
Second-Generation Antimitotics in Cancer Clinical Trials




Abstract
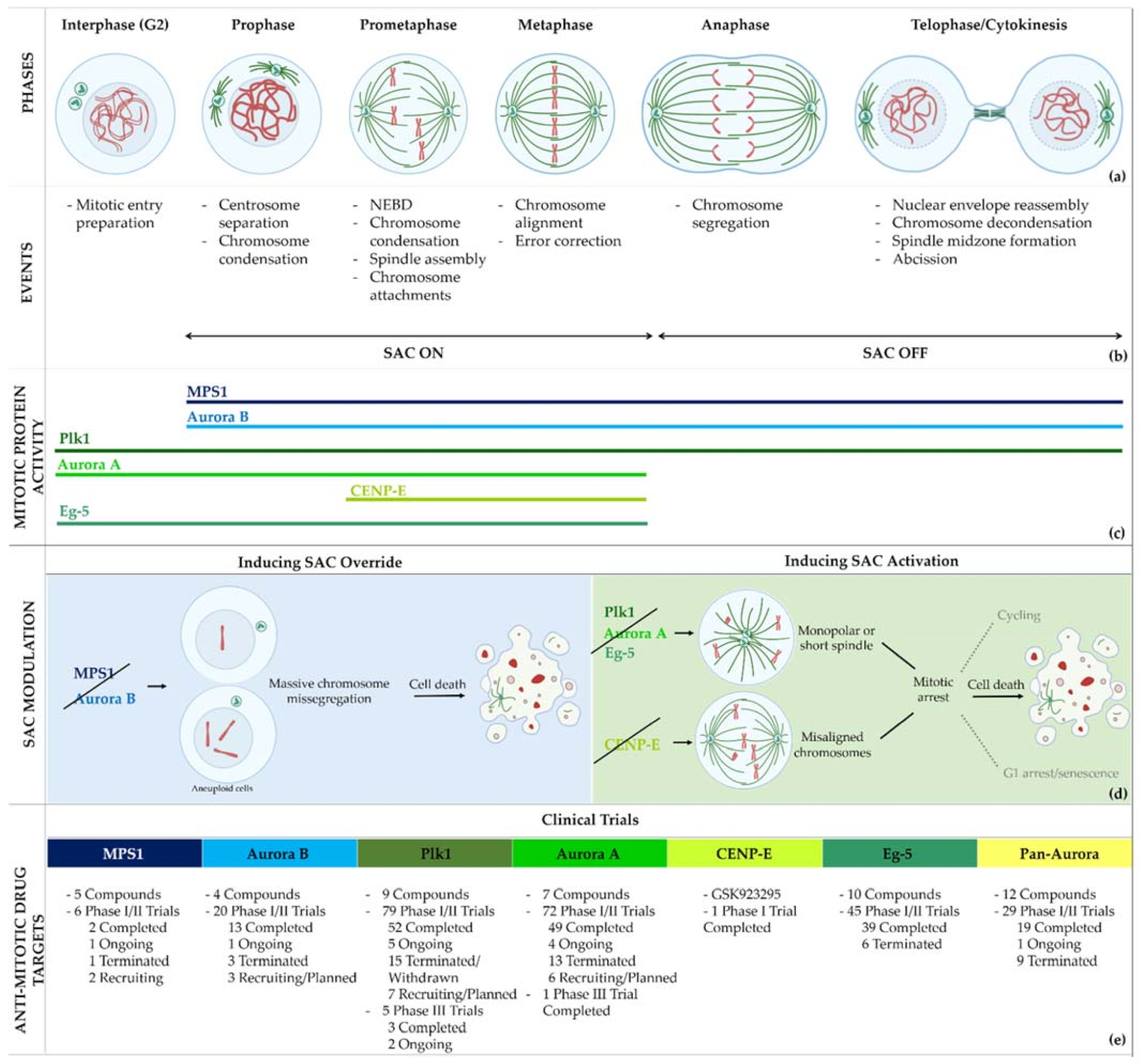
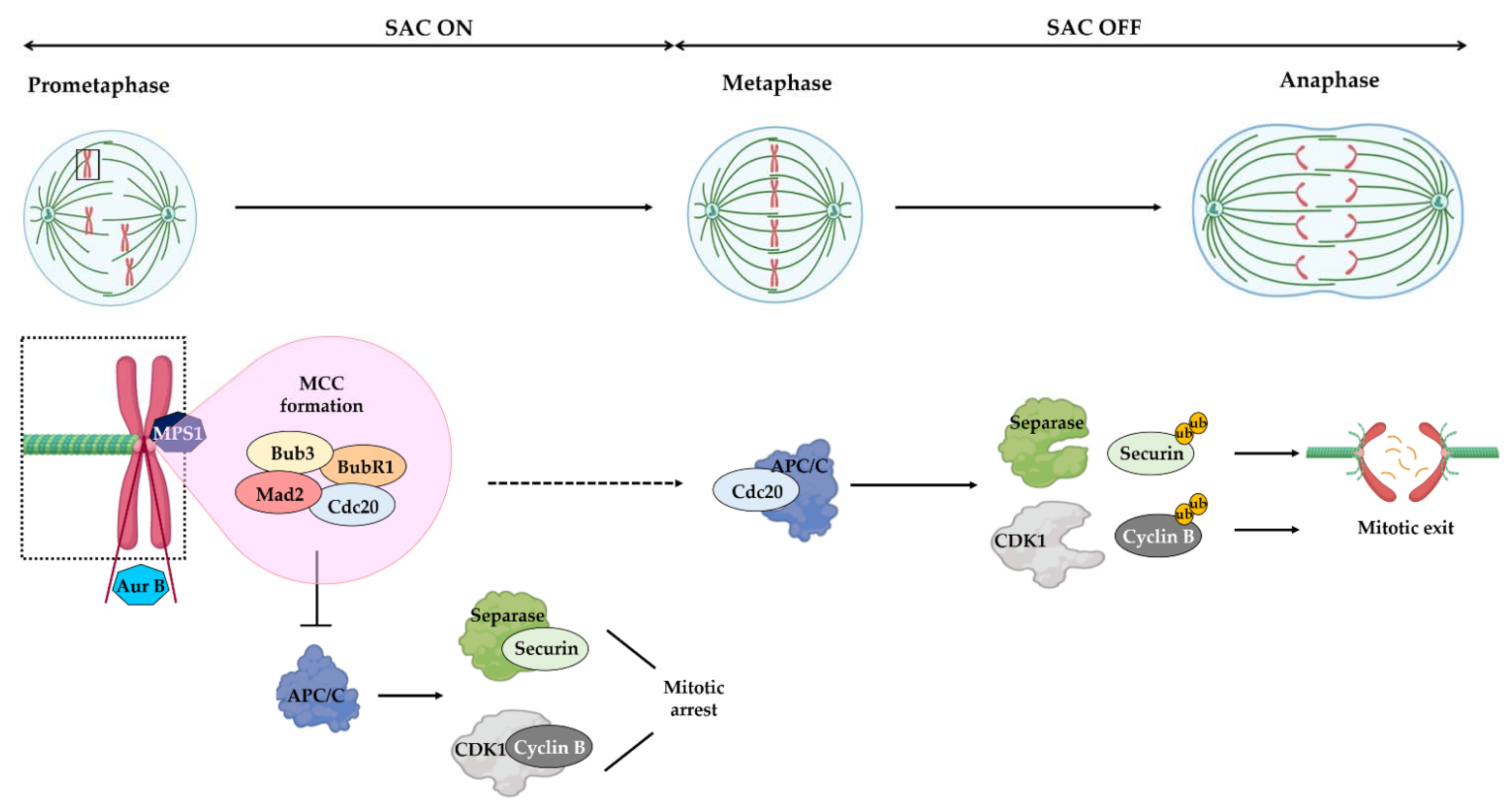
:1. Introduction
2. Limitations of Current Microtubule-Targeting Agents
3. Second-Generation Antimitotics in Clinical Trials
3.1. Mps1
3.1.1. BAY 1217389 and BAY1161909
3.1.2. S 81694
3.1.3. BOS 172722
3.1.4. CFI-402257

{kind=link}
{kind=link}
| Compound | Clinical Trials | Current Status | Conditions | Interventions | Outcomes 2 | Ref. |
|---|---|---|---|---|---|---|
BAY 1217389 | NCT02366949 | Phase I Completed | Advanced Solid Malignancies | Combination with Paclitaxel | Not published | - |
BAY 1161909 (Empesertib) | NCT02138812 | Phase I Terminated | Advanced Solid Malignancies | Combination with Paclitaxel | Best response was PR | [22] |
| S 81694 Structure Undisclosed | NCT03411161 | Phase I Phase II Completed | Metastatic Breast Cancer, Metastatic Triple-Negative Breast Cancer | Combination with Paclitaxel | Not published | - |
BOS 172722 | NCT03328494 | Phase I Ongoing | Advanced Nonhematologic Malignancies | Combination with Paclitaxel | - | - |
CFI-402257 | NCT03568422 | Phase I Phase II Recruiting | Breast Cancer | Combination with Paclitaxel | - | - |
| NCT02792465 | Phase I, Recruiting | Breast Cancer, Advanced Solid Malignancies | Monotherapy Combination with Fulvestrant | - | - |
3.2. Plk1
3.2.1. BI 2536 and BI6727 (Volasertib)
3.2.2. ON 01910.Na (Rigosertib)
3.2.3. GSK461364
3.2.4. MK-1496
3.2.5. TAK-960
3.2.6. NMS-1286937 (Onvansertib)
3.2.7. TKM-080301
3.2.8. CYC 140
| Compound | Clinical Trials | Current Status | Conditions | Interventions | Outcomes 2 | Refs. |
|---|---|---|---|---|---|---|
BI 2536 | 11 Clinical trials | Phase I/II 10 Completed 1 Terminated | Advanced Solid Tumors, Acute Myeloid Leukemia, Non-Hodgkin’s Lymphoma | Monotherapy | Best responses were CR for acute myeloid leukemia and non-Hodgkin’s lymphoma; PR for non-small-cell lung cancer, pancreatic cancer, acute myeloid leukemia, and non-Hodgkin’s lymphoma | [40,41,42,43,44] |
| Combination with Pemetrexed and Gemcitabine | Best response was PR for adenocarcinoma and squamous cell carcinoma | |||||
BI 6727 (Volasertib) | 26 Clinical trials | 25 Phase I/II 12 Completed 4 Terminated 6 Withdrawn 3 Ongoing | Acute Myeloid Leukemia Pediatric Patients with Advanced Cancers, Myelodysplastic Syndromes, Non-Hodgkin’s Lymphoma, Urothelial, Ovarian, Lung Pancreatic, Colorectal and Prostate Cancer | Monotherapy | Best responses were CR for acute myeloid leukemia; PR for non-small cell lung cancer, melanoma, ovarian cancer, acute myeloid leukemia, gastric cancer, and urothelial cancer | [46,47,48,49,50,51,52,53,54,55] |
| Combination with Cytarabine, Pemetrexed, Azacitidine, Afatinib, Decitabine, Daunorubicin, Nintedanib, Mitoxantrone and Itraconazole | Best responses were CR for breast cancer in combination with Nintedanib, PR for non-small-lung cancer in combination with Pemetrexed, Nintedanib, Afatinib, and Carboplatin, PR for head and neck carcinoma in combination with Afatinib, PR for undifferentiated follicular dendritic reticulum cell sarcoma and differentiated follicular dendritic reticulum cell retroperitoneal sarcoma in combination with Cisplatin, PR for differentiated hypopharynx carcinoma in combination with Carboplatin | |||||
| 1 Phase III Ongoing | Acute Myeloid Leukemia | Combination with Cytarabine | - | |||
ON 01910.Na (Rigosertib) | 36 Clinical trials | 32 Phase I/II 24 Completed 1 Terminated 2 Withdrawn 3 Recruiting/ Planned 2 Ongoing | Refractory Leukemia, Myelodysplastic Syndrome, Squamous Cell Carcinoma Acute/Chronic Myeloid Leukemia, Ovarian and Lung Cancer, Advanced Solid Tumors | Monotherapy | Best response was PR for non-Hodgkin’s lymphoma, thymic cancer, and pancreatic ductal adenocarcinoma | [57,58,59,60,61,72,73] |
| Combination with Nivolumab, Cisplatin, Azacitidine, Oxaplatin, Gemcitabine and Irinotecan | Best responses were CR and mCR for myelodysplastic syndrome in combination with Azacitidine | |||||
| 4 Phase III 3 Completed 1 Ongoing | Metastatic Pancreatic Adenocarcinoma, Myelodysplastic Syndromes | Monotherapy | Best responses were SD and mCR for myelodysplastic syndromes | |||
| Combination with Gemcitabine | Best response was PR for pancreatic adenocarcinoma | |||||
GSK461364 | NCT00536835 | Phase I Completed | Non-Hodgkin Lymphoma, Advanced Solid Malignancies | Monotherapy | Best response was SD for esophageal and ovarian cancers and endometrial carcinoma | [63] |
MK-1496 | NCT00880568 | Phase I Completed | Advanced Solid Tumors | Monotherapy | Best response was PR for parotid gland carcinoma and small cell lung cancer | [64] |
TAK-960 | NCT01179399 | Phase I Terminated | Advanced Nonhematologic Malignancies | Monotherapy | Discontinued strategically by sponsor due to lack of efficacy | - |
NMS-1286937/Onvansertib | 4 Clinical trials | Phase I/II 1 Completed 3 Recruiting | Advanced or Metastatic Solid Tumors | Monotherapy | Best response was SD for colorectal cancer, pancreatic carcinoma with a K-RAS mutation, head and neck squamous cell carcinoma, and basal cell carcinoma | [68] |
| Acute Myeloid Leukemia, Metastatic Prostate Cancer, Metastatic Colorectal Cancer with a KRAS mutation | Combination with Decitabine, Cytarabine, Abiraterone, Prednisone, FOLFIRI and Bevacizumab | - | ||||
| TKM-080301 (siRNA) | 3 Clinical trials | Phase I/II Completed | Hepatocellular Carcinoma, Colorectal, Pancreas, Breast and Ovarian Cancer with Hepatic Metastases, Adrenocortical Carcinoma, Neuroendocrine Tumors | Monotherapy | Best response was PR for adrenocortical carcinoma | [69,70] |
CYC 140 | NCT03884829 | Phase I Recruiting | Acute Myeloid Leukemia, Myelodysplastic Syndromes, Acute Lymphoblastic Leukemia | Monotherapy | - | - |
4. Aurora Kinases
4.1. Pan-Aurora Inhibitors
4.1.1. ABT-348 (Ilorasertib)
4.1.2. AS703569/MSC1992371A (Cenisertib)
4.1.3. VX-680 (Tozasertib)
4.1.4. BI-847325
4.1.5. AT9283
4.1.6. AMG-900
4.1.7. PHA739358 (Danusertib)
4.1.8. SNS-314 Mesylate
4.1.9. TAK-901
4.1.10. CYC116
4.1.11. GSK1070916 (NIM-900)
4.1.12. PF-03814735
| Compound | Clinical Trials | Current Status | Conditions | Interventions | Outcomes 2 | Refs. |
|---|---|---|---|---|---|---|
ABT-348 (Ilorasertib) | 4 Clinical trials | Phase I/II 3 Completed 1 Ongoing | Solid Tumors, Advanced Hematological Malignancies | Monotherapy | Best response was PR for basal cell carcinoma, and adenocarcinoma | [83,84] |
| Combination with Carboplatin, Docetaxel and Azacitidine | Best responses were CR, PR, and CRi for acute myeloid leukemia in combination with Azacitidine | |||||
AS703569/MSC1992371A (Cenisertib) | 3 Clinical trials | Phase I/II 2 Completed 1 Terminated | Solid Tumors, Advanced Malignancies | Monotherapy | Best response was CR for acute myeloid leukemia and CRi for acute lymphoid leukemia with Philadelphia chromosome | [86,87,88] |
| Combination with Gemcitabine | Best response was PR for non-small-cell lung cancer and hepatocellular carcinoma | |||||
VX-680/MK-0457 (Tozasertib) | 6 Clinical trials | Phase I/II 1 Completed 5 Terminated | Chronic Myeloid Leukemia, Philadelphia Chromosome-Positive, Acute Lymphoblastic Leukemia, Non-Small-Cell Lung Carcinoma, Advanced Solid Tumors | Monotherapy Combination with Dasatinib | Best response was one CR for chronic myeloid leukemia with BCR–ABL T315I mutation phenotype as monotherapy | [90,91,110] |
BI-847325 | NCT01324830 | Phase I Completed | Solid Tumors | Monotherapy | Best response was PR for esophageal cancer | [93] |
AT9283 | 5 Clinical trials | Phase I/II 4 Completed 1 Terminated | Multiple Myeloma, Acute Leukemia, Myelofibrosis, Advanced or metastatic solid tumors, Non-Hodgkin’s lymphoma, Solid Tumors | Monotherapy | Best response was PR for nervous system primitive neuroectodermal tumor and squamous cell carcinoma | [95,96,97] |
AMG-900 | NCT00858377 | Phase I Completed | Advanced Solid Tumors | Monotherapy | Best response was PR for clear-cell endometrial cancer | [99] |
| NCT01380756 | Phase I Completed | Acute Myeloid Leukemia | Monotherapy | Best response was CRi | [100] | |
PHA739358, (Danusertib) | NCT00766324 | Phase II Completed | Metastatic Hormone Refractory Prostate Cancer | Monotherapy | The best response was SD | [102] |
| NCT00872300 | Phase II Terminated | Multiple Myeloma | - | - | ||
SNS-314 Mesylate | NCT00519662 | Phase I Completed | Advanced Solid Tumors | Monotherapy | Best response was SD | [104] |
TAK-901 | NCT00935844 | Phase I Completed | Advanced Solid Tumors Lymphoma | Monotherapy | Not published | - |
| NCT00807677 | Phase I Completed | Advanced Hematologic Malignancies | ||||
CYC116 | NCT00560716 | Phase I Terminated | Advanced Solid Tumors | Monotherapy | - | - |
GSK1070916/NMI-900  | NCT01118611 | Phase I Completed | Advanced Solid Tumors | Monotherapy | Not published | - |
PF-03814735 | NCT00424632 | Phase I Completed | Advanced Solid Tumors | Monotherapy | Best response was SD for non-small-cell lung cancer, melanoma, renal cell carcinoma, and neuroendocrine tumor | [109] |
4.2. Aurora B inhibitors
4.2.1. AZD1152 (Barasertib)
4.2.2. BI-831266
4.2.3. BI-811283
4.2.4. Chiauranib
| Compound | Clinical Trials | Current Status | Conditions | Interventions | Outcomes 2 | Refs. |
|---|---|---|---|---|---|---|
AZD1152 (Barasertib) | 10 Clinical trials | Phase I/II 8 Completed 2 Terminated | Lymphoma, Advanced Solid Tumors, Acute Myeloid Leukemia | Monotherapy | Best response was CR for acute myeloid leukemia, and PR for lymphoma and acute myeloid leukemia | [112,113,114,115,116,117,118,119] |
| Combination with low dose of Cytarabine, Venetoclax, Azacitidine | Best response was CR and PR for acute myeloid leukemia in combination with low dose of Cytarabine | |||||
BI-831266 | NCT00756223 | Phase I Completed | Advanced Solid Tumors | Monotherapy | Best response was PR for cervical cancer | [121] |
| BI-811283 (Structure Undisclosed) | NCT00701324 | Phase I Completed | Solid Tumors | Monotherapy | Best response was SD | [123] |
| NCT00632749 | Phase II Completed | Acute Myeloid Leukemia | Combination with Cytarabine | Best responses were CR, PR, and CRi | [124] | |
Chiauranib | 7 Clinical trials | Phase I/II 2 Completed 1 Terminated 3 Recruiting/ Planned 1 Ongoing | Ovarian Cancer, Non-Hodgkin’s Lymphoma, Hepatocellular Carcinoma, Small Cell Lung Cancer, Other Advanced Solid Tumors | Monotherapy | Best response was SD | [126] |
4.3. Aurora A inhibitors
4.3.1. MLN8237 (Alisertib)
4.3.2. ENMD-2076
4.3.3. LY3295668 (Erbumine)
4.3.4. MLN8054
4.3.5. MK-5108 (VX-689)
4.3.6. TAS-119
4.3.7. KW-2449
| Compound | Clinical Trials | Current Status | Conditions | Interventions | Outcomes 2 | Refs. |
|---|---|---|---|---|---|---|
MLN8237 (Alisertib) | 53 Clinical trials | Phase I/II 38 completed 7 terminated 4 ongoing 4 recruiting | Advanced Hematological Malignancies, Advanced Solid Tumors, Lymphoma, Prostate Cancer, Ovarian, Fallopian Tube and Peritoneal Carcinoma, Leiomyosarcoma, Neuroblastoma, Acute Myeloid Leukemia, Mantle Cell Lymphoma, Burkitt’s Lymphoma, Breast Carcinoma, Glioma, Non-Hodgkin’s Lymphoma, Bladder Cancer, Non-Small-Cell Lung Cancer, Mesothelioma, Small-Cell Lung Cancer Adenocarcinoma, Melanoma, Head and Neck Squamous Cell Carcinoma, EGFR-mutant Lung Cancer, Rhabdoid Tumor | Monotherapy | Best responses were CR for neuroblastoma, Wilms tumor, non-Hodgkin’s lymphoma, and acute myeloid leukemia; PR for lymphoma, multiple myeloma, angiosarcoma, ovarian cancer, prostate cancer, hepatoblastoma, neuroblastoma, non-Hodgkin’s lymphoma, and acute myeloid leukemia | [128,129,130,131,132,133,134,135,136,137,138, 139,140,141,143,144,144,145,146,147,148] |
| Combination with Pazopanib, Bortezomib, Docetaxel, Irinotecan, Temozolomide, Daunorubicin, Idarubicin, Cytarabine, Rituximab, Vincristine, Paclitaxel, Oxaliplatin, Leucovorin, Fluorouracil, Esomeprazole, Rifampin, Fulvestrant, Itraconazole, Idarubicin, Cytarabine, Vorinostat, MLN0128, Abiraterone Acetate, Prednisone, Erlotinib, Gemcitabine, Pembrolizumab, Osimertinib, Romidepsin, Bortezomib | Best responses were CR for multiple myeloma in combination with Bortezomib, ovarian cancer in combination with Paclitaxel acute myeloid leukemia in combination with Daunorubicin, Idarubicin, and Cytarabine, refractory aggressive B-cell lymphoma in combination with Rituximab and Vincristine, breast cancer in combination with Paclitaxel, small-cell lung cancer in combination with Paclitaxel; PR for urothelial cancer in combination with Paclitaxel, breast cancer in combination with Pazopanib, Fulvestrant and Paclitaxel, mesothelioma in combination with Pazopanib, multiple myeloma in combination with Bortezomib, angiosarcoma in combination with Docetaxel, castration-resistant prostate cancer in combination with Docetaxel, neuroblastoma in combination with Temozolomide, acute myeloid leukemia in combination with Daunorubicin, Idarubicin, and Cytarabine, refractory aggressive B-cell lymphoma in combination with Rituximab and Vincristine, ovarian cancer in combination with Paclitaxel, colon cancer in combination with Oxaliplatin, Leucovorin, and Fluorouracil, small-cell lung cancer in combination with Paclitaxel | |||||
MLN8237 (Alisertib) | NCT01482962 | Phase III Completed | Relapsed/Refractory Peripheral T-Cell Lymphoma | Monotherapy | Best responses were CR and PR | [142] |
ENMD-2076 | 8 Clinical trials | Phase I/II Completed | Soft Tissue Sarcoma, Ovarian Cancer, Triple-Negative Breast Cancer, Relapsed or Refractory Hematological Malignancies, Advanced Fibrolamellar Carcinoma, Multiple Myeloma, Advanced Malignancies | Monotherapy | Best response was PR for ovarian cancer, triple-negative breast cancer, advanced fibrolamellar carcinoma, undifferentiated pleomorphic sarcoma, and angiosarcoma; and CRi for relapsed or refractory hematological malignancies | [150,151,152,153,154,155,156,167] |
LY3295668 (Erbumine) | 4 Clinical trials | Phase I/II 2 Completed 1 Recruiting 1 Planned | Small-Cell Lung Cancer, Metastatic Breast Cancer, Neuroblastoma, Solid Tumors | Monotherapy Combination with Topotecan and Cyclophosphamide | Best response was SD | [157] |
MLN8054 | NCT00249301 | Phase I Terminated | Solid Tumors | Monotherapy | Best response was SD | [159] |
| NCT00652158 | Phase I Terminated | Advanced Malignancies | Monotherapy | [160] | ||
MK-5108/VX-689 | NCT00543387 | Phase I Completed | Solid Tumors | Monotherapy | Best response was SD | [162] |
| Combination with Docetaxel | Best response was PR | |||||
| TAS-119 (Structure Undisclosed) | NCT02448589 | Phase I Terminated | Advanced Solid Tumors | Monotherapy | Best response was SD | [164] |
| NCT02134067 | Phase I Terminated | Combination with Paclitaxel | Best response was PR for ovarian/fallopian tube cancers | [165] | ||
KW-2449 | NCT00346632 | Phase I Terminated | Acute Leukemias Myelodysplastic Syndromes—Chronic Myelogenous Leukemia | Monotherapy | Terminated due to suboptimal dosing schedule | - |
| NCT00779480 | Phase I Terminated | Acute Myelogenous Leukemia | Monotherapy | Failure to demonstrate a tolerable dose that had potential for efficacy |
5. CENP-E Kinesin
| Compound | Clinical Trials | Current Status | Conditions | Interventions | Outcomes 2 | Ref. |
|---|---|---|---|---|---|---|
GSK923295 | NCT00504790 | Phase I Completed | Refractory Cancer | Monotherapy | Best response was PR for urothelial carcinoma | [177] |
6. Eg-5 Kinesin
6.1. SB-715992 (Ispinesib) and SB-743921
6.2. ARRY-250 (Filanesib)
6.3. ALN-VSP02
6.4. Litronesib
6.5. EMD 534085
6.6. SC-205
6.7. AZD4877
6.8. ARQ 621
6.9. MK-0731
| Compound | Clinical Trials | Current Status | Conditions | Interventions | Outcomes 2 | Refs. |
|---|---|---|---|---|---|---|
SB-715992 (Ispinesib) | 16 Clinical trials | Phase I/II 14 Completed 2 Terminated | Metastatic Prostate, Kidney and Colorectal Cancer, Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck, Breast, Ovarian and Liver Cancer, Melanoma, Hodgkin’s or Non-Hodgkin’s Lymphoma, Acute Leukemia Advanced Myelodysplastic Syndromes | Monotherapy | Best response was a PR for ovarian and breast cancer | [190,191,192,193,194,195,196,197] |
| Combination with Docetaxel, Capecitabine and Carboplatin | Best response was SD | |||||
SB-743921 | NCT00136513 | Phase I Completed | Solid Tumors | Monotherapy | Best response was PR for cholangiocarcinoma | [199] |
| NCT00343564 | Phase I Phase II Completed | Non-Hodgkin Lymphoma and Hodgkin Lymphoma | Monotherapy | Best response was PR for non-Hodgkin lymphoma | [200] | |
ARRY-520 (Filanesib) | 8 Clinical trials | Phase I/II Completed | Multiple Myeloma, Advanced/Refractory Myeloid Leukemia, Advanced Solid Tumors | Monotherapy | Best response was a PR for multiple myeloma | [202,203,204,205,206,207] |
| Combination with Pomalidomide, Dexamethasone, Carfilzomib, Filgrastim, Bortezomib, Carfilzomib | Best responses were a CR for multiple myeloma in combination with Pomalidomide, Bortezomib, Dexamethasone, and Filgrastim; PR for multiple myeloma in combination with Bortezomib, Pomalidomide, Dexamethasone, Filgrastim, and Carfilzomib | |||||
| ALN-VSP02 (siRNA) | NCT01158079 | Phase I Completed | Solid Tumors | Monotherapy | Best response was CR for endometrial cancer with multiple hepatic metastases | [209] |
| NCT00882180 | Phase I Completed | |||||
LY2523355 (Litronesib) | 7 Clinical trials | Phase I/II 6 Completed 1 Terminated | Metastatic Breast Cancer, Metastatic and/or Advanced Cancer, Acute Leukemia, Small Cell Lung Cancer | Monotherapy, Combination with Pegfilgrastim, and Filgrastim | Best response was PR for non-small-cell lung cancer, ovarian and neuroendocrine carcinomas, and breast cancer in combination with Pegfilgrastim | [211,212] |
EMD 534085 | Unknown | Phase I | Advanced Solid Tumors, Lymphoma | Monotherapy | Best response was SD | [214] |
| 4SC-205 (Structure Undisclosed) | NCT01065025 | Phase I Completed | Advanced Malignancies | Monotherapy | Best response was SD | [215] |
AZD4877 | 6 Clinical trials | Phase I/II 3 Completed 3 Terminated | Bladder Cancer, Transitional Cell Bladder Cancer, Urethra Cancer, Ureter Cancer, Renal Pelvis Cancer, Acute Myelogenous Leukemia, Non-Hodgkin Lymphoma | Monotherapy | Best response was SD for non-Hodgkin lymphoma, acute myeloid leukemia, and urothelial cancer | [217,218,219,220,225] |
ARQ 621 | NCT00825487 | Phase I Completed | Metastatic Solid Tumors, Refractory/Relapsed Hematologic Malignancies | Monotherapy | Best response was SD | [222] |
MK-0731 | NCT00104364 | Phase I Completed | Advanced Solid Malignancies | Monotherapy | Best response was SD for non-small-cell lung cancer, cervical and ovarian cancer | [224] |
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compton, D.A. Mechanisms of Aneuploidy. Curr. Opin. Cell Biol. 2011, 23, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.; Barbosa, J.; Nascimento, A.V.; Faria, J.; Reis, R.; Bousbaa, H. Monitoring the fidelity of mitotic chromosome segregation by the spindle assembly checkpoint. Cell Prolif. 2011, 44, 391–400. [Google Scholar] [CrossRef]
- Van Vuuren, R.J.; Visagie, M.H.; Theron, A.E.; Joubert, A.M. Antimitotic drugs in the treatment of cancer. Cancer Chemother. Pharm. 2015, 76, 1101–1112. [Google Scholar] [CrossRef] [Green Version]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef]
- Matson, D.R.; Stukenberg, P.T. Spindle Poisons and Cell Fate: A Tale of Two Pathways. Mol. Interv. 2011, 11, 141. [Google Scholar] [CrossRef]
- Brito, D.A.; Rieder, C.L. Mitotic Checkpoint Slippage in Humans Occurs via Cyclin B Destruction in the Presence of an Active Checkpoint. Curr. Biol. 2006, 16, 1194–1200. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, Y.; Inoue, T. Antiproliferative fate of the tetraploid formed after mitotic slippage and its promotion; a novel target for cancer therapy based on microtubule poisons. Molecules 2016, 21, 663. [Google Scholar] [CrossRef] [Green Version]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef] [Green Version]
- Henriques, A.C.; Ribeiro, D.; Pedrosa, J.; Sarmento, B.; Silva, P.M.A.; Bousbaa, H. Mitosis inhibitors in anticancer therapy: When blocking the exit becomes a solution. Cancer Lett. 2019, 440, 64–81. [Google Scholar] [CrossRef]
- Lauzé, E.; Stoelcker, B.; Luca, F.C.; Weiss, E.; Schutz, A.R.; Winey, M. Yeast spindle pole body duplication gene MPS1 encodes an essential dual specificity protein kinase. EMBO J. 1995, 14, 1655–1663. [Google Scholar] [CrossRef]
- Ji, Z.; Gao, H.; Jia, L.; Li, B.; Yu, H. A sequential multi-target Mps1 phosphorylation cascade promotes spindle checkpoint signaling. Elife 2017, 6. [Google Scholar] [CrossRef]
- Liu, X.; Winey, M. The MPS1 family of protein kinases. Annu. Rev. Biochem. 2012, 81, 561–585. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Zhang, F.; Huang, C.-J.; Liao, J.; Han, Y.; Hao, P.; Chu, Y.; Lu, X.; Li, W.; Yu, H.; et al. Mps1 regulates spindle morphology through MCRS1 to promote chromosome alignment. Mol. Biol. Cell 2019, 30, 1060–1068. [Google Scholar] [CrossRef]
- Maciejowski, J.; Drechsler, H.; Grundner-Culemann, K.; Ballister, E.R.; Rodriguez-Rodriguez, J.-A.; Rodriguez-Bravo, V.; Jones, M.J.K.; Foley, E.; Lampson, M.A.; Daub, H.; et al. Mps1 Regulates Kinetochore-Microtubule Attachment Stability via the Ska Complex to Ensure Error-Free Chromosome Segregation. Dev. Cell 2017, 41, 143–156. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Wang, A.; Lin, J.; Wu, L.; Zhang, H.; Yang, X.; Wan, X.; Miao, R.; Sang, X.; Zhao, H. Mps1/TTK: A novel target and biomarker for cancer. J. Drug Target. 2017, 25, 112–118. [Google Scholar] [CrossRef]
- Jemaà, M.; Galluzzi, L.; Kepp, O.; Senovilla, L.; Brands, M.; Boemer, U.; Koppitz, M.; Lienau, P.; Prechtl, S.; Schulze, V.; et al. Characterization of novel MPS1 inhibitors with preclinical anticancer activity. Cell Death Differ. 2013, 20, 1532–1545. [Google Scholar] [CrossRef]
- Kwiatkowski, N.; Jelluma, N.; Filippakopoulos, P.; Soundararajan, M.; Manak, M.S.; Kwon, M.; Choi, H.G.; Sim, T.; Deveraux, Q.L.; Rottmann, S.; et al. Small-molecule kinase inhibitors provide insight into Mps1 cell cycle function. Nat. Chem. Biol. 2010, 6, 359–368. [Google Scholar] [CrossRef] [Green Version]
- Janssen, A.; Kops, G.J.P.L.; Medema, R.H. Elevating the frequency of chromosome mis-segregation as a strategy to kill tumor cells. Proc. Natl. Acad. Sci. USA 2009, 106, 19108–19113. [Google Scholar] [CrossRef] [Green Version]
- Wengner, A.M.; Siemeister, G.; Koppitz, M.; Schulze, V.; Kosemund, D.; Klar, U.; Stoeckigt, D.; Neuhaus, R.; Lienau, P.; Bader, B.; et al. Novel Mps1 Kinase Inhibitors with Potent Antitumor Activity. Mol. Cancer Ther. 2016, 15, 583–592. [Google Scholar] [CrossRef] [Green Version]
- Lorusso, P.; Chawla, S.P.; Bendell, J.; Shields, A.F.; Shapiro, G.; Rajagopalan, P.; Cyris, C.; Bruns, I.; Mei, J.; Souza, F.; et al. First-in-human study of the monopolar spindle 1 (Mps1) kinase inhibitor BAY 1161909 in combination with paclitaxel in subjects with advanced malignancies. Ann. Oncol. 2018, 29, viii138. [Google Scholar] [CrossRef]
- Colombo, R.; Caldarelli, M.; Giorgini, M.L.; Degrassi, A.; Ciomei, M.; Pezzetta, D.; Ballinari, D.; Montagnoli, A.; Pesenti, E.; Donati, D.; et al. Abstract 2097: Targeting aneuploidy with NMS-P153, a tight binder inhibitor of the spindle assembly checkpoint MPS1 (TTK) kinase. Cancer Res. 2013, 73, 2097. [Google Scholar] [CrossRef]
- Colombo, R.; Burbridge, M.; Rodriguez, M.; Cantero, F.; Caldarelli, M.; Giorgini, M.L.; Sola, F.; Ballinari, D.; Ciomei, M.; Bosotti, R.; et al. Abstract 1638: Preclinical characterization of the novel TTK kinase inhibitor S81694 for the treatment of triple negative breast cancer. Cancer Res. 2015, 75, 1638. [Google Scholar] [CrossRef]
- Woodward, H.L.; Innocenti, P.; Cheung, K.-M.J.; Hayes, A.; Roberts, J.; Henley, A.T.; Faisal, A.; Mak, G.W.-Y.; Box, G.; Westwood, I.M.; et al. Introduction of a Methyl Group Curbs Metabolism of Pyrido[3,4- d]pyrimidine Monopolar Spindle 1 (MPS1) Inhibitors and Enables the Discovery of the Phase 1 Clinical Candidate N 2-(2-Ethoxy-4-(4-methyl-4 H-1,2,4-triazol-3-yl)phenyl)-6-methyl- N 8-neopentylp. J. Med. Chem. 2018, 61, 8226–8240. [Google Scholar] [CrossRef]
- Anderhub, S.J.; Mak, G.W.-Y.; Gurden, M.D.; Faisal, A.; Drosopoulos, K.; Walsh, K.; Woodward, H.L.; Innocenti, P.; Westwood, I.M.; Naud, S.; et al. High Proliferation Rate and a Compromised Spindle Assembly Checkpoint Confers Sensitivity to the MPS1 Inhibitor BOS172722 in Triple-Negative Breast Cancers. Mol. Cancer Ther. 2019, 18, 1696–1707. [Google Scholar] [CrossRef] [Green Version]
- Mason, J.M.; Wei, X.; Fletcher, G.C.; Kiarash, R.; Brokx, R.; Hodgson, R.; Beletskaya, I.; Bray, M.R.; Mak, T.W. Functional characterization of CFI-402257, a potent and selective Mps1/TTK kinase inhibitor, for the treatment of cancer. Proc. Natl. Acad. Sci. USA 2017, 114, 3127–3132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; Chen, Z.; Kawakami, M.; Chen, Y.; Roszik, J.; Mustachio, L.M.; Kurie, J.M.; Villalobos, P.; Lu, W.; Behrens, C.; et al. Tyrosine threonine kinase inhibition eliminates lung cancers by augmenting apoptosis and polyploidy. Mol. Cancer. Ther. 2019, 18, 1775–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szymiczek, A.; Carbone, M.; Pastorino, S.; Napolitano, A.; Tanji, M.; Minaai, M.; Pagano, I.; Mason, J.M.; Pass, H.I.; Bray, M.R.; et al. Inhibition of the spindle assembly checkpoint kinase Mps-1 as a novel therapeutic strategy in malignant mesothelioma. Oncogene 2017, 36, 6501–6507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Boussac, H.; Bruyer, A.; Jourdan, M.; Maes, A.; Robert, N.; Gourzones, C.; Vincent, L.; Seckinger, A.; Cartron, G.; Hose, D.; et al. Kinome expression profiling to target new therapeutic avenues in multiple myeloma. Haematologica 2020, 105, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Bruinsma, W.; Raaijmakers, J.A.; Medema, R.H. Switching Polo-like kinase-1 on and off in time and space. Trends Biochem. Sci. 2012, 37, 534–542. [Google Scholar] [CrossRef]
- Colicino, E.G.; Hehnly, H. Regulating a key mitotic regulator, polo-like kinase 1 (PLK1). Cytoskeleton 2018, 75, 481–494. [Google Scholar] [CrossRef] [Green Version]
- Combes, G.; Alharbi, I.; Braga, L.G.; Elowe, S. Playing polo during mitosis: PLK1 takes the lead. Oncogene 2017, 36, 4819–4827. [Google Scholar] [CrossRef]
- Wang, H.; Tao, Z.; Feng, M.; Li, X.; Deng, Z.; Zhao, G.; Yin, H.; Pan, T.; Chen, G.; Feng, Z.; et al. Dual PLK1 and STAT3 inhibition promotes glioblastoma cells apoptosis through MYC. Biochem. Biophys. Res. Commun. 2020, 533, 368–375. [Google Scholar] [CrossRef]
- Ueda, A.; Oikawa, K.; Fujita, K.; Ishikawa, A.; Sato, E.; Ishikawa, T.; Kuroda, M.; Kanekura, K. Therapeutic potential of PLK1 inhibition in triple-negative breast cancer. Lab. Investig. 2019, 99, 1275–1286. [Google Scholar] [CrossRef]
- Pajtler, K.W.; Sadowski, N.; Ackermann, S.; Althoff, K.; Schönbeck, K.; Batzke, K.; Sch, S.; Odersky, A.; Heukamp, L.; Astrahantseff, K.; et al. The GSK461364 PLK1 inhibitor exhibits strong antitumoral activity in preclinical neuroblastoma models. Oncotarget 2016, 8, 6730–6741. [Google Scholar] [CrossRef]
- Gutteridge, R.E.A.; Ndiaye, M.A.; Liu, X.; Ahmad, N. Plk1 inhibitors in cancer therapy: From laboratory to clinics. Mol. Cancer Ther. 2016, 15, 1427–1435. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Sun, Q.; Wang, X. PLK1, A Potential Target for Cancer Therapy. Transl. Oncol. 2016, 10, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Lénárt, P.; Petronczki, M.; Steegmaier, M.; Di Fiore, B.; Lipp, J.J.; Hoffmann, M.; Rettig, W.J.; Kraut, N.; Peters, J.-M. The Small-Molecule Inhibitor BI 2536 Reveals Novel Insights into Mitotic Roles of Polo-like Kinase 1. Curr. Biol. 2007, 17, 304–315. [Google Scholar] [CrossRef] [Green Version]
- Sebastian, M.; Reck, M.; Waller, C.F.; Kortsik, C.; Frickhofen, N.; Schuler, M.; Fritsch, H.; Gaschler-Markefski, B.; Hanft, G.; Munzert, G.; et al. The efficacy and safety of BI 2536, a novel Plk-1 inhibitor, in patients with stage IIIB/IV non-small cell lung cancer who had relapsed after, or failed, chemotherapy: Results from an open-label, randomized phase ii clinical trial. J. Thorac. Oncol. 2010, 5, 1060–1067. [Google Scholar] [CrossRef]
- Ellis, P.M.; Chu, Q.S.; Leighl, N.; Laurie, S.A.; Fritsch, H.; Gaschler-Markefski, B.; Gyorffy, S.; Munzert, G. A phase i open-label dose-escalation study of intravenous BI 2536 together with pemetrexed in previously treated patients with non-small-cell lung cancer. Clin. Lung Cancer 2013, 14, 19–27. [Google Scholar] [CrossRef]
- Mross, K.; Dittrich, C.; Aulitzky, W.E.; Strumberg, D.; Schutte, J.; Schmid, R.M.; Hollerbach, S.; Merger, M.; Munzert, G.; Fleischer, F.; et al. A randomised phase II trial of the Polo-like kinase inhibitor BI 2536 in chemo-nave patients with unresectable exocrine adenocarcinoma of the pancreas-a study within the Central European Society Anticancer Drug Research (CESAR) collaborative network. Br. J. Cancer 2012, 107, 280–286. [Google Scholar] [CrossRef] [Green Version]
- Müller-Tidow, C.; Bug, G.; Lübbert, M.; Krämer, A.; Krauter, J.; Valent, P.; Nachbaur, D.; Berdel, W.E.; Ottmann, O.G.; Fritsch, H.; et al. A randomized, open-label, phase I/II trial to investigate the maximum tolerated dose of the Polo-like kinase inhibitor BI 2536 in elderly patients with refractory/relapsed acute myeloid leukaemia. Br. J. Haematol. 2013, 163, 214–222. [Google Scholar] [CrossRef]
- Vose, J.M.; Friedberg, J.W.; Waller, E.K.; Cheson, B.D.; Juvvigunta, V.; Fritsch, H.; Petit, C.; Munzert, G.; Younes, A. The Plk1 inhibitor BI 2536 in patients with refractory or relapsed non-Hodgkin lymphoma: A phase I, open-label, single dose-escalation study. Leuk. Lymphoma 2013, 54, 708–713. [Google Scholar] [CrossRef]
- Rudolph, D.; Steegmaier, M.; Hoffmann, M.; Grauert, M.; Baum, A.; Quant, J.; Haslinger, C.; Garin-Chesa, P.; Adolf, G.R. BI 6727, a polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin. Cancer Res. 2009, 15, 3094–3102. [Google Scholar] [CrossRef] [Green Version]
- Schöffski, P.; Awada, A.; Dumez, H.; Gil, T.; Bartholomeus, S.; Wolter, P.; Taton, M.; Fritsch, H.; Glomb, P.; Munzert, G. A phase I, dose-escalation study of the novel Polo-like kinase inhibitor volasertib (BI 6727) in patients with advanced solid tumours. Eur. J. Cancer 2012, 48, 179–186. [Google Scholar] [CrossRef]
- Nokihara, H.; Yamada, Y.; Fujiwara, Y.; Yamamoto, N.; Wakui, H.; Nakamichi, S.; Kitazono, S.; Inoue, K.; Harada, A.; Taube, T.; et al. Phase I trial of volasertib, a Polo-like kinase inhibitor, in Japanese patients with advanced solid tumors. Investig. New Drugs 2016, 34, 66–74. [Google Scholar] [CrossRef]
- Lin, C.-C.; Su, W.-C.; Yen, C.-J.; Hsu, C.-H.; Su, W.-P.; Yeh, K.-H.; Lu, Y.-S.; Cheng, A.-L.; Huang, D.C.-L.; Fritsch, H.; et al. A phase I study of two dosing schedules of volasertib (BI 6727), an intravenous polo-like kinase inhibitor, in patients with advanced solid malignancies. Br. J. Cancer 2014, 110, 2434–2440. [Google Scholar] [CrossRef] [Green Version]
- Stadler, W.M.; Vaughn, D.J.; Sonpavde, G.; Vogelzang, N.J.; Tagawa, S.T.; Petrylak, D.P.; Rosen, P.; Lin, C.-C.; Mahoney, J.; Modi, S.; et al. An open-label, single-arm, phase 2 trial of the polo-like kinase inhibitor volasertib (BI 6727) in patients with locally advanced or metastatic urothelial cancer. Cancer 2014, 120, 976–982. [Google Scholar] [CrossRef] [Green Version]
- Pujade-Lauraine, E.; Selle, F.; Weber, B.; Ray-Coquard, I.-L.; Vergote, I.; Sufliarsky, J.; Del Campo, J.M.; Lortholary, A.; Lesoin, A.; Follana, P.; et al. Volasertib Versus Chemotherapy in Platinum-Resistant or -Refractory Ovarian Cancer: A Randomized Phase II Groupe des Investigateurs Nationaux pour l’Etude des Cancers de l’Ovaire Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 706–713. [Google Scholar] [CrossRef]
- Ellis, P.M.; Leighl, N.B.; Hirsh, V.; Reaume, M.N.; Blais, N.; Wierzbicki, R.; Sadrolhefazi, B.; Gu, Y.; Liu, D.; Pilz, K.; et al. A Randomized, Open-Label Phase II Trial of Volasertib as Monotherapy and in Combination With Standard-Dose Pemetrexed Compared With Pemetrexed Monotherapy in Second-Line Treatment for Non-Small-Cell Lung Cancer. Clin. Lung Cancer 2015, 16, 457–465. [Google Scholar] [CrossRef] [PubMed]
- De Braud, F.; Cascinu, S.; Spitaleri, G.; Pilz, K.; Clementi, L.; Liu, D.; Sikken, P.; De Pas, T. A phase I, dose-escalation study of volasertib combined with nintedanib in advanced solid tumors. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2015, 26, 2341–2346. [Google Scholar] [CrossRef] [PubMed]
- Machiels, J.-P.; Peeters, M.; Herremans, C.; Surmont, V.; Specenier, P.; De Smet, M.; Pilz, K.; Strelkowa, N.; Liu, D.; Rottey, S. A phase I study of volasertib combined with afatinib, in advanced solid tumors. Cancer Chemother. Pharm. Ther. 2015, 76, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Awada, A.; Dumez, H.; Aftimos, P.G.; Costermans, J.; Bartholomeus, S.; Forceville, K.; Berghmans, T.; Meeus, M.-A.; Cescutti, J.; Munzert, G.; et al. Phase I trial of volasertib, a Polo-like kinase inhibitor, plus platinum agents in solid tumors: Safety, pharmacokinetics and activity. Investig. New Drugs 2015, 33, 611–620. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Yamauchi, T.; Kiyoi, H.; Sakura, T.; Hata, T.; Ando, K.; Watabe, A.; Harada, A.; Taube, T.; Miyazaki, Y.; et al. Phase I trial of volasertib, a Polo-like kinase inhibitor, in Japanese patients with acute myeloid leukemia. Cancer Sci. 2015, 106, 1590–1595. [Google Scholar] [CrossRef]
- Gumireddy, K.; Reddy, M.V.R.; Cosenza, S.C.; Nathan, R.B.; Baker, S.J.; Papathi, N.; Jiang, J.; Holland, J.; Reddy, E.P. ON01910 a non-ATP-competitive small molecule inhibitor of Plk1, is a potent anticancer agent. Cancer Cell 2005, 7, 275–286. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.W.; Messersmith, W.A.; Dy, G.K.; Weekes, C.D.; Whitworth, A.; Ren, C.; Maniar, M.; Wilhelm, F.; Eckhardt, S.G.; Adjei, A.A.; et al. Phase I study of Rigosertib, an inhibitor of the phosphatidylinositol 3-kinase and Polo-like kinase 1 pathways, combined with gemcitabine in patients with solid tumors and pancreatic cancer. Clin. Cancer Res. 2012, 18, 2048–2055. [Google Scholar] [CrossRef] [Green Version]
- O’Neil, B.H.; Scott, A.J.; Ma, W.W.; Cohen, S.J.; Aisner, D.L.; Menter, A.R.; Tejani, M.A.; Cho, J.K.; Granfortuna, J.; Coveler, L.; et al. A phase II/III randomized study to compare the efficacy and safety of rigosertib plus gemcitabine versus gemcitabine alone in patients with previously untreated metastatic pancreatic cancer. Ann. Oncol. 2015, 26, 1923–1929. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Fenaux, P.; Al-Kali, A.; Baer, M.R.; Sekeres, M.A.; Roboz, G.J.; Gaidano, G.; Scott, B.L.; Greenberg, P.; Platzbecker, U.; et al. Rigosertib versus best supportive care for patients with high-risk myelodysplastic syndromes after failure of hypomethylating drugs (ONTIME): A randomised, controlled, phase 3 trial. Lancet Oncol. 2016, 17, 496–508. [Google Scholar] [CrossRef]
- Al-Kali, A.; Hiwase, D.; Baer, M.R.; Greenberg, P.; Shortt, J.; Collins, R.; Steensma, D.P.; Verma, A.; Roboz, G.J.; Shammo, J.M.; et al. Relationship of bone marrow blast (BMBL) response to overall survival (OS) in a multicenter study of rigosertib (Rigo) in patients (pts) with myelodysplastic syndrome (MDS) with excess blasts progressing on or after treatment with a hypomethylating agent. JCO 2017, 35, 7056. [Google Scholar] [CrossRef]
- Navada, S.C.; Garcia-Manero, G.; OdchimarReissig, R.; Pemmaraju, N.; Alvarado, Y.; Ohanian, M.N.; John, R.B.; Demakos, E.P.; Zbyszewski, P.S.; Maniar, M.; et al. Rigosertib in combination with azacitidine in patients with myelodysplastic syndromes or acute myeloid leukemia: Results of a phase 1 study. Leuk Res. 2020, 94, 106369. [Google Scholar] [CrossRef]
- Gilmartin, A.G.; Bleam, M.R.; Richter, M.C.; Erskine, S.G.; Kruger, R.G.; Madden, L.; Hassler, D.F.; Smith, G.K.; Gontarek, R.R.; Courtney, M.P.; et al. Distinct concentration-dependent effects of the polo-like kinase 1-specific inhibitor GSK461364A, including differential effect on apoptosis. Cancer Res. 2009, 69, 6969–6977. [Google Scholar] [CrossRef] [Green Version]
- Olmos, D.; Barker, D.; Sharma, R.; Brunetto, A.T.; Yap, T.A.; Taegtmeyer, A.B.; Barriuso, J.; Medani, H.; Degenhardt, Y.Y.; Allred, A.J.; et al. Phase I study of GSK461364, a specific and competitive Polo-like kinase 1 inhibitor, in patients with advanced solid malignancies. Clin. Cancer Res. An. Off. J. Am. Assoc. Cancer Res. 2011, 17, 3420–3430. [Google Scholar] [CrossRef] [Green Version]
- Doi, T.; Murakami, H.; Wan, K.; Miki, M.; Kotani, H.; Sakamoto, N.; Yamamoto, N.; Ohtsu, A. A first-in-human phase I dose-escalation study of MK-1496, first-in-class orally available novel PLK1 inhibitor, in patients with advanced solid tumors. JCO 2011, 29, 3012. [Google Scholar] [CrossRef]
- Nie, Z.; Feher, V.; Natala, S.; McBride, C.; Kiryanov, A.; Jones, B.; Lam, B.; Liu, Y.; Kaldor, S.; Stafford, J.; et al. Discovery of TAK-960: An orally available small molecule inhibitor of polo-like kinase 1 (PLK1). Bioorganic. Med. Chem. Lett. 2013, 23, 3662–3666. [Google Scholar] [CrossRef]
- Hikichi, Y.; Honda, K.; Hikami, K.; Miyashita, H.; Kaieda, I.; Murai, S.; Uchiyama, N.; Hasegawa, M.; Kawamoto, T.; Sato, T.; et al. TAK-960, a novel, orally available, selective inhibitor of polo-like kinase 1, shows broad-spectrum preclinical antitumor activity in multiple dosing regimens. Mol. Cancer Ther. 2012, 11, 700–709. [Google Scholar] [CrossRef] [Green Version]
- Valsasina, B.; Beria, I.; Alli, C.; Alzani, R.; Avanzi, N.; Ballinari, D.; Cappella, P.; Caruso, M.; Casolaro, A.; Ciavolella, A.; et al. NMS-P937, an orally available, specific small-molecule polo-like kinase 1 inhibitor with antitumor activity in solid and hematologic malignancies. Mol. Cancer Ther. 2012, 11, 1006–1016. [Google Scholar] [CrossRef] [Green Version]
- Weiss, G.J.; Jameson, G.; Von Hoff, D.D.; Valsasina, B.; Davite, C.; Di Giulio, C.; Fiorentini, F.; Alzani, R.; Carpinelli, P.; Di Sanzo, A.; et al. Phase I dose escalation study of NMS-1286937, an orally available Polo-Like Kinase 1 inhibitor, in patients with advanced or metastatic solid tumors. Investig. New Drugs 2018, 36, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Demeure, M.J.; Armaghany, T.; Ejadi, S.; Ramanathan, R.K.; Elfiky, A.; Strosberg, J.R.; Smith, D.C.; Whitsett, T.; Liang, W.S.; Sekar, S.; et al. A phase I/II study of TKM-080301, a PLK1 -targeted RNAi in patients with adrenocortical cancer (ACC). JCO 2016, 34, 2547. [Google Scholar] [CrossRef]
- El Dika, I.; Lim, H.Y.; Yong, W.P.; Lin, C.-C.; Yoon, J.-H.; Modiano, M.; Freilich, B.; Choi, H.J.; Chao, T.-Y.; Kelley, R.K.; et al. An Open-Label, Multicenter, Phase I, Dose Escalation Study with Phase II Expansion Cohort to Determine the Safety, Pharmacokinetics, and Preliminary Antitumor Activity of Intravenous TKM-080301 in Subjects with Advanced Hepatocellular Carcinoma. Oncologist 2019, 24, 747. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://cyclacel.com/research_programs_oncology_cyc140.shtml (accessed on 8 May 2021).
- Navada, S.C.; Fruchtman, S.M.; Odchimar-Reissig, R.; Demakos, E.P.; Petrone, M.E.; Zbyszewski, P.S.; Holland, J.F.; Silverman, L.R. A phase 1/2 study of rigosertib in patients with myelodysplastic syndromes (MDS) and MDS progressed to acute myeloid leukemia. Leuk Res. 2018, 64, 10–16. [Google Scholar] [CrossRef]
- Komrokji, R.S.; Raza, A.; Lancet, J.E.; Ren, C.; Taft, D.; Maniar, M.; Wilhelm, F.; List, A.F. Phase I clinical trial of oral rigosertib in patients with myelodysplastic syndromes. Br. J. Haematol. 2013, 162, 517–524. [Google Scholar] [CrossRef]
- King, S.I.; Purdie, C.A.; Bray, S.E.; Quinlan, P.R.; Jordan, L.B.; Thompson, A.M.; Meek, D.W. Immunohistochemical detection of Polo-like kinase-1 (PLK1) in primary breast cancer is associated with TP53 mutation and poor clinical outcome. Breast Cancer Res. 2012, 14, R40. [Google Scholar] [CrossRef] [Green Version]
- Guan, R.; Tapang, P.; Leverson, J.D.; Albert, D.; Giranda, V.L.; Luo, Y. Small Interfering RNA–Mediated Polo-Like Kinase 1 Depletion Preferentially Reduces the Survival of p53-Defective, Oncogenic Transformed Cells and Inhibits Tumor Growth in Animals. Cancer Res. 2005, 65, 2698–2704. [Google Scholar] [CrossRef] [Green Version]
- Bolanos-Garcia, V.M. Aurora kinases. Int. J. Biochem. Cell Biol. 2005, 37, 1572–1577. [Google Scholar] [CrossRef]
- Willems, E.; Dedobbeleer, M.; Digregorio, M.; Lombard, A.; Lumapat, P.N.; Rogister, B. The functional diversity of Aurora kinases: A comprehensive review. Cell Div. 2018, 13, 7. [Google Scholar] [CrossRef] [Green Version]
- Girdler, F.; Gascoigne, K.E.; Eyers, P.A.; Hartmuth, S.; Crafter, C.; Foote, K.M.; Keen, N.J.; Taylor, S.S. Validating Aurora B as an anti-cancer drug target. J. Cell Sci. 2006, 119, 3664–3675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauf, S.; Cole, R.W.; LaTerra, S.; Zimmer, C.; Schnapp, G.; Walter, R.; Heckel, A.; van Meel, J.; Rieder, C.L.; Peters, J.-M. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J. Cell Biol. 2003, 161, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Yan, L.; Torres, R.; Gong, X.; Bian, H.; Marugán, C.; Boehnke, K.; Baquero, C.; Hui, Y.-H.; Chapman, S.C.; et al. Aurora A-Selective Inhibitor LY3295668 Leads to Dominant Mitotic Arrest, Apoptosis in Cancer Cells, and Shows Potent Preclinical Antitumor Efficacy. Mol. Cancer Ther. 2019, 18, 2207–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaestner, P.; Stolz, A.; Bastians, H. Determinants for the efficiency of anticancer drugs targeting either Aurora-A or Aurora-B kinases in human colon carcinoma cells. Mol. Cancer Ther. 2009, 8, 2046–2056. [Google Scholar] [CrossRef] [Green Version]
- Glaser, K.B.; Li, J.; Marcotte, P.A.; Magoc, T.J.; Guo, J.; Reuter, D.R.; Tapang, P.; Wei, R.-Q.; Pease, L.J.; Bui, M.H.; et al. Preclinical characterization of ABT-348, a kinase inhibitor targeting the aurora, vascular endothelial growth factor receptor/platelet-derived growth factor receptor, and Src kinase families. J. Pharm. Exp. Ther. 2012, 343, 617–627. [Google Scholar] [CrossRef]
- Maitland, M.L.; Piha-Paul, S.; Falchook, G.; Kurzrock, R.; Nguyen, L.; Janisch, L.; Karovic, S.; McKee, M.; Hoening, E.; Wong, S.; et al. Clinical pharmacodynamic/exposure characterisation of the multikinase inhibitor ilorasertib (ABT-348)in a phase 1 dose-escalation trial. Br. J. Cancer 2018, 118, 1042–1050. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Tibes, R.; Kadia, T.; Kantarjian, H.; Arellano, M.; Knight, E.A.; Xiong, H.; Qin, Q.; Munasinghe, W.; Roberts-Rapp, L.; et al. Phase 1 dose escalation trial of ilorasertib, a dual Aurora/VEGF receptor kinase inhibitor, in patients with hematologic malignancies. Investig. New Drugs 2015, 33, 870–880. [Google Scholar] [CrossRef] [Green Version]
- Peter, B.; Bibi, S.; Eisenwort, G.; Wingelhofer, B.; Berger, D.; Stefanzl, G.; Blatt, K.; Herrmann, H.; Hadzijusufovic, E.; Hoermann, G.; et al. Drug-induced inhibition of phosphorylation of STAT5 overrides drug resistance in neoplastic mast cells. Leukemia 2018, 32, 1016–1022. [Google Scholar] [CrossRef]
- Mita, M.; Gordon, M.; Rejeb, N.; Gianella-Borradori, A.; Jego, V.; Mita, A.; Sarantopoulos, J.; Sankhala, K.; Mendelson, D. A phase l study of three different dosing schedules of the oral aurora kinase inhibitor MSC1992371A in patients with solid tumors. Target. Oncol. 2014, 9, 215–224. [Google Scholar] [CrossRef]
- Raymond, E.; Alexandre, J.; Faivre, S.; Goldwasser, F.; Besse-Hammer, T.; Gianella-Borradori, A.; Jego, V.; Trandafir, L.; Rejeb, N.; Awada, A. A phase I schedule dependency study of the aurora kinase inhibitor MSC1992371A in combination with gemcitabine in patients with solid tumors. Investig. New Drugs 2014, 32, 94–103. [Google Scholar] [CrossRef]
- Graux, C.; Sonet, A.; Maertens, J.; Duyster, J.; Greiner, J.; Chalandon, Y.; Martinelli, G.; Hess, D.; Heim, D.; Giles, F.J.; et al. A phase I dose-escalation study of MSC1992371A, an oral inhibitor of aurora and other kinases, in advanced hematologic malignancie. Leuk Res. 2013, 37, 1100–1106. [Google Scholar] [CrossRef] [Green Version]
- Harrington, E.A.; Bebbington, D.; Moore, J.; Rasmussen, R.K.; Ajose-Adeogun, A.O.; Nakayama, T.; Graham, J.A.; Demur, C.; Hercend, T.; Diu-Hercend, A.; et al. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat. Med. 2004, 10, 262–267. [Google Scholar] [CrossRef]
- Traynor, A.M.; Hewitt, M.; Liu, G.; Flaherty, K.T.; Clark, J.; Freedman, S.J.; Scott, B.B.; Leighton, A.M.; Watson, P.A.; Zhao, B.; et al. Phase I dose escalation study of MK-0457, a novel Aurora kinase inhibitor, in adult patients with advanced solid tumors. Cancer Chemother. Pharm. 2011, 67, 305–314. [Google Scholar] [CrossRef] [Green Version]
- Giles, F.J.; Swords, R.T.; Nagler, A.; Hochhaus, A.; Ottmann, O.G.; Rizzieri, D.A.; Talpaz, M.; Clark, J.; Watson, P.; Xiao, A.; et al. MK-0457, an Aurora kinase and BCR-ABL inhibitor, is active in patients with BCR-ABL T315I leukemia. Leukemia 2013, 27, 113–117. [Google Scholar] [CrossRef]
- Sini, P.; Gürtler, U.; Zahn, S.K.; Baumann, C.; Rudolph, D.; Baumgartinger, R.; Strauss, E.; Haslinger, C.; Tontsch-Grunt, U.; Waizenegger, I.C.; et al. Pharmacological profile of BI 847325, an orally bioavailable, ATP-competitive inhibitor of MEK and Aurora kinases. Mol. Cancer Ther. 2016, 15, 2388–2398. [Google Scholar] [CrossRef] [Green Version]
- Schöffski, P.; Aftimos, P.; Dumez, H.; Deleporte, A.; De Block, K.; Costermans, J.; Billiet, M.; Meeus, M.-A.; Lee, C.; Schnell, D.; et al. A phase I study of two dosing schedules of oral BI 847325 in patients with advanced solid tumors. Cancer Chemother Pharm. 2016, 77, 99–108. [Google Scholar] [CrossRef]
- Howard, S.; Berdini, V.; Boulstridge, J.A.; Carr, M.G.; Cross, D.M.; Curry, J.; Devine, L.A.; Early, T.R.; Fazal, L.; Gill, A.L.; et al. Fragment-based discovery of the pyrazol-4-yl urea (AT9283), a multitargeted kinase inhibitor with potent aurora kinase activity. J. Med. Chem 2009, 52, 379–388. [Google Scholar] [CrossRef]
- Dent, S.F.; Gelmon, K.A.; Chi, K.N.; Jonker, D.J.; Wainman, N.; Capier, C.A.; Chen, E.X.; Lyons, J.F.; Seymour, L. NCIC CTG IND.181: Phase i study of AT9283 given as a weekly 24 hour infusion in advanced malignancies. Investig. New Drugs 2013, 31, 1522–1529. [Google Scholar] [CrossRef] [PubMed]
- Foran, J.; Ravandi, F.; Wierda, W.; Garcia-Manero, G.; Verstovsek, S.; Kadia, T.; Burger, J.; Yule, M.; Langford, G.; Lyons, J.; et al. A phase i and pharmacodynamic study of AT9283, a small-molecule inhibitor of aurora kinases in patients with relapsed/refractory leukemia or myelofibrosis. Clin. Lymphoma Myeloma Leuk. 2014, 14, 223–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, L.; Marshall, L.V.; Pearson, A.D.J.; Morland, B.; Elliott, M.; Campbell-Hewson, Q.; Makin, G.; Halford, S.E.R.; Acton, G.; Ross, P.; et al. A phase i trial of AT9283 (a selective inhibitor of aurora kinases) in children and adolescents with solid tumors: A cancer research UK study. Clin. Cancer Res. 2015, 21, 267–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payton, M.; Bush, T.L.; Chung, G.; Ziegler, B.; Eden, P.; McElroy, P.; Ross, S.; Cee, V.J.; Deak, H.L.; Hodous, B.L.; et al. Preclinical evaluation of AMG 900, a novel potent and highly selective pan-aurora kinase inhibitor with activity in taxane-resistant tumor cell lines. Cancer Res. 2010, 70, 9846–9854. [Google Scholar] [CrossRef] [Green Version]
- Carducci, M.; Shaheen, M.; Markman, B.; Hurvitz, S.; Mahadevan, D.; Kotasek, D.; Goodman, O.B.; Rasmussen, E.; Chow, V.; Juan, G.; et al. A phase 1, first-in-human study of AMG 900, an orally administered pan-Aurora kinase inhibitor, in adult patients with advanced solid tumors. Investig. New Drugs 2018, 36, 1060–1071. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Schuster, M.W.; Jain, N.; Advani, A.; Jabbour, E.; Gamelin, E.; Rasmussen, E.; Juan, G.; Anderson, A.; Chow, V.F.; et al. A phase 1 study of AMG 900, an orally administered pan-aurora kinase inhibitor, in adult patients with acute myeloid leukemia. Am. J. Hematol. 2017, 92, 660–667. [Google Scholar] [CrossRef] [Green Version]
- Carpinelli, P.; Ceruti, R.; Giorgini, M.L.; Cappella, P.; Gianellini, L.; Croci, V.; Degrassi, A.; Texido, G.; Rocchetti, M.; Vianello, P.; et al. PHA-739358, a potent inhibitor of Aurora kinases with a selective target inhibition profile relevant to cancer. Mol. Cancer Ther. 2007, 6, 3158–3168. [Google Scholar] [CrossRef] [Green Version]
- Meulenbeld, H.J.; Bleuse, J.P.; Vinci, E.M.; Raymond, E.; Vitali, G.; Santoro, A.; Dogliotti, L.; Berardi, R.; Cappuzzo, F.; Tagawa, S.T.; et al. Randomized phase II study of danusertib in patients with metastatic castration-resistant prostate cancer after docetaxel failure. BJU Int. 2013, 111, 44–52. [Google Scholar] [CrossRef]
- Oslob, J.D.; Romanowski, M.J.; Allen, D.A.; Baskaran, S.; Bui, M.; Elling, R.A.; Flanagan, W.M.; Fung, A.D.; Hanan, E.J.; Harris, S.; et al. Discovery of a potent and selective aurora kinase inhibitor. Bioorg. Med. Chem Lett. 2008, 18, 4880–4884. [Google Scholar] [CrossRef]
- Robert, F.; Verschraegen, C.; Hurwitz, H.; Uronis, H.; Advani, R.; Chen, A.; Taverna, P.; Wollman, M.; Fox, J.; Michelson, G. A phase I trial of sns-314, a novel and selective pan-aurora kinase inhibitor, in advanced solid tumor patients. JCO 2009, 27, 2536. [Google Scholar] [CrossRef]
- Farrell, P.; Shi, L.; Matuszkiewicz, J.; Balakrishna, D.; Hoshino, T.; Zhang, L.; Elliott, S.; Fabrey, R.; Lee, B.; Halkowycz, P.; et al. Biological characterization of TAK-901, an investigational, novel, multitargeted Aurora B kinase inhibitor. Mol. Cancer Ther. 2013, 12, 460–470. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Midgley, C.A.; Scaërou, F.; Grabarek, J.B.; Griffiths, G.; Jackson, W.; Kontopidis, G.; McClue, S.J.; McInnes, C.; Meades, C.; et al. Discovery of N-Phenyl-4-(thiazol-5-yl)pyrimidin-2-amine aurora kinase inhibitors. J. Med. Chem. 2010, 53, 4367–4378. [Google Scholar] [CrossRef] [Green Version]
- Adams, N.D.; Adams, J.L.; Burgess, J.L.; Chaudhari, A.M.; Copeland, R.A.; Donatelli, C.A.; Drewry, D.H.; Fisher, K.E.; Hamajima, T.; Hardwicke, M.A.; et al. Discovery of GSK1070916, a potent and selective inhibitor of aurora B/C kinase. J. Med. Chem. 2010, 53, 3973–4001. [Google Scholar] [CrossRef]
- Jani, J.P.; Arcari, J.; Bernardo, V.; Bhattacharya, S.K.; Briere, D.; Cohen, B.D.; Coleman, K.; Christensen, J.G.; Emerson, E.O.; Jakowski, A.; et al. PF-03814735, an orally bioavailable small molecule aurora kinase inhibitor for cancer therapy. Mol. Cancer Ther. 2010, 9, 883–894. [Google Scholar] [CrossRef] [Green Version]
- Schöffski, P.; Jones, S.F.; Dumez, H.; Infante, J.R.; Van Mieghem, E.; Fowst, C.; Gerletti, P.; Xu, H.; Jakubczak, J.L.; English, P.A.; et al. Phase I, open-label, multicentre, dose-escalation, pharmacokinetic and pharmacodynamic trial of the oral aurora kinase inhibitor PF-03814735 in advanced solid tumours. Eur. J. Cancer 2011, 47, 2256–2264. [Google Scholar] [CrossRef]
- Seymour, J.F.; Kim, D.W.; Rubin, E.; Haregewoin, A.; Clark, J.; Watson, P.; Hughes, T.; Dufva, I.; Jimenez, J.L.; Mahon, F.-X.; et al. A phase 2 study of MK-0457 in patients with BCR-ABL T315I mutant chronic myelogenous leukemia and philadelphia chromosome-positive acute lymphoblastic leukemia. Blood Cancer J. 2014, 4, e238. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Ikezoe, T.; Nishioka, C.; Tasaka, T.; Taniguchi, A.; Kuwayama, Y.; Komatsu, N.; Bandobashi, K.; Togitani, K.; Koeffler, H.P.; et al. AZD1152, a novel and selective aurora B kinase inhibitor, induces growth arrest, apoptosis, and sensitization for tubulin depolymerizing agent or topoisomerase II inhibitor in human acute leukemia cells in vitro and in vivo. Blood 2007, 110, 2034–2040. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, G.K.; Carvajal, R.D.; Midgley, R.; Rodig, S.J.; Stockman, P.K.; Ataman, O.; Wilson, D.; Das, S.; Shapiro, G.I. Phase i study of barasertib (AZD1152), a selective inhibitor of Aurora B kinase, in patients with advanced solid tumors. Investig. New Drugs 2013, 31, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Boss, D.S.; Witteveen, P.O.; van der Sar, J.; Lolkema, M.P.; Voest, E.E.; Stockman, P.K.; Ataman, O.; Wilson, D.; Das, S.; Schellens, J.H. Clinical evaluation of AZD1152, an i.v. inhibitor of Aurora B kinase, in patients with solid malignant tumors. Ann. Oncol. 2011, 22, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Löwenberg, B.; Muus, P.; Ossenkoppele, G.; Rousselot, P.; Cahn, J.-Y.; Ifrah, N.; Martinelli, G.; Amadori, S.; Berman, E.; Sonneveld, P.; et al. Phase 1/2 study to assess the safety, efficacy, and pharmacokinetics of barasertib (AZD1152) in patients with advanced acute myeloid leukemia. Blood 2011, 118, 6030–6036. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, K.; Yokozawa, T.; Sakura, T.; Watanabe, T.; Fujisawa, S.; Yamauchi, T.; Uike, N.; Ando, K.; Kihara, R.; Tobinai, K.; et al. A phase I study to assess the safety, pharmacokinetics and efficacy of barasertib (AZD1152), an Aurora B kinase inhibitor, in Japanese patients with advanced acute myeloid leukemia. Leuk. Res. 2011, 35, 1384–1389. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Martinelli, G.; Jabbour, E.J.; Quintás-Cardama, A.; Ando, K.; Bay, J.-O.; Wei, A.; Gröpper, S.; Papayannidis, C.; Owen, K.; et al. Stage i of a phase 2 study assessing the efficacy, safety, and tolerability of barasertib (AZD1152) versus low-dose cytosine arabinoside in elderly patients with acute myeloid leukemia. Cancer 2013, 119, 2611–2619. [Google Scholar] [CrossRef]
- Dennis, M.; Davies, M.; Oliver, S.; D’Souza, R.; Pike, L.; Stockman, P. Phase i study of the Aurora B kinase inhibitor barasertib (AZD1152) to assess the pharmacokinetics, metabolism and excretion in patients with acute myeloid leukemi. Cancer Chemother. Pharm. 2012, 70, 461–469. [Google Scholar] [CrossRef] [Green Version]
- Kantarjian, H.M.; Sekeres, M.A.; Ribrag, V.; Rousselot, P.; Garcia-Manero, G.; Jabbour, E.J.; Owen, K.; Stockman, P.K.; Oliver, S.D. Phase I study assessing the safety and tolerability of barasertib (AZD1152) with low-dose cytosine arabinoside in elderly patients with AML. Clin. Lymphoma Myeloma Leuk. 2013, 13, 559–567. [Google Scholar] [CrossRef] [Green Version]
- Collins, G.P.; Eyre, T.A.; Linton, K.M.; Radford, J.; Vallance, G.D.; Soilleux, E.; Hatton, C. A phase II trial of AZD1152 in relapsed/refractory diffuse large B-cell lymphoma. Br. J. Haematol. 2015, 170, 886–890. [Google Scholar] [CrossRef]
- Available online: https://opnme.com/molecules/aurb-bi831266 (accessed on 1 July 2021).
- Dittrich, C.; Fridrik, M.A.; Koenigsberg, R.; Lee, C.; Goeldner, R.-G.; Hilbert, J.; Greil, R. A phase 1 dose escalation study of BI 831266, an inhibitor of Aurora kinase B, in patients with advanced solid tumors. Investig. New Drugs 2015, 33, 409–422. [Google Scholar] [CrossRef] [Green Version]
- Tontsch-Grunt, U.; Gürtler, U.; Zahn, S.K.; Boehmelt, G.; Jarvis, M.; Adolf, G.R.; Solca, F. Abstract 1080: Molecular and cellular pharmacology of BI 811283, a potent inhibitor of Aurora B kinase. Cancer Res. 2010, 70, 1080. [Google Scholar] [CrossRef]
- Mross, K.; Richly, H.; Frost, A.; Scharr, D.; Nokay, B.; Graeser, R.; Lee, C.; Hilbert, J.; Goeldner, R.-G.; Fietz, O.; et al. A phase I study of BI 811283, an Aurora B kinase inhibitor, in patients with advanced solid tumors. Cancer Chemother. Pharm. 2016, 78, 405–417. [Google Scholar] [CrossRef] [Green Version]
- Döhner, H.; Müller-Tidow, C.; Lübbert, M.; Fiedler, W.; Krämer, A.; Westermann, J.; Bug, G.; Schlenk, R.F.; Krug, U.; Goeldner, R.-G.; et al. A phase I trial investigating the Aurora B kinase inhibitor BI 811283 in combination with cytarabine in patients with acute myeloid leukaemia. Br. J. Haematol. 2019, 185, 583–587. [Google Scholar] [CrossRef]
- Zhou, Y.; Shan, S.; Li, Z.; Xin, L.; Pan, D.; Yang, Q.; Liu, Y.; Yue, X.; Liu, X.; Gao, J.; et al. CS2164, a novel multi-target inhibitor against tumor angiogenesis, mitosis and chronic inflammation with anti-tumor potency. Cancer Sci. 2017, 108, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Yang, L.; Hao, X.; Liu, Y.; Zhang, J.; Ning, Z.; Shi, Y. Phase I dose-escalation study of chiauranib, a novel angiogenic, mitotic, and chronic inflammation inhibitor, in patients with advanced solid tumors. J. Hematol. Oncol. 2019, 12, 9. [Google Scholar] [CrossRef]
- Manfredi, M.G.; Ecsedy, J.A.; Chakravarty, A.; Silverman, L.; Zhang, M.; Hoar, K.M.; Stroud, S.G.; Chen, W.; Shinde, V.; Huck, J.J.; et al. Characterization of alisertib (MLN8237), an investigational small-molecule inhibitor of Aurora A kinase using novel in vivo pharmacodynamic assays. Clin. Cancer Res. 2011, 17, 7614–7624. [Google Scholar] [CrossRef] [Green Version]
- Dees, E.C.; Cohen, R.B.; von Mehren, M.; Stinchcombe, T.E.; Liu, H.; Venkatakrishnan, K.; Manfredi, M.; Fingert, H.; Burris, H.A.; Infante, J.R. Phase I study of aurora A kinase inhibitor MLN8237 in advanced solid tumors: Safety, pharmacokinetics, pharmacodynamics, and bioavailability of two oral formulations. Clin. Cancer Res. 2012, 18, 4775–4784. [Google Scholar] [CrossRef] [Green Version]
- Matulonis, U.A.; Sharma, S.; Ghamande, S.; Gordon, M.S.; Del Prete, S.A.; Ray-Coquard, I.; Kutarska, E.; Liu, H.; Fingert, H.; Zhou, X.; et al. Phase II study of MLN8237 (alisertib), an investigational Aurora A kinase inhibitor, in patients with platinum-resistant or -refractory epithelial ovarian, fallopian tube, or primary peritoneal carcinoma. Gynecol. Oncol. 2012, 127, 63–69. [Google Scholar] [CrossRef]
- Dickson, M.A.; Mahoney, M.R.; Tap, W.D.; D’Angelo, S.P.; Keohan, M.L.; Van Tine, B.A.; Agulnik, M.; Horvath, L.E.; Nair, J.S.; Schwartz, G.K. Phase II study of MLN8237 (Alisertib) in advanced/metastatic sarcoma. Ann. Oncol. 2016, 27, 1855–1860. [Google Scholar] [CrossRef]
- Beltran, H.; Oromendia, C.; Danila, D.C.; Montgomery, B.; Hoimes, C.; Szmulewitz, R.Z.; Vaishampayan, U.; Armstrong, A.J.; Stein, M.; Pinski, J.; et al. A Phase II Trial of the Aurora Kinase a Inhibitor Alisertib for Patients with Castration-resistant and Neuroendocrine Prostate Cancer: Efficacy and Biomarkers. Clin. Cancer Res. 2019, 25, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Falchook, G.; Coleman, R.L.; Roszak, A.; Behbakht, K.; Matulonis, U.; Ray-Coquard, I.; Sawrycki, P.; Duska, L.R.; Tew, W.; Ghamande, S.; et al. Alisertib in Combination with Weekly Paclitaxel in Patients With Advanced Breast Cancer or Recurrent Ovarian Cancer: A Randomized Clinical Trial. JAMA Oncol. 2019, 5, e183773. [Google Scholar] [CrossRef] [Green Version]
- Necchi, A.; Lo Vullo, S.; Mariani, L.; Raggi, D.; Giannatempo, P.; Calareso, G.; Togliardi, E.; Crippa, F.; Di Genova, N.; Perrone, F.; et al. An open-label, single-arm, phase 2 study of the Aurora kinase a inhibitor alisertib in patients with advanced urothelial cancer. Investig. New Drugs 2016, 34, 236–242. [Google Scholar] [CrossRef]
- Owonikoko, T.K.; Niu, H.; Nackaerts, K.; Csoszi, T.; Ostoros, G.; Mark, Z.; Baik, C.; Joy, A.A.; Chouaid, C.; Jaime, J.C.; et al. Randomized Phase II Study of Paclitaxel plus Alisertib versus Paclitaxel plus Placebo as Second-Line Therapy for SCLC: Primary and Correlative Biomarker Analyses. J. Thorac. Oncol. 2020, 15, 274–287. [Google Scholar] [CrossRef] [Green Version]
- Graff, J.N.; Higano, C.S.; Hahn, N.M.; Taylor, M.H.; Zhang, B.; Zhou, X.; Venkatakrishnan, K.; Leonard, E.J.; Sarantopoulos, J. Open-label, multicenter, phase 1 study of alisertib (MLN8237), an aurora A kinase inhibitor, with docetaxel in patients with solid tumors. Cancer 2016, 122, 2524–2533. [Google Scholar] [CrossRef]
- Haddad, T.C.; D’Assoro, A.; Suman, V.; Opyrchal, M.; Peethambaram, P.; Liu, M.C.; Goetz, M.P.; Ingle, J.N. Phase I trial to evaluate the addition of alisertib to fulvestrant in women with endocrine-resistant, ER+ metastatic breast cancer. Breast Cancer Res. Treat. 2018, 168, 639–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goff, L.W.; Azad, N.S.; Stein, S.; Whisenant, J.G.; Koyama, T.; Vaishampayan, U.; Hochster, H.; Connolly, R.; Weise, A.; LoRusso, P.M.; et al. Phase I study combining the aurora kinase a inhibitor alisertib with mFOLFOX in gastrointestinal cancer. Investig. New Drugs 2019, 37, 315–322. [Google Scholar] [CrossRef] [PubMed]
- DuBois, S.G.; Mosse, Y.P.; Fox, E.; Kudgus, R.A.; Reid, J.M.; McGovern, R.; Groshen, S.; Bagatell, R.; Maris, J.M.; Twist, C.J.; et al. Phase II Trial of Alisertib in Combination with Irinotecan and Temozolomide for Patients with Relapsed or Refractory Neuroblastoma. Clin. Cancer Res. 2018, 24, 6142–6149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, H.A.; Fischer, J.H.; Venepalli, N.K.; Danciu, O.C.; Christian, S.; Russell, M.J.; Liu, L.C.; Zacny, J.P.; Dudek, A.Z. Phase I Study of Aurora A Kinase Inhibitor Alisertib (MLN8237) in Combination With Selective VEGFR Inhibitor Pazopanib for Therapy of Advanced Solid Tumors. Am. J. Clin. Oncol 2019, 42, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.R.; Shea, T.C.; Goy, A.; Berdeja, J.G.; Reeder, C.B.; McDonagh, K.T.; Zhou, X.; Danaee, H.; Liu, H.; Ecsedy, J.A.; et al. Phase I study of MLN8237—investigational Aurora A kinase inhibitor—in relapsed/refractory multiple myeloma, non-Hodgkin lymphoma and chronic lymphocytic leukemia. Investig. New Drugs 2014, 32, 489–499. [Google Scholar] [CrossRef] [Green Version]
- Barr, P.M.; Li, H.; Spier, C.; Mahadevan, D.; LeBlanc, M.; Ul Haq, M.; Huber, B.D.; Flowers, C.R.; Wagner-Johnston, N.D.; Horwitz, S.M.; et al. Phase II Intergroup Trial of Alisertib in Relapsed and Refractory Peripheral T-Cell Lymphoma and Transformed Mycosis Fungoides: SWOG 1108. J. Clin. Oncol. 2015, 33, 2399–2404. [Google Scholar] [CrossRef]
- O’Connor, O.A.; Özcan, M.; Jacobsen, E.D.; Roncero, J.M.; Trotman, J.; Demeter, J.; Masszi, T.; Pereira, J.; Ramchandren, R.; Beaven, A.; et al. Randomized phase III study of alisertib or investigator’s choice (selected single agent) in patients with relapsed or refractory peripheral T-cell lymphoma. J. Clin. Oncol. 2019, 37, 613–623. [Google Scholar] [CrossRef]
- Friedberg, J.W.; Mahadevan, D.; Cebula, E.; Persky, D.; Lossos, I.; Agarwal, A.B.; Jung, J.; Burack, R.; Zhou, X.; Leonard, E.J.; et al. Phase II study of alisertib, a selective Aurora A kinase inhibitor, in relapsed and refractory aggressive B- and T-cell non-Hodgkin lymphomas. J. Clin. Oncol. 2014, 32, 44–50. [Google Scholar] [CrossRef]
- Kelly, K.R.; Friedberg, J.W.; Park, S.I.; McDonagh, K.; Hayslip, J.; Persky, D.; Ruan, J.; Puvvada, S.; Rosen, P.; Iyer, S.P.; et al. Phase I Study of the Investigational Aurora A Kinase Inhibitor Alisertib plus Rituximab or Rituximab/Vincristine in Relapsed/Refractory Aggressive B-cell Lymphoma. Clin. Cancer Res. 2018, 24, 6150–6159. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, A.; Kumar, S.; Hofmeister, C.; Laubach, J.; Vij, R.; Dueck, A.; Gano, K.; Stewart, A.K. A Phase Ib Study of the combination of the Aurora Kinase Inhibitor Alisertib (MLN8237) and Bortezomib in Relapsed Multiple Myeloma. Br. J. Haematol. 2016, 174, 323–325. [Google Scholar] [CrossRef] [Green Version]
- Brunner, A.M.; Blonquist, T.M.; DeAngelo, D.J.; McMasters, M.; Fell, G.; Hermance, N.M.; Winer, E.S.; Lindsley, R.C.; Hobbs, G.S.; Amrein, P.C.; et al. Alisertib plus induction chemotherapy in previously untreated patients with high-risk, acute myeloid leukaemia: A single-arm, phase 2 trial. Lancet Haematol. 2020, 7, e122–e133. [Google Scholar] [CrossRef]
- Mossé, Y.P.; Lipsitz, E.; Fox, E.; Teachey, D.T.; Maris, J.M.; Weigel, B.; Adamson, P.C.; Ingle, M.A.; Ahern, C.H.; Blaney, S.M. Pediatric phase I trial and pharmacokinetic study of MLN8237, an investigational oral selective small-molecule inhibitor of Aurora kinase A: A Children’s Oncology Group Phase I Consortium study. Clin. Cancer Res. 2012, 18, 6058–6064. [Google Scholar] [CrossRef] [Green Version]
- Mossé, Y.P.; Fox, E.; Teachey, D.T.; Reid, J.M.; Safgren, S.L.; Carol, H.; Lock, R.B.; Houghton, P.J.; Smith, M.A.; Hall, D.; et al. A Phase II Study of Alisertib in Children with Recurrent/Refractory Solid Tumors or Leukemia: Children’s Oncology Group Phase I and Pilot Consortium (ADVL0921). Clin. Cancer Res. 2019, 25, 3229–3238. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, G.C.; Brokx, R.D.; Denny, T.A.; Hembrough, T.A.; Plum, S.M.; Fogler, W.E.; Sidor, C.F.; Bray, M.R. ENMD-2076 is an orally active kinase inhibitor with antiangiogenic and antiproliferative mechanisms of action. Mol. Cancer Ther. 2011, 10, 126–137. [Google Scholar] [CrossRef] [Green Version]
- Diamond, J.R.; Bastos, B.R.; Hansen, R.J.; Gustafson, D.L.; Eckhardt, S.G.; Kwak, E.L.; Pandya, S.S.; Fletcher, G.C.; Pitts, T.M.; Kulikowski, G.N.; et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of ENMD-2076, a novel angiogenic and aurora kinase inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2011, 17, 849–860. [Google Scholar] [CrossRef] [Green Version]
- Matulonis, U.A.; Lee, J.; Lasonde, B.; Tew, W.P.; Yehwalashet, A.; Matei, D.; Behbakht, K.; Grothusen, J.; Fleming, G.; Lee, N.K.; et al. ENMD-2076, an oral inhibitor of angiogenic and proliferation kinases, has activity in recurrent, platinum resistant ovarian cancer. Eur. J. Cancer 2013, 49, 121–131. [Google Scholar] [CrossRef]
- Lheureux, S.; Tinker, A.; Clarke, B.; Ghatage, P.; Welch, S.; Weberpals, J.I.; Dhani, N.C.; Butler, M.O.; Tonkin, K.; Tan, Q.; et al. A Clinical and Molecular Phase II Trial of Oral ENMD-2076 in Ovarian Clear Cell Carcinoma (OCCC): A Study of the Princess Margaret Phase II Consortium. Clin. Cancer Res. 2018, 24, 6168–6174. [Google Scholar] [CrossRef] [Green Version]
- Veitch, Z.; Zer, A.; Loong, H.; Salah, S.; Masood, M.; Gupta, A.; Bradbury, P.A.; Hogg, D.; Wong, A.; Kandel, R.; et al. A phase II study of ENMD-2076 in advanced soft tissue sarcoma (STS). Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Diamond, J.R.; Eckhardt, S.G.; Pitts, T.M.; van Bokhoven, A.; Aisner, D.; Gustafson, D.L.; Capasso, A.; Sams, S.; Kabos, P.; Zolman, K.; et al. A phase II clinical trial of the Aurora and angiogenic kinase inhibitor ENMD-2076 for previously treated, advanced, or metastatic triple-negative breast cancer. Breast Cancer Res. 2018, 20, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abou-Alfa, G.K.; Mayer, R.; Venook, A.P.; O’Neill, A.F.; Beg, M.S.; LaQuaglia, M.; Kingham, P.T.; Kobos, R.; Basturk, O.; Brennan, C.; et al. Phase II Multicenter, Open-Label Study of Oral ENMD-2076 for the Treatment of Patients with Advanced Fibrolamellar Carcinoma. Oncologist 2020, 25, e1837–e1845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yee, K.W.L.; Chen, H.-W.T.; Hedley, D.W.; Chow, S.; Brandwein, J.; Schuh, A.C.; Schimmer, A.D.; Gupta, V.; Sanfelice, D.; Johnson, T.; et al. A phase I trial of the aurora kinase inhibitor, ENMD-2076, in patients with relapsed or refractory acute myeloid leukemia or chronic myelomonocytic leukemia. Investig. New Drugs 2016, 34, 614–624. [Google Scholar] [CrossRef]
- Chu, Q.; Bouganim, N.; Fortier, C.; Zaknoen, S.; Stille, J.R.; Kremer, J.D.; Yuen, E.; Hui, Y.-H.; Peña, A.; de la Lithio, A.; et al. Abstract CT083: A Phase I/II study of aurora kinase A inhibitor, LY3295668 erbumine (AK-01): Safety as monotherapy in patients with locally advanced or metastatic solid tumors. Cancer Res. 2019, 79, CT083. [Google Scholar] [CrossRef]
- Manfredi, M.G.; Ecsedy, J.A.; Meetze, K.A.; Balani, S.K.; Burenkova, O.; Chen, W.; Galvin, K.M.; Hoar, K.M.; Huck, J.J.; LeRoy, P.J.; et al. Antitumor activity of MLN8054, an orally active small-molecule inhibitor of Aurora A kinase. Proc. Natl. Acad. Sci. USA 2007, 104, 4106–4111. [Google Scholar] [CrossRef] [Green Version]
- Dees, E.C.; Infante, J.R.; Cohen, R.B.; O’Neil, B.H.; Jones, S.; von Mehren, M.; Danaee, H.; Lee, Y.; Ecsedy, J.; Manfredi, M.; et al. Phase 1 study of MLN8054, a selective inhibitor of Aurora A kinase in patients with advanced solid tumors. Cancer Chemother. Pharm. 2011, 67, 945–954. [Google Scholar] [CrossRef] [Green Version]
- Macarulla, T.; Cervantes, A.; Elez, E.; Rodríguez-Braun, E.; Baselga, J.; Roselló, S.; Sala, G.; Blasco, I.; Danaee, H.; Lee, Y.; et al. Phase I study of the selective Aurora A kinase inhibitor MLN8054 in patients with advanced solid tumors: Safety, pharmacokinetics, and pharmacodynamics. Mol. Cancer Ther. 2010, 9, 2844–2852. [Google Scholar] [CrossRef] [Green Version]
- Shimomura, T.; Hasako, S.; Nakatsuru, Y.; Mita, T.; Ichikawa, K.; Kodera, T.; Sakai, T.; Nambu, T.; Miyamoto, M.; Takahashi, I.; et al. MK-5108, a highly selective Aurora-A kinase inhibitor, shows antitumor activity alone and in combination with docetaxel. Mol. Cancer Ther. 2010, 9, 157–166. [Google Scholar] [CrossRef] [Green Version]
- Amin, M.; Minton, S.E.; LoRusso, P.M.; Krishnamurthi, S.S.; Pickett, C.A.; Lunceford, J.; Hille, D.; Mauro, D.; Stein, M.N.; Wang-Gillam, A.; et al. A phase I study of MK-5108, an oral aurora a kinase inhibitor, administered both as monotherapy and in combination with docetaxel, in patients with advanced or refractory solid tumors. Investig. New Drugs 2016, 34, 84–95. [Google Scholar] [CrossRef] [Green Version]
- Miura, A.; Sootome, H.; Fujita, N.; Suzuki, T.; Fukushima, H.; Mizuarai, S.; Masuko, N.; Ito, K.; Hashimoto, A.; Uto, Y.; et al. TAS-119, a novel selective Aurora A and TRK inhibitor, exhibits antitumor efficacy in preclinical models with deregulated activation of the Myc, β-Catenin, and TRK pathways. Investig. New Drugs 2021. [Google Scholar] [CrossRef]
- Robbrecht, D.G.J.; Lopez, J.; Calvo, E.; He, X.; Hiroshi, H.; Soni, N.; Cook, N.; Dowlati, A.; Fasolo, A.; Moreno, V.; et al. A first-in-human phase 1 and pharmacological study of TAS-119, a novel selective Aurora A kinase inhibitor in patients with advanced solid tumours. Br. J. Cancer 2020, 1–8. [Google Scholar] [CrossRef]
- Cardin, D.B.; Park, H.; Diamond, J.R.; Drilon, A.E.; VerMeulen, W.L.; He, X.; Hirai, H.; Soni, N.; Berlin, J. Phase I study of the Aurora A kinase (AurA) inhibitor TAS-119 with paclitaxel (P) in advanced solid tumors. JCO 2019, 37, 3031. [Google Scholar] [CrossRef]
- Shiotsu, Y.; Kiyoi, H.; Ishikawa, Y.; Tanizaki, R.; Shimizu, M.; Umehara, H.; Ishii, K.; Mori, Y.; Ozeki, K.; Minami, Y.; et al. KW-2449, a novel multikinase inhibitor, suppresses the growth of leukemia cells with FLT3 mutations or T315I-mutated BCR/ABL translocation. Blood 2009, 114, 1607–1617. [Google Scholar] [CrossRef] [Green Version]
- Farag, S.; Zhang, S.; Suvannasankha, A.; Liang, J.; O’Bryant, R.; Lisa, W.; Gupta, S.; Bray, M.; Sidor, C.F.; Abonour, R. Clinical Activity of a Novel Multiple Tyrosine Kinase and Aurora Kinase Inhibitor, ENMD-2076, Against Multiple Myeloma: Interim Phase I Trial Results. Blood 2010, 116, 1957. [Google Scholar] [CrossRef]
- Warner, S.L.; Munoz, R.M.; Stafford, P.; Koller, E.; Hurley, L.H.; Hoff, D.D.; Von Han, H. Comparing Aurora A and Aurora B as molecular targets for growth inhibition of pancreatic cancer cells. Mol. Cancer Ther. 2006, 5, 2450–2458. [Google Scholar] [CrossRef] [Green Version]
- Carpinelli, P.; Moll, J. Aurora kinase inhibitors: Identification and preclinical validation of their biomarkers. Expert Opin. Targets 2008, 12, 69–80. [Google Scholar] [CrossRef]
- Ohashi, A.; Ohori, M.; Iwai, K. Motor activity of centromere-associated protein-E contributes to its localization at the center of the midbody to regulate cytokinetic abscission. Oncotarget 2016, 7, 79964–79980. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.-W.; Zhong, N.; Xiao, Y.; She, Z.-Y. Mechanisms of kinesin-7 CENP-E in kinetochore-microtubule capture and chromosome alignment during cell division. Biol. Cell 2019, 111, 143–160. [Google Scholar] [CrossRef]
- Mao, Y.; Desai, A.; Cleveland, D.W. Microtubule capture by CENP-E silences BubR1-dependent mitotic checkpoint signaling. J. Cell Biol. 2005, 170, 873–880. [Google Scholar] [CrossRef] [Green Version]
- Kung, P.-P.; Martinez, R.; Zhu, Z.; Zager, M.; Blasina, A.; Rymer, I.; Hallin, J.; Xu, M.; Carroll, C.; Chionis, J.; et al. Chemogenetic evaluation of the mitotic kinesin CENP-E reveals a critical role in triple-negative breast cancer. Mol. Cancer Ther. 2014, 13, 2104–2115. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, A.; Ohori, M.; Iwai, K.; Nambu, T.; Miyamoto, M.; Kawamoto, T.; Okaniwa, M. A novel time-dependent CENP-E inhibitor with potent antitumor activity. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Tanudji, M.; Shoemaker, J.; L’Italien, L.; Russell, L.; Chin, G.; Schebye, X.M. Gene silencing of CENP-E by small interfering RNA in HeLa cells leads to missegregation of chromosomes after a mitotic delay. Mol. Biol. Cell 2004, 15, 3771–3781. [Google Scholar] [CrossRef]
- Wood, K.W.; Lad, L.; Luo, L.; Qian, X.; Knight, S.D.; Nevins, N.; Brejc, K.; Sutton, D.; Gilmartin, A.G.; Chua, P.R.; et al. Antitumor activity of an allosteric inhibitor of centromere-associated protein-E. Proc. Natl. Acad. Sci. USA 2010, 107, 5839–5844. [Google Scholar] [CrossRef] [Green Version]
- Chung, V.; Heath, E.I.; Schelman, W.R.; Johnson, B.M.; Kirby, L.C.; Lynch, K.M.; Botbyl, J.D.; Lampkin, T.A.; Holen, K.D. First-time-in-human study of GSK923295, a novel antimitotic inhibitor of centromere-associated protein e (CENP-E), in patients with refractory cancer. Cancer Chemother. Pharm. 2012, 69, 733–741. [Google Scholar] [CrossRef]
- Sawin, K.E.; LeGuellec, K.; Philippe, M.; Mitchison, T.J. Mitotic spindle organization by a plus-end-directed microtubule motor. Nature 1992, 359, 540–543. [Google Scholar] [CrossRef]
- Blangy, A.; Lane, H.A.; D’Hérin, P.; Harper, M.; Kress, M.; Nigg, E.A. Phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell 1995, 83, 1159–1169. [Google Scholar] [CrossRef] [Green Version]
- Weil, D.; Garçon, L.; Harper, M.; Duménil, D.; Dautry, F.; Kress, M. Targeting the kinesin Eg5 to monitor siRNA transfection in mammalian cells. Biotechniques 2002, 33, 1244–1248. [Google Scholar] [CrossRef]
- Nakai, R.; Iida, S.; Takahashi, T.; Tsujita, T.; Okamoto, S.; Takada, C.; Akasaka, K.; Ichikawa, S.; Ishida, H.; Kusaka, H.; et al. K858, a novel inhibitor of mitotic kinesin Eg5 and antitumor agent, induces cell death in cancer cells. Cancer Res. 2009, 69, 3901–3909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Yu, H.; Huo, L.; Liu, J.; Li, M.; Zhou, J. Validating the mitotic kinesin Eg5 as a therapeutic target in pancreatic cancer cells and tumor xenografts using a specific inhibitor. Biochem. Pharm. Ther. 2008, 76, 169–178. [Google Scholar] [CrossRef]
- Liu, L.; Liu, X.; Mare, M.; Dumont, A.S.; Zhang, H.; Yan, D.; Xiong, Z. Overexpression of Eg5 correlates with high grade astrocytic neoplasm. J. Neurooncol 2016, 126, 77–80. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, N.; Li, J.; Kong, J.; Guan, X.; Wang, X. Eg5 Overexpression Is Predictive of Poor Prognosis in Hepatocellular Carcinoma Patients. Dis. Markers 2017, 2017. [Google Scholar] [CrossRef] [Green Version]
- Ding, S.; Xing, N.; Lu, J.; Zhang, H.; Nishizawa, K.; Liu, S.; Yuan, X.; Qin, Y.; Liu, Y.; Ogawa, O.; et al. Overexpression of Eg5 predicts unfavorable prognosis in non-muscle invasive bladder urothelial carcinoma. Int. J. Urol. 2011, 18, 432–438. [Google Scholar] [CrossRef]
- Liu, M.; Wang, X.; Yang, Y.; Li, D.; Ren, H.; Zhu, Q.; Chen, Q.; Han, S.; Hao, J.; Zhou, J. Ectopic expression of the microtubule-dependent motor protein Eg5 promotes pancreatic tumourigenesis. J. Pathol. 2010, 221, 221–228. [Google Scholar] [CrossRef]
- Blagden, S.P.; Molife, L.R.; Seebaran, A.; Payne, M.; Reid, A.H.M.; Protheroe, A.S.; Vasist, L.S.; Williams, D.D.; Bowen, C.; Kathman, S.J.; et al. A phase I trial of ispinesib, a kinesin spindle protein inhibitor, with docetaxel in patients with advanced solid tumours. Br. J. Cancer 2008, 98, 894–899. [Google Scholar] [CrossRef] [Green Version]
- Rodon, J.; Till, E.; Patnaik, A.; Takimoto, C.; Beeram, M.; Williams, D.; Bowen, C.; Hodge, J.; Dar, M.; Toclher, A. 640 POSTER Phase I study of ispinesib (SB-715992), a kinesin spindle protein inhibitor, in combination with capecitabine in patients with advanced solid tumors. Eur. J. Cancer Suppl. 2006, 4, 193. [Google Scholar] [CrossRef]
- Jones, S.F.; Plummer, E.R.; Burris, H.A.; Razak, A.R.; Meluch, A.A.; Bowen, C.J.; Williams, D.H.; Hodge, J.P.; Dar, M.M.; Calvert, A.H. Phase I study of ispinesib in combination with carboplatin in patients with advanced solid tumors. JCO 2006, 24, 2027. [Google Scholar] [CrossRef]
- Beer, T.M.; Goldman, B.; Synold, T.W.; Ryan, C.W.; Vasist, L.S.; Van Veldhuizen, P.J.; Dakhil, S.R.; Lara, P.N.; Drelichman, A.; Hussain, M.H.A.; et al. Southwest Oncology Group phase II study of ispinesib in androgen-independent prostate cancer previously treated with taxanes. Clin. Genitourin Cancer 2008, 6, 103–109. [Google Scholar] [CrossRef]
- Tang, P.A.; Siu, L.L.; Chen, E.X.; Hotte, S.J.; Chia, S.; Schwarz, J.K.; Pond, G.R.; Johnson, C.; Colevas, A.D.; Synold, T.W.; et al. Phase II study of ispinesib in recurrent or metastatic squamous cell carcinoma of the head and neck. Investig. New Drugs 2008, 26, 257–264. [Google Scholar] [CrossRef]
- Lee, C.W.; Bélanger, K.; Rao, S.C.; Petrella, T.M.; Tozer, R.G.; Wood, L.; Savage, K.J.; Eisenhauer, E.A.; Synold, T.W.; Wainman, N.; et al. A phase II study of ispinesib (SB-715992) in patients with metastatic or recurrent malignant melanoma: A National Cancer Institute of Canada Clinical Trials Group trial. Investig. New Drugs 2008, 26, 249–255. [Google Scholar] [CrossRef]
- Knox, J.J.; Gill, S.; Synold, T.W.; Biagi, J.J.; Major, P.; Feld, R.; Cripps, C.; Wainman, N.; Eisenhauer, E.; Seymour, L. A phase II and pharmacokinetic study of SB-715992, in patients with metastatic hepatocellular carcinoma: A study of the National Cancer Institute of Canada Clinical Trials Group (NCIC CTG IND.168). Investig. New Drugs 2008, 26, 265–272. [Google Scholar] [CrossRef]
- Lee, R.T.; Beekman, K.E.; Hussain, M.; Davis, N.B.; Clark, J.I.; Thomas, S.P.; Nichols, K.F.; Stadler, W.M. A University of Chicago consortium phase II trial of SB-715992 in advanced renal cell cance. Clin. Genitourin. Cancer 2008, 6, 21–24. [Google Scholar] [CrossRef] [Green Version]
- Shahin, M.S.; Braly, P.; Rose, P.; Malpass, T.; Bailey, H.; Alvarez, R.D.; Hodge, J.; Bowen, C.; Buller, R. A phase II, open-label study of ispinesib (SB-715992) in patients with platinum/taxane refractory or resistant relapsed ovarian cancer. JCO 2007, 25, 5562. [Google Scholar] [CrossRef]
- Gomez, H.L.; Philco, M.; Pimentel, P.; Kiyan, M.; Monsalvo, M.L.; Conlan, M.G.; Saikali, K.G.; Chen, M.M.; Seroogy, J.J.; Wolff, A.A.; et al. Phase I dose-escalation and pharmacokinetic study of ispinesib, a kinesin spindle protein inhibitor, administered on days 1 and 15 of a 28-day schedule in patients with no prior treatment for advanced breast cancer. Anticancer Drugs 2012, 23, 335–341. [Google Scholar] [CrossRef]
- Souid, A.-K.; Dubowy, R.L.; Ingle, A.M.; Conlan, M.G.; Sun, J.; Blaney, S.M.; Adamson, P.C. A pediatric phase I trial and pharmacokinetic study of ispinesib: A Children’s Oncology Group phase I consortium study. Pediatr. Blood Cancer 2010, 55, 1323–1328. [Google Scholar] [CrossRef] [Green Version]
- Jackson, J.R.; Gilmartin, A.; Dhanak, D.; Knight, S.; Parrish, C.; Luo, L.; Sutton, D.; Caulder, E.; Diamond, M.; Giardiniere, M.; et al. A second generation KSP inhibitor, SB-743921, is a highly potent and active therapeutic in preclinical models of cancer. Clin. Cancer Res. 2006, 12, B11. [Google Scholar]
- Holen, K.D.; Belani, C.P.; Wilding, G.; Ramalingam, S.; Volkman, J.L.; Ramanathan, R.K.; Vasist, L.S.; Bowen, C.J.; Hodge, J.P.; Dar, M.M.; et al. A first in human study of SB-743921, a kinesin spindle protein inhibitor, to determine pharmacokinetics, biologic effects and establish a recommended phase II dose. Cancer Chemother. Pharm. 2011, 67, 447–454. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, O.A.; Gerecitano, J.; Van Deventer, H.; Hainsworth, J.; Zullo, K.M.; Saikali, K.; Seroogy, J.; Wolff, A.; Escandón, R. The addition of granulocyte-colony stimulating factor shifts the dose limiting toxicity and markedly increases the maximum tolerated dose and activity of the kinesin spindle protein inhibitor SB-743921 in patients with relapsed or refractory lymphoma: R. Leuk. Lymphoma 2015, 56, 2585–2591. [Google Scholar] [CrossRef]
- Lemieux, C.; Dewolf, W.; Voegtli, W.; Delisle, R.; Laird, E.; Wallace, E.; Woessner, R.; Corrette, C.; Allen, S.; Hans, J.; et al. ARRY-520, a novel, highly selective KSP inhibitor with potent anti-proliferative activity. Proc. Am. Assoc. Cancer Res. 2007, 48, 5590. [Google Scholar]
- LoRusso, P.M.; Goncalves, P.H.; Casetta, L.; Carter, J.A.; Litwiler, K.; Roseberry, D.; Rush, S.; Schreiber, J.; Simmons, H.M.; Ptaszynski, M.; et al. First-in-human phase 1 study of filanesib (ARRY-520), a kinesin spindle protein inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2015, 33, 440–449. [Google Scholar] [CrossRef]
- Khoury, H.J.; Garcia-Manero, G.; Borthakur, G.; Kadia, T.; Foudray, M.C.; Arellano, M.; Langston, A.; Bethelmie-Bryan, B.; Rush, S.; Litwiler, K.; et al. A phase 1 dose-escalation study of ARRY-520, a kinesin spindle protein inhibitor, in patients with advanced myeloid leukemias. Cancer 2012, 118, 3556–3564. [Google Scholar] [CrossRef] [Green Version]
- Shah, J.J.; Kaufman, J.L.; Zonder, J.A.; Cohen, A.D.; Bensinger, W.I.; Hilder, B.; Rush, S.; Walker, D.; Tunquist, B.; Litwiler, K.; et al. A Phase 1/2 study of filanesib alone and in combination with low-dose dexamethasone in relapsed/refractory multiple myeloma. Cancer 2017, 123, 4617–4630. [Google Scholar] [CrossRef]
- Chari, A.; Htut, M.; Zonder, J.A.; Fay, J.W.; Jakubowiak, A.J.; Levy, J.B.; Lau, K.; Burt, S.M.; Tunquist, B.J.; Hilder, B.W.; et al. A phase 1 dose-escalation study of filanesib plus bortezomib and dexamethasone in patients with recurrent/refractory multiple myeloma. Cancer 2016, 122, 3327–3335. [Google Scholar] [CrossRef]
- Lee, H.C.; Shah, J.J.; Feng, L.; Manasanch, E.E.; Lu, R.; Morphey, A.; Crumpton, B.; Patel, K.K.; Wang, M.L.; Alexanian, R.; et al. A phase 1 study of filanesib, carfilzomib, and dexamethasone in patients with relapsed and/or refractory multiple myeloma. Blood Cancer J. 2019, 9, 80. [Google Scholar] [CrossRef] [Green Version]
- Ocio, E.M.; Motlló, C.; Rodríguez-Otero, P.; Martínez-López, J.; Cejalvo, M.J.; Martín-Sánchez, J.; Bladé, J.; García-Malo, M.D.; Dourdil, M.V.; García-Mateo, A.; et al. Filanesib in combination with pomalidomide and dexamethasone in refractory MM patients: Safety and efficacy, and association with alpha 1-acid glycoprotein (AAG) levels. Phase Ib/II Pomdefil clinical trial conducted by the Spanish MM group. Br. J. Haematol. 2021, 192, 522–530. [Google Scholar] [CrossRef]
- Toudjarska, I.; Judge, A.; Buck, T.; McClintock, K.; Jong, S.; de Ambegia, E.; Brodsky, J.; Akinc, A.; Racie, T.; Jeffs, L.; et al. Abstract B204: Development of ALN-VSP: An RNAi therapeutic for liver malignancies. Mol. Cancer Ther. 2009, 8, B204. [Google Scholar] [CrossRef] [Green Version]
- Tabernero, J.; Shapiro, G.I.; LoRusso, P.M.; Cervantes, A.; Schwartz, G.K.; Weiss, G.J.; Paz-Ares, L.; Cho, D.C.; Infante, J.R.; Alsina, M.; et al. First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov. 2013, 3, 406–417. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.S.; Fan, L.; Van Horn, R.D.; Nakai, R.; Ohta, Y.; Akinaga, S.; Murakata, C.; Yamashita, Y.; Yin, T.; Credille, K.M.; et al. A novel Eg5 inhibitor (LY2523355) causes mitotic arrest and apoptosis in cancer cells and shows potent antitumor activity in xenograft tumor models. Mol. Cancer Ther. 2015, 14, 2463–2472. [Google Scholar] [CrossRef] [Green Version]
- Infante, J.R.; Patnaik, A.; Verschraegen, C.F.; Olszanski, A.J.; Shaheen, M.; Burris, H.A.; Tolcher, A.W.; Papadopoulos, K.P.; Beeram, M.; Hynes, S.M.; et al. Two Phase 1 dose-escalation studies exploring multiple regimens of litronesib (LY2523355), an Eg5 inhibitor, in patients with advanced cancer. Cancer Chemother. Pharm. 2017, 79, 315–326. [Google Scholar] [CrossRef]
- Wakui, H.; Yamamoto, N.; Kitazono, S.; Mizugaki, H.; Nakamichi, S.; Fujiwara, Y.; Nokihara, H.; Yamada, Y.; Suzuki, K.; Kanda, H.; et al. A phase 1 and dose-finding study of LY2523355 (litronesib), an Eg5 inhibitor, in Japanese patients with advanced solid tumors. Cancer Chemother. Pharm. 2014, 74, 15–23. [Google Scholar] [CrossRef]
- Schiemann, K.; Finsinger, D.; Zenke, F.; Amendt, C.; Knöchel, T.; Bruge, D.; Buchstaller, H.-P.; Emde, U.; Stähle, W.; Anzali, S. The discovery and optimization of hexahydro-2H-pyrano[3,2-c]quinolines (HHPQs) as potent and selective inhibitors of the mitotic kinesin-5. Bioorg. Med. Chem. Lett. 2010, 20, 1491–1495. [Google Scholar] [CrossRef]
- Hollebecque, A.; Deutsch, E.; Massard, C.; Gomez-Roca, C.; Bahleda, R.; Ribrag, V.; Bourgier, C.; Lazar, V.; Lacroix, L.; Gazzah, A.; et al. A phase I, dose-escalation study of the Eg5-inhibitor EMD 534085 in patients with advanced solid tumors or lymphoma. Investig. New Drugs 2013, 31, 1530–1538. [Google Scholar] [CrossRef]
- Mross, K.B.; Scharr, D.; Richly, H.; Frost, A.; Bauer, S.; Krauss, B.; Krauss, R.; Mais, A.; Hauns, B.; Hentsch, B.; et al. First-in-human study of 4SC-205 (AEGIS), a novel oral inhibitor of Eg5 kinesin spindle protein. JCO 2014, 32, 2564. [Google Scholar] [CrossRef]
- Theoclitou, M.-E.; Aquila, B.; Block, M.H.; Brassil, P.J.; Castriotta, L.; Code, E.; Collins, M.P.; Davies, A.M.; Deegan, T.; Ezhuthachan, J.; et al. Discovery of (+)-N-(3-aminopropyl)-N-[1-(5-benzyl-3-methyl-4-oxo-[1,2]thiazolo[5,4-d]pyrimidin-6-yl)-2-methylpropyl]-4-methylbenzamide (AZD4877), a kinesin spindle protein inhibitor and potential anticancer agent. J. Med. Chem. 2011, 54, 6734–6750. [Google Scholar] [CrossRef]
- Esaki, T.; Seto, T.; Ariyama, H.; Arita, S.; Fujimoto, C.; Tsukasa, K.; Kometani, T.; Nosaki, K.; Hirai, F.; Yagawa, K. Phase I study to assess the safety, Tolerability and pharmacokinetics of AZD4877 in japanese patients with solid tumors. Arch. Drug Inf. 2011, 4, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Infante, J.R.; Kurzrock, R.; Spratlin, J.; Burris, H.A.; Eckhardt, S.G.; Li, J.; Wu, K.; Skolnik, J.M.; Hylander-Gans, L.; Osmukhina, A.; et al. A Phase I study to assess the safety, tolerability, and pharmacokinetics of AZD4877, an intravenous Eg5 inhibitor in patients with advanced solid tumors. Cancer Chemother. Pharm. 2012, 69, 165–172. [Google Scholar] [CrossRef]
- Jones, R.; Vuky, J.; Elliott, T.; Mead, G.; Arranz, J.A.; Chester, J.; Chowdhury, S.; Dudek, A.Z.; Müller-Mattheis, V.; Grimm, M.-O.; et al. Phase II study to assess the efficacy, safety and tolerability of the mitotic spindle kinesin inhibitor AZD4877 in patients with recurrent advanced urothelial cancer. Investig. New Drugs 2013, 31, 1001–1007. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Padmanabhan, S.; Stock, W.; Tallman, M.S.; Curt, G.A.; Li, J.; Osmukhina, A.; Wu, K.; Huszar, D.; Borthukar, G.; et al. Phase I/II multicenter study to assess the safety, tolerability, pharmacokinetics and pharmacodynamics of AZD4877 in patients with refractory acute myeloid leukemia. Investig. New Drugs 2012, 30, 1107–1115. [Google Scholar] [CrossRef] [Green Version]
- Jeay, S.; Ali, S.; Chen, C.-R.; Uppalapati, U.; Senator, D.; Chan, T.; France, D.; Ashwell, M. Discovery of a novel Eg5 kinesin inhibitor, ARQ 621, with potent antitumor activity while sparing bone marrow-derived cells. Cancer Res. 2008, 68, 656. [Google Scholar]
- Chen, L.C.; Rosen, L.S.; Iyengar, T.; Goldman, J.W.; Savage, R.; Kazakin, J.; Chan, T.C.K.; Schwartz, B.E.; Abbadessa, G.; Von Hoff, D.D. First-in-human study with ARQ 621, a novel inhibitor of Eg5: Final results from the solid tumors cohort. JCO 2011, 29, 3076. [Google Scholar] [CrossRef]
- Cox, C.D.; Garbaccio, R.M. Discovery of allosteric inhibitors of kinesin spindle protein (KSP) for the treatment of taxane-refractory cancer: MK-0731 and analogs. Anticancer Agents Med. Chem. 2010, 10, 697–712. [Google Scholar] [CrossRef] [PubMed]
- Holen, K.; DiPaola, R.; Liu, G.; Tan, A.R.; Wilding, G.; Hsu, K.; Agrawal, N.; Chen, C.; Xue, L.; Rosenberg, E.; et al. A phase I trial of MK-0731, a kinesin spindle protein (KSP) inhibitor, in patients with solid tumors. Investig. New Drugs 2012, 30, 1088–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerecitano, J.F.; Stephenson, J.J.; Lewis, N.L.; Osmukhina, A.; Li, J.; Wu, K.; You, Z.; Huszar, D.; Skolnik, J.M.; Schwartz, G.K. A Phase I trial of the kinesin spindle protein (Eg5) inhibitor AZD4877 in patients with solid and lymphoid malignancies. Investig. New Drugs 2013, 31, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Mitchison, T.J. The proliferation rate paradox in antimitotic chemotherapy. Mol. Biol. Cell 2012, 23, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Bennett, A.; Sloss, O.; Topham, C.; Nelson, L.; Tighe, A.; Taylor, S.S. Inhibition of Bcl-xL sensitizes cells to mitotic blockers, but not mitotic drivers. Open Biol. 2016, 6, 160134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gascoigne, K.E.; Taylor, S.S. Cancer Cells Display Profound Intra- and Interline Variation following Prolonged Exposure to Antimitotic Drugs. Cancer Cell 2008, 14, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, K.A.; Malhotra, M.; Curtin, C.M.; O’ Brien, F.J.; O’ Driscoll, C.M. Life in 3D is never flat: 3D models to optimise drug delivery. J. Control. Release 2015, 215, 39–54. [Google Scholar] [CrossRef]
- Pinto, B.; Henriques, A.C.; Silva, P.M.A.; Bousbaa, H. Three-Dimensional Spheroids as In vitro Preclinical Models for Cancer Research. Pharmaceutics 2020, 12, 1186. [Google Scholar] [CrossRef]
- Sgorla, D.; Bunhak, É.J.; Cavalcanti, O.A.; Fonte, P.; Sarmento, B. Exploitation of lipid-polymeric matrices at nanoscale for drug delivery applications. Expert Opin. Drug. Deliv. 2016, 13, 1301–1309. [Google Scholar] [CrossRef]
- Lv, J.-F.; Hu, L.; Zhuo, W.; Zhang, C.-M.; Zhou, H.-H.; Fan, L. Epigenetic alternations and cancer chemotherapy response. Cancer Chemother. Pharm. 2016, 77, 673–684. [Google Scholar] [CrossRef]
- Mountzios, G.; Dimopoulos, M.-A.; Soria, J.-C.; Sanoudou, D.; Papadimitriou, C.A. Histopathologic and genetic alterations as predictors of response to treatment and survival in lung cancer: A review of published data. Crit. Rev. Oncol. Hematol. 2010, 75, 94–109. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Novais, P.; Silva, P.M.A.; Amorim, I.; Bousbaa, H. Second-Generation Antimitotics in Cancer Clinical Trials. Pharmaceutics 2021, 13, 1011. https://doi.org/10.3390/pharmaceutics13071011
Novais P, Silva PMA, Amorim I, Bousbaa H. Second-Generation Antimitotics in Cancer Clinical Trials. Pharmaceutics. 2021; 13(7):1011. https://doi.org/10.3390/pharmaceutics13071011
Chicago/Turabian StyleNovais, Pedro, Patrícia M. A. Silva, Isabel Amorim, and Hassan Bousbaa. 2021. "Second-Generation Antimitotics in Cancer Clinical Trials" Pharmaceutics 13, no. 7: 1011. https://doi.org/10.3390/pharmaceutics13071011