunohistofluorescence
2.9. TUNEL Staining Assay
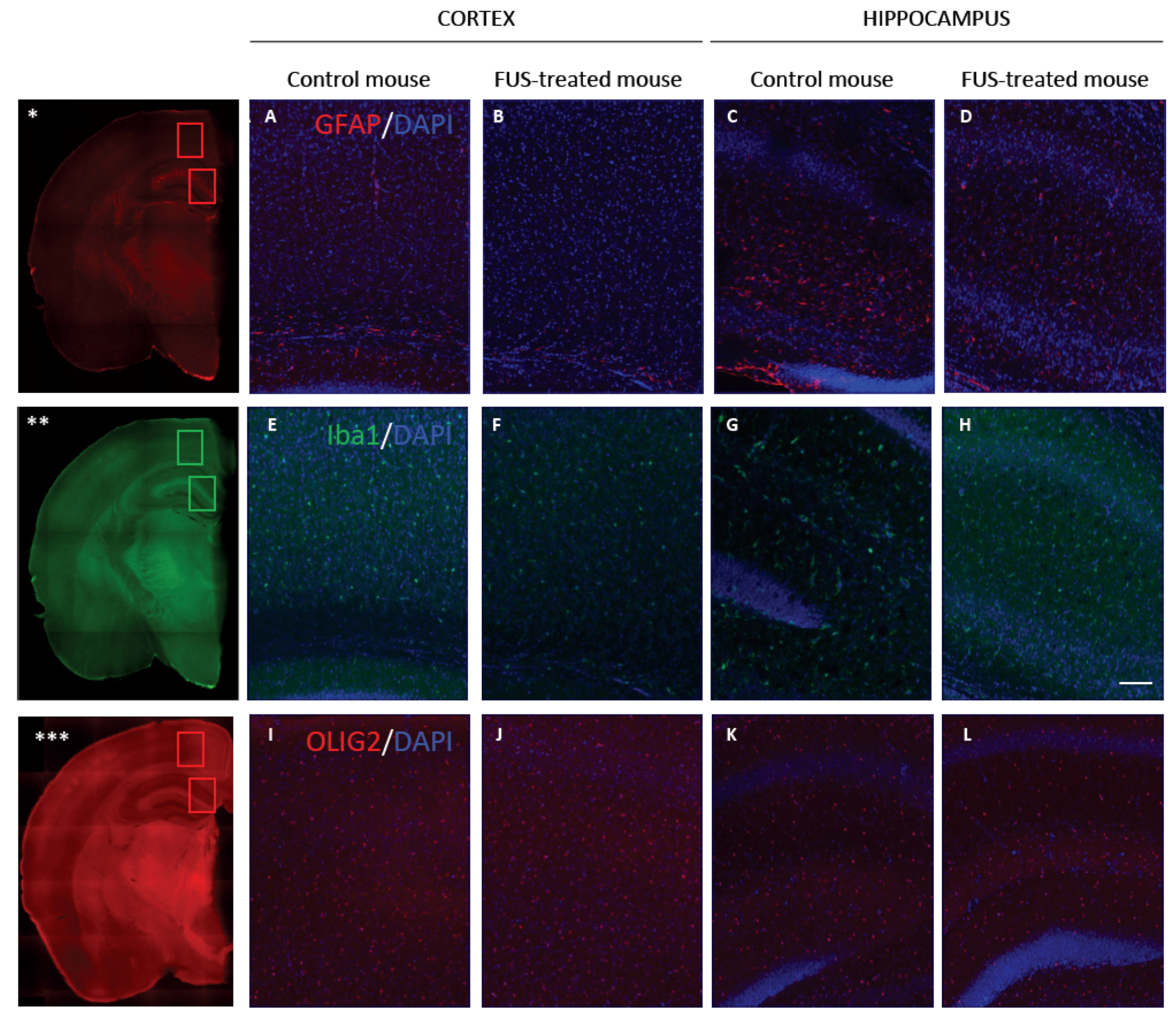
2.10. Iba1, GFAP and Olig2 Immunohistofluorescence
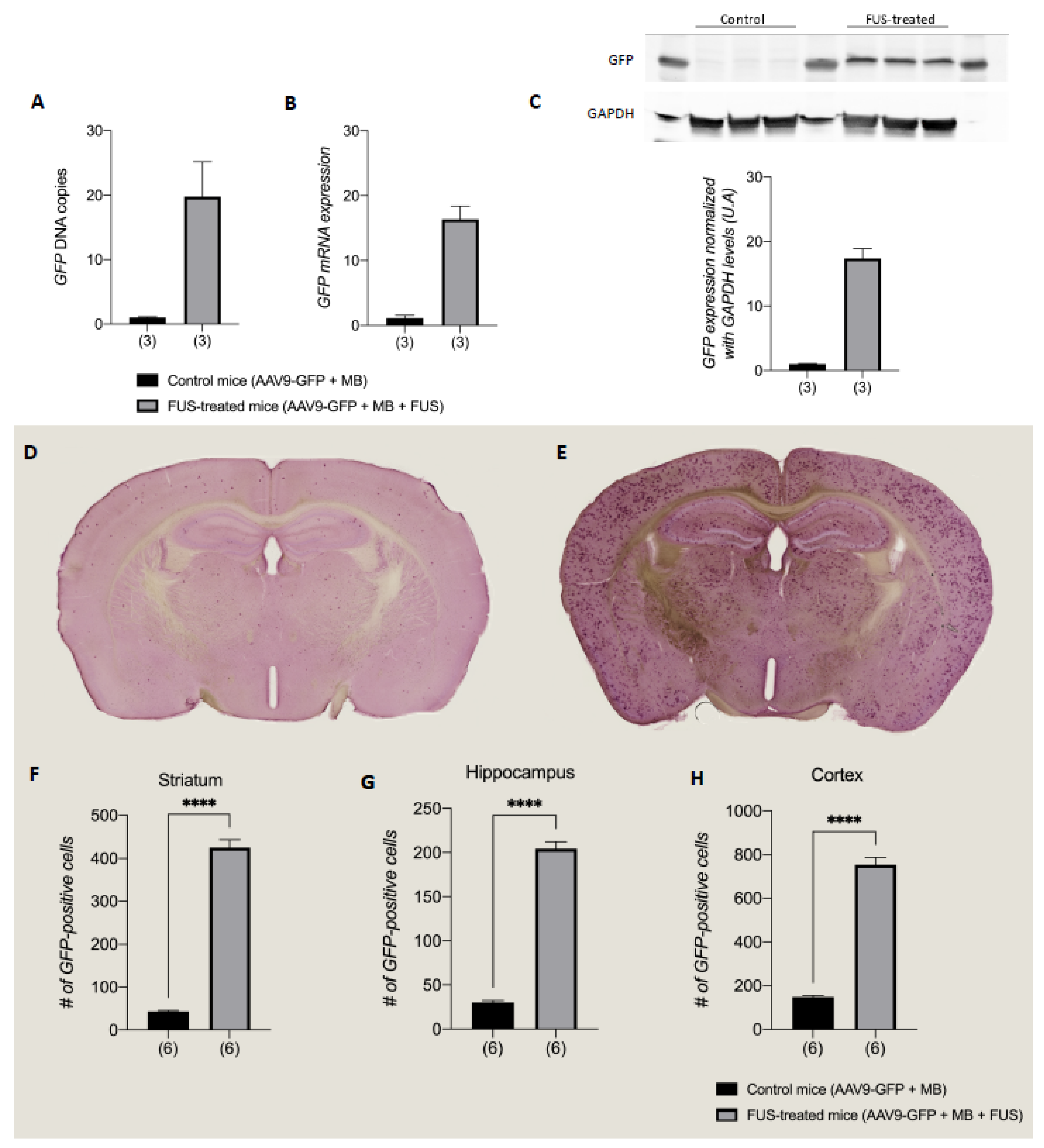
2.11. GFP Immunohistochemistry
2.12. DNA Extraction and Quantitative Real-Time qPCR
2.13. RNA Extraction, Reverse Transcription, and Quantitative Real-Time qPCR
2.14. Western Blotting
2.15. Statistical Analysis
3. Results
3.1. Ultrasound Transmission through the Skull
3.2. Short-Term Safety after FUS-Treatment
3.3. FUS-Treatment Homogeneously Increased AAV9 Vector Transduction in the Brain
3.4. Long-Term Safety after FUS Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Pardridge, W.M. Blood-brain barrier delivery. Drug Discov. Today 2007, 12, 54–61. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Ingusci, S.; Verlengia, G.; Soukupova, M.; Zucchini, S.; Simonato, M. Gene therapy tools for brain diseases. Front. Pharmacol. 2019, 10, 724. [Google Scholar] [CrossRef] [PubMed]
- Piguet, F.; de Saint Denis, T.; Audouard, E.; Beccaria, K.; André, A.; Wurtz, G.; Schatz, R.; Alves, S.; Sevin, C.; Zerah, M.; et al. The challenge of gene therapy for neurological diseases: Strategies and tools to achieve efficient delivery to the central nervous system. Hum. Gene Ther. 2021, 32, 349–374. [Google Scholar] [CrossRef]
- Zhu, D.; Schieferecke, A.J.; Lopez, P.A.; Schaffer, D.V. Adeno-Associated Virus Vector for Central Nervous System Gene Therapy. Trends Mol. Med. 2021, 27, 524–537. [Google Scholar] [CrossRef]
- Fu, H.; Dirosario, J.; Killedar, S.; Zaraspe, K.; McCarty, D.M. Correction of neurological disease of mucopolysaccharidosis IIIB in adult mice by rAAV9 trans-blood-brain barrier gene delivery. Mol. Ther. 2011, 19, 1025–1033. [Google Scholar] [CrossRef]
- Piguet, F.; Alves, S.; Cartier, N. Clinical gene therapy for neurodegenerative diseases: Past, present, and future. Hum. Gene Ther. 2017, 28, 988–1003. [Google Scholar] [CrossRef]
- Meijer, D.H.; Maguire, C.A.; LeRoy, S.G.; Sena-Esteves, M. Controlling brain tumor growth by intraventricular administration of an AAV vector encoding IFN-beta. Cancer Gene Ther. 2009, 16, 664–671. [Google Scholar] [CrossRef]
- Samaranch, L.; Salegio, E.A.; San Sebastian, W.; Kells, A.P.; Bringas, J.R.; Forsayeth, J.; Bankiewicz, K.S. Strong cortical and spinal cord transduction after AAV7 and AAV9 delivery into the cerebrospinal fluid of nonhuman primates. Hum. Gene Ther. 2013, 24, 526–532. [Google Scholar] [CrossRef] [Green Version]
- Hocquemiller, M.; Giersch, L.; Audrain, M.; Parker, S.; Cartier, N. Adeno-Associated Virus-Based Gene Therapy for CNS Diseases. Hum. Gene Ther. 2016, 27, 478–496. [Google Scholar] [CrossRef] [Green Version]
- Hynynen, K.; McDannold, N.; Vykhodtseva, N.; Jolesz, F.A. Noninvasive MR imaging-guided focal opening of the blood-brain barrier in rabbits. Radiology 2001, 220, 640–646. [Google Scholar] [CrossRef]
- Thévenot, E.; Jordão, J.F.; O’Reilly, M.A.; Markham, K.; Weng, Y.-Q.; Foust, K.D.; Kaspar, B.K.; Hynynen, K.; Aubert, I. Targeted delivery of self-complementary adeno-associated virus serotype 9 to the brain, using magnetic resonance imaging-guided focused ultrasound. Hum. Gene Ther. 2012, 23, 1144–1155. [Google Scholar] [CrossRef] [PubMed]
- Dauba, A.; Delalande, A.; Kamimura, H.A.S.; Conti, A.; Larrat, B.; Tsapis, N.; Novell, A. Recent Advances on Ultrasound Contrast Agents for Blood-Brain Barrier Opening with Focused Ultrasound. Pharmaceutics 2020, 12, 1125. [Google Scholar] [CrossRef]
- Dasgupta, A.; Liu, M.; Ojha, T.; Storm, G.; Kiessling, F.; Lammers, T. Ultrasound-mediated drug delivery to the brain: Principles, progress and prospects. Drug Discov. Today. Technol. 2016, 20, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.-H.; Wei, K.-C.; Huang, C.-Y.; Wen, C.-J.; Yen, T.-C.; Liu, C.-L.; Lin, Y.-T.; Chen, J.-C.; Shen, C.-R.; Liu, H.-L. Noninvasive and targeted gene delivery into the brain using microbubble-facilitated focused ultrasound. PLoS ONE 2013, 8, e57682. [Google Scholar] [CrossRef] [Green Version]
- Stavarache, M.A.; Petersen, N.; Jurgens, E.M.; Milstein, E.R.; Rosenfeld, Z.B.; Ballon, D.J.; Kaplitt, M.G. Safe and stable noninvasive focal gene delivery to the mammalian brain following focused ultrasound. J. Neurosurg. 2018, 130, 989–998. [Google Scholar] [CrossRef] [Green Version]
- Alonso, A.; Reinz, E.; Leuchs, B.; Kleinschmidt, J.; Fatar, M.; Geers, B.; Lentacker, I.; Hennerici, M.G.; de Smedt, S.C.; Meairs, S. Focal Delivery of AAV2/1-transgenes Into the Rat Brain by Localized Ultrasound-induced BBB Opening. Mol. Ther. Nucleic Acids 2013, 2, e73. [Google Scholar] [CrossRef]
- Keon, M.; Musrie, B.; Dinger, M.; Brennan, S.E.; Santos, J.; Saksena, N.K. Destination amyotrophic lateral sclerosis. Front. Neurol. 2021, 12, 596006. [Google Scholar] [CrossRef]
- Molcho, L.; Ben-Zur, T.; Barhum, Y.; Angel, A.; Glat, M.; Offen, D. Combined gene therapy to reduce the neuronal damage in the mouse model of focal ischemic injury. J. Mol. Neurosci. 2018, 66, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Madsen, E.L.; Dong, F.; Frank, G.R.; Garra, B.S.; Wear, K.A.; Wilson, T.; Zagzebski, J.A.; Miller, H.L.; Shung, K.K.; Wang, S.H.; et al. Interlaboratory comparison of ultrasonic backscatter, attenuation, and speed measurements. J. Ultrasound Med. 1999, 18, 615–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayuso, E.; Mingozzi, F.; Bosch, F. Production, purification and characterization of adeno-associated vectors. Curr. Gene Ther. 2010, 10, 423–436. [Google Scholar] [CrossRef]
- Matagne, V.; Ehinger, Y.; Saidi, L.; Borges-Correia, A.; Barkats, M.; Bartoli, M.; Villard, L.; Roux, J.-C. A codon-optimized Mecp2 transgene corrects breathing deficits and improves survival in a mouse model of Rett syndrome. Neurobiol. Dis. 2017, 99, 1–11. [Google Scholar] [CrossRef]
- Talu, E.; Powell, R.L.; Longo, M.L.; Dayton, P.A. Needle size and injection rate impact microbubble contrast agent population. Ultrasound Med. Biol. 2008, 34, 1182–1185. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Brenner, M. Role of GFAP in CNS injuries. Neurosci. Lett. 2014, 565, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Miles, D.K.; Hoang, T.; Shi, J.; Hurlock, E.; Kernie, S.G.; Lu, Q.R. The basic helix-loop-helix transcription factor olig2 is critical for reactive astrocyte proliferation after cortical injury. J. Neurosci. 2008, 28, 10983–10989. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Chen, Y.; Cai, W.-H.; Hurlock, E.C.; Wu, H.; Kernie, S.G.; Parada, L.F.; Lu, Q.R. A crucial role for Olig2 in white matter astrocyte development. Development 2007, 134, 1887–1899. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Calle, R.; Vicente-Rodríguez, M.; Gramage, E.; Pita, J.; Pérez-García, C.; Ferrer-Alcón, M.; Uribarri, M.; Ramos, M.P.; Herradón, G. Pleiotrophin regulates microglia-mediated neuroinflammation. J. Neuroinflam. 2017, 14, 46. [Google Scholar] [CrossRef] [Green Version]
- Matagne, V.; Borloz, E.; Ehinger, Y.; Saidi, L.; Villard, L.; Roux, J.-C. Severe offtarget effects following intravenous delivery of AAV9-MECP2 in a female mouse model of Rett syndrome. Neurobiol. Dis. 2020, 149, 105235. [Google Scholar] [CrossRef]
- Szablowski, J.O.; Harb, M. Focused Ultrasound Induced Blood-Brain Barrier Opening for Targeting Brain Structures and Evaluating Chemogenetic Neuromodulation. J. Vis. Exp. 2020, 166, e61352. [Google Scholar]
- Magnin, R.; Rabusseau, F.; Salabartan, F.; Mériaux, S.; Aubry, J.-F.; Le Bihan, D.; Dumont, E.; Larrat, B. Magnetic resonance-guided motorized transcranial ultrasound system for blood-brain barrier permeabilization along arbitrary trajectories in rodents. J. Ther. Ultrasound 2015, 3, 22. [Google Scholar] [CrossRef] [Green Version]
- Tran, V.L.; Novell, A.; Tournier, N.; Gerstenmayer, M.; Schweitzer-Chaput, A.; Mateos, C.; Jego, B.; Bouleau, A.; Nozach, H.; Winkeler, A.; et al. Impact of blood-brain barrier permeabilization induced by ultrasound associated to microbubbles on the brain delivery and kinetics of cetuximab: An immunoPET study using 89Zr-cetuximab. J. Control Release 2020, 328, 304–312. [Google Scholar] [CrossRef]
- Guy, J.; Hendrich, B.; Holmes, M.; Martin, J.E.; Bird, A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 2001, 27, 322–326. [Google Scholar] [CrossRef]
- Gerstenmayer, M.; Fellah, B.; Magnin, R.; Selingue, E.; Larrat, B. Acoustic Transmission Factor through the Rat Skull as a Function of Body Mass, Frequency and Position. Ultrasound Med. Biol. 2018, 44, 2336–2344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinoshita, M.; McDannold, N.; Jolesz, F.A.; Hynynen, K. Noninvasive localized delivery of Herceptin to the mouse brain by MRI-guided focused ultrasound-induced blood-brain barrier disruption. Proc. Natl. Acad. Sci. USA 2006, 103, 11719–11723. [Google Scholar] [CrossRef] [Green Version]
- Hynynen, K.; McDannold, N.; Sheikov, N.A.; Jolesz, F.A.; Vykhodtseva, N. Local and reversible blood-brain barrier disruption by noninvasive focused ultrasound at frequencies suitable for trans-skull sonications. Neuroimage 2005, 24, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Dheen, S.T.; Kaur, C.; Ling, E.-A. Microglial activation and its implications in the brain diseases. Curr. Med. Chem. 2007, 14, 1189–1197. [Google Scholar] [CrossRef]
- Kempuraj, D.; Thangavel, R.; Selvakumar, G.P.; Zaheer, S.; Ahmed, M.E.; Raikwar, S.P.; Zahoor, H.; Saeed, D.; Natteru, P.A.; Iyer, S.; et al. Brain and peripheral atypical inflammatory mediators potentiate neuroinflammation and neurodegeneration. Front. Cell Neurosci. 2017, 11, 216. [Google Scholar] [CrossRef] [PubMed]
- Reuter-Rice, K.; Eads, J.K.; Berndt, S.B.; Bennett, E. Chapter6 state of the science of pediatric traumatic brain injury: Biomarkers and gene association studies. Annu. Rev. Nurs. Res. 2015, 33, 185–217. [Google Scholar] [CrossRef] [Green Version]
- McMahon, D.; Bendayan, R.; Hynynen, K. Acute effects of focused ultrasound-induced increases in blood-brain barrier permeability on rat microvascular transcriptome. Sci. Rep. 2017, 7, 45657. [Google Scholar] [CrossRef]
- Gorick, C.M.; Mathew, A.S.; Garrison, W.J.; Thim, E.A.; Fisher, D.G.; Copeland, C.A.; Song, J.; Klibanov, A.L.; Miller, G.W.; Price, R.J. Sonoselective transfection of cerebral vasculature without blood-brain barrier disruption. Proc. Natl. Acad. Sci. USA 2020, 117, 5644–5654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartlett, J.S.; Wilcher, R.; Samulski, R.J. Infectious entry pathway of adeno-associated virus and adeno-associated virus vectors. J. Virol. 2000, 74, 2777–2785. [Google Scholar] [CrossRef] [Green Version]
- Sattin, D.; Leonardi, M.; Picozzi, M. The autonomic nervous system and the brainstem: A fundamental role or the background actors for consciousness generation? Hypothesis, evidence, and future directions for rehabilitation and theoretical approaches. Brain Behav. 2020, 10, e01474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammond, S.L.; Leek, A.N.; Richman, E.H.; Tjalkens, R.B. Cellular selectivity of AAV serotypes for gene delivery in neurons and astrocytes by neonatal intracerebroventricular injection. PLoS ONE 2017, 12, e0188830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehtinen, M.K.; Bjornsson, C.S.; Dymecki, S.M.; Gilbertson, R.J.; Holtzman, D.M.; Monuki, E.S. The choroid plexus and cerebrospinal fluid: Emerging roles in development, disease, and therapy. J. Neurosci. 2013, 33, 17553–17559. [Google Scholar] [CrossRef] [PubMed]
- Mrowczynski, O.D.; Langan, S.T.; Rizk, E.B. Intra-cerebrospinal fluid antibiotics to treat central nervous system infections: A review and update. Clin. Neurol. Neurosurg. 2018, 170, 140–158. [Google Scholar] [CrossRef] [PubMed]
- Passini, M.A.; Wolfe, J.H. Widespread gene delivery and structure-specific patterns of expression in the brain after intraventricular injections of neonatal mice with an adeno-associated virus vector. J. Virol. 2001, 75, 12382–12392. [Google Scholar] [CrossRef] [Green Version]
- Davidson, B.L.; Stein, C.S.; Heth, J.A.; Martins, I.; Kotin, R.M.; Derksen, T.A.; Zabner, J.; Ghodsi, A.; Chiorini, J.A. Recombinant adeno-associated virus type 2, 4, and 5 vectors: Transduction of variant cell types and regions in the mammalian central nervous system. Proc. Natl. Acad. Sci. USA 2000, 97, 3428–3432. [Google Scholar] [CrossRef]
- Van Esch, H.; Bauters, M.; Ignatius, J.; Jansen, M.; Raynaud, M.; Hollanders, K.; Lugtenberg, D.; Bienvenu, T.; Jensen, L.R.; Gecz, J.; et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am. J. Hum. Genet. 2005, 77, 442–453. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | n | AAV9-GFP + MB | FUS | Experiments | ||
|---|---|---|---|---|---|---|
| Safety | Short term (24 h) | FUS-treated mice | 3 | ✔ | ✔ | H&E staining TUNEL staining assay Cleaved caspase-3 Immunohistofluorescence |
| Control mice | 3 | ✔ | ||||
| Long term (1 month) | FUS-treated mice | 3 | ✔ | ✔ | Iba1 immunohistofluorescence GFAP immunohistofluorescence Olig2 immunohistofluorescence | |
| Control mice | 3 | ✔ | ||||
| Efficacy | Long term (1 month) | FUS-treated mice | 3 | ✔ | ✔ | GFP DNA quantification (qPCR) GFP mRNA quantification (RT-qPCR) GFP protein quantification (WB) |
| Control mice | 3 | ✔ | ||||
| FUS-treated mice | 6 | ✔ | ✔ | GFP immunohistochemistry Positive cells counted | ||
| Control mice | 6 | ✔ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Felix, M.-S.; Borloz, E.; Metwally, K.; Dauba, A.; Larrat, B.; Matagne, V.; Ehinger, Y.; Villard, L.; Novell, A.; Mensah, S.; et al. Ultrasound-Mediated Blood-Brain Barrier Opening Improves Whole Brain Gene Delivery in Mice. Pharmaceutics 2021, 13, 1245. https://doi.org/10.3390/pharmaceutics13081245
Felix M-S, Borloz E, Metwally K, Dauba A, Larrat B, Matagne V, Ehinger Y, Villard L, Novell A, Mensah S, et al. Ultrasound-Mediated Blood-Brain Barrier Opening Improves Whole Brain Gene Delivery in Mice. Pharmaceutics. 2021; 13(8):1245. https://doi.org/10.3390/pharmaceutics13081245
Chicago/Turabian StyleFelix, Marie-Solenne, Emilie Borloz, Khaled Metwally, Ambre Dauba, Benoit Larrat, Valerie Matagne, Yann Ehinger, Laurent Villard, Anthony Novell, Serge Mensah, and et al. 2021. "Ultrasound-Mediated Blood-Brain Barrier Opening Improves Whole Brain Gene Delivery in Mice" Pharmaceutics 13, no. 8: 1245. https://doi.org/10.3390/pharmaceutics13081245