
Combined Use of Structure Analysis, Studies of Molecular Association in Solution, and Molecular Modelling to Understand the Different Propensities of Dihydroxybenzoic Acids to Form Solid Phases


Abstract
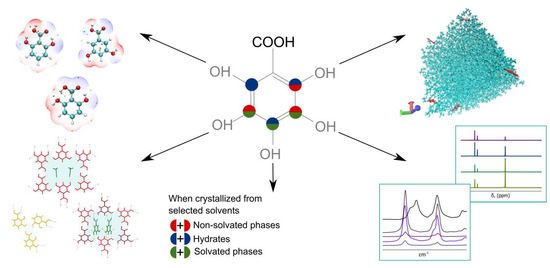
:
1. Introduction
2. Methods and Materials
2.1. Materials, Crystallization Experiments, and Crystal Form Identification
2.2. Ab Initio Calculations
2.3. Association Studies by Using FT-IR and NMR Spectroscopy
2.4. Molecular Dynamics (MD) Simulations
3. Results and Discussion
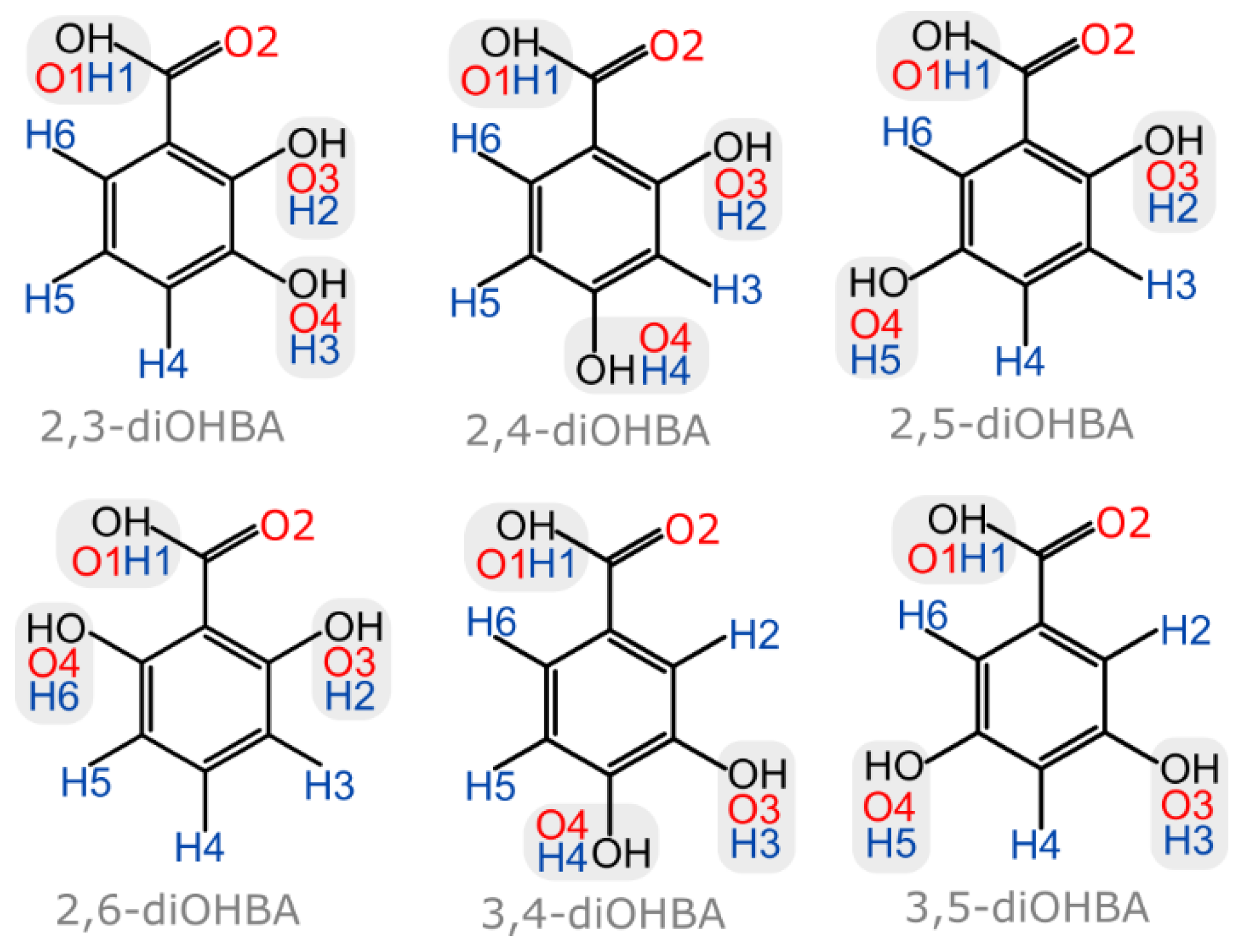
3.1. Crystal Form Screening and Crystal Structure Evaluation
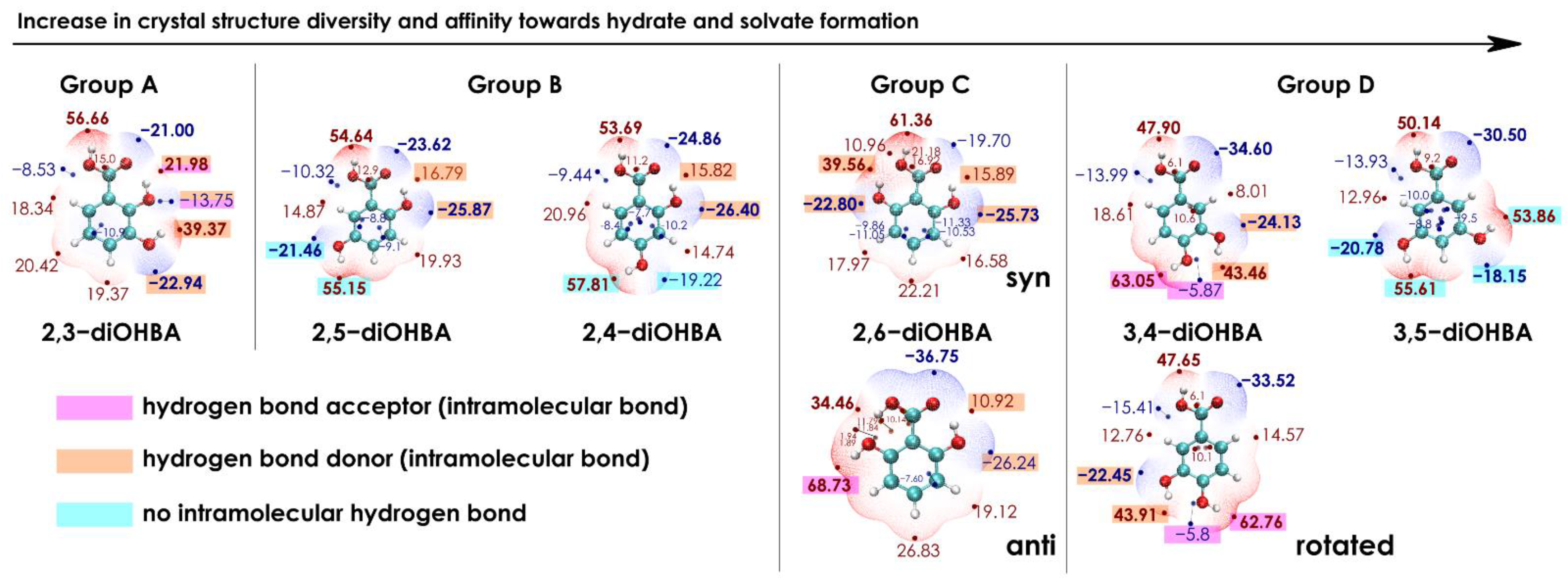
3.2. Analysis of the Electrostatic Potential of diOHBA
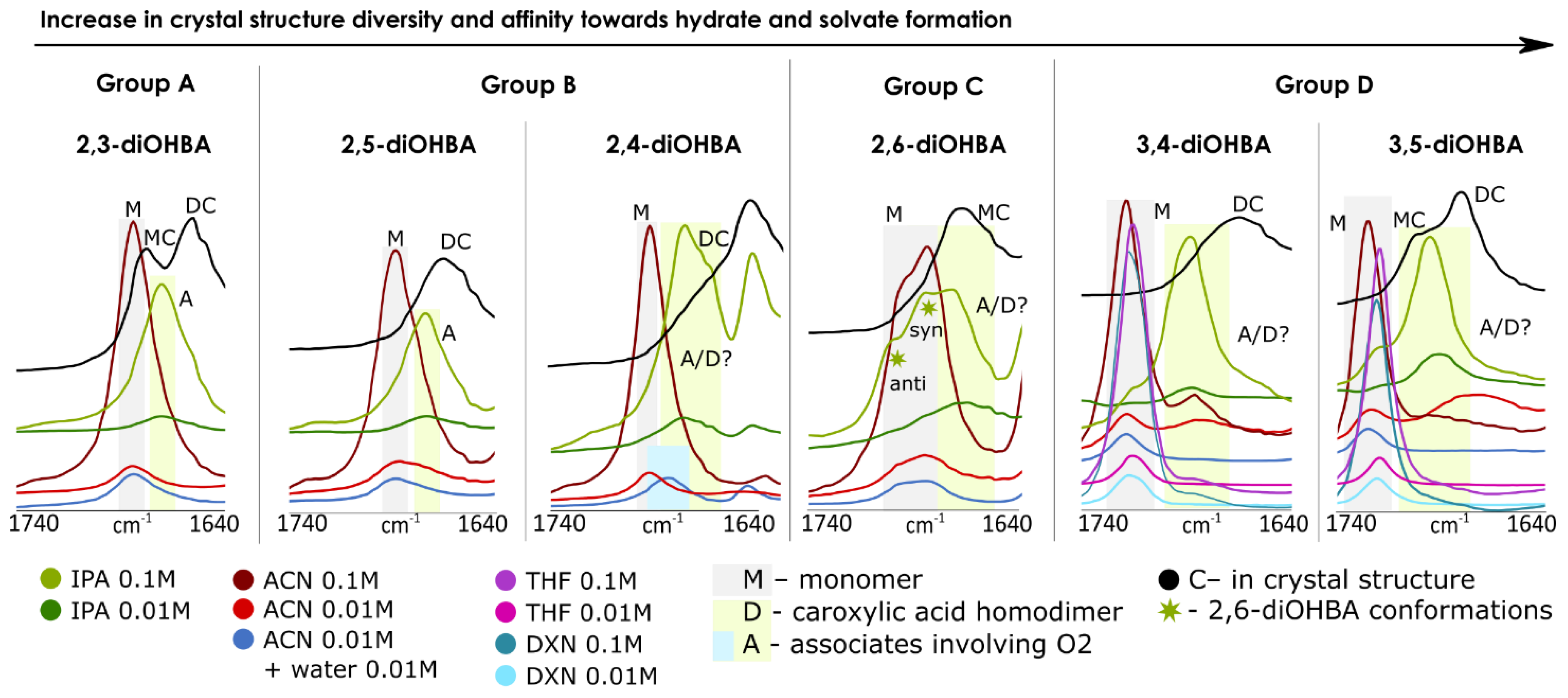
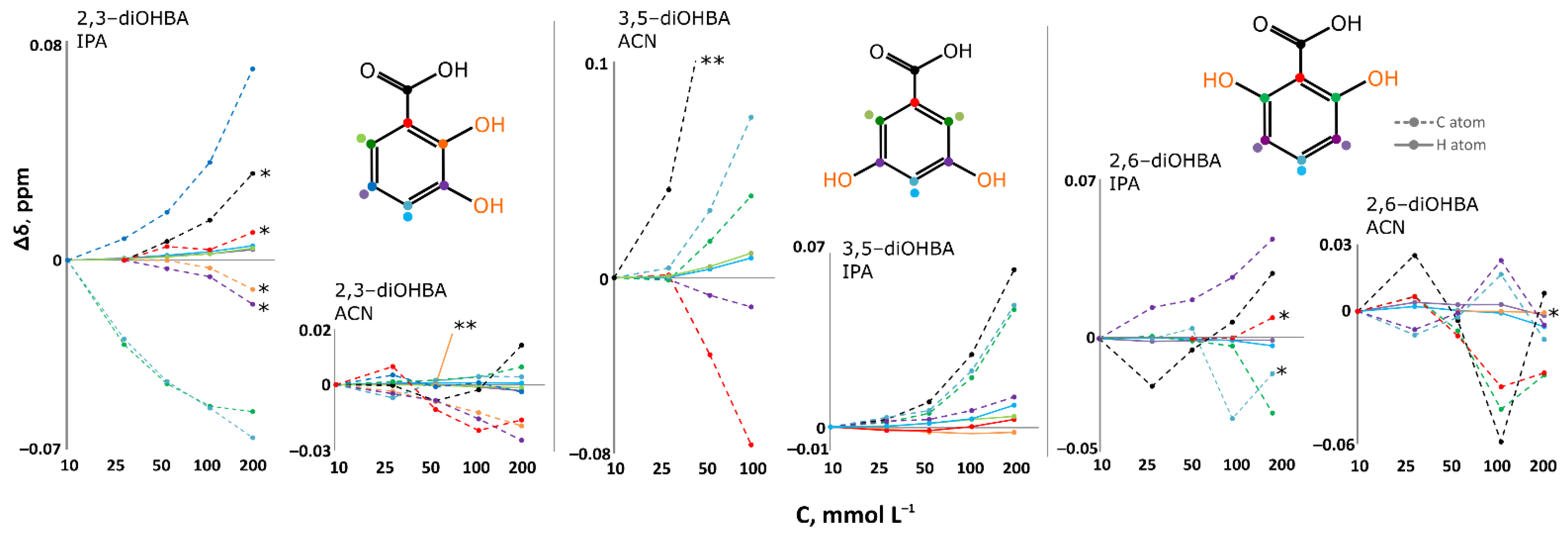
3.3. FTIR, 1H, 13C NMR Spectroscopy Studies of Association of diOHBAs in Solutions
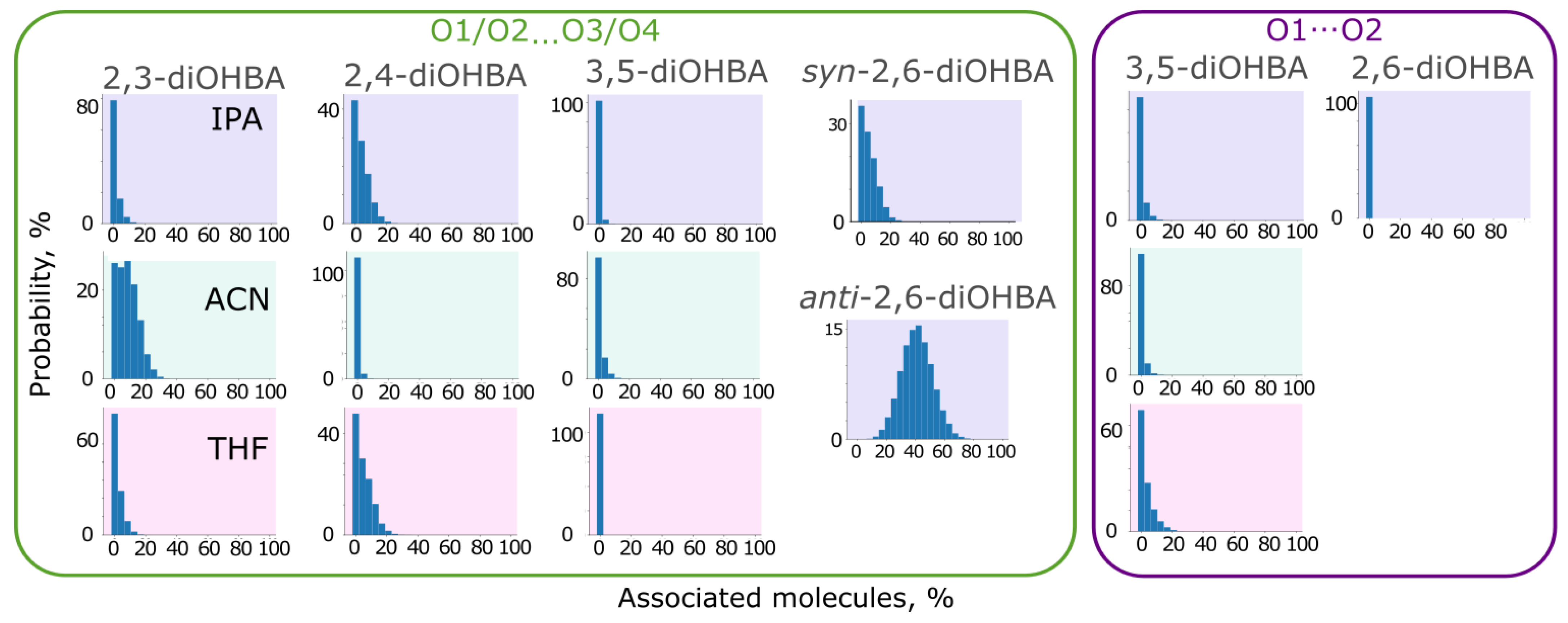
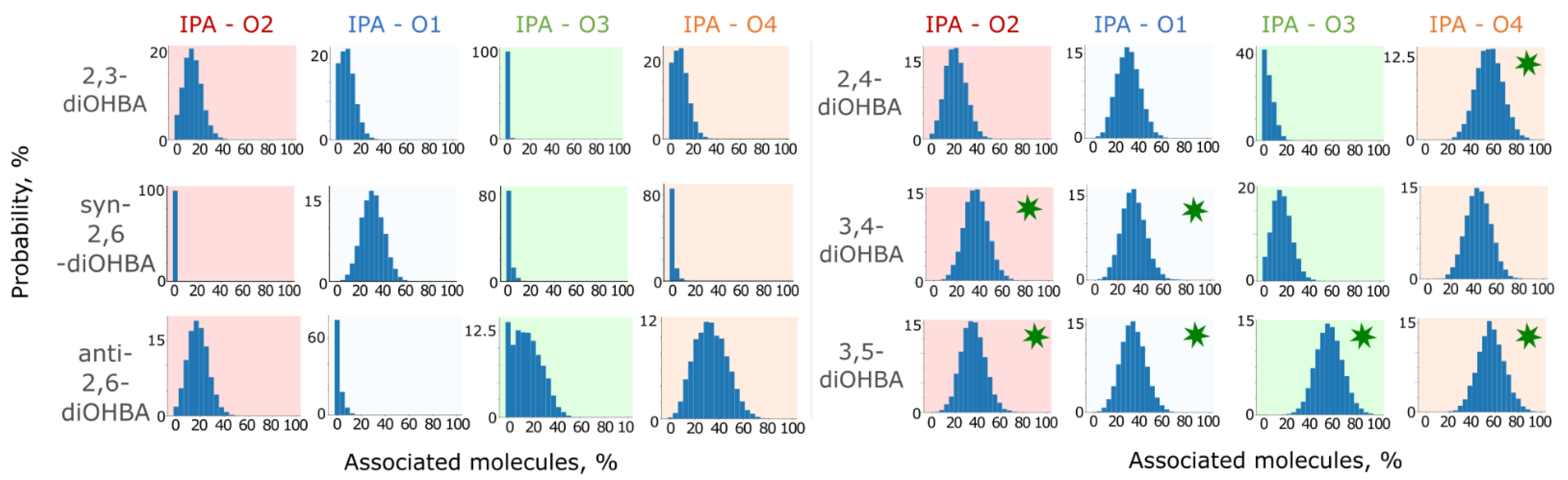
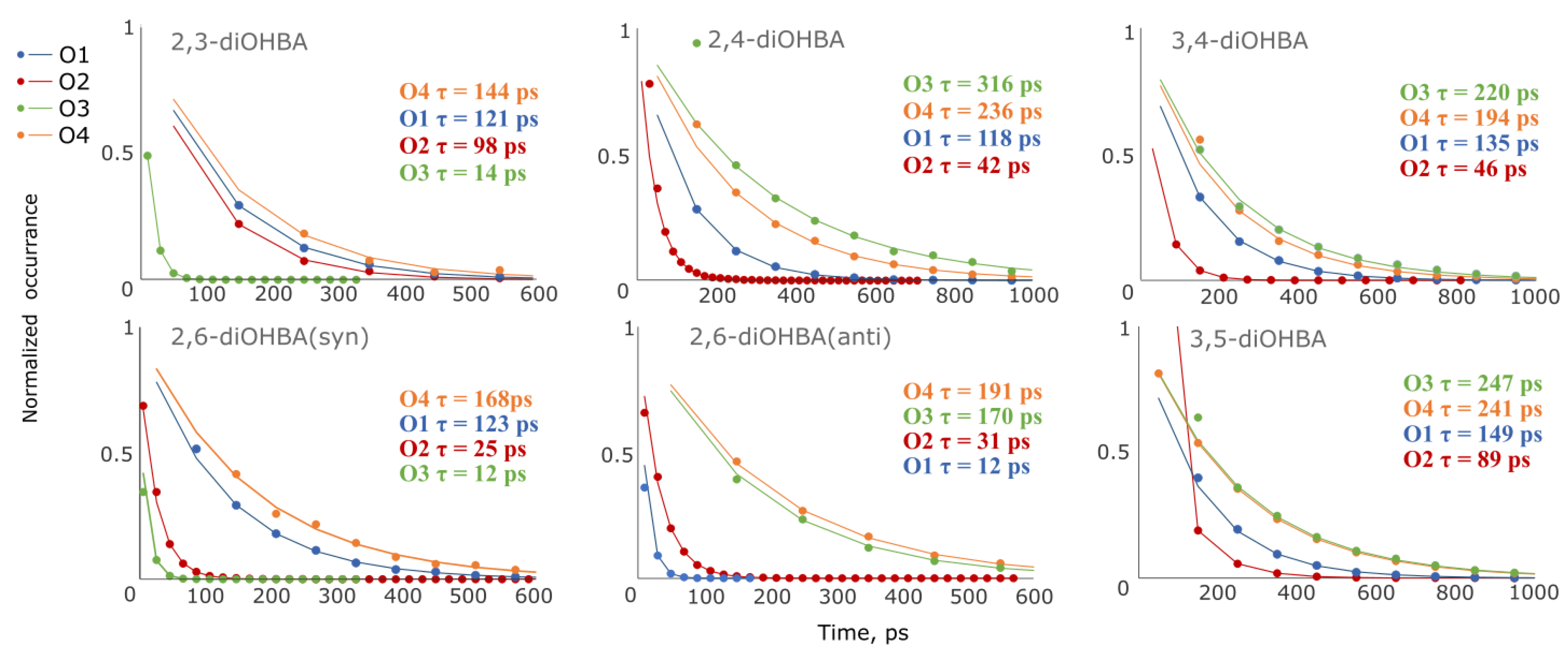
3.4. Studies of Association of diOHBAs in Solutions Using Molecular Dynamics (MD) Simulations
3.5. Summary of the Link Between Solvate Formation, Crystal Structures, and Association in Solution
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cruz-Cabeza, A.J.; Reutzel-Edens, S.M.; Bernstein, J. Facts and fictions about polymorphism. Chem. Soc. Rev. 2015, 49, 8619–8635. [Google Scholar] [CrossRef] [PubMed]
- Healy, A.M.; Worku, Z.A.; Kumar, D.; Madi, A.M. Pharmaceutical solvates, hydrates and amorphous forms: A special emphasis on cocrystals. Adv. Drug Deliv. Rev. 2017, 117, 25–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corpinot, M.K.; Bučar, D.K. A Practical Guide to the Design of Molecular Crystals. Cryst. Growth Des. 2019, 19, 1426–1453. [Google Scholar] [CrossRef] [Green Version]
- Bhardwaj, R.M.; Price, L.S.; Price, S.L.; Reutzel-Edens, S.M.; Miller, G.J.; Oswald, I.D.H.; Johnston, B.F.; Florence, A.J. Exploring the experimental and computed crystal energy landscape of olanzapine. Cryst. Growth Des. 2013, 13, 1602–1617. [Google Scholar] [CrossRef]
- Braun, D.E.; Ardid-Candel, M.; D’Oria, E.; Karamertzanis, P.G.; Arlin, J.B.; Florence, A.J.; Jones, A.G.; Price, S.L. Racemic naproxen: A multidisciplinary structural and thermodynamic comparison with the enantiopure form. Cryst. Growth Des. 2011, 12, 1602–1617. [Google Scholar] [CrossRef]
- Hulme, A.T.; Price, S.L.; Tocher, D.A. A new polymorph of 5-fluorouracil found following computational crystal structure predictions. J. Am. Chem. Soc. 2005. [Google Scholar] [CrossRef] [PubMed]
- Price, S.L.; Braun, D.E.; Reutzel-Edens, S.M. Can computed crystal energy landscapes help understand pharmaceutical solids? Chem. Commun. 2016, 52, 7065–7077. [Google Scholar] [CrossRef]
- Braun, D.E.; Karamertzanis, P.G.; Price, S.L. Which, if any, hydrates will crystallise? Predicting hydrate formation of two dihydroxybenzoic acids. Chem. Commun. 2011, 47, 5443–5445. [Google Scholar] [CrossRef] [Green Version]
- Braun, D.E.; Griesser, U.J. Why Do Hydrates (Solvates) Form in Small Neutral Organic Molecules? Exploring the Crystal Form Landscapes of the Alkaloids Brucine and Strychnine. Cryst. Growth Des. 2016, 16, 6405–6418. [Google Scholar] [CrossRef] [Green Version]
- Xin, D.; Gonnella, N.C.; He, X.; Horspool, K. Solvate Prediction for Pharmaceutical Organic Molecules with Machine Learning. Cryst. Growth Des. 2019, 52, 7065–7077. [Google Scholar] [CrossRef]
- Musil, F.; De, S.; Yang, J.; Campbell, J.E.; Day, G.M.; Ceriotti, M. Machine learning for the structure-energy-property landscapes of molecular crystals. Chem. Sci. 2018, 9, 1289–1300. [Google Scholar] [CrossRef] [Green Version]
- Takieddin, K.; Khimyak, Y.Z.; Fábián, L. Prediction of Hydrate and Solvate Formation Using Statistical Models. Cryst. Growth Des. 2016, 16, 70–81. [Google Scholar] [CrossRef] [Green Version]
- Cole, J.C.; Raithby, P.R.; Taylor, R. Prior Likelihoods and Space-Group Preferences of Solvates. Cryst. Growth Des. 2021, 21, 1178–1189. [Google Scholar] [CrossRef]
- Werner, J.E.; Swift, J.A. Organic solvates in the Cambridge Structural Database. CrystEngComm 2021, 23, 1555–1565. [Google Scholar] [CrossRef]
- Cruz-Cabeza, A.J.; Feeder, N.; Davey, R.J. Open questions in organic crystal polymorphism. Commun Chem 2020. [Google Scholar] [CrossRef]
- Aminpour, M.; Montemagno, C.; Tuszynski, J.A. An overview of molecular modeling for drug discovery with specific illustrative examples of applications. Molecules 2019, 1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheraga, H.A.; Khalili, M.; Liwo, A. Protein-folding dynamics: Overview of molecular simulation techniques. Annu. Rev. Phys. Chem. 2007, 58, 57–83. [Google Scholar] [CrossRef] [Green Version]
- Jalili, S.; Amani, P. Molecular dynamics simulation study of solvation effects of water and trifluoroethanol on gamma-aminobutyric acid (GABA). J. Mol. Liq. 2014, 197, 27–34. [Google Scholar] [CrossRef]
- Salvalaglio, M.; Perego, C.; Giberti, F.; Mazzotti, M.; Parrinello, M. Molecular-dynamics simulations of urea nucleation from aqueous solution. Proc. Natl. Acad. Sci. USA 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polêto, M.D.; Grisci, B.I.; Dorn, M.; Verli, H. ConfID: An analytical method for conformational characterization of small molecules using molecular dynamics trajectories. Bioinformatics 2020, 36, 3576–3577. [Google Scholar] [CrossRef]
- Boothroyd, S.; Kerridge, A.; Broo, A.; Buttar, D.; Anwar, J. Why Do Some Molecules Form Hydrates or Solvates? Cryst. Growth Des. 2018, 18, 1903–1908. [Google Scholar] [CrossRef]
- Bērziņš, A.; Kons, A.; Saršūns, K.; Belyakov, S.; Actiņš, A. On the rationalization of formation of solvates: Experimental and computational study of solid forms of several nitrobenzoic acid derivatives. Cryst. Growth Des. 2020, 20, 5767–5784. [Google Scholar] [CrossRef]
- Bērziņš, A.; Zvaniņa, D.; Trimdale, A.; Bērziņš, A.; Zvaniņa, D.; Trimdale, A. Detailed Analysis of Packing Efficiency Allows Rationalization of Solvate Formation Propensity for Selected Structurally Similar Organic Molecules. Cryst. Growth Des. 2018, 18, 2040–2045. [Google Scholar] [CrossRef]
- Braun, D.E.; McMahon, J.A.; Koztecki, L.H.; Price, S.L.; Reutzel-Edens, S.M. Contrasting polymorphism of related small molecule drugs correlated and guided by the computed crystal energy landscape. Cryst. Growth Des. 2014, 14, 2056–2072. [Google Scholar] [CrossRef]
- Case, D.H.; Srirambhatla, V.K.; Guo, R.; Watson, R.E.; Price, L.S.; Polyzois, H.; Cockcroft, J.K.; Florence, A.J.; Tocher, D.A.; Price, S.L. Successful Computationally Directed Templating of Metastable Pharmaceutical Polymorphs. Cryst. Growth Des. 2018, 18, 5322–5331. [Google Scholar] [CrossRef] [Green Version]
- Horneffer, V.; Dreisewerd, K.; Lüdemann, H.C.; Hillenkamp, F.; Läge, M.; Strupat, K. Is the incorporation of analytes into matrix crystals a prerequisite for matrix-assisted laser desorption/ionization mass spectrometry? A study of five positional isomers of dihydroxybenzoic acid. Int. J. Mass Spectrom. 1999. [Google Scholar] [CrossRef]
- Bērziņš, A.; Actiņš, A. Why Do Chemically Similar Pharmaceutical Molecules Crystallize in Different Structures: A Case of Droperidol and Benperidol. Cryst. Growth Des. 2016, 16, 1643–1653. [Google Scholar] [CrossRef]
- McGregor, L.; Rychkov, D.A.; Coster, P.L.; Day, S.; Drebushchak, V.A.; Achkasov, A.F.; Nichol, G.S.; Pulham, C.R.; Boldyreva, E.V. A new polymorph of metacetamol. CrystEngComm 2015, 17, 6183–6192. [Google Scholar] [CrossRef] [Green Version]
- Trotta, J.T.; Zeidan, T.A.; Tilak, P.A.; Foxman, B.M.; Almarsson, Ö.; Oliveira, M.A.; Chiarella, R.A.; Hickey, M.B.; Remenar, J.F. Aripiprazole and Dehydro-Aripiprazole Solid Solutions: Crystalline Combinations of Drug and Active Metabolite in Tailored Compositions. Cryst. Growth Des. 2020, 20, 3944–3956. [Google Scholar] [CrossRef]
- Srirambhatla, V.K.; Guo, R.; Price, S.L.; Florence, A.J. Isomorphous template induced crystallisation: A robust method for the targeted crystallisation of computationally predicted metastable polymorphs. Chem. Commun. 2016, 52, 7384–7386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeidan, T.A.; Trotta, J.T.; Tilak, P.A.; Oliveira, M.A.; Chiarella, R.A.; Foxman, B.M.; Almarsson, Ö.; Hickey, M.B. An unprecedented case of dodecamorphism: The twelfth polymorph of aripiprazole formed by seeding with its active metabolite. CrystEngComm 2016, 18, 1486–1488. [Google Scholar] [CrossRef]
- Okabe, N.; Kyoyama, H. 2,3-Dihydroxybenzoic acid. Acta Crystallogr. Sect. E Struct. Reports Online 2001. [Google Scholar] [CrossRef]
- Haisa, M.; Kashino, S.; Hanada, S.-I.; Tanaka, K.; Okazaki, S.; Shibagaki, M. The structures of 2-hydroxy-5-methylbenzoic acid and dimorphs of 2,5-dihydroxybenzoic acid. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1982. [Google Scholar] [CrossRef]
- Braun, D.E.; Karamertzanis, P.G.; Arlin, J.B.; Florence, A.J.; Kahlenberg, V.; Tocher, D.A.; Griesser, U.J.; Price, S.L. Solid-state forms of β-resorcylic acid: How exhaustive should a polymorph screen be? Cryst. Growth Des. 2011, 11, 210–220. [Google Scholar] [CrossRef]
- Gdaniec, M.; Gilski, M.; Denisov, G.S. γ-Resorcylic acid, its monohydrate and its pyridinium complex. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1994. [Google Scholar] [CrossRef]
- MacGillivray, L.R.; Zaworotko, M.J. Crystal and molecular structure of 2,6-dihydroxybenzoic acid. J. Chem. Crystallogr. 1994. [Google Scholar] [CrossRef]
- Mazurek, J.; Dova, E.; Helmond, R. 3,4-Dihydroxybenzoic acid acetonitrile solvate at 120 K. Acta Crystallogr. Sect. E Struct. Reports Online 2007. [Google Scholar] [CrossRef]
- Sarma, B.; Sanphui, P.; Nangia, A. Polymorphism in isomeric dihydroxybenzoic acids. Cryst. Growth Des. 2010, 10, 2388–2399. [Google Scholar] [CrossRef]
- Varughese, S.; Desiraju, G.R. Using water as a design element in crystal engineering. Host-guest compounds of hydrated 3,5-dihydroxybenzoic acid. Cryst. Growth Des. 2010, 10, 4184–4196. [Google Scholar] [CrossRef]
- Bērziņš, A.; Trimdale, A.; Kons, A.; Zvaniņa, D. On the formation and desolvation mechanism of organic molecule solvates: A structural study of methyl cholate solvates. Cryst. Growth Des. 2017, 17, 5712–5724. [Google Scholar] [CrossRef]
- Galabov, B.; Bobadova-Parvanova, P.; Ilieva, S.; Dimitrova, V. The electrostatic potential at atomic sites as a reactivity index in the hydrogen bond formation. Proc. J. Mol. Struct.: THEOCHEM 2003, 25, 101–112. [Google Scholar] [CrossRef]
- Bajpai, A.; Scott, H.S.; Pham, T.; Chen, K.J.; Space, B.; Lusi, M.; Perry, M.L.; Zaworotko, M.J. Towards an understanding of the propensity for crystalline hydrate formation by molecular compounds. IUCrJ 2016. [Google Scholar] [CrossRef] [PubMed]
- Davey, R.J.; Dent, G.; Mughal, R.K.; Parveen, S. Concerning the relationship between structural and growth synthons in crystal nucleation: Solution and crystal chemistry of carboxylic acids as revealed through IR spectroscopy. Cryst. Growth Des. 2006, 6, 1788–1796. [Google Scholar] [CrossRef]
- Hansen, P.E.; Spanget-Larsen, J. NMR and IR investigations of strong intramolecular hydrogen bonds. Molecules 2017, 552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobrovs, R.; Drunka, L.; Auzins, A.A.; Jaudzems, K.; Salvalaglio, M. Polymorph-Selective Role of Hydrogen Bonding and π-π Stacking in p-Aminobenzoic Acid Solutions. Cryst. Growth Des. 2020, 21, 436–448. [Google Scholar] [CrossRef]
- Frisch, Æ.; Plata, R.E.; Singleton, D.A. Gaussian 09W Reference. J. Am. Chem. Soc. 2009, 137, 3811–3826. [Google Scholar] [CrossRef] [Green Version]
- Di Tommaso, D. The molecular self-association of carboxylic acids in solution: Testing the validity of the link hypothesis using a quantum mechanical continuum solvation approach. CrystEngComm 2013, 15, 6564–6577. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017. [Google Scholar] [CrossRef] [Green Version]
- Kashinski, D.O.; Chase, G.M.; Nelson, R.G.; Di Nallo, O.E.; Scales, A.N.; Vanderley, D.L.; Byrd, E.F.C. Harmonic Vibrational Frequencies: Approximate Global Scaling Factors for TPSS, M06, and M11 Functional Families Using Several Common Basis Sets. J. Phys. Chem. A 2017, 121, 2265–2273. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general Amber force field. J. Comput. Chem. 2004. [Google Scholar] [CrossRef] [PubMed]
- AMBER Amber 2019 Reference Manual. 2019. Available online: https://ambermd.org/doc12/Amber19.pdf (accessed on 15 May 2020).
- Caleman, C.; Van Maaren, P.J.; Hong, M.; Hub, J.S.; Costa, L.T.; Van Der Spoel, D. Force field benchmark of organic liquids: Density, enthalpy of vaporization, heat capacities, surface tension, isothermal compressibility, volumetric expansion coefficient, and dielectric constant. J. Chem. Theory Comput. 2012, 8, 61–74. [Google Scholar] [CrossRef]
- Van der Spoel, D.; van Maaren, P.J.; Caleman, C. GROMACS molecule & liquid database. Bioinformatics 2012. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007. [Google Scholar] [CrossRef] [Green Version]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Bonomi, M.; Bussi, G.; Camilloni, C.; Tribello, G.A.; Banáš, P.; Barducci, A.; Bernetti, M.; Bolhuis, P.G.; Bottaro, S.; Branduardi, D.; et al. Promoting transparency and reproducibility in enhanced molecular simulations. Nat. Methods 2019. [Google Scholar] [CrossRef] [Green Version]
- Adam, M.S.; Gutmann, M.J.; Leech, C.K.; Middlemiss, D.S.; Parkin, A.; Thomas, L.H.; Wilson, C.C. Stability and cooperativity of hydrogen bonds in dihydroxybenzoic acids. New J. Chem. 2010, 34, 85–91. [Google Scholar] [CrossRef]
- Parkin, A.; Adam, M.; Cooper, R.I.; Middlemiss, D.S.; Wilson, C.C. Structure and hydrogen bonding in 2,4-dihydroxybenzoic acid at 90, 100, 110 and 150 K; a theoretical and single-crystal X-ray diffraction study. Acta Crystallogr. Sect. B Struct. Sci. 2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sridhar, B. Synthon preference in a hydrated β-resorcylic acid structure and its cocrystal with thymine. Acta Crystallogr. Sect. C Struct. Chem. 2015. [Google Scholar] [CrossRef] [PubMed]
- Price, C.P.; Glick, G.D.; Matzger, A.J. Dissecting the behavior of a promiscuous solvate former. Angew. Chemie—Int. Ed. 2006. [Google Scholar] [CrossRef]
- Jensen, T.T.; Hall, C.L.; Potticary, J.; Andrusenko, I.; Gemmi, M.; Hall, S.R. An experimental and computational study into the crystallisation propensity of 2nd generation sulflower. Chem. Commun. 2019, 55, 14586–14589. [Google Scholar] [CrossRef]
- Rychkov, D.A.; Hunter, S.; Kovalskii, V.Y.; Lomzov, A.A.; Pulham, C.R.; Boldyreva, E.V. Towards an understanding of crystallization from solution. DFT studies of multi-component serotonin crystals. Comput. Theor. Chem. 2016, 1088, 52–61. [Google Scholar] [CrossRef]
- Davey, R.J.; Schroeder, S.L.M.; Ter Horst, J.H. Nucleation of organic crystals—A molecular perspective. Angew. Chemie—Int. Ed. 2013. [Google Scholar] [CrossRef]
- Bux, K.; Moin, S.T. Solvation of cholesterol in different solvents: A molecular dynamics simulation study. Phys. Chem. Chem. Phys. 2020. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | THF | DXN | ACN | IPA | Water | Group |
|---|---|---|---|---|---|---|
| Compound | ||||||
| 2,3-diOHBA | I | I | I | I | I | Group A |
| 2,5-diOHBA | II | II | II | I + II | II | Group B |
| 2,4-diOHBA | I/II | II + I | II | II | HH | |
| 2,6-diOHBA | MH/ II + MH | MH/ II + MH | MH+II | II | MH | Group C |
| 3,4-diOHBA | I + MH I + MH II | S0.5DXN | SACN + MH II /SACN | MH I | MH I | Group D |
| 3,5-diOHBA | S0.25THFMH /S0.5THF | SDXN | SACN | I + HH | MH I |
| Ortho-Substituted diOHBA | Non-Ortho-Substituted diOHBA |
|---|---|
| Crystal form landscape | |
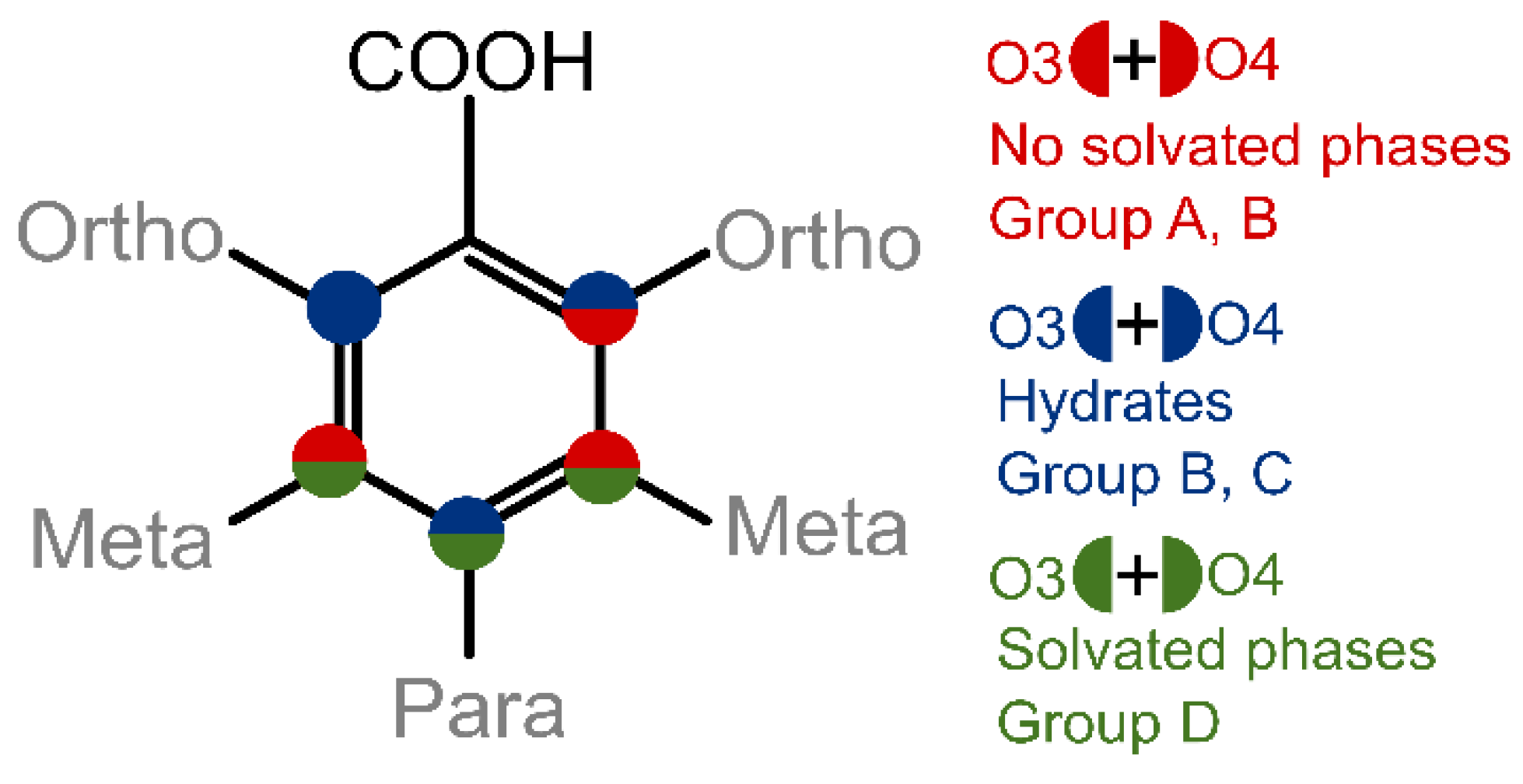
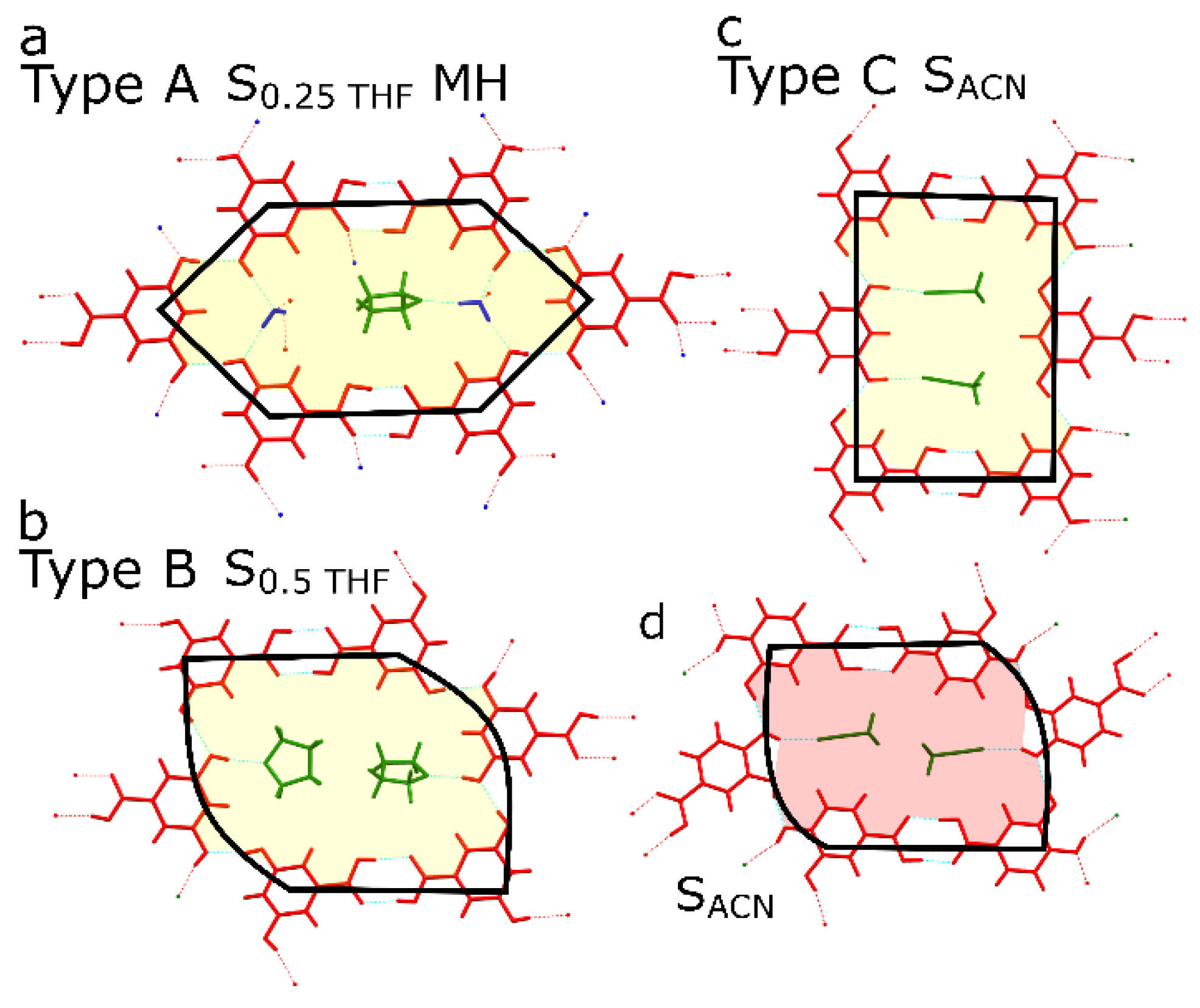
| Low propensity to form solvates; can be divided into Group A, Group B (for both groups mostly the most stable polymorph was obtained) and Group C (prone to form hydrate). | Readily forms hydrates and solvates; nonsolvated phases are complicated to obtain in crystallization (Group D compounds). |
| Crystal structure evaluation | |
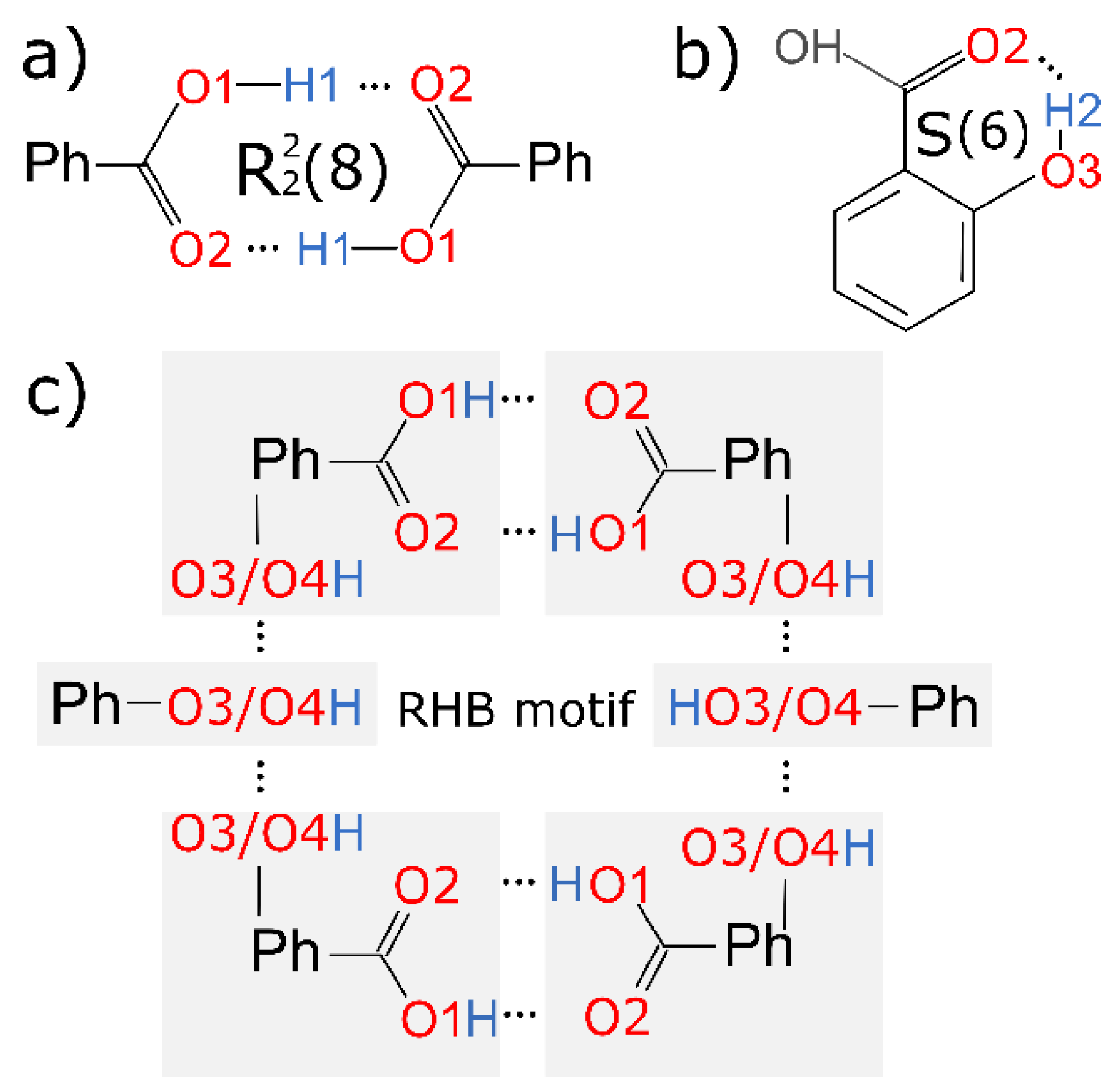
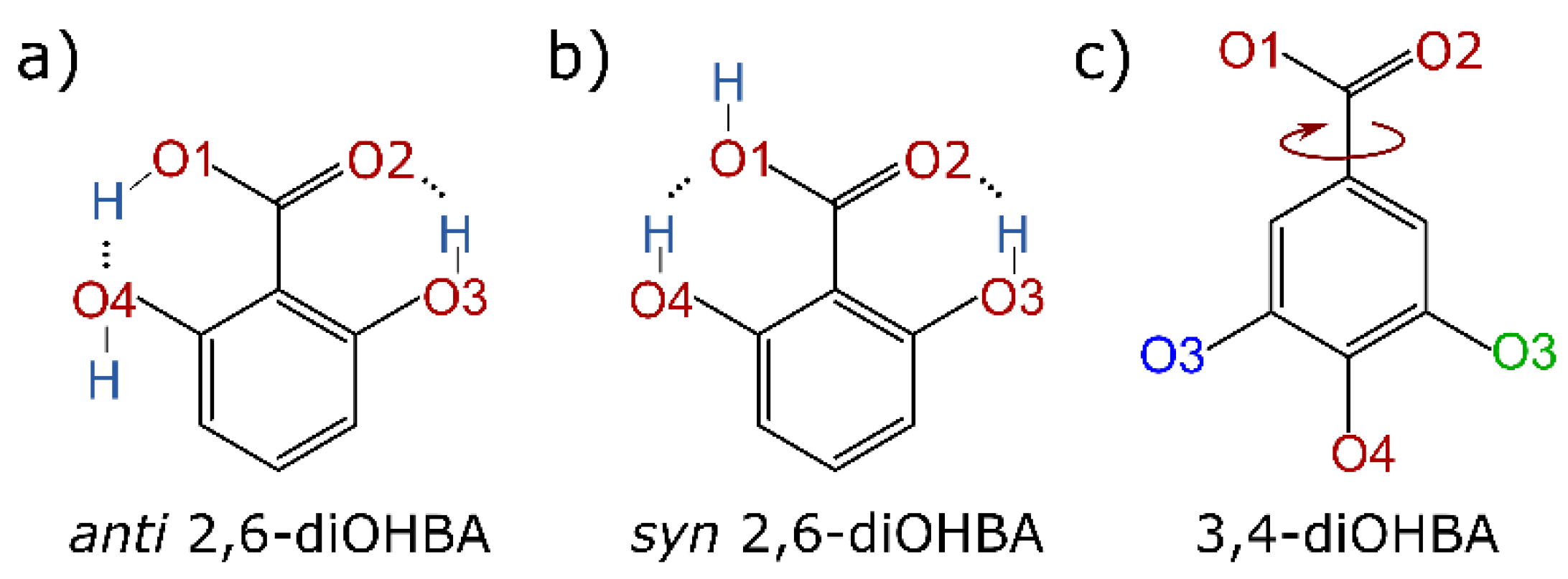
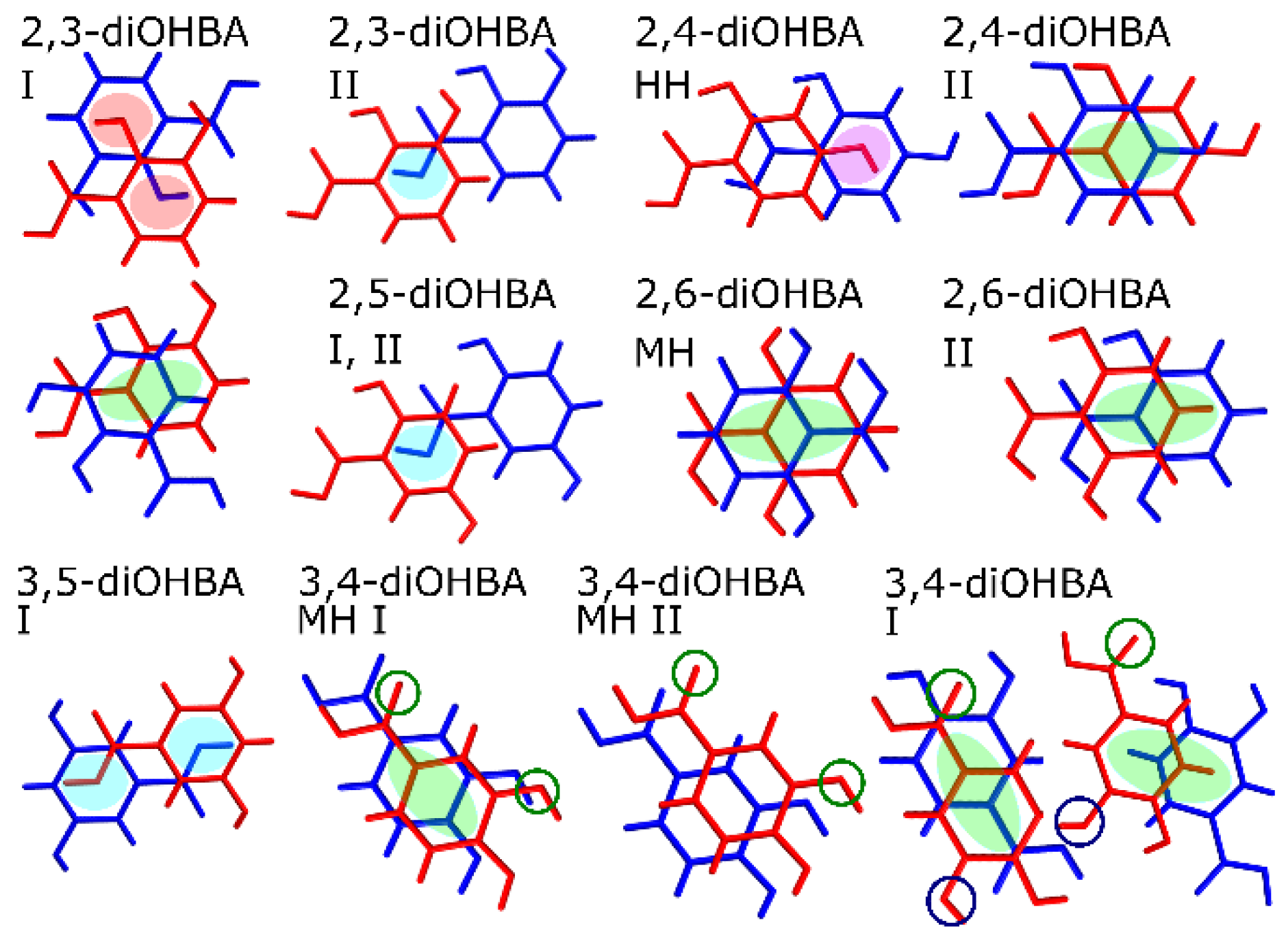
| |
| If able, phenolic hydroxyl groups form infinite hydrogen-bonded chains, which stabilize the structures and the incorporation of guest molecules in the structure is hindered. | Ring-like hydrogen bond motifs are essential for solvate formation. In case of 3,5-diOHBA, guest molecules stabilize the structures. |
| Electrostatic potential surfaces of diOHBA | |
| Intramolecular bond O2…H–O3 causes an uneven distribution of ESP extrema in the molecule. | ESP extrema values on phenolic hydroxy and carboxyl groups does not significantly differ, ESP extrema are evenly distributed across the molecule. |
| Spectroscopy studies of association | |
| |
| Carboxyl group involving associates are present only in 2-propanol solutions. | Carboxyl group involving associates are present in acetonitrile and 2-propanol solutions. |
| Molecular dynamics simulations | |
| |
| In simulations, the most abundant are the carboxyl group–phenolic hydroxyl group associates, followed by the phenolic hydroxyl group associates. The intramolecular bond O3–H…O2 heavily affects the abundance of associates formed by the hydrogen bond between the phenolic hydroxyl group (O3) and solvent molecules. | In simulations, the most abundant are carboxyl group self-associates. The probability of the formation of a phenolic hydroxyl group–solvent interaction is considerably higher than that exhibited by ortho-substituted diOHBAs. The intramolecular bond between the phenolic hydroxyl groups has almost no effect on the probability of association with the solvent. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trimdale, A.; Mishnev, A.; Bērziņš, A. Combined Use of Structure Analysis, Studies of Molecular Association in Solution, and Molecular Modelling to Understand the Different Propensities of Dihydroxybenzoic Acids to Form Solid Phases. Pharmaceutics 2021, 13, 734. https://doi.org/10.3390/pharmaceutics13050734
Trimdale A, Mishnev A, Bērziņš A. Combined Use of Structure Analysis, Studies of Molecular Association in Solution, and Molecular Modelling to Understand the Different Propensities of Dihydroxybenzoic Acids to Form Solid Phases. Pharmaceutics. 2021; 13(5):734. https://doi.org/10.3390/pharmaceutics13050734
Chicago/Turabian StyleTrimdale, Aija, Anatoly Mishnev, and Agris Bērziņš. 2021. "Combined Use of Structure Analysis, Studies of Molecular Association in Solution, and Molecular Modelling to Understand the Different Propensities of Dihydroxybenzoic Acids to Form Solid Phases" Pharmaceutics 13, no. 5: 734. https://doi.org/10.3390/pharmaceutics13050734