
Cyclodextrins in Antiviral Therapeutics and Vaccines

 ,
,  , , and
, , and
Abstract
:
1. Introduction
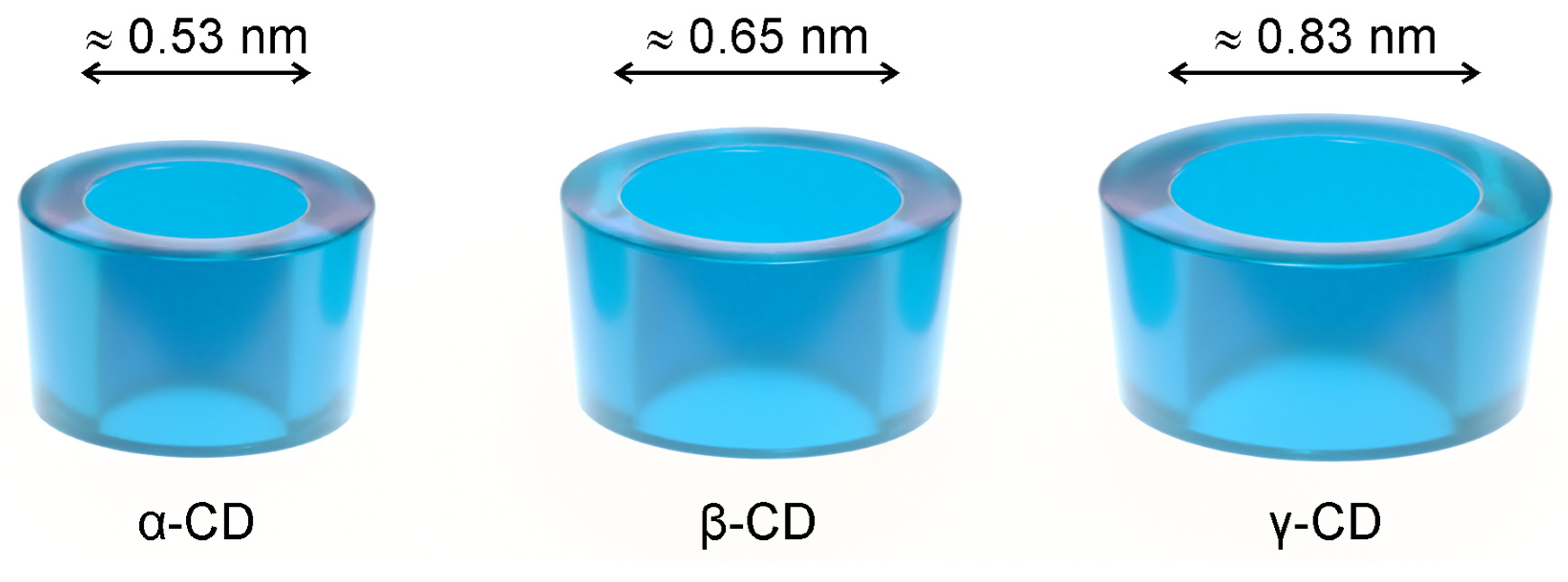
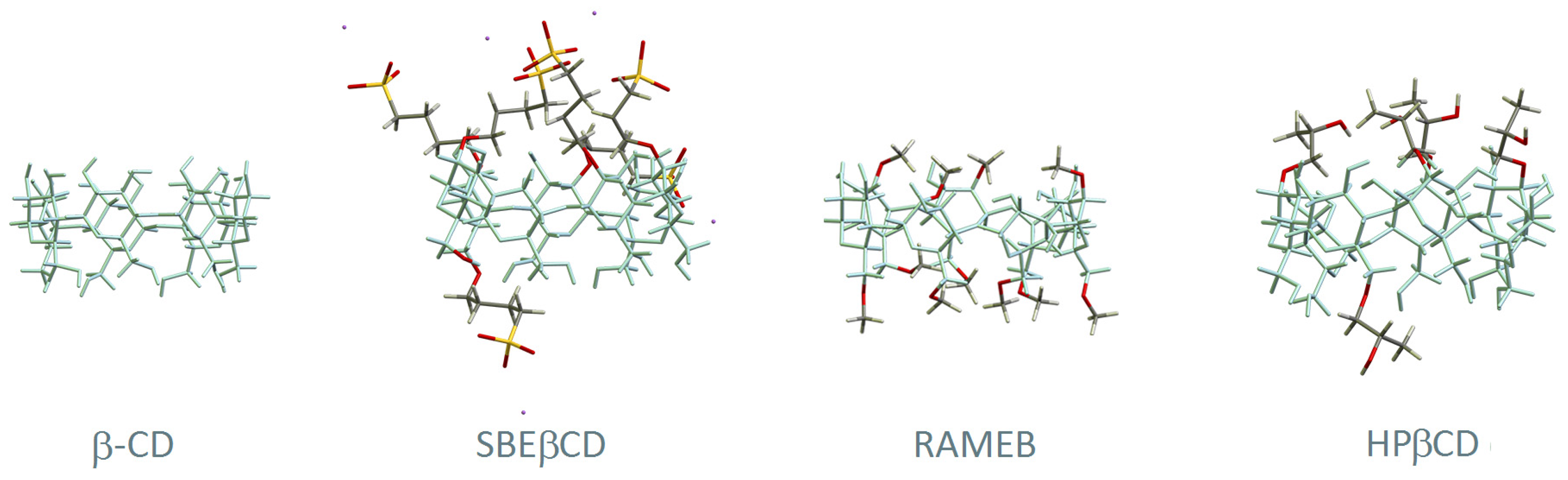
2. Cyclodextrins and Antiviral Drugs
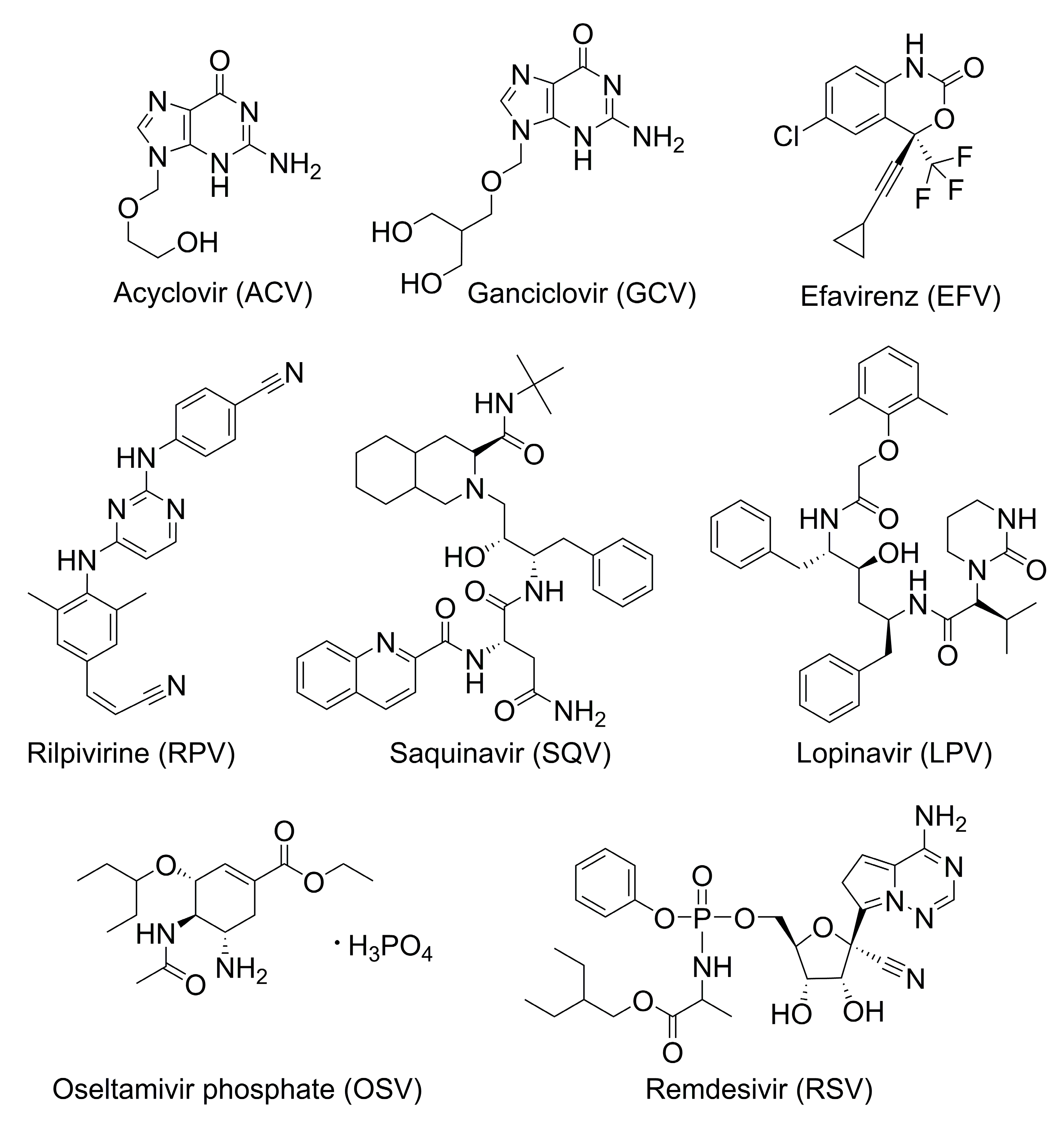
2.1. Acyclovir
2.2. Ganciclovir
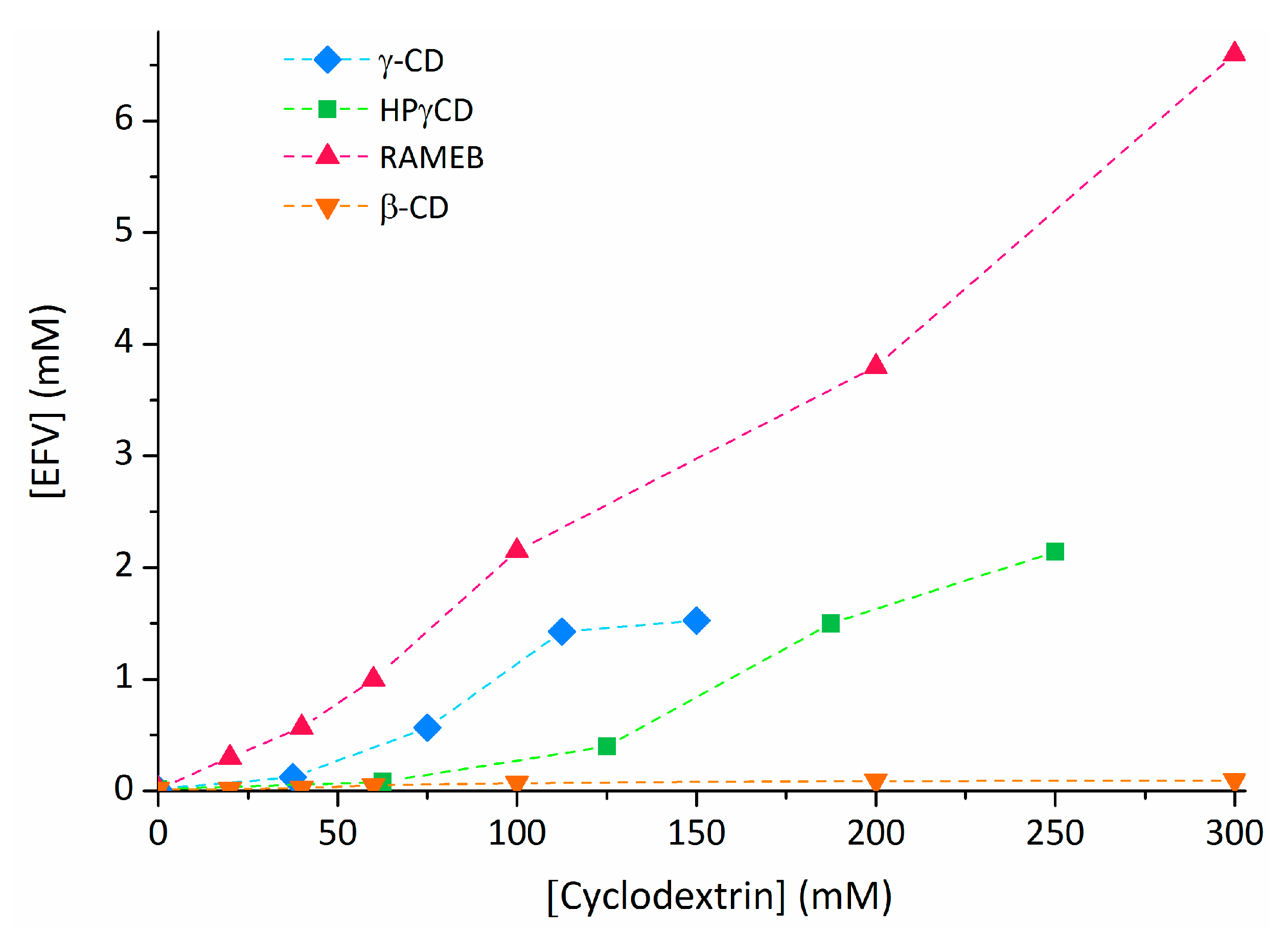
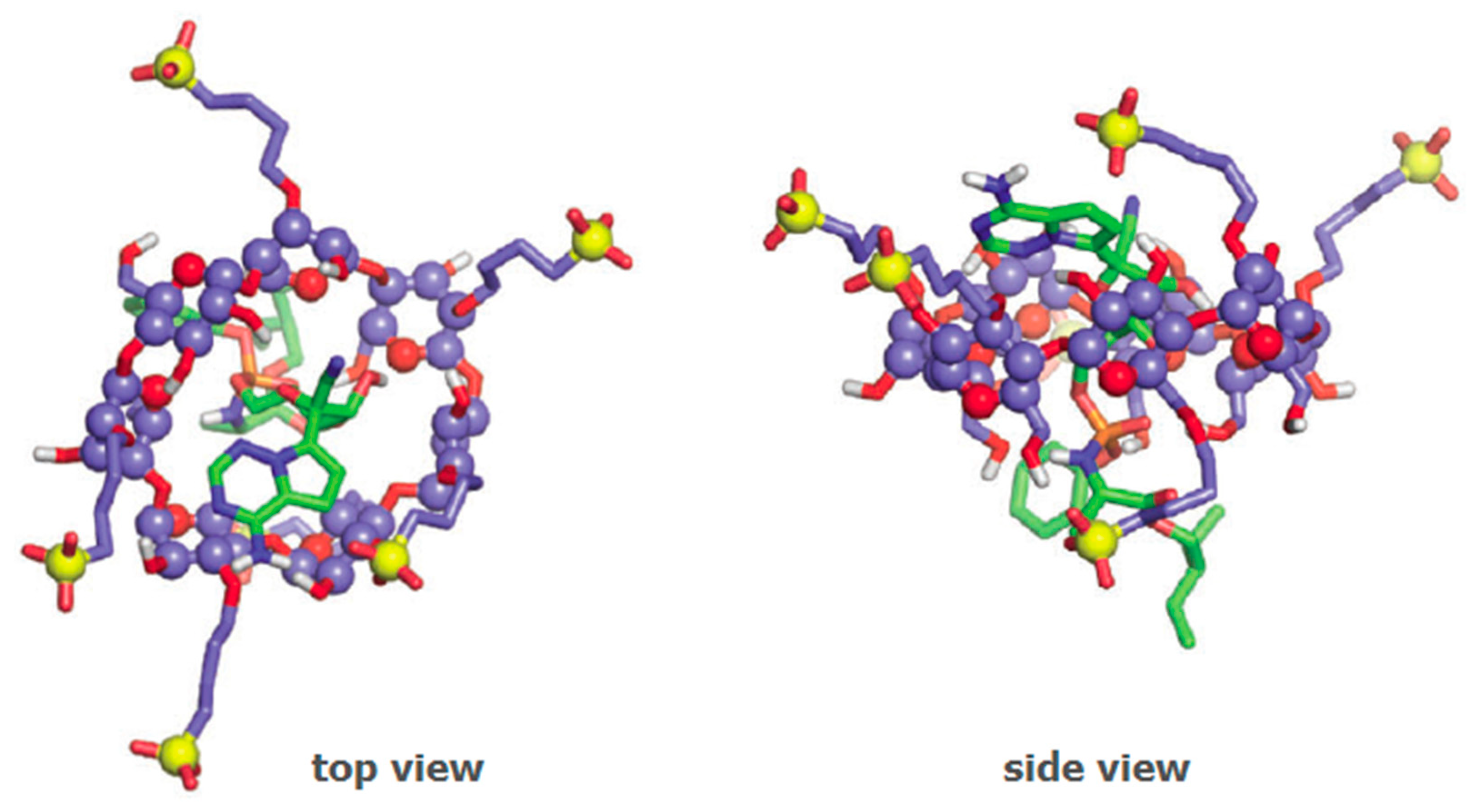
2.3. Efavirenz
2.4. Rilpivirine
2.5. Saquinavir
2.6. Lopinavir
2.7. Oseltamivir
2.8. Remdesivir
3. Cyclodextrins in Vaccines
3.1. Cyclodextrins as Vaccine Cryopreservatives: The Example of ad26.cov2.s
3.2. Cyclodextrins as Vaccine Adjuvants
3.2.1. Porcine Circovirus Vaccine
3.2.2. Human Influenza Vaccine
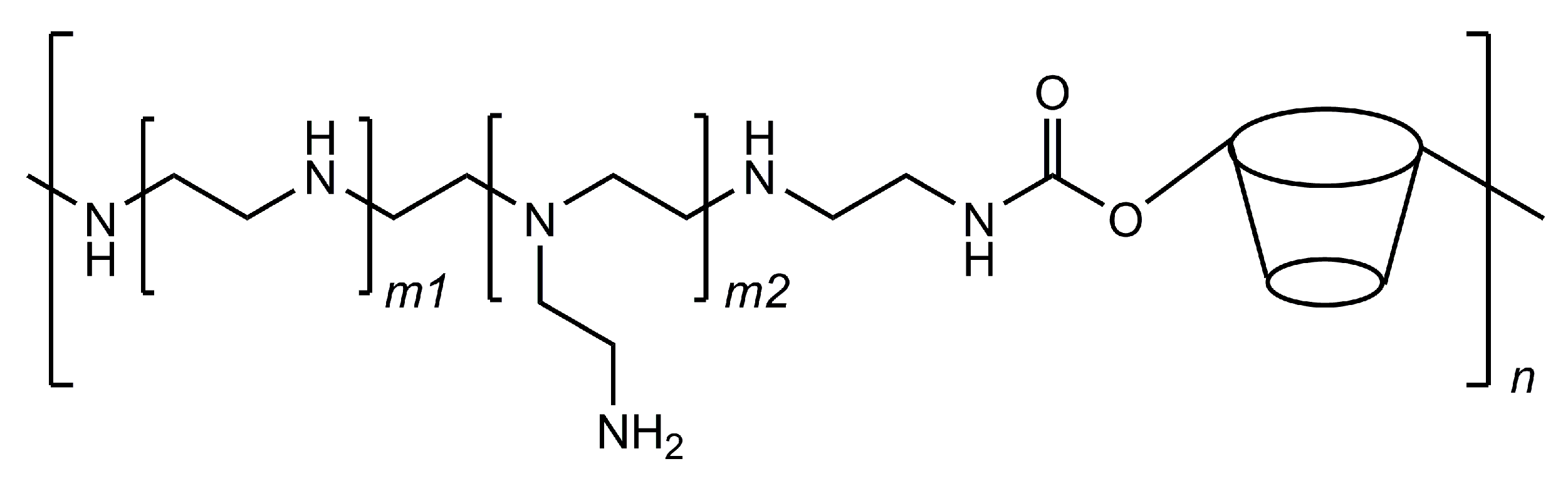
3.3. Cyclodextrins in mRNA Vaccines—A Future Trend?
4. Cyclodextrins as New Antiviral Agents
4.1. Cyclodextrins against Influenza
4.2. Cyclodextrins against the Dengue Virus
4.3. Cyclodextrins against Hepatitis C
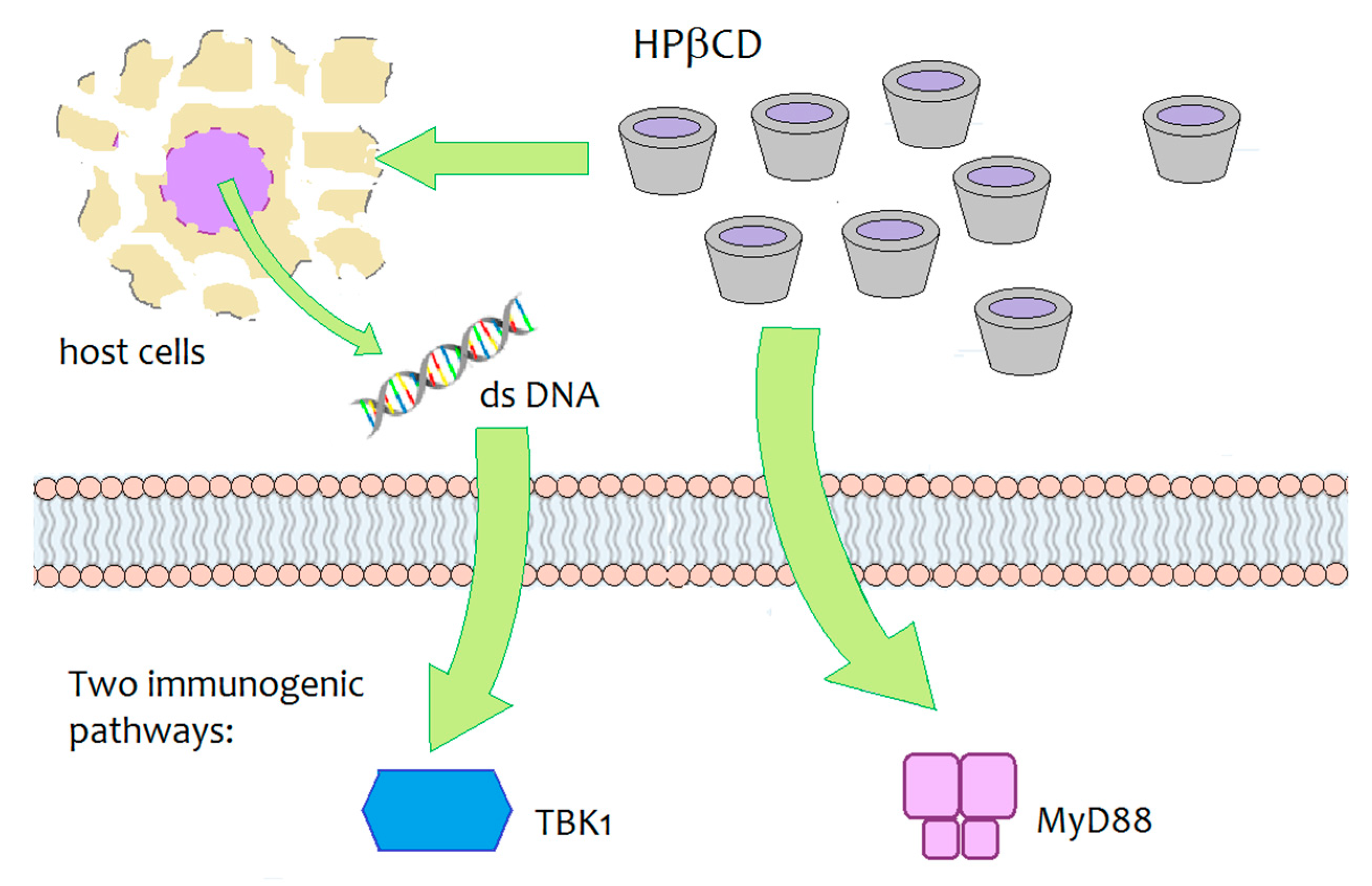
4.4. HPβCD against HIV
4.5. Labial Herpes Management with Cyclodextrins
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| ACV | acyclovir |
| API | active pharmaceutical ingredient |
| BCS | biopharmaceutical classification system |
| CD | cyclodextrin |
| CHMP | committee for medicinal products for human use |
| DIMEB | heptakis-2,6-di-O-methyl-β-CD |
| DMEM | Dulbecco’s modified eagle medium |
| DSC | differential scanning calorimetry |
| FDA | US Food and Drugs Administration |
| EFV | efavirenz |
| ELISA | enzyme-linked immunosorbent assay |
| EMA | European Medicines Agency |
| EMEM | Eagle’s minimum essential medium |
| GI | gastrointestinal |
| GCV | ganciclovir |
| HCMV | human cytomegalovirus |
| HCV | hepatitis C virus |
| HPβCD | (2-hydroxyl)propyl-β-CD |
| HIV | human immunodeficiency virus |
| HSV | herpex simplex virus |
| IgG | immunoglobulin G |
| LPV | lopinavir |
| MERS | Middle Eastern respiratory syndrome |
| MLK | montelukast sodium |
| m.o.i. | multiplicity of infection |
| MV | measles virus |
| MyD88 | myeloid differentiation primary response protein 88 |
| NS-1 | non-structural protein 1 |
| OSV | oseltamivir phosphate (TamifluTM) |
| PAA | poly(amidoamine) |
| PBS | phosphate buffer saline |
| PEG | polyethyleneglycol |
| PEI | polyethyleneimine |
| RBV | ribavirin |
| RAMEB | randomly methylated β-CD |
| RPV | rilpivirine |
| RSV | remdesivir |
| RTV | ritonavir |
| SARS | severe acute respiratory syndrome |
| SBEβCD | sulfobutylether-β-cyclodextrin (Captisol®) |
| SD | spray-dried |
| SL-β-CD | sulfolipo-β-CD |
| SLS | sodium lauryl sulphate |
| SQV | saquinavir |
| TBK1 | TANK-binding kinase 1 Th2, type 2 T-helper |
| TRIMEB | heptakis-2,3,6-tris-O-methyl β-CD |
| VZV | varicella-zoster virus |
References
- Luo, G.; Gao, S.-J. Global health concerns stirred by emerging viral infections. J. Med. Virol. 2020, 92, 399–400. [Google Scholar] [CrossRef] [PubMed]
- Roth, G.A.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788. [Google Scholar] [CrossRef] [Green Version]
- Fouchier, R.A.M.; Kuiken, T.; Schutten, M.; van Amerongen, G.; van Doornum, G.J.J.; van den Hoogen, B.G.; Peiris, M.; Lim, W.; Stöhr, K.; Osterhaus, A.D.M.E. Koch’s postulates fulfilled for SARS virus. Nature 2003, 423, 240. [Google Scholar] [CrossRef] [PubMed]
- Vijaykrishna, D.; Poon, L.L.M.; Zhu, H.C.; Ma, S.K.; Li, O.T.W.; Cheung, C.L.; Smith, G.J.D.; Peiris, J.S.M.; Guan, Y. Reassortment of pandemic H1N1/2009 influenza A virus in swine. Science 2010, 328, 1529. [Google Scholar] [CrossRef] [Green Version]
- Zumla, A.; Hui, D.S.; Perlman, S. Middle East respiratory syndrome. Lancet 2015, 386, 995–1007. [Google Scholar] [CrossRef] [Green Version]
- Baseler, L.; Chertow, D.S.; Johnson, K.M.; Feldmann, H.; Morens, D.M. The pathogenesis of Ebola virus disease. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 387–418. [Google Scholar] [CrossRef]
- Connors, M.D.; Graham, B.S.; Lane, H.C.; Fauci, A.S. SARS-CoV-2 Vaccines: Much accomplished, much to learn. Ann. Intern. Med. 2021. Epub ahead of print. [Google Scholar] [CrossRef]
- Anthony, S.J.; Epstein, J.H.; Murray, K.A.; Navarrete-Macias, I.; Zambrana-Torrelio, C.M.; Solovyov, A.; Ojeda-Flores, R.; Arrigo, N.C.; Islam, A.; Khan, S.A.; et al. A strategy to estimate unknown viral diversity in mammals. mBio 2013, 4, e00598–e00613. [Google Scholar] [CrossRef] [Green Version]
- García-Serradilla, M.; Risco, C.; Pacheco, B. Drug repurposing for new, efficient, broad spectrum antivirals. Virus Res. 2019, 264, 22–31. [Google Scholar] [CrossRef]
- Andersen, P.I.; Ianevski, A.; Lysvand, H.; Vitkauskiene, A.; Oksenych, V.; Bjørås, M.; Telling, K.; Lutsar, I.; Dumpis, U.; Irie, Y.; et al. Discovery and development of safe-in-man broad-spectrum antiviral agents. Int. J. Infect. Dis. 2020, 93, 268–276. [Google Scholar] [CrossRef]
- Lei, C.; Qian, K.; Li, T.; Zhang, S.; Fu, W.; Ding, M.; Hu, S. Neutralization of SARS-CoV-2 spike pseudotyped virus by recombinant ACE2-Ig. Nat. Commun. 2020, 11, 2070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warren, T.K.; Jordan, R.; Lo, M.K.; Ray, A.S.; Mackman, R.L.; Soloveva, V.; Siegel, D.; Perron, M.; Bannister, R.; Hui, H.C.; et al. Therapeutic efficacy of the small molecule GS-5734 against ebola virus in rhesus monkeys. Nature 2016, 531, 381–385. [Google Scholar] [CrossRef]
- Li, Y.F.; Li, Y.; Reed, B.; Shen, X.; Sun, D.; Zhou, S. Cyclodextrin affects distinct tissue drug disposition as a novel drug delivery vehicle. Ann. Med. Med. Res. 2019, 2, 1021. [Google Scholar]
- Carrier, R.L.; Miller, L.A.; Ahmed, I. The utility of cyclodextrins for enhancing oral bioavailability. J. Control. Release 2007, 123, 78–99. [Google Scholar] [CrossRef]
- Fenyvesi, É. Approved pharmaceutical products containing cyclodextrins. Cyclodext. News 2013, 26, 1–4. Available online: https://cyclolab.hu/userfiles/cdn_2013_feb.pdf (accessed on 7 February 2021).
- European Medicines Agency. Background Review for Cyclodextrins Used as Excipients; EMA: London, UK, 2014; Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Report/2014/12/WC500177936.pdf (accessed on 8 February 2021).
- Buschmann, H.-J.; Schollmeyer, E. Applications of cyclodextrins in cosmetic products: A review. J. Cosmet. Sci. 2002, 53, 185–191. [Google Scholar]
- Braga, S.S.; Pais, J. Getting under the skin: Cyclodextrin inclusion for the controlled delivery of active substances to the dermis. In Design of Nanostructures for Versatile Therapeutic Applications, 1st ed.; Grumezescu, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; Chapter 10; pp. 407–449. [Google Scholar]
- Barbosa, J.S.; Almeida Paz, F.A.; Braga, S.S. Montelukast medicines of today and tomorrow: From molecular pharmaceutics to technological formulations. Drug Deliv. 2016, 23, 3257–3265. [Google Scholar] [CrossRef] [Green Version]
- Del Valle, E.M. Cyclodextrins and their uses: A review. Process. Biochem. 2004, 39, 1033–1046. [Google Scholar] [CrossRef]
- Agrawal, R.; Gupta, V. Cyclodextrins—A Review on pharmaceutical application for drug delivery. Int. J. Pharm. Front. Res. 2012, 2, 95–112. [Google Scholar]
- Kroes, R.; Verger, P.; Larsen, J.C. Safety Evaluation of Certain Food Additives (α-Cyclodextrin-Addendum); WHO Food Additives Series; WHO: Geneva, Switzerland, 2006; Volume 54, pp. 3–15. [Google Scholar]
- Pollit, F.D. Safety Evaluation of Certain Food Additives (β-Cyclodextrin); WHO Food Additives Series; WHO: Geneva, Switzerland, 1996; Volume 35, pp. 257–268. [Google Scholar]
- Abbott, P.J. JEFCA 55th Meeting: Safety Evaluation of Certain Food Additives and Contaminants (γ-Cyclodextrin); WHO Food Additives Series; WHO: Geneva, Switzerland, 2000; Volume 44, p. 969. [Google Scholar]
- Agency Response Letter Gras Notice GRN No. 155; Office of Food Additive Safety, Center for Food Safety and Applied Nutrition, US Food and Drug Administration: Baltimore, MD, USA, 2004.
- Agency Response Letter Gras Notice GRN No. 74; Office of Food Additive Safety, Center for Food Safety and Applied Nutrition, US Food and Drug Administration: Baltimore, MD, USA, 2001.
- Agency Response Letter Gras Notice GRN No. 46; Office of Food Additive Safety, Center for Food Safety and Applied Nutrition, US Food and Drug Administration: Baltimore, MD, USA, 2000.
- Irie, T.; Otagiri, M.; Sunada, M.; Uekama, K.; Ohtani, Y.; Yamada, Y.; Sugiyama, Y. Cyclodextrin-induced hemolysis and shape changes of human erythrocytes in vitro. J. Pharm. Dyn. 1982, 5, 741–744. [Google Scholar] [CrossRef] [Green Version]
- Ohtani, Y.; Irie, T.; Uekama, K.; Fukunaga, K.; Pitha, J. Differential effects of α-, β- and γ-cyclodextrins on human erythrocytes. Eur. J. Biochem. 1989, 186, 17–22. [Google Scholar] [CrossRef]
- Nitalikar, M.M.; Sakarkar, D.M.; Jain, P.V. The cyclodextrins: A review. J. Curr. Pharm. Res. 2012, 10, 1–6. [Google Scholar]
- Uekama, K.; Hirayama, F.; Irie, T. Pharmaceutical uses of Cyclodextrin Derivatives. In High Performance Biomaterials, A Comprehensive Guide to Medical and Pharmaceutical Applications, 1st ed.; Szycher, M., Ed.; Technomic: Lancaster, PA, USA, 1991; pp. 789–806. [Google Scholar]
- Excipients in Vaccines per 0.5 mL Dose. Available online: https://www.vaccinesafety.edu/components-Excipients.htm (accessed on 24 February 2021).
- Szente, L.; Singhal, A.; Domokos, A.; Song, B. Cyclodextrins: Assessing the impact of cavity size, occupancy, and substitutions on cytotoxicity and cholesterol homeostasis. Molecules 2018, 23, 1228. [Google Scholar] [CrossRef] [Green Version]
- Kiss, T.; Fenyvesi, F.; Bácskay, I.; Váradi, J.; Fenyvesi, É.; Iványi, R.; Szente, L.; Tósaki, Á.; Vecsernyé, M. Evaluation of the cytotoxicity of β-cyclodextrin derivatives: Evidence for the role of cholesterol extraction. Eur. J. Pharm. Sci. 2010, 40, 376–380. [Google Scholar] [CrossRef]
- Captisol. Available online: https://www.captisol.com/technology/history (accessed on 17 September 2019).
- Lin, P.; Torres, G.; Tyring, S.K. Changing paradigms in dermatology: Antivirals in dermatology. Clin. Dermatol. 2003, 21, 426–446. [Google Scholar] [CrossRef]
- Spruance, S.L.; Nett, R.; Marbury, T.; Wolff, R.; Johnson, J.; Spaulding, T.; The Acyclovir Cream Study Group. Acyclovir cream for treatment of herpes simplex labialis: Results of two randomized, double-blind, vehicle-controlled, multicenter clinical trials. Antimicrob. Agents Chemother. 2002, 46, 2238–2243. [Google Scholar] [CrossRef] [Green Version]
- Arnal, J.; Gonzalez-Alvarez, I.; Bermejo, M.; Amidon, G.L.; Juninger, H.E.; Kopp, S.; Midha, K.K.; Shah, V.P.; Stavchansky, S.; Dressman, J.B.; et al. Biowaiver monographs for immediate release solid oral dosage forms: Aciclovir. J. Pharm. Sci. 2008, 97, 5061–5073. [Google Scholar] [CrossRef] [Green Version]
- Karpe, M.; Mali, N.; Kadam, V. Formulation development and evaluation of acyclovir orally disintegrating tablets. J. Appl. Pharm. Sci. 2012, 2, 101–105. [Google Scholar]
- Rossel, C.P.; Carreño, J.S.; Rodríguez-Baeza, M.; Alderete, J.B. Inclusion complex of the antiviral drug acyclovir with cyclodextrin in aqueous solution and in solid phase. Quím. Nova 2000, 23, 749–752. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, P.B.; Dandagi, P.; Udupa, N.; Gopal, S.V.; Jain, S.S.; Vasanth, S.G. Controlled release polymeric ocular delivery of acyclovir. Pharm. Dev. Technol. 2010, 15, 369–378. [Google Scholar] [CrossRef]
- Tomar, V.; Garud, N.; Kannojia, P.; Garud, A.; Jain, N.; Singh, N. Enhancement of solubility of acyclovir by solid dispersion and inclusion complexation methods. Pharm. Lett. 2010, 2, 341–352. [Google Scholar]
- Luengo, J.; Aránguiz, T.; Sepúlveda, J.; Hernández, L.; von Plessing, C. Preliminary pharmacokinetic study of different preparations of acyclovir with β-cyclodextrin. J. Pharm. Sci. 2002, 91, 2593–2598. [Google Scholar] [CrossRef]
- Nair, A.B.; Attimarad, M.; Al-Dhubiab, B.E.; Wadhwa, J.; Harsha, S.; Ahmed, M. Enhanced oral bioavailability of acyclovir by inclusion complex using hydroxypropyl-β-cyclodextrin. Drug Deliv. 2014, 21, 540–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bencini, M.; Ranucci, E.; Ferruti, P.; Trotta, F.; Donalisio, M.; Cornaglia, M.; Lembo, D.; Cavalli, R. Preparation and in vitro evaluation of the antiviral activity of the Acyclovir complex of a β-cyclodextrin/poly(amidoamine) copolymer. J. Control. Release 2008, 126, 17–25. [Google Scholar] [CrossRef]
- Piperno, A.; Zagami, R.; Cordaro, A.; Pennisi, R.; Musarra-Pizzo, M.; Scala, A.; Sciortino, M.T.; Mazzaglia, A. Exploring the entrapment of antiviral agents in hyaluronic acid-cyclodextrin conjugates. J. Incl. Phenom. Macrocycl. 2019, 93, 33–40. [Google Scholar] [CrossRef]
- Cytovene. Available online: https://www.rxlist.com/cytovene-drug.htm (accessed on 18 November 2020).
- Benjamin, E.J.; Firestone, B.A.; Bergstrom, R.; Fass, M.; Massey, I.; Tsina, I.; Lin, Y.Y.T. Selection of a derivative of the antiviral agent 9-[1,3(dihydroxy)-2-(propoxy)-methyl]guanine (DHPG) with improved oral absorption. Pharm. Res. 1987, 4, 120–125. [Google Scholar] [CrossRef]
- Ganciclovir Sodium. Available online: https://go.drugbank.com/salts/DBSALT000309 (accessed on 19 November 2020).
- Janoly-Dumenil, A.; Rouvet, I.; Bleyzac, N.; Morfin, F.; Zabot, M.T.; Tode, M.A. Pharmacodynamic model of ganciclovir antiviral effect and toxicity for lymphoblastoid cells suggests a new dosing regimen to treat cytomegalovirus infection. Antimicrob. Agents Chemother. 2012, 56, 3732–3738. [Google Scholar] [CrossRef] [Green Version]
- Nicolazzi, C.; Abdou, S.; Collomb, J.; Marsura, A.; Finance, C. Effect of the complexation with cyclodextrins on the in vitro antiviral activity of ganciclovir against human cytomegalovirus. Bioorg. Med. Chem. 2001, 9, 275–282. [Google Scholar] [CrossRef]
- Nicolazzi, C.; Venard, V.; Le Faou, A.; Finance, C. In vitro antiviral efficacy of the ganciclovir complexed with β-cyclodextrin on human cytomegalovirus clinical strains. Antivir. Res. 2002, 54, 121–127. [Google Scholar] [CrossRef]
- Tirucherai, G.S.; Mitra, A.K. Effect of hydroxypropyl beta cyclodextrin complexation on aqueous solubility, stability, and corneal permeation of acyl ester prodrugs of ganciclovir. AAPS Pharm. Sci. Tech. 2003, 4, 45. [Google Scholar] [CrossRef] [Green Version]
- Braga, S.S.; Lysenko, K.; El-Saleh, F.; Almeida Paz, F.A. Cyclodextrin-efavirenz complexes investigated by solid state and solubility studies. Proceedings 2021, 78, 15. [Google Scholar] [CrossRef]
- Cristofoletti, R.; Nair, A.; Abrahamsson, B.; Groot, D.W.; Koop, S.; Langguth, P.; Polli, J.E.; Shah, V.P.; Dressman, J. Biowaiver monographs for immediate release solid oral dosage forms: Efavirenz. J. Pharm. Sci. 2012, 102, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Sathigari, S.; Chadha, G.; Lee, Y.H.P.; Wright, N.; Parsons, D.L.; Rangari, V.K.; Fasina, O.; Babu, R.J. Physicochemical characterization of efavirenz-cyclodextrin inclusion complexes. AAPS Pharm. Sci. Tech. 2009, 10, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Shown, I.; Banerjee, S.; Ramchandran, A.V.; Geckeler, K.E.; Murthy, C.N. Synthesis of cyclodextrin and sugar-based oligomers for the efavirenz drug delivery. Macromol. Symp. 2010, 287, 51–59. [Google Scholar] [CrossRef]
- Braga, S.S.; El-Saleh, F.; Lysenko, K.; Paz, F.A.A. Inclusion compound of efavirenz and γ-cyclodextrin: Solid state studies and effect on solubility. Molecules 2021, 26, 519. [Google Scholar] [CrossRef] [PubMed]
- Vieira, A.C.C.; Ferreira Fontes, D.A.; Chaves, L.L.; Alves, L.D.S.; de Freitas Neto, J.L.; de la Roca Soares, M.F.; Soares-Sobrinho, J.L.; Rolim, L.A.; Rolim-Neto, P.J. Multicomponent systems with cyclodextrins and hydrophilic polymers for the delivery of efavirenz. Carbohydr. Polym. 2015, 130, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Chowdary, K.P.R.; Naresh, A. Formulation development of efavirenz tablets employing β cyclodextrin-PVP K30-SLS: A factorial study. J. Appl. Pharm. Sci. 2011, 1, 130–134. [Google Scholar]
- Rao, M.R.P.; Chaudhari, J.; Trotta, F.; Caldera, F. Investigation of cyclodextrin-based nanosponges for solubility and bioavailability enhancement of rilpivirine. AAPS Pharm. Sci. Tech. 2018, 19, 2358–2369. [Google Scholar] [CrossRef]
- Australian Public Assessment Report for Rilpivirine. Available online: https://www.tga.gov.au/sites/default/files/auspar-rilpivirine-120327.pdf (accessed on 11 February 2021).
- Srivani, S.; Kumar, Y.A.; Rao, N.G.R. Enhancement of solubility of rilpivirine by inclusion complexation with cyclodextrins. Int. J. Pharm. Sci. Drug Res. 2018, 10, 31–38. [Google Scholar] [CrossRef]
- Zainuddin, R.; Zaheer, Z.; Sangshetti, J.N.; Momin, M. Enhancement of oral bioavailability of anti-HIV drug rilpivirine HCl through nanosponge formulation. Drug Dev. Ind. Pharm. 2017, 43, 2076–2084. [Google Scholar] [CrossRef]
- Ford, J.; Khoo, S.H.; Back, D.J. The intracellular pharmacology of antiretroviral protease inhibitors. J. Antimicrob. Chemother. 2004, 54, 982–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchanan, C.M.; Buchanan, N.L.; Edgar, K.J.; Little, J.L.; Ramsey, M.G.; Ruble, K.M.; Wacher, V.J.; Wempe, M.F. Pharmacokinetics of saquinavir after intravenous and oral dosing of saquinavir: Hydroxybutenyl-β-cyclodextrin formulations. Biomacromolecules 2008, 9, 305–313. [Google Scholar] [CrossRef]
- Pathak, S.M.; Musmade, P.; Dengle, S.; Karthik, A.; Bhat, K.; Udupa, N. Enhanced oral absorption of saquinavir with methyl-beta-cyclodextrin—Preparation and in vitro and in vivo evaluation. Eur. J. Pharm. Sci. 2010, 41, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Branham, M.L.; Moyo, T.; Govender, T. Preparation and solid-state characterization of ball milled saquinavir mesylate for solubility enhancement. Eur. J. Pharm. Biopharm. 2012, 80, 194–202. [Google Scholar] [CrossRef]
- Boudad, H.; Legrand, P.; Lebas, G.; Cheron, M.; Duchêne, D.; Ponchel, G. Combined hydroxypropyl-β-cyclodextrin and poly(alkylcyanoacrylate) nanoparticles intended for oral administration of saquinavir. Int. J. Pharm. 2001, 218, 113–124. [Google Scholar] [CrossRef]
- Mahajan, H.S.; Pingale, M.H.; Agrawal, K.M. Solubility and dissolution enhancement of saquinavir mesylate by inclusion complexation technique. J. Incl. Phenom. Macrocycl. Chem. 2013, 76, 467–472. [Google Scholar] [CrossRef]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A Trial of lopinavir–ritonavir in adults hospitalized with severe covid-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Gérard, A.; Romani, S.; Fresse, A.; Viard, D.; Parassol, N.; Granvuillemin, A.; Chouchana, L.; Rocher, F.; Drici, M.D. French Network of Pharmacovigilance Centers. “Off-label” use of hydroxychloroquine, azithromycin, lopinavir-ritonavir and chloroquine in COVID-19: A survey of cardiac adverse drug reactions by the French Network of Pharmacovigilance Centers. Therapies 2020, 75, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Goyal, G.; Vavia, P.R. Complexation approach for fixed dose tablet formulation of lopinavir and ritonavir: An anomalous relationship between stability constant, dissolution rate and saturation solubility. J. Incl. Phenom. Macrocycl. Chem. 2012, 73, 75–85. [Google Scholar] [CrossRef]
- Sakuma, S.; Matsumoto, S.; Ishizuka, N.; Mohri, K.; Fukushima, M.; Ohba, C.; Kawakami, K. Enhanced boosting of oral absorption of lopinavir through electrospray coencapsulation with ritonavir. J. Pharm. Sci. 2015, 104, 2977–2985. [Google Scholar] [CrossRef] [Green Version]
- Martínez, E.; Domingo, P.; Galindo, M.J.; Milinkovic, A.; Arroyo, J.A.; Baldoví, F.; Larrousse, M.; León, A.; de Lazzari, E.; Gatell, J.M. Risk of metabolic abnormalities in patients infected with HIV receiving antiretroviral therapy that contains Lopinavir-Ritonavir. Clin. Infect. Dis. 2004, 38, 1017–1023. [Google Scholar] [CrossRef]
- Adeoye, O.; Conceição, J.; Serra, P.A.; da Silva, A.B.; Duarte, N.; Guedes, R.C.; Corvo, M.C.; Aguiar-Ricardo, A.; Jicsinszky, L.; Casimiro, T.; et al. Cyclodextrin solubilization and complexation of antiretroviral drug lopinavir: In silico prediction; Effects of derivatization, molar ratio and preparation method. Carbohydr. Polym. 2020, 227, 115287. [Google Scholar] [CrossRef] [PubMed]
- Oo, C.; Snell, P.; Barret, J.; Dorr, A.; Liu, B.; Wilding, I. Pharmacokinetics and delivery of the anti-influenza prodrug oseltamivir to the small intestine and colon using site-specific delivery capsules. Int. J. Pharm. 2003, 257, 297–299. [Google Scholar] [CrossRef]
- Molecule of the Week: Oseltamivir Phosphate. Americal Chemical Society. 12 February 2018. Available online: https://www.acs.org/content/acs/en/molecule-of-the-week/archive/o/oseltamivir-phosphate.html (accessed on 8 February 2021).
- Sevukarajan, M.; Bachala, T.; Nair, R. Novel inclusion complexs of oseltamivir phosphate-with β cyclodextrin: Physico-chemical characterization. J. Pharm. Sci. Res. 2010, 2, 583–589. [Google Scholar]
- Li, C. Improved Oseltamivir Phosphate Medicinal Composition. Chinese Patent CN102068425A, 25 October 2011. [Google Scholar]
- Grein, J.; Ohmagari, N.; Shin, D.; Diaz, G.; Asperges, E.; Castagna, A.; Feldt, T.; Green, G.; Green, M.L.; Lescure, F.-X.; et al. Compassionate use of remdesivir for patients with severe Covid-19. N. Engl. J. Med. 2020, 382, 2327–2336. [Google Scholar] [CrossRef] [PubMed]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the treatment of Covid-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Summary on Compassionate Use—Remdesivir Gilead. Procedure No. EMEA/H/K/005622/CU. 3 April 2020. Available online: https://www.ema.europa.eu/en/documents/other/summary-compassionate-use-remdesivir-gilead_en.pdf (accessed on 9 February 2021).
- Food and Drug Administration. Coronavirus (COVID-19) Update: FDA Issues Emergency Use Authorization for Potential COVID-19 Treatment. FDA News; 1 May 2020. Available online: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-issues-emergency-use-authorization-potential-covid-19-treatment (accessed on 9 February 2021).
- Food and Drug Administration. Remdesivir Prescribing Information. Drugsdata at FDA; October 2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/214787Orig1s000lbl.pdf (accessed on 9 February 2021).
- Pipkin, J.; Antle, V.; García-Fondiño, R. Application of Captisol® Technology to Enable the Formulation of Remdesivir in Treating COVID-19. Drug Dev. Deliv. 2020, 20, 42–50. Available online: https://drug-dev.com/formulation-forum-application-of-captisol-technology-to-enable-the-formulation-of-remdesivir-in-treating-covid-19/ (accessed on 16 February 2021).
- Janssen COVID-19 Vaccine. Available online: https://www.fda.gov/emergency-preparedness-and-response/coronavirus-disease-2019-covid-19/janssen-covid-19-vaccine (accessed on 11 March 2021).
- EMA Recommends COVID-19 Vaccine Janssen for Authorisation in the EU. Available online: https://www.ema.europa.eu/en/news/ema-recommends-covid-19-vaccine-janssen-authorisation-eu (accessed on 11 March 2021).
- Label: JANSSEN COVID-19 VACCINE—ad26.cov2.s Injection, Suspension. Available online: https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=14a822ff-f353-49f9-a7f2-21424b201e3b (accessed on 24 February 2021).
- Adriaansen, J.; Hesselink, R.W. Methods for Preventing Surface-Induced Degradation of Viruses Using Cyclodextrins. Canadian Patent CA3001050A1, 13 April 2017. [Google Scholar]
- Braga, S.S. Cyclodextrins: Emerging medicines of the new millennium. Biomolecules 2019, 9, 801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilgers, L.A.T.; Lejeune, G.; Nicolas, I.; Fochesato, M.; Boon, B. Sulfolipo-cyclodextrin in squalane-in-water as a novel and safe vaccine adjuvant. Vaccine 1999, 17, 219–228. [Google Scholar] [CrossRef]
- Romera, S.A.; Hilgers, L.A.T.; Puntel, M.; Zamorano, P.I.; Alcon, V.L.; Santos, M.J.; Viera, J.B.; Borca, M.V.; Sadir, A.M. Adjuvant effects of sulfolipo-cyclodextrin in a squalane-in-water and water-in-mineral oil emulsions for BHV-1 vaccines in cattle. Vaccine 2001, 19, 132–141. [Google Scholar] [CrossRef]
- European Medicines Agency. Suvaxyn PCV Product Information. EMEA/V/C/000149-R/0028. Available online: https://www.ema.europa.eu/en/medicines/veterinary/EPAR/suvaxyn-pcv#product-information-section (accessed on 10 February 2021).
- Onishi, M.; Ozasa, K.; Kobiyama, K.; Ohata, K.; Kitano, M.; Taniguchi, K.; Homma, T.; Kobayashi, M.; Sato, A.; Katakai, Y.; et al. Hydroxypropyl-β-cyclodextrin spikes local inflammation that induces Th2 cell and T follicular helper cell responses to the coadministered antigen. J. Immunol. 2015, 194, 2673–2682. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.K.; Yun, C.H.; Han, S.H. Induction of dendritic cell maturation and activation by a potential adjuvant, 2-hydroxypropyl-β-cyclodextrin. Front. Immunol. 2016, 7, 435. [Google Scholar] [CrossRef] [Green Version]
- A Phase 1 Study of Hydroxypropyl-beta-cyclodextrin(HP-beta-CyD)-Adjuvanted Influenza Split Vaccine. Available online: https://rctportal.niph.go.jp/en/detail?trial_id=UMIN000028530 (accessed on 9 February 2021).
- Kusakabe, T.; Ozasa, K.; Kobari, S.; Momota, M.; Kishishita, N.; Kobiyama, K.; Kuroda, E.; Ishii, K.J. Intranasal hydroxypropyl-β-cyclodextrin-adjuvanted influenza vaccine protects against sub-heterologous virus infection. Vaccine 2016, 34, 3191–3198. [Google Scholar] [CrossRef]
- Reichmuth, A.M.; Oberli, M.A.; Jaklenec, A.; Langer, R.; Blankschtein, D. mRNA vaccine delivery using lipid nanoparticles. Ther. Deliv. 2016, 7, 319–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiser, J. Temperature concerns could slow the rollout of new coronavirus vaccines. Science 2020. Available online: https://www.sciencemag.org/news/2020/11/temperature-concerns-could-slow-rollout-new-coronavirus-vaccines (accessed on 12 February 2021). [CrossRef]
- Haley, R.M.; Gottardi, R.; Langer, R.; Mitchell, M.J. Cyclodextrins in drug delivery: Applications in gene and combination therapy. Drug Deliv. Transl. Res. 2020, 10, 661–677. [Google Scholar] [CrossRef]
- Tan, L.; Zheng, T.; Li, M.; Zhong, X.; Tang, Y.; Qin, M.; Sun, X. Optimization of an mRNA vaccine assisted with cyclodextrin-polyethyleneimine conjugates. Drug Deliv. Transl. Res. 2020, 10, 678–689. [Google Scholar] [CrossRef]
- Davis, M.E.; Zuckerman, J.E.; Choi, C.H.J.; Seligson, D.; Tolcher, A.; Alabi, C.A.; Yen, Y.; Heidel, J.D.; Ribas, A. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 2010, 464, 1067–1070. [Google Scholar] [CrossRef] [PubMed]
- Orphanet, Orphan Designation—USA. Available online: https://www.orpha.net/consor/cgi-bin/Drugs_Search.php?lng=EN&data_id=88421&search=Drugs_Search_Simple&data_type=Status&Typ=Sub (accessed on 8 September 2019).
- Orphan Designation EU/3/13/1124. Available online: https://www.ema.europa.eu/en/medicines/human/orphan-designations/eu3131124 (accessed on 8 September 2019).
- Zimmer, S.; Grebe, A.; Bakke, S.S.; Bode, N.; Halvorsen, B.; Ulas, T.; Skjelland, M.; De Nardo, D.; Labzin, L.I.; Kerksiek, A.; et al. Cyclodextrin promotes atherosclerosis regression via macrophage reprogramming. Sci. Transl. Med. 2016, 8, 333ra50. [Google Scholar] [CrossRef] [Green Version]
- Bakke, S.S.; Aune, M.H.; Niyonzima, N.; Pilely, K.; Ryan, L.; Skjelland, M.; Garred, P.; Aukrust, P.; Halvorsen, B.; Latz, E.; et al. Cyclodextrin reduces cholesterol crystal–induced inflammation by modulating complement activation. J. Immunol. 2017, 199, 2910–2920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castagne, D.; Fillet, M.; Delattre, L.; Evrard, B.; Nusgens, B.; Piel, B. Study of the cholesterol extraction capacity of β-cyclodextrin and its derivatives, relationships with their effects on endothelial cell viability and on membrane models. J. Incl. Phenom. Macrocycl. Chem. 2009, 63, 225–231. [Google Scholar] [CrossRef]
- Barman, S.; Nayak, D.P. Lipid raft disruption by cholesterol depletion enhances influenza A virus budding from MDCK cells. J. Virol. 2007, 81, 12169–12178. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Whittaker, G.R. Role for influenza virus envelope cholesterol in virus entry and infection. J. Virol. 2003, 77, 12543–12551. [Google Scholar] [CrossRef] [Green Version]
- Verma, D.K.; Gupta, D.; Lal, S.K. Host lipid rafts play a major role in binding and endocytosis of Influenza A virus. Viruses 2018, 10, 650. [Google Scholar] [CrossRef] [Green Version]
- Xiao, S.; Si, L.; Tian, Z.; Jiao, P.; Fan, Z.; Meng, K.; Zhou, X.; Wang, H.; Xu, R.; Han, X.; et al. Pentacyclic triterpenes grafted on CD cores to interfere with influenza virus entry: A dramatic multivalent effect. Biomaterials 2016, 78, 74–85. [Google Scholar] [CrossRef]
- Tian, Z.; Si, L.; Meng, K.; Zhou, X.; Zhang, Y.; Zhou, D.; Xiao, S. Inhibition of influenza virus infection by multivalent pentacyclic triterpene-functionalized per-O-methylated cyclodextrin conjugates. Eur. J. Med. Chem. 2017, 134, 133–139. [Google Scholar] [CrossRef]
- Lee, C.J.; Lin, H.R.; Liao, C.L.; Lin, Y.L. Cholesterol effectively blocks entry of flavivirus. J. Virol. 2008, 82, 6470–6480. [Google Scholar] [CrossRef] [Green Version]
- Puerta-Guardo, H.; Mosso, C.; Medina, F.; Liprandi, F.; Ludert, J.E.; del Angel, R.M. Antibody-dependent enhancement of dengue virus infection in U937 cells requires cholesterol-rich membrane microdomains. J. Gen. Virol. 2010, 91, 394–403. [Google Scholar] [CrossRef]
- Carro, A.C.; Damonte, E.B. Requirement of cholesterol in the viral envelope for dengue virus infection. Virus Res. 2013, 174, 78–87. [Google Scholar] [CrossRef]
- Alcalá, A.C.; Hernández-Bravo, R.; Medina, F.; Coll, D.S.; Zambrano, J.L.; del Angel, R.M.; Ludert, J.E. The dengue virus non-structural protein 1 (NS1) is secreted from infected mosquito cells via a non-classical caveolin-1-dependent pathway. J. Gen. Virol. 2017, 98, 2088–2099. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, W.B.; Zou, Z.Y.; Hu, Z.H.; Fan, Q.S.; Xiong, J. Hepatitis C virus entry into macrophages/monocytes mainly depends on the phagocytosis of macrophages. Digest. Dis. Sci. 2019, 64, 1226–1237. [Google Scholar] [CrossRef]
- Shanmugam, S.; Saravanabalaji, D.; Yi, M. Detergent-resistant membrane association of NS2 and E2 during hepatitis C virus replication. J. Virol. 2015, 89, 4562–4574. [Google Scholar] [CrossRef] [Green Version]
- Xiao, S.; Wang, Q.; Yu, F.; Peng, Y.Y.; Yang, M.; Sollogoub, M.; Sinaÿ, P.; Zhang, Y.M.; Zhang, L.H.; Zhou, D.M. Conjugation of cyclodextrin with fullerene as a new class of HCV entry inhibitors. Bioorg. Med. Chem. 2012, 20, 5616–5622. [Google Scholar] [CrossRef]
- Liao, Z.; Graham, D.R.; Hildreth, J.E. Lipid rafts and HIV pathogenesis: Virion-associated cholesterol is required for fusion and infection of susceptible cells. AIDS Res. Hum. Retrovir. 2003, 19, 675–687. [Google Scholar] [CrossRef]
- Graham, D.R.M.; Chertova, E.; Hilburn, J.M.; Arthur, L.O.; Hildreth, J.E.K. Cholesterol depletion of human immunodeficiency virus type 1 and simian immunodeficiency virus with β-cyclodextrin inactivates and permeabilizes the virions: Evidence for virion-associated lipid rafts. J. Virol. 2003, 77, 8237–8248. [Google Scholar] [CrossRef] [Green Version]
- Khanna, K.V.; Whaley, K.J.; Zeitlin, L.; Moench, T.R.; Mehrazar, K.; Cone, R.A.; Liao, Z.; Hildreth, J.E.; Hoen, T.E.; Shultz, L.; et al. Vaginal transmission of cell-associated HIV-1 in the mouse is blocked by a topical, membrane-modifying agent. J. Clin. Investig. 2002, 109, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, Z.; Compton, L.; Michael Piatak, M., Jr.; Lu, D.; Alvord, W.G.; Lubomirski, M.S.; Hildreth, J.E.K.; Lifson, J.D.; Miller, C.J.; KewalRamani, V.N. Incomplete protection against simian immunodeficiency virus vaginal transmission in rhesus macaques by a topical antiviral agent revealed by repeat challenges. J. Virol. 2008, 82, 6591–6599. [Google Scholar] [CrossRef] [Green Version]
- Senti, G.; Iannaccone, R.; Graf, N.; Felder, M.; Tay, F.; Kündig, T. A Randomized, double-blind, placebo-controlled study to test the efficacy of topical 2-hydroxypropyl-beta-cyclodextrin in the prophylaxis of recurrent herpes labialis. Dermatology 2013, 226, 247–252. [Google Scholar] [CrossRef]
- Jones, S.T.; Cagno, V.; Janeček, M.; Ortiz, D.; Gasilova, N.; Piret, J.; Gasbarri, M.; Constant, D.A.; Han, Y.; Vuković, L.; et al. Modified cyclodextrins as broad-spectrum antivirals. Sci. Adv. 2020, 6, eaax9318. [Google Scholar] [CrossRef] [Green Version]
- Pharmacology Review 20-966. Sporanox (Itraconazole) Injection. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/99/20-966_SPORANOX%20INJECTION%2010MG%20PER%20ML_PHARMR.PDF (accessed on 12 February 2021).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cyclodextrin | Pharmaceutical Dosage Forms | ADI/Max Dose | Toxicity Concerns | ||||
|---|---|---|---|---|---|---|---|
| Oral | Nasal | Ocular | Dermal | Parenteral | |||
| α-CD | — | — | — | — | ✓ | none | haemolytic |
| β-CD | ✓ | — | ✓ | ✓ | not allowed | 5 mg/kg per day | nephrotoxic; haemolytic |
| γ-CD | ✓ | — | — | ✓ | — | none | haemolytic |
| HPβCD | ✓ | — | ✓ | ✓ | ✓ | n.s. | — |
| HPγCD | not allowed | ✓ | ✓ | not allowed | 1.5% (w/v) | — | |
| DIMEB | — | — | — | — | ✓ | n.s. | hepatotoxic |
| RAMEB | not allowed | ✓ | ✓ | not allowed | n.s. | nephrotoxic; haemolytic | |
| SBEβCD | ✓ | — | — | — | ✓ | none | — |
| Viral Strain | Ganciclovir | β-CD:GCV | γ-CD:GCV | ||
|---|---|---|---|---|---|
| IC50 (μM) 1 | IC50 (μM) | Inc. Ratio 2 | IC50 (μM) | Inc. Ratio | |
| AD169 | 2.70 ± 0.55 | 0.20 ± 0.05 | 13.5 | 0.30 ± 0.01 | 9.0 |
| RCL-1 | 14.50 ± 2.50 | 1.60 ± 0.12 | 9.1 | — | — |
| 1558 | 3.25 ± 0.62 | 0.20 ± 0.06 | 16.2 | — | — |
| 539 | 6.45 ± 0.82 | 2.50 ± 0.51 | 2.6 | — | — |
| 731 | 6.70 ± 0.55 | 5.80 ± 0.51 | 1.1 | — | — |
| 2288 | 18.25 ± 2.25 | 0.75 ± 0.80 | 24.3 | — | — |
| Guest Drug | Host | H:G Ratio | Kapp (M−1) a | Benefits | Ref |
|---|---|---|---|---|---|
| Acyclovir | β-CD | 1:1 | — | ↑ Solubility by c.a. 1.9-fold. ↑ Bioavailability by c.a. 1.2-fold | [40,41,43] |
| Acyclovir | β-CD | 5:1 | — | ↑ Dissolution: 1.5 and 1.3 folds in HCl 0.1 N and PBS (pH 7.4) | [42] |
| Acyclovir | HPβCD | 1:1 | 758 ± 7 | Affords 100% drug dissolution in HCl 0.1 N ↑ Bioavailability by 1.6 fold regarding control (a 1:1 physical mixture of ACV and HPβCD) | [44] |
| Acyclovir | HPβCD | 5:1 | — | ↑ Dissolution: 1.5 and 1.45 folds in HCl 0.1 N and PBS (pH 7.4) | [42] |
| Ganciclovir | β-CD | 1:1 | 4976 b | (not tested) | [51] |
| Ganciclovir | β-CD | 10:1 | — | ↑ In vitro antiviral potency Active against drug-resistant viral strains | [51,52] |
| Ganciclovir | γ-CD | 10:1 | — | ↑ In vitro antiviral potency Active against drug-resistant viral strains | [51] |
| Ganciclovir dibutyrate diester | HPβCD | 10:1 | 106.7 c | ↑ In vitro corneal permeation by c.a. 2.6-fold (in a solution containing 5% HPβCD) | [53] |
| Efavirenz | β-CD | 1:1 | 288 d | ↑ Dissolution rate at 180 min by c.a. four-fold, that is, it was around 44% | [56] |
| Efavirenz | HPβCD | 1:1 | 469 d | Dissolution rate increased to 60% at as early as 50 min and it remained at 60% until the end of the test (180 min) | [56] |
| Efavirenz | RAMEB | 1:1 | 1073 d | ↑ Dissolution rate at 180 min by c.a. six-fold, that is, it was around 60% | [56] |
| Efavirenz | γ-CD | 3:2 | — | ↑ Solubility | [54,58] |
| Rilpivirine | β-CD | 2:1 | — | ↑ Dissolution in acidic medium | [63] |
| Saquinavir | β-CD | — | 4086 | ↑ Dissolution rate at 60 min by c.a. two-fold | [69,70] |
| Saquinavir | RAMEB | 3:1 e | 6148 f | 100% dissolution rate | [67] |
| Saquinavir mesylate | RAMEB | 3:2 e | — | 100% dissolution rate ↑ Oral bioavailability: earlier tmax and Cmax raised 10-fold | [67] |
| Lopinavir | HPβCD | 1:1 | 443.9 | ↑ Solubility 50% dissolution at 120 min from the kneaded product | [73] |
| Lopinavir | RAMEB | 1:1 | 582.9 | ↑ Solubility 30% dissolution at 120 min from the kneaded product | [73] |
| Lopinavir | γ-CD | 1:1 | 305.0 | ↑ Solubility 55% dissolution at 120 min from the kneaded product 7% dissolution at 60 min from the spray-dried (SD) product | [73,77] |
| Lopinavir | HPγCD | 1:1 | — | ↑ Solubility 14% dissolution at 60 min from the SD product | [77] |
| Lopinavir | (HP)17γCD | 1:1 | — | ↑ Solubility 33% dissolution at 60 min from the SD product | [77] |
| Oseltamivir | β-CD | 1:1 | — | Taste-masking effect | [79] |
| Remdesivir | SBEβCD | 14:1 g | — | Enables the preparation of an injectable formulation | [85] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Braga, S.S.; Barbosa, J.S.; Santos, N.E.; El-Saleh, F.; Paz, F.A.A. Cyclodextrins in Antiviral Therapeutics and Vaccines. Pharmaceutics 2021, 13, 409. https://doi.org/10.3390/pharmaceutics13030409
Braga SS, Barbosa JS, Santos NE, El-Saleh F, Paz FAA. Cyclodextrins in Antiviral Therapeutics and Vaccines. Pharmaceutics. 2021; 13(3):409. https://doi.org/10.3390/pharmaceutics13030409
Chicago/Turabian StyleBraga, Susana Santos, Jéssica S. Barbosa, Nádia E. Santos, Firas El-Saleh, and Filipe A. Almeida Paz. 2021. "Cyclodextrins in Antiviral Therapeutics and Vaccines" Pharmaceutics 13, no. 3: 409. https://doi.org/10.3390/pharmaceutics13030409