1. Introduction
The rational use of our arsenal of anti-infective drugs provides the basis of a successful treatment of bacterial infections. Unfortunately, the emergence of resistance challenges this paradigm. Treatment of lower respiratory tract infections (LRTI), currently one of the leading infectious causes of death worldwide [
1] and mainly caused by
S. pneumoniae [
2], is highly affected by antibiotic resistance [
1]. Due to the lack of new antibiotics, it is urgently required to investigate innovative treatment options such as stimulating the innate immune system [
3].
One frequently used drug to treat LRTI is amoxicillin (AMX), a well-established beta-lactam antibiotic classified as essential medicine by the World Health Organisation [
4]. To sustain the drug’s effectiveness, the immunomodulatory characteristics of monophosphoryl lipid A (MPLA) were recently studied in combination with AMX [
5]. MPLA, a toll-like receptor 4 (TLR4) agonist with a favorable safety profile [
6], is already licensed as adjuvant with immunomodulatory characteristics for vaccines (Fendrix
®, hepatitis B [
7]); Cervarix
®, human papilloma virus [
8]). Based on in vivo data in a murine infection model, Casilag et al. proposed that MPLA in monotherapy has the ability to stimulate the immune system, resulting in higher efficacy in terms of reduced bacterial numbers at the target sites lung and spleen as well as increased survival [
5]. These effects were even more pronounced in combination with AMX [
5]. However, the mechanisms of these interactions are still unknown.
By bridging knowledge from drug concentrations to bacterial numbers and ultimately survival, this study aims to integrate knowledge from multiple levels of generated data and addresses a more detailed understanding of the underlying drug interactions. To gain quantitative and mechanistic insights into the combination, it is favorable to analyze pharmacokinetic (PK) and pharmacodynamic (PD) characteristics. To accomplish this, nonlinear mixed-effects (NLME) modelling in terms of PK/PD modelling can be employed to study experimentally obtained results beyond a common descriptive analysis, allowing to amalgamate information on PK and PD levels, to integrate multiple study data and to translate results into a clinical setting within one approach. Several examples in a preclinical or clinical setting have shown the advantages of this approach [
9,
10,
11,
12,
13,
14,
15]: PK and PD, in terms of drug concentrations and bacterial numbers or survival, are analyzed simultaneously over the entire time in a coherent framework.
The objective of this study was to evaluate the preclinical PK/PD relationship of a novel experimental antibiotic and immunostimulatory combination regimen and quantitatively define its efficacy and interactions by applying pharmacometric PK/PD modelling. Using NLME modelling not only allows to link information from drug concentrations and bacterial numbers but also enables the link to survival data. Thereby, we aimed to associate overall survival to respective model-predicted parameters by performing time-to-event (TTE) analysis.
3. Results
Pharmacometric Modelling
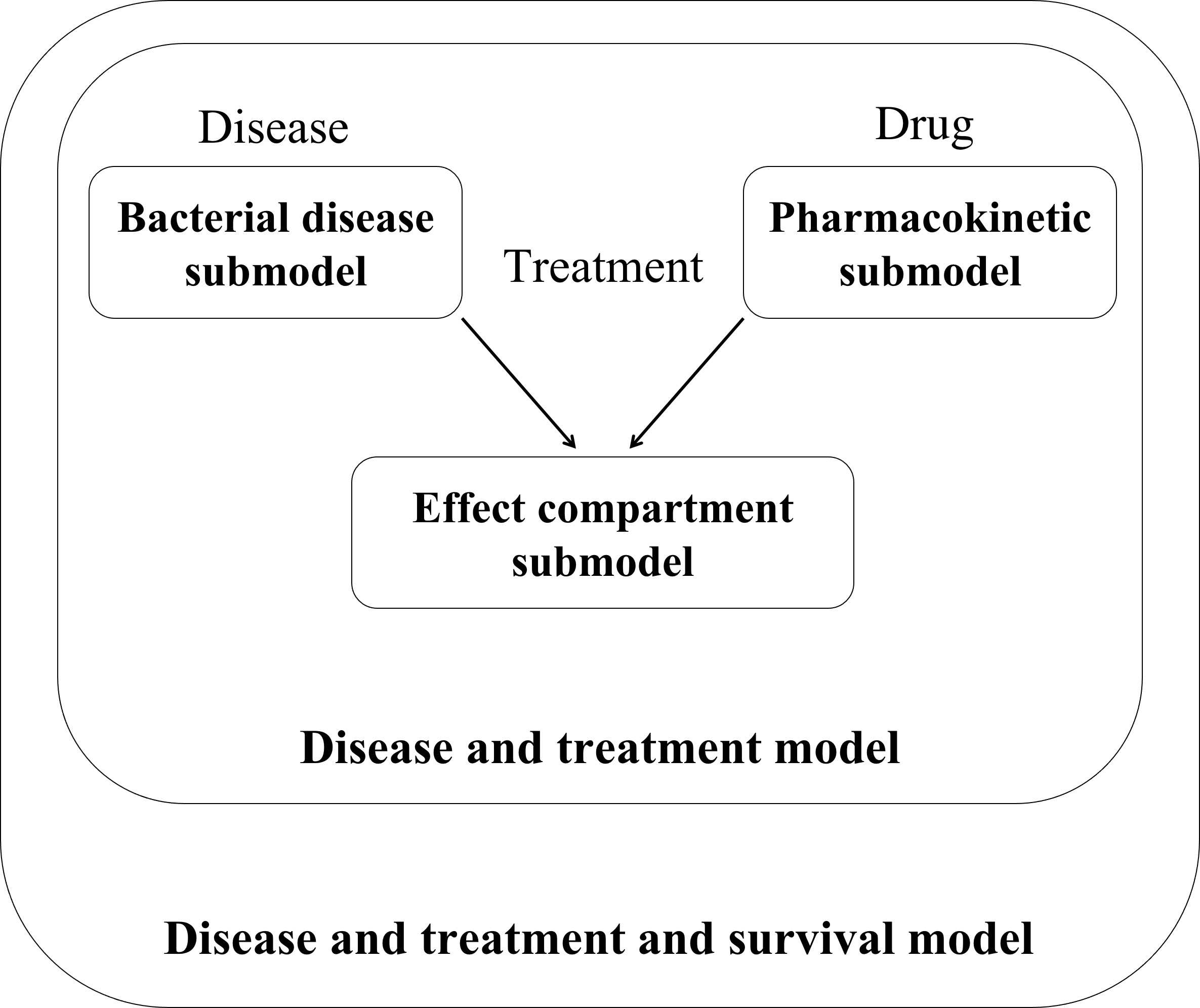
The separately developed PK, bacterial disease, effect compartment and disease and treatment submodels were combined to a “pharmacometric PK/PD model” (
Figure 1;
Table 1;
Supplementary Section S3).
The final structural “PK submodel” (
Figure 1, left) for AMX comprised 106 quantified murine serum concentrations (15.1% below the lower limit of quantification (LLOQ = 0.01 µg/mL), successful in-study validation with acceptable accuracy and precision) with one to two samples per individual mouse. Within this study, total AMX concentrations were quantified and used for further analyses given the relatively low and linear protein binding (17% [
20]). The submodel consisted of a two-compartment model with first-order absorption including a lag time as well as first-order linear elimination and a proportional residual unexplained variability (RUV) model and was able to describe the general trend of the AMX concentration-time profile. Given the observed shift of the maximum serum concentration (C
max) of AMX from 0.167 h in monotherapy to 0.5 h in a combined treatment approach (
Figure S1), the integration of MPLA coadministration on the AMX clearance depending on the AMX dose as covariate significantly increased the predictive performance of the model as assessed by standard model evaluation techniques. Indeed, MPLA combined with the highest AMX dose (14 mg/kg) decreased the clearance of AMX by 40.9% (73.3 mL/h) compared to monotherapy; at low dose (0.4 mg/kg), the effect was negligible (–1.17%, 123 mL/h). The mouse type (RjOrl:Swiss, Balb/cJRj) did not significantly have an impact on the model performance and, hence, was not included.
The proposed “bacterial disease submodel” (
Figure 1, right), characterized by natural growth and treatment-unrelated killing and natural death kinetics, described bacterial growth of intranasally administered
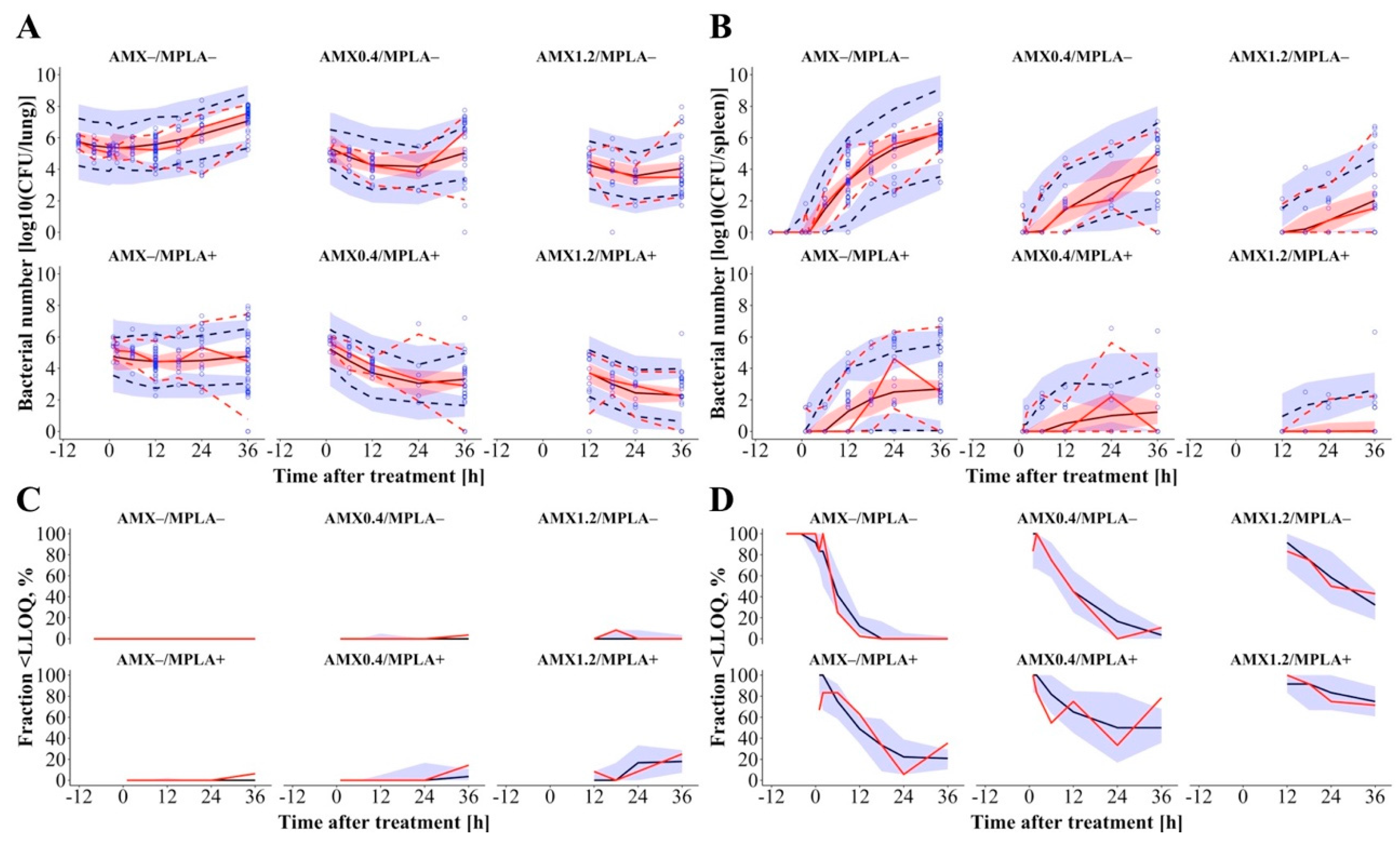
S. pneumoniae in lung with bacterial growth after a slight short-term reduction in bacterial numbers adequately (
Figure 2A, “AMX-/MPLA-“ study group). By use of a transit compartment model, of which the number of compartments was optimized to be 23, the transit of bacteria from lung to spleen was described. In the spleen, the bacterial input was modelled as a transit of bacteria from the lung without any outflow leading to accumulation of bacteria after more than 12 h after infection as seen in the original study data. The PK and the bacterial disease model were linked via two separate effect compartments in lung and spleen assuming a transfer of AMX to the respective organs (
Figure 1, middle).
According to the reported results of the murine infection model [
5], AMX monotherapy decreased bacterial numbers in the lung up to 24 h and a dose-dependent regrowth was observed afterwards, whereas MPLA-treated animals displayed more or less stable bacterial numbers after initial bacterial killing and the combination of both drugs led to the highest reduction in bacterial numbers (Symbols in
Figure 2A, “AMX+/MPLA-“, “AMX-/MPLA+“ and “AMX+/MPLA+“ study groups). In the spleen, bacterial numbers increased after more than 2 h showing comparable patterns regarding the efficacy of mono- versus combination therapies as in the lung for all treatment groups (Symbols in
Figure 2B). These trends were captured in the “disease and treatment submodel”, where the effect of AMX was best characterized in lung by a sigmoidal maximum effect (E
max) model as drug-dependent bacterial killing process. The concentration-effect relationship was steep (Hill factor = 20) indicating that AMX was only efficacious as long as AMX concentrations in lung were above the AMX concentration to achieve half maximum effect (EC
50 = 0.0109 µg/mL). In the spleen, the AMX killing effect was described by a power model. The effect of MPLA on bacteria in the lung was implemented as its ability to enhance the efficacy of the treatment-unrelated killing and natural death effects on bacteria. Contrarily, the effect of MPLA in spleen was included as separate killing process. A potential PD interaction of AMX and MPLA was further analyzed to define potential synergy or antagonism. However, the results clearly indicated an additive effect of AMX and MPLA and did not hint at any synergism or antagonism. Final model parameter estimates described the underlying PD processes reliably with acceptable relative standard errors (RSE<46.2%) and based on performed bootstrap analyses (
Table 1) and VPC adequately captured measured AMX serum concentrations (
Figure S2) and bacterial numbers in lung and spleen (
Figure 2) above and below the LLOQ (general trend, variability, and fraction, respectively). Here, PK parameters were fixed to the final parameters of the PK submodel, although a simultaneous modelling approach of PK and PD was aimed for but excluded due to (i) plausibility (PK and PD were not determined within one animal) and (ii) to improve model stability. Further results of all model specifications are given in
Supplementary Section S3.
AMX as monotherapy at the highest investigated dose (1.2 mg/kg, PD studies) and MPLA reduced model-predicted bacterial numbers in the lung by 3.03 and 1.71 log10 colony forming units (CFU)/lung, respectively, compared to natural growth after 36 h (
Figure S3, left). The combination also showed a bactericidal effect with a reduction of 4.77 log10 CFU/lung. In the spleen, comparable characteristics were observed: Here, the maximum effect of total bacterial elimination was determined for doses of 0.4 and 1.2 mg/kg AMX with MPLA coadministration (
Figure S3, right).
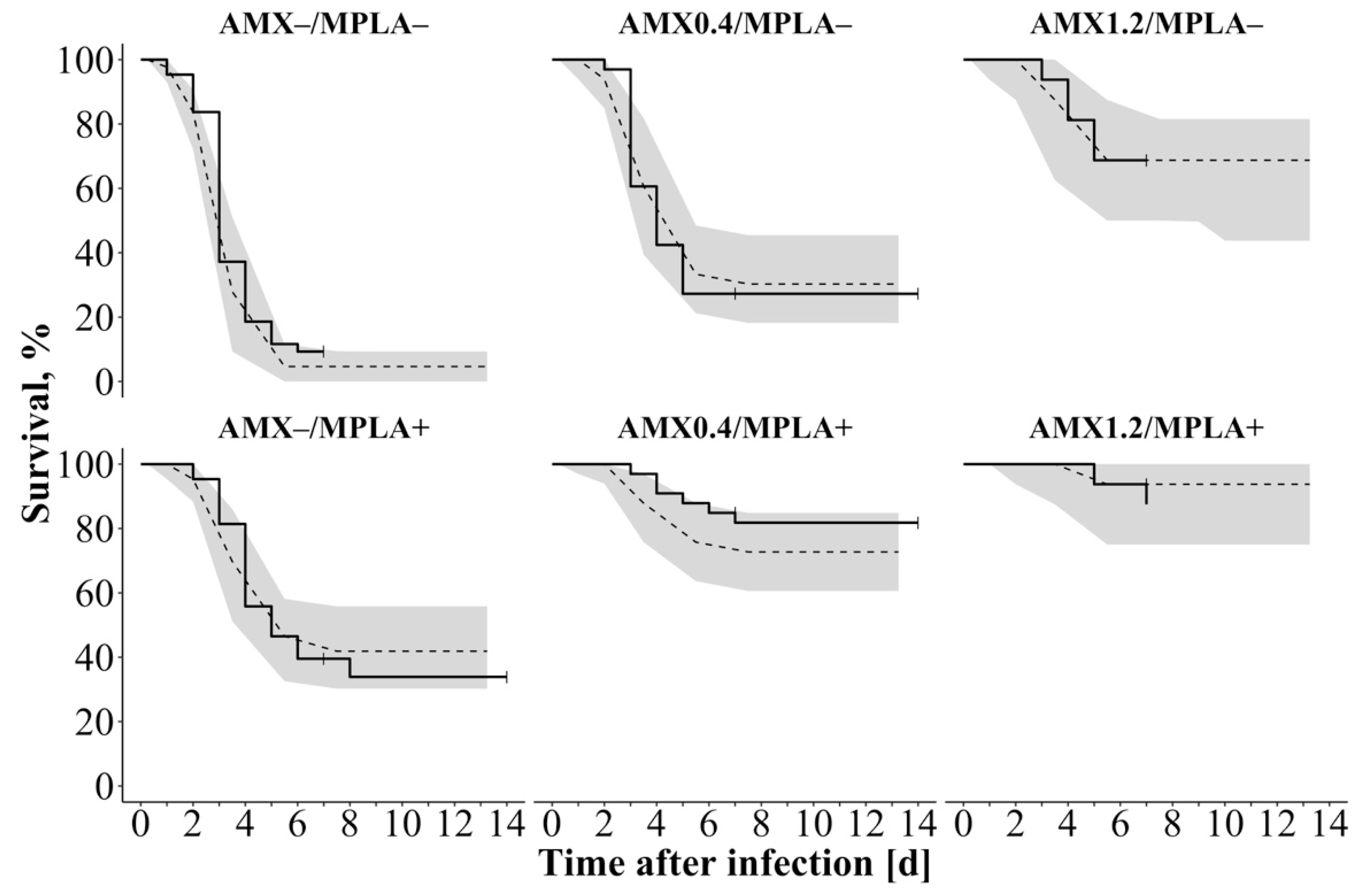
Survival studies of infected mice revealed the highest mortality for untreated mice, whereas combined treated mice had highest survival rates [
5]. The “TTE model” revealed the best predictive performance of the survival data by using a surge function to describe the hazard being mostly present between day 3 and 6. Combining %T
>MIC in serum and coadministration with MPLA displayed the best relation and plausibility between PK, PD, and survival, when included exponentially as covariates on the overall hazard: In all study groups, survival was predicted reliably except for mice treated with 0.40 mg/kg AMX and MPLA. Here, survival was slightly overestimated, but as median observed survival was still within the 90% CI of the median simulated survival, the predictions were thus considered appropriate. Other investigated covariates, e.g., the model-predicted bacterial number in spleen after 36 h after treatment, were excluded due to the simplicity of %T
>MIC, although a comparably good predictability was observed. VPC (
Figure 3) as well as a bootstrap analysis (
Table 2) supported this structure with stable parameter estimates.
A high hazard was primarily present between day 1 and 6 with the highest maximum hazard between day 3 and 5 justifying a surge function (
Figure S4). A larger %T>MIC of AMX as well as presence of MPLA decreased the overall hazard and thereby increased survival: The overall hazard of untreated mice was 3.71-fold higher than for MPLA-treated mice. Most important, cotreatment with MPLA was able to reduce the hazard of AMX-treated mice at maximum dose by a factor of 4.00. After 14 d, survival was increased 1.35-fold from 66.7% for AMX monotherapy at highest dose to 90.3% in a combined treatment approach due to the immune system stimulation effect of MPLA: Coadministration of MPLA (T
>MIC > 3.25 h) reduced the required T
>MIC to reach survival >95% by 1.23 h compared to AMX monotherapy at highest dose (T
>MIC > 4.48 h).
4. Discussion
Aiming to develop novel therapeutic approaches to overcome the emergence of bacterial resistance, the PK/PD relationship of the innovative treatment option of the antibiotic AMX and the TLR4 agonist MPLA was exploited. Here, pharmacometric approaches were comprehensively investigated as useful tools to further quantitatively interpret in vivo defined outcomes.
Experimental data indicated beneficial effects of AMX, when coadministered by MPLA to stimulate the immune system, but lacked a more pronounced mechanistic and quantitative understanding of involved processes and interactions [
5]. Our pooled pharmacometric analysis of various studies integrated several information starting from PK in terms of drug concentrations to PD in terms of bacterial numbers and survival to provide quantitative and mechanistic insights into this treatment option against pneumonia. On basis of the comprehensive analysis of the immune system stimulating effects of MPLA and the antibiotic effects of AMX by Casilag et al. [
5], we demonstrated that MPLA has the ability to stimulate the immune system and additively enhanced the AMX activity on top of a PK interaction. We stepwise integrated more data allowing reliable estimation of PK/PD relationships that were successfully linked to overall survival employing easily accessible clinical parameters (%T
>MIC). This analysis highlights the necessity to amalgamate multiple levels of generated data to gain detailed information of underlying interactions and physiological processes.
In our “PK submodel”, we found that MPLA substantially reduced the AMX clearance depending on the AMX dose. Although no pharmacokinetic studies analyzing the combination of AMX and MPLA have been published, an analogous pharmacometric model has been reported by Moine et al. in mice being treated with AMX subcutaneously [
21] supporting plausibility of the “PK submodel”. The elimination of AMX monotherapy with a clearance of 124 mL/h was higher than reported creatinine clearance values in mice [
22,
23] indicating tubular secretion of AMX, most probably by organic anion transporters [
24]. This is also in agreement with described elimination processes of AMX being rarely non-renal [
20]. MPLA is also partly excreted renally [
6] and one can hypothesize that MPLA reduces tubular secretion of AMX (PK interaction) competitively at high AMX concentrations. Such a mechanism may also result in a potential PK interaction in humans. Therefore, a clinical dose finding study and drug–drug interaction study would need to be conducted to evaluate the relevance in humans. The identified PK interaction is limited given the fact that only a standard dose of MPLA, that was chosen based on prior studies [
5], and two different doses of AMX were studied, and hence the PK interaction was simplified by a linear relationship, which, e.g., could also have been exponential or sigmoidal. Still, the PK interaction did not quantitatively explain the PD results after combination therapy, especially at the investigated relatively low doses of the PD study (0.2–1.2 mg/kg) that only displayed negligible PK interaction compared to the highest dose of the PK study (14 mg/kg). Consequently, a PD interaction was investigated by analyzing bacterial numbers on top of the PK interaction.
The “bacterial disease submodel” in the lung consisted of a first-order growth process being influenced by a delay in onset of bacterial growth probably because bacteria needed to adjust to the new environment. The high model-predicted number of transit compartments characterized the manifold involved processes needed for the transit of bacteria from lung to spleen and displayed the physiological relevance: Several membranes have to be passed by bacteria to enter the blood stream and ultimately reach the spleen. The implemented bacterial elimination contained not only natural death of bacteria, but also killing effects attributable to the immune system that were predicted to be present consistently over time after initial stimulation by the bacteria. One limitation of our analysis is that we were not able to distinguish these processes within the model, which could, e.g., be investigated in immunodeficient mice in a next step. Nevertheless, bacterial growth outperformed the killing processes over time leading to a higher bacterial burden and, subsequently, reduced survival rates.
Analyzing the “disease and treatment submodel”, the killing effects of AMX and MPLA were successfully characterized. Separate first-order rate constants for the effect delay of AMX in lung and spleen were determined. These empirically derived differences seem to be plausible given the physiological differences in the AMX transfer from serum to lung and spleen as a manifold of processes are involved, e.g., different membranes, transporters, or blood flow. AMX bacterial killing in the lung was best described by on–off kinetics with the EC
50 as threshold being related to effect compartment concentrations, whereas MPLA increased the immune system attributable killing effects constantly over time, showing highest killing in combination with AMX. Although the AMX effect was rather expected to be drug than organ specific, the effects of AMX needed to be modelled differently between lung and spleen. The different situation in the lung compared to the spleen, where a slower equilibrium formation with serum (
Figure S5) led to longer lasting effects and missing observations attributable to killing effects of the immune system, required a more simplified implementation in the spleen and led to a missing bacterial outflow from the spleen compartment. Here, future studies would benefit from quantifying drug concentrations at the target sites to describe these processes more physiologically. The model-predicted efficacy of AMX being associated with a high time of antibiotic concentrations above the EC
50 (T
>EC50) correlated well with the %T
>MIC, the main PK/PD parameter used for the usually time-dependent beta-lactam antibiotics. %T
>MIC as an easily accessible clinical parameter in addition to simple binary MPLA implementation was able to capture murine survival after infection.
In this preclinical setting, mice received only a single dose of AMX. Our results suggest that AMX affects bacteria within the first phase of infection, mainly visible only for a limited period, whereas MPLA at the same time stimulates the immune system in a sustained manner. Unfortunately, the PK of MPLA was not monitored due to missing sensitivity of investigated MPLA assays, and, hence, no response surface analysis [
10,
25] was feasible to comprehensively investigate possible further PD interactions over time. However, the immune system stimulating characteristics of MPLA may be advantageous in a clinical framework.
This work adds weight to the use of pharmacometric PK/PD modelling and subsequent survival analysis being useful tools to support and further quantitatively interpret experimentally defined outcomes of in vivo studies and to evaluate the effectiveness of the proposed combination comprehensively. Despite certain limitations such as the variability in the experimental data or the ethical limitation of only one sample per individual mouse in terms of limited blood volume (PK) or physiological limitations (PD) that only allowed to analyze the in vivo data with caution due to missing intraindividual variability especially for PD observations, valuable quantitative insights into the combination were achieved by the use of NLME modelling taking all these different individual observations into account.
The herein outlined approach contributes to reducing the use of animals in preclinical drug development studies, since various scenarios, e.g., investigating different dosing regimens, can be simulated (a priori). In a next step, the PK/PD-TTE model is apt to be translated into a clinical setting for the clinical development program bringing this promising combination therapy closer to patients as translation to clinics of comparable approaches based on immune stimulation has already been proposed [
3]. However, due to the use of separate mice for PK, PD and immunostimulation experiments, translating the current results to humans is challenging. To accomplish this step, additional concentration-effect data, or dose-response data for MPLA would be needed. Therefore, further studies investigating adjunct use of immunostimulatory compounds should use i) different doses of the immunomodulator, ii) measure PK and PD simultaneously at least in some experiments, and iii) use multiple experimental systems such as different animal species, immunocompetent and -deficient animals or in vitro set-ups tailored to specific endpoints of interest. Still, the safe use of MPLA in humans and the augmented innate antimicrobial immunity might establish a basis for a successful translation as adjunct therapy to conventional antibiotic treatments.
In conclusion, we demonstrated that combining antibiotics with immunostimulatory drugs may serve as a promising approach to augment the effect of antibiotics. Leveraging a pharmacometric PK/PD analysis approach, the performed analysis showed the ability of MPLA to stimulate the immune system and additively enhance the efficacy of the antibiotic AMX in terms of bacterial burden and survival. Further studies with other antibiotics and—ultimately—evaluation of such approaches in the clinical setting are warranted.



 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}