
Clinical Advances of siRNA-Based Nanotherapeutics for Cancer Treatment


Abstract
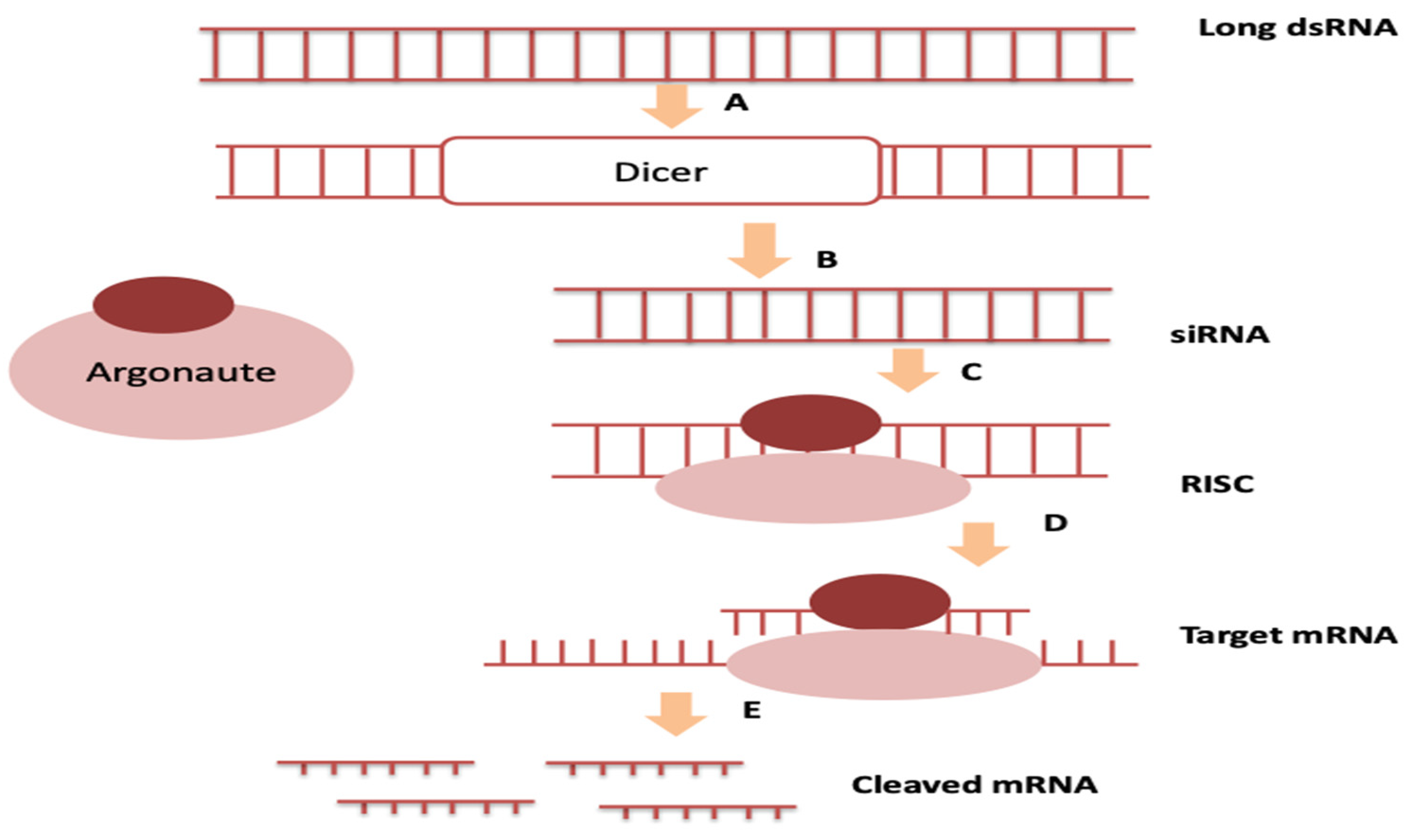
:1. Introduction
2. Investigational Phase and Study
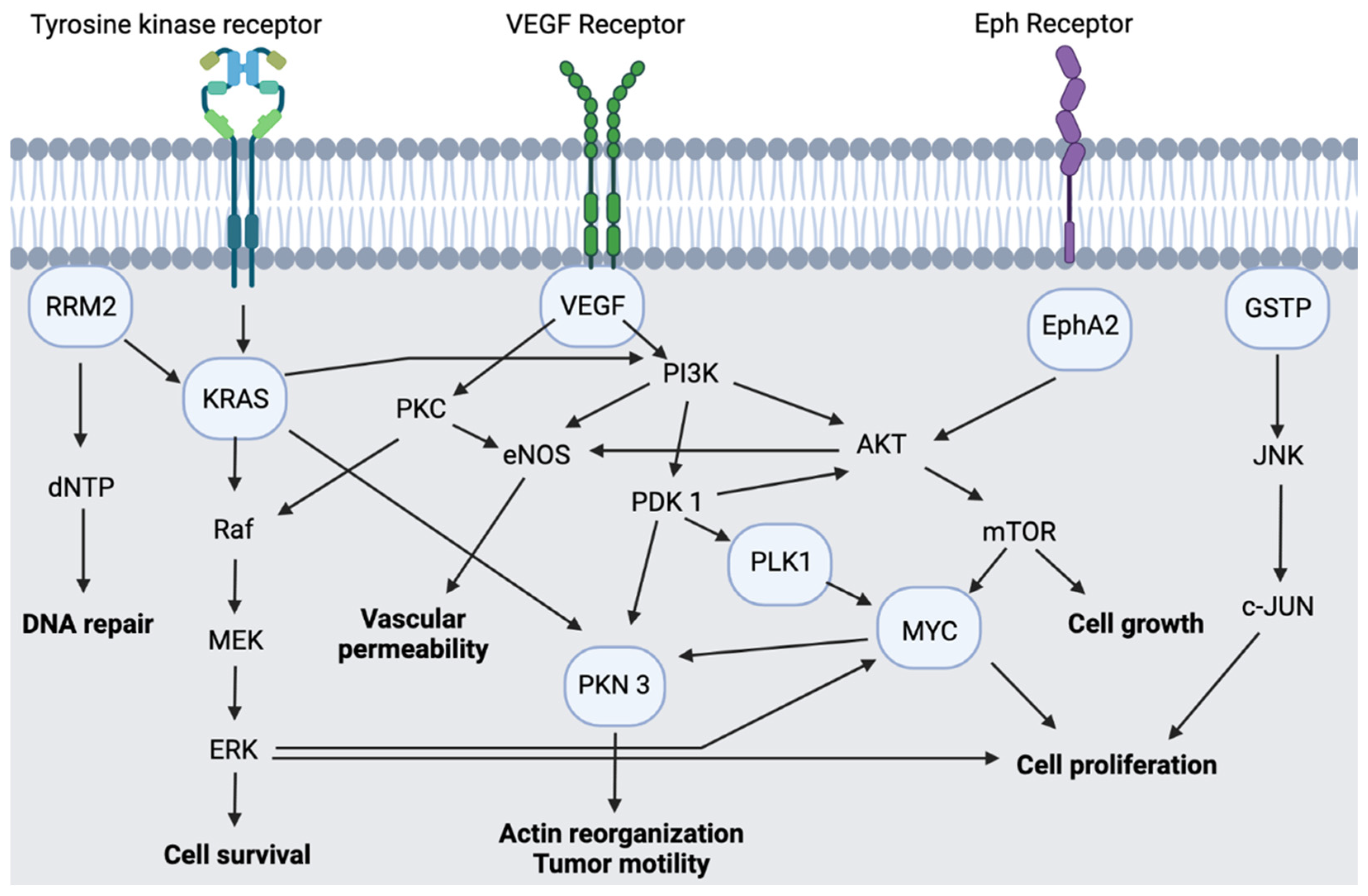
2.1. CALAA-01
2.2. ALN-VSP02
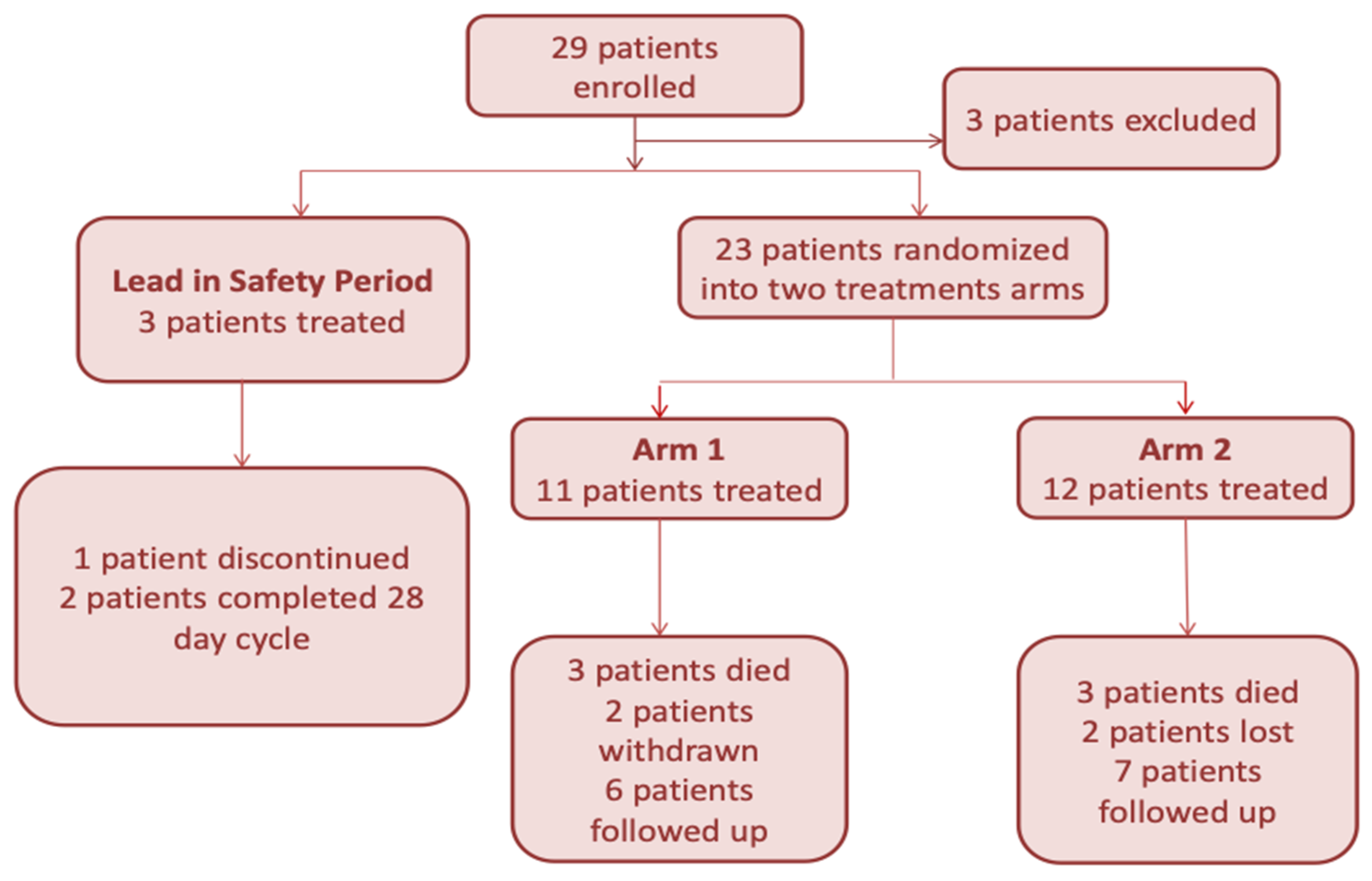
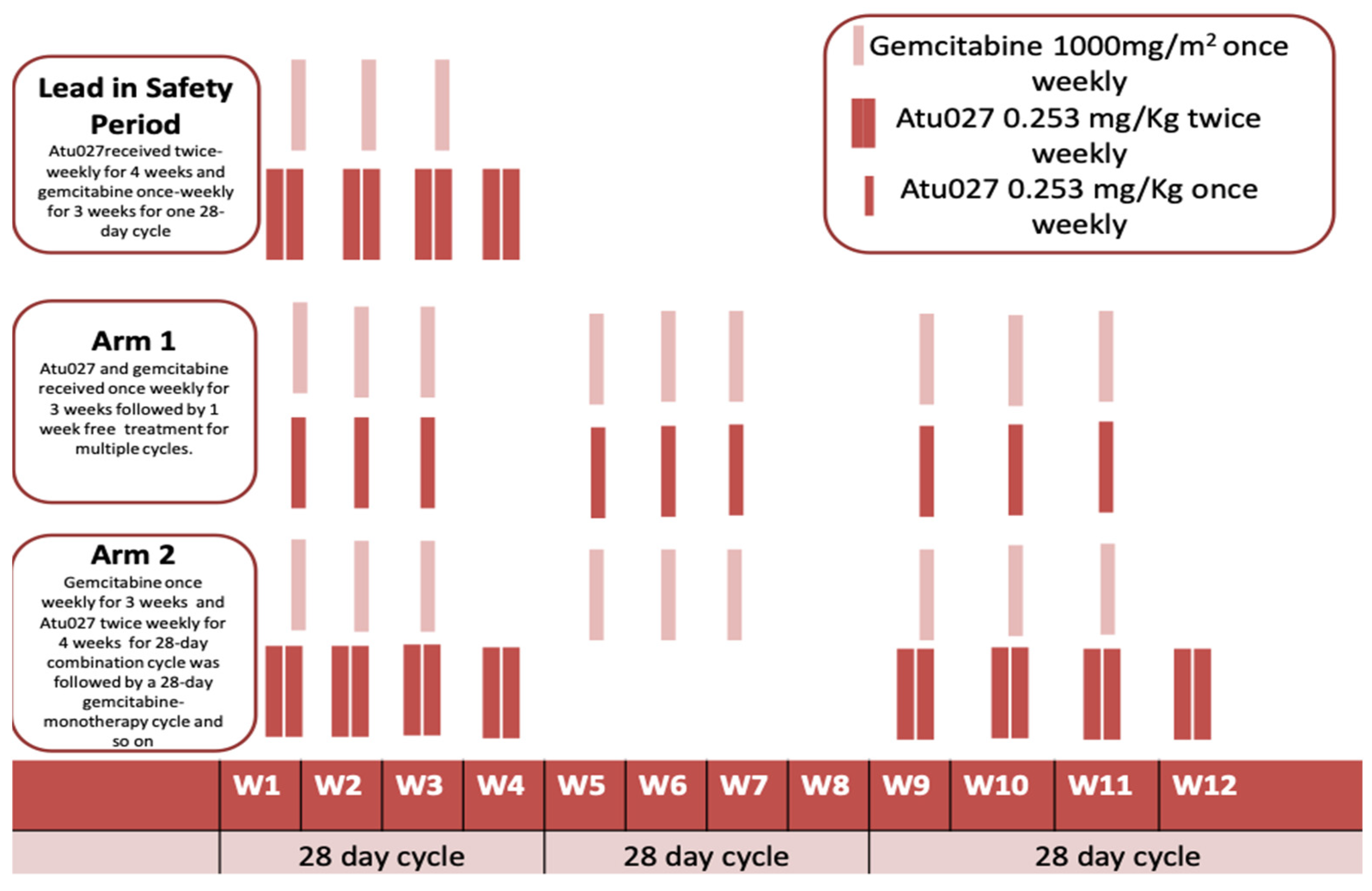
2.3. Atu027
2.4. TKM-PLK1
2.5. siG12D LODER
2.6. KRAS G12D
2.7. DCR-MYC
2.8. EphA2-siRNA-DOPC
2.9. NBF-006
3. siRNA-Mediated Nanotherapeutics in Clinical Trials
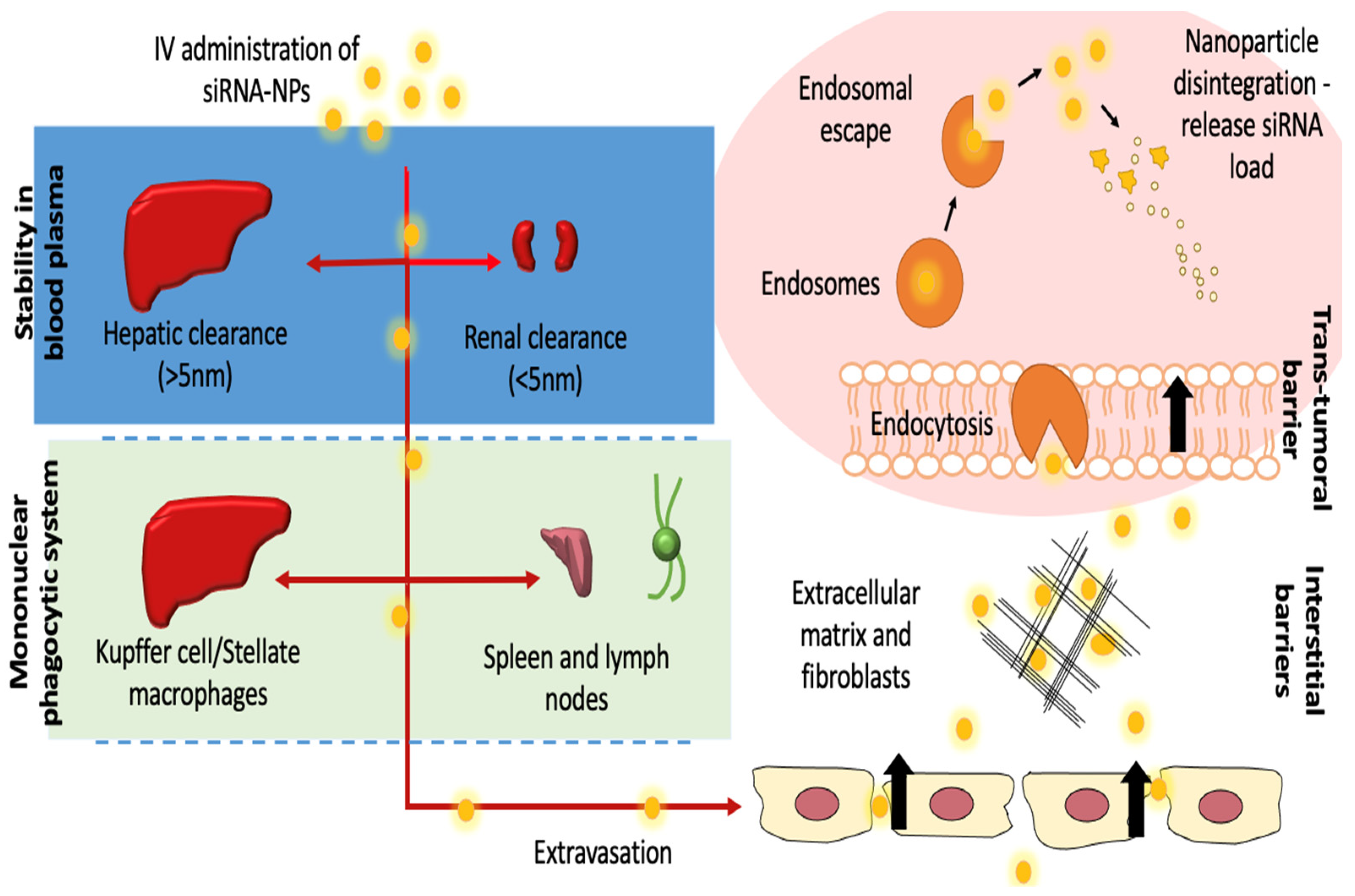
3.1. Administration and Distribution
3.2. Dosage Schedule
3.3. Pharmacokinetic Parameters
3.4. Safety Profile
3.5. Antitumor Effect and Pharmacodynamics
3.6. Challenges of Ongoing and Future Human Trials
3.7. Design Concerns for Future Development of RNAi-Mediated Anticancer Nanotherapeutics
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Conde, J.; Ambrosone, A.; Hernandez, Y.; Tian, F.; McCully, M.; Berry, C.C.; Baptista, P.; Tortiglione, C.; de la Fuente, J.M. 15 years on siRNA delivery: Beyond the State-of-the-Art on inorganic nanoparticles for RNAi therapeutics. Nano Today 2015, 10, 421–450. [Google Scholar] [CrossRef] [Green Version]
- Hattab, D.; Bakhtiar, A. Bioengineered siRNA-Based Nanoplatforms Targeting Molecular Signaling Pathways for the Treatment of Triple Negative Breast Cancer: Preclinical and Clinical Advancements. Pharmaceutics 2020, 12, 929. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Wilson, R.C.; Doudna, J.A. Molecular Mechanisms of RNA Interference. Annu. Rev. Biophys. 2013, 42, 217–239. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Kim, A.; Miyata, K.; Kataoka, K. Recent progress in development of siRNA delivery vehicles for cancer therapy. Adv. Drug Deliv. Rev. 2016, 104, 61–77. [Google Scholar] [CrossRef] [Green Version]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nat. Cell Biol. 2001, 411, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.A.; Nam, Y.S. Functional Nanostructures for Effective Delivery of Small Interfering RNA Therapeutics. Theranostics 2014, 4, 1211. [Google Scholar] [CrossRef]
- Chen, X.; Mangala, L.S.; Rodriguez-Aguayo, C.; Kong, X.; Lopez-Berestein, G.; Sood, A.K. RNA interference-based therapy and its delivery systems. Cancer Metastasis Rev. 2018, 37, 107–124. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Shi, Q.; Zhang, H.; Yang, K.; Ke, Y.; Wang, Y.; Qiao, L. Advances in the techniques and methodologies of cancer gene therapy. Discov. Med. 2019, 27, 45–55. [Google Scholar] [PubMed]
- Chen, M.; Du, Q.; Zhang, H.-Y.; Wahlestedt, C.; Liang, Z. Vector-based siRNA delivery strategies for high-throughput screening of novel target genes. J. RNAi Gene Silencing Int. J. RNA Gene Target. Res. 2005, 1, 5–11. [Google Scholar]
- Zhou, Z.; Liu, X.; Zhu, D.; Wang, Y.; Zhang, Z.; Zhou, X.; Qiu, N.; Chen, X.; Shen, Y. Nonviral cancer gene therapy: Delivery cascade and vector nanoproperty integration. Adv. Drug Deliv. Rev. 2017, 115, 115–154. [Google Scholar] [CrossRef]
- Kanasty, R.L.; Dorkin, J.R.; Vegas, A.; Anderson, D.G. Delivery materials for siRNA therapeutics. Nat. Mater. 2013, 12, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Dang, Y.; Guan, J. Nanoparticle-based drug delivery systems for cancer therapy. Smart Mater. Med. 2020, 1, 10–19. [Google Scholar] [CrossRef]
- Thakur, V.; Kutty, R.V. Recent advances in nanotheranostics for triple negative breast cancer treatment. J. Exp. Clin. Cancer Res. 2019, 38, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Arranja, A.G.; Pathak, V.; Lammers, T.; Shi, Y. Tumor-targeted nanomedicines for cancer theranostics. Pharmacol. Res. 2017, 115, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Teles, R.H.G.; Moralles, H.; Cominetti, M.R. Global trends in nanomedicine research on triple negative breast cancer: A bibliometric analysis. Int. J. Nanomed. 2018, 13, 2321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roscigno, G.; Scognamiglio, I.; Ingenito, F.; Chianese, R.V.; Palma, F.; Chan, A.; Condorelli, G. Modulating the Crosstalk between the Tumor and the Microenvironment Using SiRNA: A Flexible Strategy for Breast Cancer Treatment. Cancers 2020, 12, 3744. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Lopez-Berestein, G.; Calin, G.; Sood, A.K. RNAi Therapies: Drugging the Undruggable. Sci. Transl. Med. 2014, 6, 240ps7. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Huo, S.; Hardie, J.; Liang, X.-J.; Rotello, V.M. Progress and perspective of inorganic nanoparticle-based siRNA delivery systems. Expert Opin. Drug Deliv. 2016, 13, 547–559. [Google Scholar] [CrossRef] [Green Version]
- Parvani, J.G.; Jackson, M.W. Silencing the roadblocks to effective triple-negative breast cancer treatments by siRNA nanoparticles. Endocr. Relat. Cancer 2017, 24, R81–R97. [Google Scholar] [CrossRef] [Green Version]
- Binnemars-Postma, K.; Bansal, R.; Storm, G.; Prakash, J. Targeting the Stat6 pathway in tumor-associated macrophages reduces tumor growth and metastatic niche formation in breast cancer. FASEB J. 2018, 32, 969–978. [Google Scholar] [CrossRef] [Green Version]
- Song, W.J.; Du, J.Z.; Sun, T.M.; Zhang, P.Z.; Wang, J. Gold nanoparticles capped with polyethyleneimine for enhanced siRNA delivery. Small 2010, 6, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Tang, C.; Yin, C. Combination antitumor immunotherapy with VEGF and PIGF siRNA via systemic delivery of multi-functionalized nanoparticles to tumor-associated macrophages and breast cancer cells. Biomaterials 2018, 185, 117–132. [Google Scholar] [CrossRef]
- Egorova, A.A.; Shtykalova, S.V.; Maretina, M.A.; Sokolov, D.I.; Selkov, S.A.; Baranov, V.S.; Kiselev, A.V. Synergistic Anti-Angiogenic Effects Using Peptide-Based Combinatorial Delivery of siRNAs Targeting VEGFA, VEGFR1, and Endoglin Genes. Pharmaceutics 2019, 11, 261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, H.J.; Lee, Y.R.; Kang, D.; Lee, H.C.; Seo, H.R.; Ryu, J.K.; Kim, Y.N.; Ko, Y.G.; Park, H.J.; Lee, J.S. Endothelial cells under therapy-induced senescence secrete CXCL11, which increases aggressiveness of breast cancer cells. Cancer Lett. 2020, 490, 100–110. [Google Scholar] [CrossRef]
- Zuckerman, J.E.; Gritli, I.; Tolcher, A.; Heidel, J.D.; Lim, D.; Morgan, R.; Chmielowski, B.; Ribas, A.; Davis, M.E.; Yen, Y. Correlating animal and human phase Ia/Ib clinical data with CALAA-01, a targeted, polymer-based nanoparticle containing siRNA. Proc. Natl. Acad. Sci. USA 2014, 111, 11449–11454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.A.; Amin, A.R.; Wang, X.; Zuckerman, J.E.; Choi, C.H.J.; Zhou, B.; Wang, D.; Nannapaneni, S.; Koenig, L.; Chen, Z.; et al. Systemic delivery of siRNA nanoparticles targeting RRM2 suppresses head and neck tumor growth. J. Control Release 2012, 159, 384–392. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.E.; Zuckerman, J.E.; Choi, C.H.J.; Seligson, D.; Tolcher, A.; Alabi, C.A.; Yen, Y.; Heidel, J.D.; Ribas, A. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nat. Cell Biol. 2010, 464, 1067–1070. [Google Scholar] [CrossRef]
- Barba, A.A.; Bochicchio, S.; Dalmoro, A.; Lamberti, G. Lipid Delivery Systems for Nucleic-Acid-Based-Drugs: From Production to Clinical Applications. Pharmaceutics 2019, 11, 360. [Google Scholar] [CrossRef] [Green Version]
- Gollob, J.; Infante, J.R.; Shapiro, G.; Lorusso, P.; Dezube, B.J.; Heymach, J.; Cehelsky, J.; Falzone, R.; Vaishnaw, A.; Burris, H.A. Interim safety and pharmacodynamic results for ALN-VSP02, a novel RNAi therapeutic for solid tumors with liver involvement. J. Clin. Oncol. 2010, 28, a3042. [Google Scholar] [CrossRef]
- Cervantes, A.; Alsina, M.; Tabernero, J.; Infante, J.R.; Lorusso, P.; Shapiro, G.; Paz-Ares, L.G.; Falzone, R.; Hill, J.; Cehelsky, J.; et al. Phase I dose-escalation study of ALN-VSP02, a novel RNAi therapeutic for solid tumors with liver involvement. J. Clin. Oncol. 2011, 29 (Suppl. 15), 3025. [Google Scholar] [CrossRef]
- Tabernero, J.; Shapiro, G.I.; Lorusso, P.M.; Cervantes, A.; Schwartz, G.K.; Weiss, G.J.; Paz-Ares, L.; Cho, D.C.; Infante, J.R.; Alsina, M.; et al. First-in-Humans Trial of an RNA Interference Therapeutic Targeting VEGF and KSP in Cancer Patients with Liver Involvement. Cancer Discov. 2013, 3, 406–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aleku, M.; Schulz, P.; Keil, O.; Santel, A.; Schaeper, U.; Dieckhoff, B.; Janke, O.; Endruschat, J.; Durieux, B.; Röder, N.; et al. Atu027, a liposomal small interfering RNA formulation targeting protein kinase N3, inhibits cancer progression. Cancer Res. 2008, 68, 9788–9798. [Google Scholar] [CrossRef] [Green Version]
- Strumberg, D.; Schultheis, B.; Santel, A.; Gebhardt, F.; Meyer-Sabellek, W.; Keil, O.; Giese, K.; Kaufmann, J. Antimetastatic Activity of Atu027, a Liposomal Sirna Formulation, Targeting Protein Kinase N3: Final Results of a Phase I Study. Ann. Oncol. 2013, 24, i7. [Google Scholar] [CrossRef] [Green Version]
- Strumberg, D.; Schultheis, B.; Traugott, U.; Vank, C.; Santel, A.; Keil, O.; Giese, K.; Kaufmann, J.; Drevs, J. Phase I clinical development of Atu027, a siRNA formulation targeting PKN3 in patients with advanced solid tumors. Int. J. Clin. Pharmacol. Ther. 2012, 50, 76. [Google Scholar] [CrossRef]
- Schultheis, B.; Strumberg, D.; Santel, A.; Vank, C.; Gebhardt, F.; Keil, O.; Lange, C.; Giese, K.; Kaufmann, J.; Khan, M.; et al. First-in-Human Phase I Study of the Liposomal RNA Interference Therapeutic Atu027 in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2014, 32, 4141–4148. [Google Scholar] [CrossRef]
- Russo, S.; Saif, M.W. 2016 Gastrointestinal Cancers Symposium: Update on pancreatic cancer. Ann. Gastroenterol. 2016, 29, 238. [Google Scholar] [CrossRef] [Green Version]
- Schultheis, B.; Strumberg, D.; Kuhlmann, J.; Wolf, M.; Link, K.; Seufferlein, T.; Kaufmann, J.; Gebhardt, F.; Bruyniks, N.; Pelzer, U. A phase Ib/IIa study of combination therapy with gemcitabine and Atu027 in patients with locally advanced or metastatic pancreatic adenocarcinoma. J. Clin. Oncol. 2016, 34 (Suppl. 4), 385.–content. [Google Scholar] [CrossRef]
- Liu, X. Targeting Polo-Like Kinases: A Promising Therapeutic Approach for Cancer Treatment. Transl. Oncol. 2015, 8, 185–195. [Google Scholar] [CrossRef] [Green Version]
- Northfelt, D.W.; Hamburg, S.I.; Borad, M.J.; Seetharam, M.; Curtis, K.K.; Lee, P.; Crowell, B.; Vocila, L.; Fredlund, P.; Gilbert, M.J.; et al. A phase I dose-escalation study of TKM-080301, a RNAi therapeutic directed against polo-like kinase 1 (PLK1), in patients with advanced solid tumors: Expansion cohort evaluation of biopsy samples for evidence of pharmacodynamic effects of PLK1 inhibition. J. Clin. Oncol. 2013, 31 (Suppl. 15), TPS2621. [Google Scholar] [CrossRef]
- Ramanathan, R.K.; Hamburg, S.I.; Borad, M.J.; Seetharam, M.; Kundranda, M.N.; Lee, P.; Fredlund, P.; Gilbert, M.; Mast, C.; Semple, S.C.; et al. Abstract LB-289: A phase I dose escalation study of TKM-080301, a RNAi therapeutic directed against PLK1, in patients with advanced solid tumors. Cancer Res. 2013, 73 (Suppl. 8). [Google Scholar] [CrossRef]
- Demeure, M.J.; Armaghany, T.; Ejadi, S.; Ramanathan, R.K.; ElFiky, A.; Strosberg, J.R.; Smith, D.C.; Whitsett, T.; Liang, W.S.; Sekar, S.; et al. A phase I/II study of TKM-080301, a PLK1-targeted RNAi in patients with adrenocortical cancer (ACC). J. Clin. Oncol. 2016, 34, 2547. [Google Scholar] [CrossRef]
- Kamimura, K.; Yokoo, T.; Abe, H.; Terai, A.S. Gene Therapy for Liver Cancers: Current Status from Basic to Clinics. Cancers 2019, 11, 1865. [Google Scholar] [CrossRef] [Green Version]
- National Cancer Institute. TKM 080301 for Primary or Secondary Liver Cancer/A Phase 1 Dose Escalation Study of Hepatic Intra-Arterial Administration of TKM 080301 (Lipid Nanoparticles Containing siRNA Against the PLK1 Gene Product) in Patients With Colorectal, Pancreas, Gastric, Breast, Ovarian and Esophageal Cancers with Hepatic; National Cancer Institute: Bethesda, ML, USA, 2018.
- Abou-Alfa, G.; Yoon, J.; Modiano, M.; Ryoo, B.; Yau, T.; Freilich, B.; Knox, J.; Ly, M.; Ahmad, H.; Gahir, S.; et al. An open-label, multi-center, phase I/II, dose escalation study of IV TKM-080301 in subjects with advanced hepatocellular carcinoma. Eur. J. Cancer 2016, 69, S22. [Google Scholar] [CrossRef]
- El Dika, I.; Lim, H.Y.; Yong, W.P.; Lin, C.; Yoon, J.; Modiano, M.; Freilich, B.; Choi, H.J.; Chao, T.; Kelley, R.K.; et al. An Open-Label, Multicenter, Phase I, Dose Escalation Study with Phase II Expansion Cohort to Determine the Safety, Pharmacokinetics, and Preliminary Antitumor Activity of Intravenous TKM-080301 in Subjects with Advanced Hepatocellular Carcinoma. Oncologist 2019, 24, 747. [Google Scholar] [CrossRef] [Green Version]
- Khvalevsky, E.Z.; Gabai, R.; Rachmut, I.H.; Horwitz, E.; Brunschwig, Z.; Orbach, A.; Shemi, A.; Golan, T.; Domb, A.J.; Yavin, E.; et al. Mutant KRAS is a druggable target for pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20723–20728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramot, Y.; Rotkopf, S.; Gabai, R.M.; Khvalevsky, E.Z.; Muravnik, S.; Marzoli, G.A.; Domb, A.J.; Shemi, A.; Nyska, A. Preclinical Safety Evaluation in Rats of a Polymeric Matrix Containing an siRNA Drug Used as a Local and Prolonged Delivery System for Pancreatic Cancer Therapy. Toxicol. Pathol. 2016, 44, 856–865. [Google Scholar] [CrossRef] [Green Version]
- Golan, T.; Khvalevsky, E.Z.; Hubert, A.; Gabai, R.M.; Hen, N.; Segal, A.; Domb, A.; Harari, G.; Ben David, E.; Raskin, S.; et al. RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget 2015, 6, 24560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vakhshiteh, F.; Atyabi, F.; Ostad, S.N. Mesenchymal stem cell exosomes: A two-edged sword in cancer therapy. Int. J. Nanomed. 2019, 14, 2847–2859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiteside, T.L. Therapeutic targeting of oncogenic KRAS in pancreatic cancer by engineered exosomes. Transl. Cancer Res. 2017, 6 (Suppl. 9), S1406–S1408. [Google Scholar] [CrossRef] [PubMed]
- Mendt, M.; Kamerkar, S.; Sugimoto, H.; McAndrews, K.M.; Wu, C.C.; Gagea, M.; Yang, S.; Blanko, E.; Peng, Q.; Ma, X.; et al. Generation and testing of clinical-grade exosomes for pancreatic cancer. JCI Insight 2018, 3, e99263. [Google Scholar] [CrossRef] [PubMed]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.; Melo, S.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nat. Cell Biol. 2017, 546, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Papadopoulos, K.P.; Patnaik, A.; Rasco, D.W.; Martinez, D.; Wood, D.L.; Fielman, B.; Sharma, M.; Janisch, L.A.; Brown, B.D.; et al. Safety and activity of DCR-MYC, a first-in-class Dicer-substrate small interfering RNA (DsiRNA) targeting MYC, in a phase I study in patients with advanced solid tumors. J. Clin. Oncol. 2015, 33, 11006. [Google Scholar] [CrossRef]
- Kotelianski, V.; Zatsepin, T.S.; Kotelevtsev, Y.V. Lipid nanoparticles for targeted siRNA delivery—Going from bench to bedside. Int. J. Nanomed. 2016, 11, 3077. [Google Scholar] [CrossRef] [Green Version]
- Landen, C.N.; Chavez-Reyes, A.; Bucana, C.; Schmandt, R.; Deavers, M.T.; Lopez-Berestein, G.; Sood, A.K. Therapeutic EphA2 Gene Targeting In vivo Using Neutral Liposomal Small Interfering RNA Delivery. Cancer Res. 2005, 65, 6910–6918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, M.J.; Mitra, R.; McArthur, M.J.; Baze, W.; Barnhart, K.; Wu, S.; Rodriguez-Aguayo, C.; Zhang, X.; Coleman, R.L.; Lopez-Berestein, G.; et al. Preclinical Mammalian Safety Studies of EPHARNA (DOPC Nanoliposomal EphA2-Targeted siRNA). Mol. Cancer Ther. 2017, 16, 1114–1123. [Google Scholar] [CrossRef] [Green Version]
- Naing, A.; Lopez-Berestein, G.; Fu, S.; Tsimberidou, A.M.; Pant, S.; Piha-Paul, S.A.; Janku, F.; Hong, D.S.; Sulovic, S.; Meng, X.; et al. EphA2 gene targeting using neutral liposomal small interfering RNA (EPHARNA) delivery: A phase I clinical trial. J. Clin. Oncol. 2017, 35, TPS2604. [Google Scholar] [CrossRef]
- O’Brien, Z.; Wang, L.; Majeti, B.; Clamme, J.; Baclig, R.; Chu, J.; Fong, S.; Harborth, J.; Ibarra, J.; Yin, H.; et al. A novel lipid nanoparticle (NBF-006) encapsulating glutathione S-transferase P (GSTP) siRNA for the treatment of KRAS-driven non-small cell lung cancer. Cancer Res. 2018, 78 (Suppl. 13), 5917. [Google Scholar]
- A Study of NBF-006 in Non-Small Cell Lung, Pancreatic, or Colorectal Cancer. ClinicalTrials.gov, Identifier: NCT03819387 ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03819387 (accessed on 30 June 2021).
- Shahabipour, F.; Banach, M.; Sahebkar, A. Exosomes as nanocarriers for siRNA delivery: Paradigms and challenges. Arch. Med Sci. AMS 2016, 6, 1324. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Das, M.; Musetti, S.; Huang, L. RNA Interference-Based Cancer Drugs: The Roadblocks, and the “Delivery” of the Promise. Nucleic Acid Ther. 2019, 29, 61–66. [Google Scholar] [CrossRef]
- Zuckerman, J.E.; Davis, M.E. Clinical experiences with systemically administered siRNA-based therapeutics in cancer. Nat. Rev. Drug Discov. 2015, 14, 843–856. [Google Scholar] [CrossRef]
- A Maier, M.; Jayaraman, M.; Matsuda, S.; Liu, J.; Barros, S.; Querbes, W.; Tam, Y.K.; Ansell, S.M.; Kumar, V.; Qin, J.; et al. Biodegradable Lipids Enabling Rapidly Eliminated Lipid Nanoparticles for Systemic Delivery of RNAi Therapeutics. Mol. Ther. 2013, 21, 1570–1578. [Google Scholar] [CrossRef] [Green Version]
- Whitehead, K.A.; Dorkin, J.R.; Vegas, A.J.; Chang, P.H.; Veiseh, O.; Matthews, J.; Fenton, O.S.; Zhang, Y.; Olejnik, K.T.; Yesilyurt, V.; et al. Degradable lipid nanoparticles with predictable in vivo siRNA delivery activity. Nat. Commun. 2014, 5, 4277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, T.L.; Fellmann, C.; Lee, C.-S.; Ritchie, C.D.; Thapar, V.; Lee, L.C.; Hsu, D.J.; Grace, D.; Carver, J.O.; Zuber, J.; et al. Development of siRNA Payloads to Target KRAS-Mutant Cancer. Cancer Discov. 2014, 4, 1182–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behlke, M.A. Chemical Modification of siRNAs for In Vivo Use. Oligonucleotides 2008, 18, 305–320. [Google Scholar] [CrossRef] [Green Version]
- Sioud, M. Single-stranded small interfering RNA are more immunostimulatory than their double-stranded counterparts: A central role for 2′-hydroxyl uridines in immune responses. Eur. J. Immunol. 2006, 36, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Moyano, D.F.; Goldsmith, M.; Solfiell, D.J.; Landesman-Milo, D.; Miranda, O.R.; Peer, D.; Rotello, V.M. Nanoparticle hydrophobicity dictates immune response. J. Am. Chem. Soc. 2012, 134, 3965–3967. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, D.W.; Davis, M.E. Insights into the kinetics of siRNA-mediated gene silencing from live-cell and live-animal bioluminescent imaging. Nucleic Acids Res. 2006, 34, 322–333. [Google Scholar] [CrossRef]
- Munzone, E.; Colleoni, M. Clinical overview of metronomic chemotherapy in breast cancer. Nat. Rev. Clin. Oncol. 2015, 12, 631. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, J.E.; Hsueh, T.; Koya, R.C.; Davis, M.E.; Ribas, A. siRNA Knockdown of Ribonucleotide Reductase Inhibits Melanoma Cell Line Proliferation Alone or Synergistically with Temozolomide. J. Investig. Dermatol. 2011, 131, 453–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, L.; Yang, X.; Yin, Q.; Cai, K.; Wang, H.; Chaudhury, I.; Yao, C.; Zhou, Q.; Kwon, M.; Hartman, J.A.; et al. Investigating the optimal size of anticancer nanomedicine. Proc. Natl. Acad. Sci. USA 2014, 111, 15344–15349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adjei, I.M.; Peetla, C.; Labhasetwar, V. Heterogeneity in nanoparticles influences biodistribution and targeting. Nanomedicine 2014, 9, 267–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Pei, Y.; Zhang, H.; Wang, L.; Arrington, L.; Zhang, Y.; Glass, A.; Leone, A.M. Assessing the Heterogeneity Level in Lipid Nanoparticles for siRNA Delivery: Size-Based Separation, Compositional Heterogeneity, and Impact on Bioperformance. Mol. Pharm. 2012, 10, 397–405. [Google Scholar] [CrossRef]
- Cuellar, T.L.; Barnes, D.; Nelson, C.; Tanguay, J.; Yu, S.F.; Wen, X.; Scales, S.J.; Gesch, J.; Davis, D.; van Brabant, S.A.; et al. Systematic evaluation of antibody-mediated siRNA delivery using an industrial platform of THIOMAB–siRNA conjugates. Nucleic Acids Res. 2015, 43, 1189–1203. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapeutic Name | Indications | Target Gene/Protein | Route of Administration | Phase/Status | Reference |
|---|---|---|---|---|---|
| CALAA-01 | Cancer, Solid Tumor | RRM 2 | Systemic/IV infusion | Phase I/Terminated | [9] |
| ALN-VSP02 | Solid Tumors | VEGF, KSP | Systemic/IV infusion | Phase I/Completed | [15] |
| Mesenchymal Stromal Cells-derived iExosomes | Pancreatic Cancer | KRAS G12D Mutation | Systemic/IV infusion | Phase I/Not yet recruited | [36] |
| siRNA-EphA2-DOPC | Advanced Cancers | EphA2 | Systemi /IV infusion | Phase I/Not completed yet | [39,41] |
| Atu027 | Advanced or Metastatic Pancreatic Cancer(II), Solid Tumors(I) | PKN3 | Systemic/IV infusion | Phase II/Completed | [16,18,19] |
| TKM- PLK1 (TKM-080301) | Adrenal Cortical Carcinoma(II), Hepatocellular Carcinoma(II), Neuroendocrine Tumor(II), Solid Tumors(I) | PLK-1 | Systemic/IV infusion | Phase II/Completed | [25,28,29] |
| siG12D LODER | Pancreatic Ductal Adenocarcinoma, Pancreatic Cancer | KRAS G12D mutation | Local/Surgical implantation | Phase II/Ongoing | [32] |
| DCR-MYC (DCRM1711) | Solid Tumors, Hepatocellular Carcinoma, Multiple Myeloma, NonHodgkins Lymphoma, Pancreatic Neuroendocrine Tumors | MYC | Systemic/IV infusion | Phase II/Terminated | [37] |
| NBF-006 | Non-Small-Cell Lung, Colorectal, and Pancreatic Cancer | GSTP | Systemic/IV infusion | Phase I/Recruiting | [60] |
| Therapeutics’ Name | siRNA Characteristics | Delivery System | ||||||
|---|---|---|---|---|---|---|---|---|
| Encapsulation | Chemical Modification | Numbers of siRNA Motifs | Natural/Artificial | Type | Biodegra-Dability | Targeting | Targeting Ligand | |
| CALAA-01 | Encapsulated | Yes | Single | Artificial | Cyclo-dextrin based polymer | No | Yes | Human transferrin ligand |
| ALN-VSP02 | Encapsulated | No | Two | Artificial | LNP | No | No | - |
| siRNA-EphA2-DOPC | Encapsulated | No | Single | Artificial | DOPC LNP | No | No | - |
| Mesenchymal Stromal Cells-derived iExosomes | Encapsulated | No | Single | Natural | Exosomes | No | Yes | CD47 |
| Atu027 | Encapsulated | Yes | Single | Artificial | AtuPlex Technology | No | No | - |
| TKM-PLK1 (TKM-080301) | Encapsulated | No | Single | Artificial | LNP | No | No | - |
| siG12D LODER | Encapsulated | No | Single | Artificial | LODER polymer | Yes | No | - |
| DCR-MYC (DCRM1711) | Encapsulated | No | Single | Artificial | EnCore LNP | No | No | - |
| NBF-006 | Encapsulated | No | Single | Artificial | LNP | Yes | No | - |
| Therapeutics’ Name | Dosing Scale | Dose | Infusion Time | Dosage Frequency | Cycle | Pre-Medications | Maximum Tolerated Dose |
|---|---|---|---|---|---|---|---|
| CALAA-01 | Body surface area | 3–30 mg/m2 | 30 min | Twice weekly | 21 days | Yes | Not determined |
| ALN-VSP02 | Body weight | 0.1–1.7 mg/kg | 15 min | Once every two weeks | 28 days | Yes | Not determined |
| siRNA-EphA2-DOPC | Body Surface area | 0.45 mg/m2 | 2 h | Twice weekly | 21 days | - | The trial not recruited yet |
| Mesenchymal Stromal Cells-derived iExosomes | Body weight | Not reported | 15–20 min | Twice weekly | 14 days | - | The trial is on-going |
| Atu027 | Body weight | Phase II: 0.253 mg/kg | 4 h | Twice weekly | 28 days | No | Not determined |
| TKM- PLK1 (TKM-080301) | Body weight | Phase II: 0.6–0.75 mg/kg | 30 min | Once weekly | 28 days | Yes | 0.6mg/kg |
| siG12D LODER | - | 0.025–3 mg | - | Once monthly | - | No | Not determined |
| DCR-MYC (DCRM1711) | Body weight | 0.1–0.3 mg/kg | 2 h | Once weekly | 21 days | - | Not determined |
| NBF-006 | - | - | - | Once weekly × 4 weeks | Every 6 weeks | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hattab, D.; Gazzali, A.M.; Bakhtiar, A. Clinical Advances of siRNA-Based Nanotherapeutics for Cancer Treatment. Pharmaceutics 2021, 13, 1009. https://doi.org/10.3390/pharmaceutics13071009
Hattab D, Gazzali AM, Bakhtiar A. Clinical Advances of siRNA-Based Nanotherapeutics for Cancer Treatment. Pharmaceutics. 2021; 13(7):1009. https://doi.org/10.3390/pharmaceutics13071009
Chicago/Turabian StyleHattab, Dima, Amirah Mohd Gazzali, and Athirah Bakhtiar. 2021. "Clinical Advances of siRNA-Based Nanotherapeutics for Cancer Treatment" Pharmaceutics 13, no. 7: 1009. https://doi.org/10.3390/pharmaceutics13071009