
Nanomedicine for Gene Delivery and Drug Repurposing in the Treatment of Muscular Dystrophies

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Muscular Dystrophies Characterised by Gene Alteration
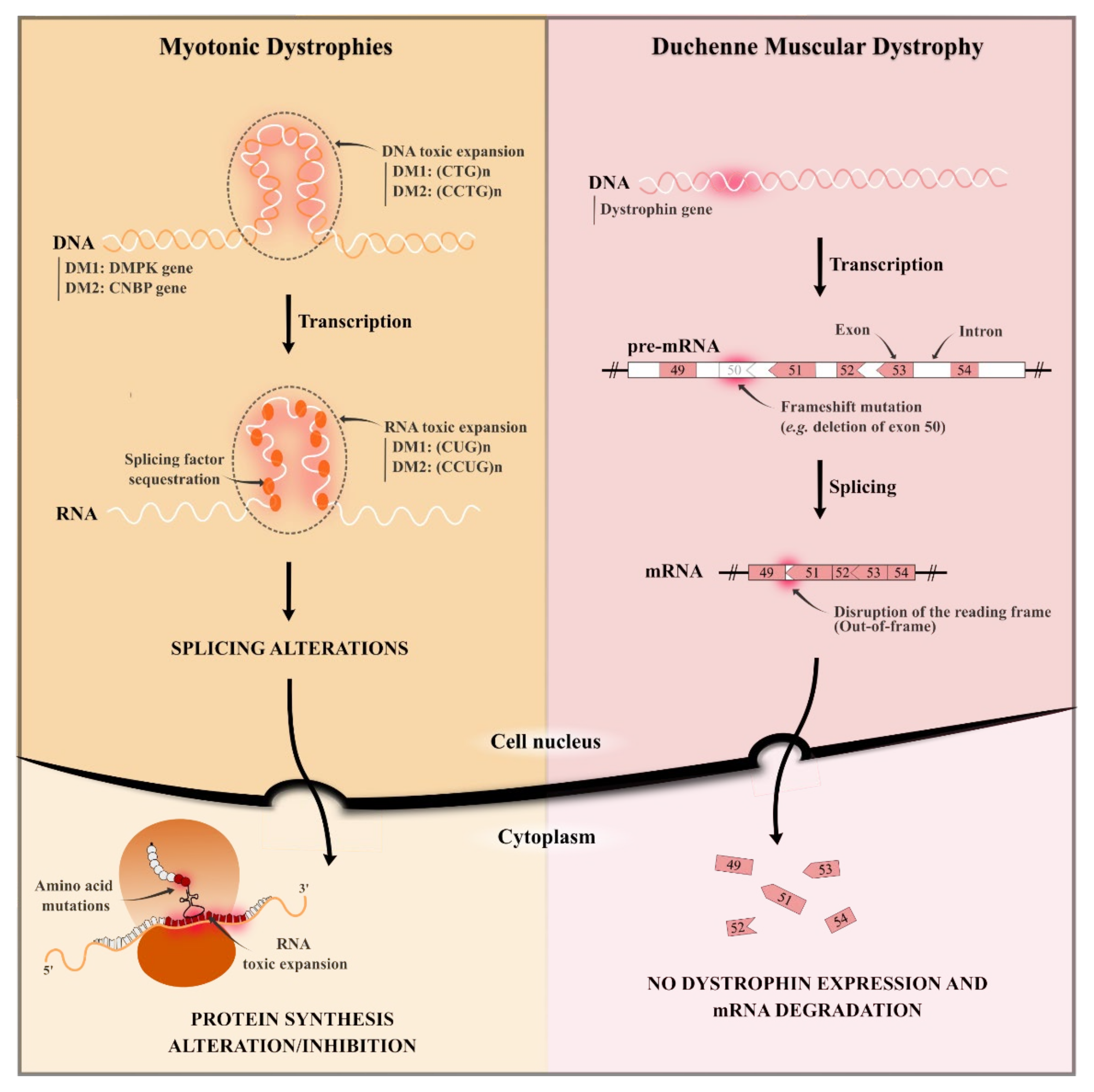
2.1. Duchenne Muscular Dystrophy
2.2. Myotonic Dystrophies (Type 1 and 2)
3. Targets and How to Reach Them: DNA and RNA
3.1. Gene Therapy and Genome Editing for MDs
3.2. Drug Repurposing
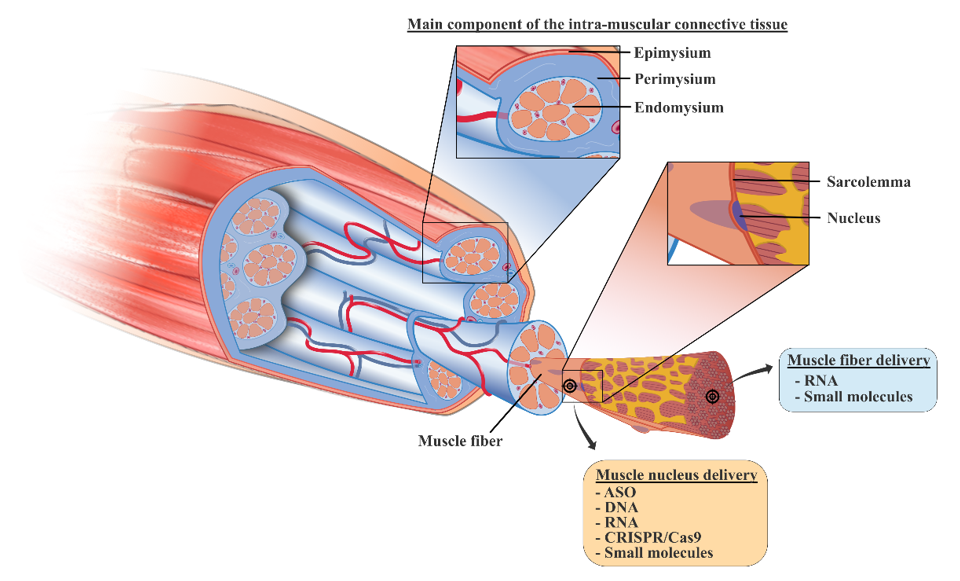
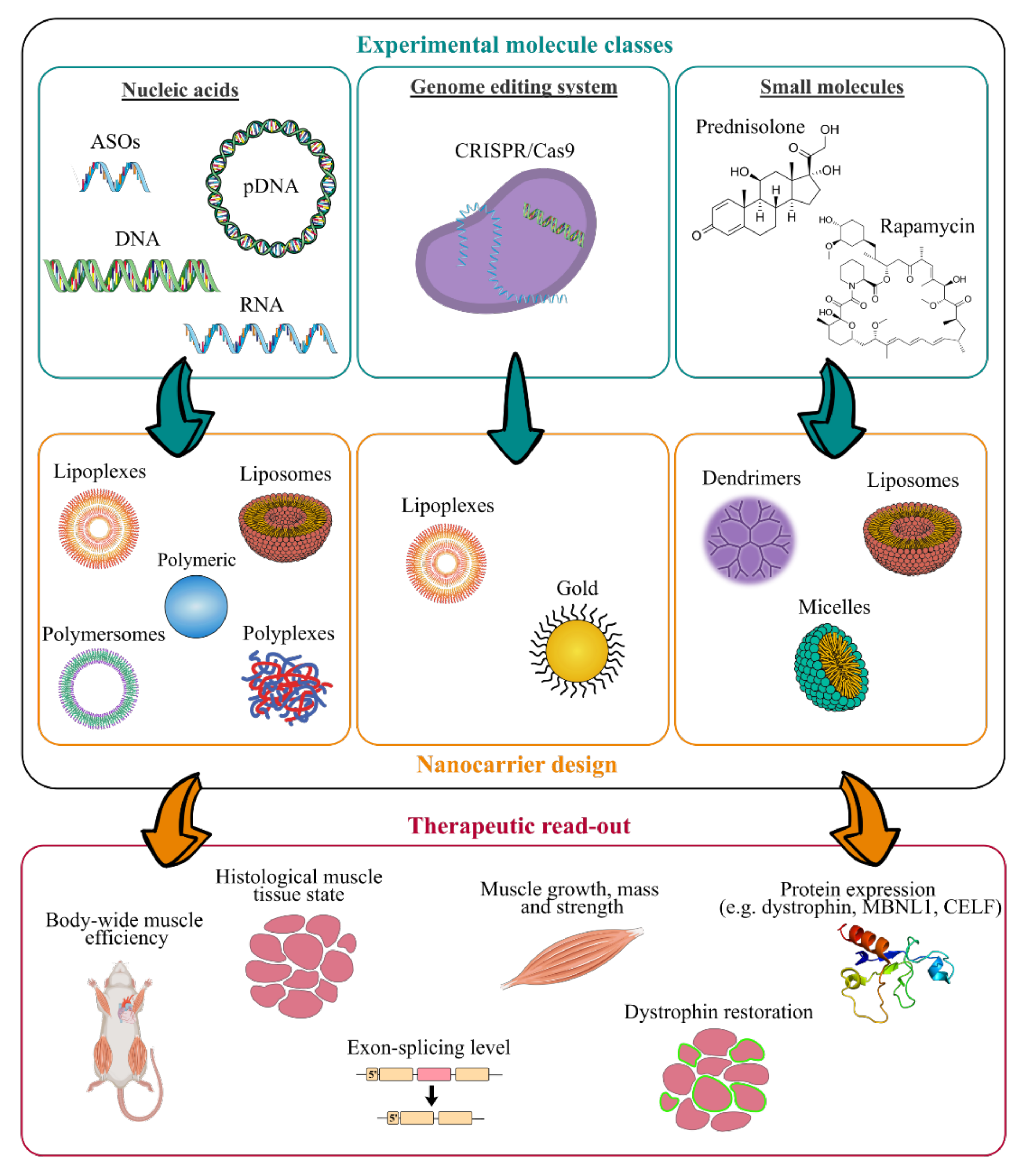
4. New Treatments based on Nanocarriers as Alternative Strategies to Facilitate Skeletal Muscle Targeting
4.1. Antisense Oligonucleotides
4.2. Oligonucleotides
4.3. Small Molecules
4.4. CRISPR/Cas9 System
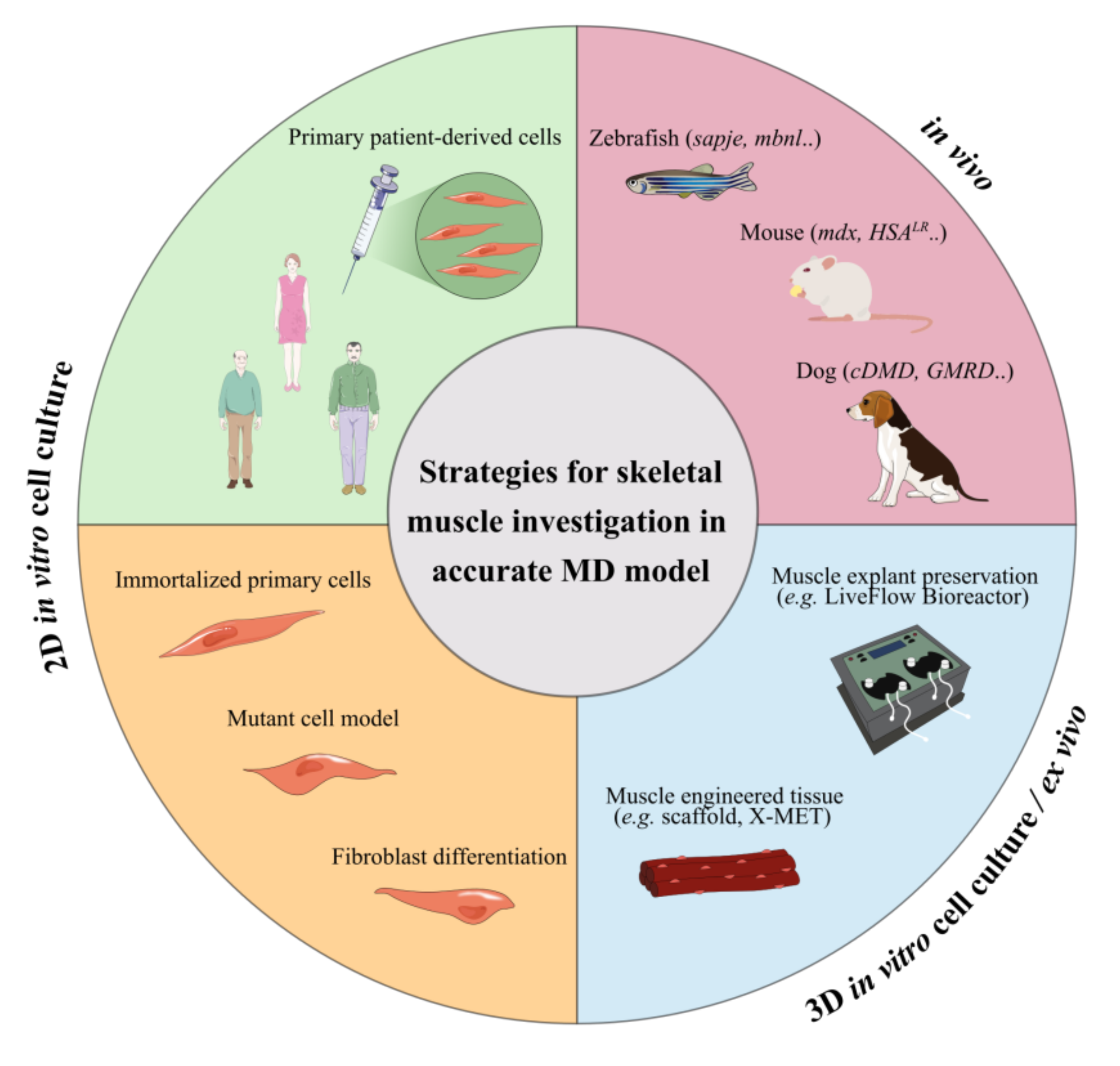
5. Limitations In In Vitro and In Vivo Testing of Novel Treatments
6. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shieh, P.B. Muscular dystrophies and other genetic myopathies. Neurol. Clin. 2013, 31, 1009–1029. [Google Scholar] [CrossRef] [PubMed]
- Theadom, A.; Rodrigues, M.; Roxburgh, R.; Balalla, S.; Higgins, C.; Bhattacharjee, R.; Jones, K.; Krishnamurthi, R.; Feigin, V. Prevalence of muscular dystrophies: A systematic literature review. Neuroepidemiology 2014, 43, 259–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercuri, E.; Bönnemann, C.G.; Muntoni, F. Muscular dystrophies. Lancet Lond. Engl. 2019, 394, 2025–2038. [Google Scholar] [CrossRef]
- Carter, J.C.; Sheehan, D.W.; Prochoroff, A.; Birnkrant, D.J. Muscular Dystrophies. Clin. Chest Med. 2018, 39, 377–389. [Google Scholar] [CrossRef]
- Johnson, N.E. Myotonic Dystrophies. Continuum 2019, 25, 1682–1695. [Google Scholar] [CrossRef] [PubMed]
- Meola, G. Clinical aspects, molecular pathomechanisms and management of myotonic dystrophies. Acta Myol. 2013, 32, 154–165. [Google Scholar]
- Messina, S.; Vita, G.L. Clinical management of Duchenne muscular dystrophy: The state of the art. Neurol. Sci. 2018, 39, 1837–1845. [Google Scholar] [CrossRef] [PubMed]
- Nio, Y.; Tanaka, M.; Hirozane, Y.; Muraki, Y.; Okawara, M.; Hazama, M.; Matsuo, T. Phosphodiesterase 4 inhibitor and phosphodiesterase 5 inhibitor combination therapy has antifibrotic and anti-inflammatory effects in mdx mice with Duchenne muscular dystrophy. FASEB J. 2017, 31, 5307–5320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanotti, S.; Bragato, C.; Zucchella, A.; Maggi, L.; Mantegazza, R.; Morandi, L.; Mora, M. Anti-fibrotic effect of pirfenidone in muscle derived-fibroblasts from Duchenne muscular dystrophy patients. Life Sci. 2016, 145, 127–136. [Google Scholar] [CrossRef]
- McDonald, C.M.; Henricson, E.K.; Abresch, R.T.; Duong, T.; Joyce, N.C.; Hu, F.; Clemens, P.R.; Hoffman, E.P.; Cnaan, A.; Gordish-Dressman, H.; et al. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne Muscular Dystrophy: A prospective cohort study. Lancet 2018, 391, 451–461. [Google Scholar] [CrossRef]
- Kao, K.-T.; Joseph, S.; Capaldi, N.; Brown, S.; Di Marco, M.; Dunne, J.; Horrocks, I.; Shepherd, S.; Ahmed, S.F.; Wong, S.C. Skeletal disproportion in glucocorticoid-treated boys with Duchenne muscular dystrophy. Eur. J. Pediatr. 2019, 178, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Mayo, A.L.; Craven, B.C.; McAdam, L.C.; Biggar, W.D. Bone health in boys with Duchenne muscular dystrophy on long-term daily deflazacort therapy. Neuromuscul. Disord. 2012, 22, 1040–1045. [Google Scholar] [CrossRef]
- Ward, L.M.; Weber, D.R. Growth, pubertal development, and skeletal health in boys with Duchenne muscular dystrophy. Curr. Opin. Endocrinol. Diabetes Obes. 2019, 26, 39–48. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Brumbaugh, D.; Case, L.E.; Clemens, P.R.; Hadjiyannakis, S.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018, 17, 251–267. [Google Scholar] [CrossRef] [Green Version]
- Fayssoil, A.; Lazarus, A.; Wahbi, K.; Ogna, A.; Nardi, O.; Lofaso, F.; Clair, B.; Orlikowski, D.; Annane, D. Cardiac implantable electronic devices in tracheotomized muscular dystrophy patients: Safety and risks. Int. J. Cardiol. 2016, 222, 975–977. [Google Scholar] [CrossRef] [PubMed]
- Bach, J.R.; Saporito, L.R.; Shah, H.R.; Sinquee, D. Decanulation of patients with severe respiratory muscle insufficiency: Efficacy of mechanical insufflation-exsufflation. J. Rehabil. Med. 2014, 46, 1037–1041. [Google Scholar] [CrossRef] [Green Version]
- Warf, M.B.; Nakamori, M.; Matthys, C.M.; Thornton, C.A.; Berglund, J.A. Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc. Natl. Acad. Sci. USA 2009, 106, 18551–18556. [Google Scholar] [CrossRef] [Green Version]
- Konieczny, P.; Selma-Soriano, E.; Rapisarda, A.S.; Fernandez-Costa, J.M.; Perez-Alonso, M.; Artero, R. Myotonic dystrophy: Candidate small molecule therapeutics. Drug Discov. Today 2017, 22, 1740–1748. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Bennett, C.F.; Cooper, T.A. RNase H-mediated degradation of toxic RNA in myotonic dystrophy type 1. Proc. Natl. Acad. Sci. USA 2012, 109, 4221–4226. [Google Scholar] [CrossRef] [Green Version]
- Verhaart, I.E.C.; Aartsma-Rus, A. Therapeutic developments for Duchenne muscular dystrophy. Nat. Rev. Neurol. 2019, 15, 373–386. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, H.; Min, Y.L.; Sanchez-Ortiz, E.; Huang, J.; Mireault, A.A.; Shelton, J.M.; Kim, J.; Mammen, P.P.A.; Bassel-Duby, R.; et al. Enhanced CRISPR-Cas9 correction of Duchenne muscular dystrophy in mice by a self-complementary AAV delivery system. Sci. Adv. 2020, 6, eaay6812. [Google Scholar] [CrossRef] [Green Version]
- Lo Scrudato, M.; Poulard, K.; Sourd, C.; Tomé, S.; Klein, A.F.; Corre, G.; Huguet, A.; Furling, D.; Gourdon, G.; Buj-Bello, A. Genome editing of expanded CTG repeats within the human DMPK gene reduces nuclear RNA foci in the muscle of DM1 mice. Mol. Ther. 2019, 27, 1372–1388. [Google Scholar] [CrossRef] [Green Version]
- Pedrini, I.; Gazzano, E.; Chegaev, K.; Rolando, B.; Marengo, A.; Kopecka, J.; Fruttero, R.; Ghigo, D.; Arpicco, S.; Riganti, C. Liposomal nitrooxy-doxorubicin: One step over caelyx in drug-resistant human cancer cells. Mol. Pharm. 2014, 11, 3068–3079. [Google Scholar] [CrossRef] [PubMed]
- Autio, K.A.; Dreicer, R.; Anderson, J.; Garcia, J.A.; Alva, A.; Hart, L.L.; Milowsky, M.I.; Posadas, E.M.; Ryan, C.J.; Graf, R.P.; et al. Safety and efficacy of BIND-014, a docetaxel nanoparticle targeting prostate-specific membrane antigen for patients with metastatic castration-resistant prostate cancer: A phase 2 clinical trial. JAMA Oncol. 2018, 4, 1344–1351. [Google Scholar] [CrossRef] [Green Version]
- Van der Meel, R.; Sulheim, E.; Shi, Y.; Kiessling, F.; Mulder, W.J.M.; Lammers, T. Smart cancer nanomedicine. Nat. Nanotechnol. 2019, 14, 1007–1017. [Google Scholar] [CrossRef]
- Wang-Gillam, A.; Hubner, R.A.; Siveke, J.T.; Von Hoff, D.D.; Belanger, B.; de Jong, F.A.; Mirakhur, B.; Chen, L.-T. NAPOLI-1 phase 3 study of liposomal irinotecan in metastatic pancreatic cancer: Final overall survival analysis and characteristics of long-term survivors. Eur. J. Cancer 2019, 108, 78–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanif, S.; Muhammad, P.; Chesworth, R.; Rehman, F.U.; Qian, R.; Zheng, M.; Shi, B. Nanomedicine-Based Immunotherapy for Central Nervous System Disorders. Acta Pharmacol. Sin. 2020, 41, 936–953. [Google Scholar] [CrossRef] [PubMed]
- You, L.; Wang, J.; Liu, T.; Zhang, Y.; Han, X.; Wang, T.; Guo, S.; Dong, T.; Xu, J.; Anderson, G.J.; et al. Targeted brain delivery of rabies virus glycoprotein 29-modified deferoxamine-loaded nanoparticles reverses functional deficits in Parkinsonian mice. ACS Nano 2018, 12, 4123–4139. [Google Scholar] [CrossRef]
- Dos Santos Tramontin, N.; da Silva, S.; Arruda, R.; Ugioni, K.S.; Canteiro, P.B.; de Bem Silveira, G.; Mendes, C.; Silveira, P.C.L.; Muller, A.P. Gold nanoparticles treatment reverses brain damage in Alzheimer’s disease model. Mol. Neurobiol. 2020, 57, 926–936. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.M.; Podojil, J.R.; Shea, L.D.; King, N.J.C.; Miller, S.D.; Getts, D.R. Overcoming challenges in treating autoimmuntity: Development of tolerogenic immune-modifying nanoparticles. Nanomedicine 2019, 18, 282–291. [Google Scholar] [CrossRef]
- Zhao, G.; Liu, A.; Zhang, Y.; Zuo, Z.-Q.; Cao, Z.-T.; Zhang, H.-B.; Xu, C.-F.; Wang, J. Nanoparticle-delivered siRNA targeting Bruton’s tyrosine kinase for rheumatoid arthritis therapy. Biomater. Sci. 2019, 7, 4698–4707. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, D.A.; Bickerton, S.; Koss, M.; Fahmy, T.M.; La Cava, A. Suppression of murine Lupus by CD4+ and CD8+ treg cells induced by T cell-targeted nanoparticles loaded with interleukin-2 and transforming growth factor β. Arthritis Rheumatol. 2019, 71, 632–640. [Google Scholar] [CrossRef]
- Fries, C.N.; Curvino, E.J.; Chen, J.-L.; Permar, S.R.; Fouda, G.G.; Collier, J.H. Advances in nanomaterial vaccine strategies to address infectious diseases impacting global health. Nat. Nanotechnol. 2020, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nanomedicine and the COVID-19 vaccines. Nat. Nanotechnol. 2020, 1. [CrossRef]
- Sahin, U.; Muik, A.; Derhovanessian, E.; Vogler, I.; Kranz, L.M.; Vormehr, M.; Baum, A.; Pascal, K.; Quandt, J.; Maurus, D.; et al. COVID-19 vaccine BNT162b1 elicits human antibody and TH1 T cell responses. Nature 2020, 586, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and safety of the MRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 2020, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.H.; Beiss, V.; Fiering, S.N.; Steinmetz, N.F. COVID-19 vaccine frontrunners and their nanotechnology design. ACS Nano 2020, 14, 12522–12537. [Google Scholar] [CrossRef]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Xin, X.; Kumar, V.; Lin, F.; Kumar, V.; Bhattarai, R.; Bhatt, V.R.; Tan, C.; Mahato, R.I. Redox-responsive nanoplatform for codelivery of miR-519c and gemcitabine for pancreatic cancer therapy. Sci. Adv. 2020, eabd6764. [Google Scholar] [CrossRef] [PubMed]
- Sasso, M.S.; Lollo, G.; Pitorre, M.; Solito, S.; Pinton, L.; Valpione, S.; Bastiat, G.; Mandruzzato, S.; Bronte, V.; Marigo, I.; et al. Low dose gemcitabine-loaded lipid nanocapsules target monocytic myeloid-derived suppressor cells and potentiate cancer immunotherapy. Biomaterials 2016, 96, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Lancet, J.E.; Uy, G.L.; Cortes, J.E.; Newell, L.F.; Lin, T.L.; Ritchie, E.K.; Stuart, R.K.; Strickland, S.A.; Hogge, D.; Solomon, S.R.; et al. CPX-351 (cytarabine and daunorubicin) liposome for injection versus conventional cytarabine plus daunorubicin in older patients with newly diagnosed secondary acute myeloid leukemia. J. Clin. Oncol. 2018, 36, 2684–2692. [Google Scholar] [CrossRef]
- Salvioni, L.; Rizzuto, M.A.; Bertolini, J.A.; Pandolfi, L.; Colombo, M.; Prosperi, D. Thirty years of cancer nanomedicine: Success, frustration, and hope. Cancers 2019, 11, 1855. [Google Scholar] [CrossRef] [Green Version]
- Gillies, A.R.; Lieber, R.L. Structure and function of the skeletal muscle extracellular matrix. Muscle Nerve 2011, 44, 318–331. [Google Scholar] [CrossRef] [Green Version]
- Yhee, J.Y.; Yoon, H.Y.; Kim, H.; Jeon, S.; Hergert, P.; Im, J.; Panyam, J.; Kim, K.; Nho, R.S. The effects of collagen-rich extracellular matrix on the intracellular delivery of glycol chitosan nanoparticles in human lung fibroblasts. Int. J. Nanomed. 2017, 12, 6089–6105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sleboda, D.A.; Stover, K.K.; Roberts, T.J. Diversity of extracellular matrix morphology in vertebrate skeletal muscle. J. Morphol. 2020, 281, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Engin, A.B.; Nikitovic, D.; Neagu, M.; Henrich-Noack, P.; Docea, A.O.; Shtilman, M.I.; Golokhvast, K.; Tsatsakis, A.M. Mechanistic understanding of nanoparticles’ interactions with extracellular matrix: The cell and immune system. Part. Fibre Toxicol. 2017, 14, 22. [Google Scholar] [CrossRef] [PubMed]
- Stylianopoulos, T.; Poh, M.Z.; Insin, N.; Bawendi, M.G.; Fukumura, D.; Munn, L.L.; Jain, R.K. Diffusion of particles in the extracellular matrix: The effect of repulsive electrostatic interactions. Biophys. J. 2010, 99, 1342–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhigaltsev, I.V.; Belliveau, N.; Hafez, I.; Leung, A.K.K.; Huft, J.; Hansen, C.; Cullis, P.R. Bottom-up design and synthesis of limit size lipid nanoparticle systems with aqueous and triglyceride cores using millisecond microfluidic mixing. Langmuir 2012, 28, 3633–3640. [Google Scholar] [CrossRef]
- Evers, M.J.W.; Kulkarni, J.A.; van der Meel, R.; Cullis, P.R.; Vader, P.; Schiffelers, R.M. State-of-the-art design and rapid-mixing production techniques of lipid nanoparticles for nucleic acid delivery. Small Methods 2018, 2, 1700375. [Google Scholar] [CrossRef]
- Feng, J.; Markwalter, C.E.; Tian, C.; Armstrong, M.; Prud’homme, R.K. Translational formulation of nanoparticle therapeutics from laboratory discovery to clinical scale. J. Transl. Med. 2019, 17, 200. [Google Scholar] [CrossRef] [PubMed]
- Ebner, D.C.; Bialek, P.; El-Kattan, A.F.; Ambler, C.M.; Tu, M. Strategies for skeletal muscle targeting in drug discovery. Curr. Pharm. Des. 2015, 21, 1327–1336. [Google Scholar] [CrossRef] [PubMed]
- Mah, J.K.; Korngut, L.; Fiest, K.M.; Dykeman, J.; Day, L.J.; Pringsheim, T.; Jette, N. A Systematic review and meta-analysis on the epidemiology of the muscular dystrophies. Can. J. Neurol. Sci. 2015, 43, 163–177. [Google Scholar] [CrossRef] [Green Version]
- Nascimento Osorio, A.; Medina Cantillo, J.; Camacho Salas, A.; Madruga Garrido, M.; Vilchez Padilla, J.J. Consensus on the diagnosis, treatment and follow-up of patients with Duchenne muscular dystrophy. Neurologia 2019, 34, 469–481. [Google Scholar] [CrossRef]
- Koenig, M.; Hoffman, E.P.; Bertelson, C.J.; Monaco, A.P.; Feener, C.; Kunkel, L.M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987, 50, 509–517. [Google Scholar] [CrossRef]
- Zhu, J.F.; Liu, H.H.; Zhou, T.; Tian, L. Novel mutation in exon 56 of the dystrophin gene in a child with Duchenne muscular dystrophy. Int. J. Mol. Med. 2013, 32, 1166–1170. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef]
- Koenig, M.; Beggs, A.H.; Moyer, M.; Scherpf, S.; Heindrich, K.; Bettecken, T.; Meng, G.; Müller, C.R.; Lindlöf, M.; Kaariainen, H.; et al. The molecular basis for Duchenne versus Becker muscular dystrophy: Correlation of severity with type of deletion. Am. J. Hum. Genet. 1989, 45, 498–506. [Google Scholar] [PubMed]
- Miyatake, S.; Mizobe, Y.; Takizawa, H.; Hara, Y.; Yokota, T.; Takeda, S.; Aoki, Y. Exon skipping therapy using phosphorodiamidate morpholino oligomers in the mdx52 mouse model of Duchenne muscular dystrophy. Methods Mol. Biol. 2018, 1687, 123–141. [Google Scholar] [CrossRef]
- Zhu, P.; Wu, F.; Mosenson, J.; Zhang, H.; He, T.-C.; Wu, W.-S. CRISPR/Cas9-mediated genome editing corrects dystrophin mutation in skeletal muscle stem cells in a mouse model of muscle dystrophy. Mol. Ther. Nucleic Acids 2017, 7, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Bird, T.D. Myotonic Dystrophy Type 1. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2020. [Google Scholar]
- Thornton, C.A. Myotonic dystrophy. Neurol. Clin. 2014, 32, 705–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, C.; Hilton-Jones, D. The myotonic dystrophies: Diagnosis and management. J. Neurol. Neurosurg. Psychiatry 2010, 81, 358–367. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.E.; Cooper, T.A. Pathogenic mechanisms of myotonic dystrophy. Biochem. Soc. Trans. 2009, 37, 1281–1286. [Google Scholar] [CrossRef]
- De Temmerman, N.; Sermon, K.; Seneca, S.; De Rycke, M.; Hilven, P.; Lissens, W.; Van Steirteghem, A.; Liebaers, I. Intergenerational instability of the expanded CTG repeat in the DMPK gene: Studies in human gametes and preimplantation embryos. Am. J. Hum. Genet. 2004, 75, 325–329. [Google Scholar] [CrossRef] [Green Version]
- Malatesta, M.; Cardani, R.; Pellicciari, C.; Meola, G. RNA transcription and maturation in skeletal muscle cells are similarly impaired in myotonic dystrophy and sarcopenia: The ultrastructural evidence. Front. Aging Neurosci. 2014, 6, 196. [Google Scholar] [CrossRef]
- Nakamori, M.; Sobczak, K.; Puwanant, A.; Welle, S.; Eichinger, K.; Pandya, S.; Dekdebrun, J.; Heatwole, C.R.; McDermott, M.P.; Chen, T.; et al. Splicing biomarkers of disease severity in myotonic dystrophy. Ann. Neurol. 2013, 74, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.W.; Urbinati, C.R.; Teng-Umnuay, P.; Stenberg, M.G.; Byrne, B.J.; Thornton, C.A.; Swanson, M.S. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 2000, 19, 4439–4448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meola, G.; Cardani, R. Myotonic dystrophies: An update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim. Biophys. Acta 2015, 1852, 594–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, S.; Dansithong, W.; Kim, D.; Rossi, J.; Webster, N.J.; Comai, L.; Reddy, S. Interaction of musleblind, CUG-BP1 and hnRNP H proteins in DM1-associated aberrant IR splicing. EMBO J. 2006, 25, 4271–4283. [Google Scholar] [CrossRef] [Green Version]
- Perdoni, F.; Malatesta, M.; Cardani, R.; Giagnacovo, M.; Mancinelli, E.; Meola, G.; Pellicciari, C. RNA/MBNL1-containing foci in myoblast nuclei from patients affected by myotonic dystrophy type 2: An immunocytochemical study. Eur. J. Histochem. 2009, 53, e18. [Google Scholar] [CrossRef] [Green Version]
- Mulders, S.A.M.; van den Broek, W.J.A.A.; Wheeler, T.M.; Croes, H.J.E.; van Kuik-Romeijn, P.; de Kimpe, S.J.; Furling, D.; Platenburg, G.J.; Gourdon, G.; Thornton, C.A.; et al. Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc. Natl. Acad. Sci. USA 2009, 106, 13915–13920. [Google Scholar] [CrossRef] [Green Version]
- Sardone, V.; Zhou, H.; Muntoni, F.; Ferlini, A.; Falzarano, M.S. Antisense oligonucleotide-based therapy for neuromuscular disease. Molecules 2017, 22, 563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanadia, R.N.; Shin, J.; Yuan, Y.; Beattie, S.G.; Wheeler, T.M.; Thornton, C.A.; Swanson, M.S. Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc. Natl. Acad. Sci. USA 2006, 103, 11748–11753. [Google Scholar] [CrossRef] [Green Version]
- Arambula, J.F.; Ramisetty, S.R.; Baranger, A.M.; Zimmerman, S.C. A simple ligand that selectively targets CUG trinucleotide repeats and inhibits MBNL protein binding. Proc. Natl. Acad. Sci. USA 2009, 106, 16068–16073. [Google Scholar] [CrossRef] [Green Version]
- Childs-Disney, J.L.; Hoskins, J.; Rzuczek, S.G.; Thornton, C.A.; Disney, M.D. Rationally designed small molecules targeting the RNA that causes myotonic dystrophy type 1 are potently bioactive. ACS Chem. Biol. 2012, 7, 856–862. [Google Scholar] [CrossRef] [Green Version]
- Udd, B.; Krahe, R. The myotonic dystrophies: Molecular, clinical, and therapeutic challenges. Lancet Neurol. 2012, 11, 891–905. [Google Scholar] [CrossRef]
- Mendell, J.R.; Rodino-Klapac, L.R.; Sahenk, Z.; Roush, K.; Bird, L.; Lowes, L.P.; Alfano, L.; Gomez, A.M.; Lewis, S.; Kota, J.; et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol. 2013, 74, 637–647. [Google Scholar] [CrossRef]
- Goemans, N.; Mercuri, E.; Belousova, E.; Komaki, H.; Dubrovsky, A.; Mcdonald, C.M.; Kraus, J.E.; Lourbakos, A.; Lin, Z.; Campion, G.; et al. A randomized placebo-controlled phase 3 trial of an antisense oligonucleotide, drisapersen, in Duchenne muscular dystrophy. Neuromuscul. Disord. 2018, 28, 4–15. [Google Scholar] [CrossRef] [Green Version]
- Goemans, N.M.; Tulinius, M.; van den Akker, J.T.; Burm, B.E.; Ekhart, P.F.; Heuvelmans, N.; Holling, T.; Janson, A.A.; Platenburg, G.J.; Sipkens, J.A.; et al. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N. Engl. J. Med. 2011, 365, 1513–1522. [Google Scholar] [CrossRef]
- Pandey, S.K.; Wheeler, T.M.; Justice, S.L.; Kim, A.; Younis, H.S.; Gattis, D.; Jauvin, D.; Puymirat, J.; Swayze, E.E.; Freier, S.M.; et al. Identification and characterization of modified antisense oligonucleotides targeting DMPK in mice and nonhuman primates for the treatment of myotonic dystrophy type 1s. J. Pharmacol. Exp. Ther. 2015, 355, 329–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, A.F.; Varela, M.A.; Arandel, L.; Holland, A.; Naouar, N.; Arzumanov, A.; Seoane, D.; Revillod, L.; Bassez, G.; Ferry, A.; et al. Peptide-conjugated oligonucleotides evoke long-lasting myotonic dystrophy correction in patient-derived cells and mice. J. Clin. Investig. 2019, 129, 4739–4744. [Google Scholar] [CrossRef] [Green Version]
- Min, Y.L.; Li, H.; Rodriguez-Caycedo, C.; Mireault, A.A.; Huang, J.; Shelton, J.M.; McAnally, J.R.; Amoasii, L.; Mammen, P.P.A.; Bassel-Duby, R.; et al. CRISPR-Cas9 corrects Duchenne muscular dystrophy exon 44 deletion mutations in mice and human cells. Sci. Adv. 2019, 5, eaav4324. [Google Scholar] [CrossRef] [Green Version]
- Amoasii, L.; Hildyard, J.C.W.; Li, H.; Sanchez-Ortiz, E.; Mireault, A.; Caballero, D.; Harron, R.; Stathopoulou, T.-R.; Massey, C.; Shelton, J.M.; et al. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science 2018, 362, 86–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotta, A. Genome editing gene therapy for Duchenne muscular dystrophy. J. Neuromuscul. Dis. 2015, 2, 343–355. [Google Scholar] [CrossRef] [Green Version]
- Amoasii, L.; Long, C.; Li, H.; Mireault, A.A.; Shelton, J.M.; Sanchez-Ortiz, E.; Mcanally, J.R.; Bhattacharyya, S.; Schmidt, F.; Grimm, D.; et al. Single-cut genome editing restores dystrophin expression in a new mouse model of muscular dystrophy. Sci. Transl. Med. 2017, 29, eaan8081. [Google Scholar] [CrossRef] [Green Version]
- Dastidar, S.; Ardui, S.; Singh, K.; Majumdar, D.; Nair, N.; Fu, Y.; Reyon, D.; Samara, E.; Gerli, M.F.M.; Klein, A.F.; et al. Efficient CRISPR/Cas9-mediated editing of trinucleotide repeat expansion in myotonic dystrophy patient-derived iPS and myogenic cells. Nucleic Acids Res. 2018, 46, 8275–8298. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.R.; Hamed, S.; Hadley, D.W.; Gropman, A.L.; Burstein, A.H.; Escolar, D.M.; Hoffman, E.P.; Fischbeck, K.H. Gentamicin treatment of Duchenne and Becker muscular dystrophy due to nonsense mutations. Ann. Neurol. 2001, 49, 706–711. [Google Scholar] [CrossRef]
- Barton-Davis, E.R.; Cordier, L.; Shoturma, D.I.; Leland, S.E.; Sweeney, H.L. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J. Clin. Investig. 1999, 104, 375–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitiello, L.; Tibaudo, L.; Pegoraro, E.; Bello, L.; Canton, M. Teaching an old molecule new tricks: Drug repositioning for Duchenne muscular dystrophy. Int. J. Mol. Sci. 2019, 20, 6053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welch, E.M.; Barton, E.R.; Zhuo, J.; Tomizawa, Y.; Friesen, W.J.; Trifillis, P.; Paushkin, S.; Patel, M.; Trotta, C.R.; Hwang, S.; et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007, 447, 87–91. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.M.; Campbell, C.; Torricelli, R.E.; Finkel, R.S.; Flanigan, K.M.; Goemans, N.; Heydemann, P.; Kaminska, A.; Kirschner, J.; Muntoni, F.; et al. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): A multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1489–1498. [Google Scholar] [CrossRef]
- Siboni, R.B.; Bodner, M.J.; Khalifa, M.M.; Docter, A.G.; Choi, J.Y.; Nakamori, M.; Haley, M.M.; Berglund, J.A. Biological efficacy and toxicity of diamidines in myotonic dystrophy type 1 models. J. Med. Chem. 2015, 58, 5770–5780. [Google Scholar] [CrossRef] [Green Version]
- Jenquin, J.R.; Coonrod, L.A.; Silverglate, Q.A.; Pellitier, N.A.; Hale, M.A.; Xia, G.; Nakamori, M.; Berglund, J.A. Furamidine rescues myotonic dystrophy type I associated mis-splicing through multiple mechanisms. ACS Chem. Biol. 2018, 13, 2708–2718. [Google Scholar] [CrossRef]
- Reddy, K.; Jenquin, J.R.; Cleary, J.D.; Berglund, J.A. Mitigating RNA toxicity in myotonic dystrophy using small molecules. Int. J. Mol. Sci. 2019, 20, 4017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenquin, J.R.; Yang, H.; Huigens III, R.W.; Nakamori, M.; Berglund, J.A. Combination treatment of erythromycin and furamidine provides additive and synergistic rescue of mis-splicing in myotonic dystrophy type 1 models. ACS Pharmacol. Transl. Sci. 2019, 2, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Bodycombe, N.E.; Haskell, K.M.; Sun, Y.L.; Wang, E.T.; Morris, C.A.; Jones, L.H.; Wood, L.D.; Pletcher, M.T. A flow cytometry-based screen identifies MBNL1 modulators that rescue splicing defects in myotonic dystrophy type I. Hum. Mol. Genet. 2017, 26, 3056–3068. [Google Scholar] [CrossRef] [Green Version]
- Naldini, L. Gene therapy returns to centre stage. Nature 2015, 526, 351–360. [Google Scholar] [CrossRef]
- Fischer, A.; Hacein-Bey-Abina, S.; Cavazzana-Calvo, M. 20 years of gene therapy for SCID. Nat. Immunol. 2010, 11, 457–460. [Google Scholar] [CrossRef] [PubMed]
- Dunbar, C.E.; High, K.A.; Joung, J.K.; Kohn, D.B.; Ozawa, K.; Sadelain, M. Gene therapy comes of age. Science 2018, 359, eaan4672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, R.A.; Miller, T.M.; Yamanaka, K.; Monia, B.P.; Condon, T.P.; Hung, G.; Lobsiger, C.S.; Ward, C.M.; McAlonis-Downes, M.; Wei, H.; et al. Antisense oligonucleotide therapy for neurodegenerative disease. J. Clin. Investig. 2006, 116, 2290–2296. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.H.; Sun, H.; Nichols, J.G.; Crooke, S.T. RNase H1-dependent antisense oligonucleotides are robustly active in directing RNA cleavage in both the cytoplasm and the nucleus. Mol. Ther. 2017, 25, 2075–2092. [Google Scholar] [CrossRef] [Green Version]
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.-H.; Shen, W.; Sun, H.; Kinberger, G.A.; Prakash, T.P.; Nichols, J.G.; Crooke, S.T. Hsp90 protein interacts with phosphorothioate oligonucleotides containing hydrophobic 2′-modifications and enhances antisense activity. Nucleic Acids Res. 2016, 44, 3892–3907. [Google Scholar] [CrossRef] [Green Version]
- He, X.-Y.; Wang, J.; Lu, D.-D.; Wang, S.-Q. Synthesis and antisense properties of 2′β-F-arabinouridine modified oligonucleotides with 4′-C-OMe substituent. Molecules 2018, 23, 2374. [Google Scholar] [CrossRef] [Green Version]
- Hagedorn, P.H.; Pontoppidan, M.; Bisgaard, T.S.; Berrera, M.; Dieckmann, A.; Ebeling, M.; Møller, M.R.; Hudlebusch, H.; Jensen, M.L.; Hansen, H.F.; et al. Identifying and avoiding off-target effects of RNase H-dependent antisense oligonucleotides in mice. Nucleic Acids Res. 2018, 46, 5366–5380. [Google Scholar] [CrossRef] [Green Version]
- Bosgra, S.; Sipkens, J.; De Kimpe, S.; Den Besten, C.; Datson, N.; Van Deutekom, J. The pharmacokinetics of 2′-O-methyl phosphorothioate antisense oligonucleotides: Experiences from developing exon skipping therapies for Duchenne muscular dystrophy. Nucleic Acid Ther. 2019, 29, 305–322. [Google Scholar] [CrossRef] [PubMed]
- Hara, Y.; Mizobe, Y.; Miyatake, S.; Takizawa, H.; Nagata, T.; Yokota, T.; Takeda, S.I.; Aoki, Y. Exon skipping using antisense oligonucleotides for laminin-alpha2-deficient muscular dystrophy. Methods Mol. Biol. 2018, 1828, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Sicinski, P.; Geng, Y.; Ryder-Cook, A.S.; Barnard, E.A.; Darlison, M.G.; Barnard, P.J. The molecular basis of muscular dystrophy in the mdx mouse: A point mutation. Science 1989, 244, 1578–1580. [Google Scholar] [CrossRef]
- Kinali, M.; Arechavala-Gomeza, V.; Feng, L.; Cirak, S.; Hunt, D.; Adkin, C.; Guglieri, M.; Ashton, E.; Abbs, S.; Nihoyannopoulos, P.; et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: A single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009, 8, 918–928. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, N.; Nagata, T.; Satou, Y.; Masuda, S.; Saito, T.; Kitagawa, H.; Komaki, H.; Takagaki, K.; Takeda, S. NS-065/NCNP-01: An antisense oligonucleotide for potential treatment of exon 53 skipping in Duchenne muscular dystrophy. Mol. Ther. Nucleic Acids 2018, 13, 442–449. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Cooper, T.A. Antisense oligonucleotides: Rising stars in eliminating RNA toxicity in myotonic dystrophy. Hum. Gene Ther. 2013, 24, 499–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huguet, A.; Medja, F.; Nicole, A.; Vignaud, A.; Guiraud-Dogan, C.; Ferry, A.; Decostre, V.; Hogrel, J.Y.; Metzger, F.; Hoeflich, A.; et al. Molecular, physiological, and motor performance defects in DMSXL mice carrying >1,000 CTG repeats from the human DM1 locus. PLoS Genet. 2012, 8, e1003043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, T.M.; Leger, A.J.; Pandey, S.K.; MacLeod, A.R.; Nakamori, M.; Cheng, S.H.; Wentworth, B.M.; Bennett, C.F.; Thornton, C.A. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature 2012, 488, 111–115. [Google Scholar] [CrossRef] [Green Version]
- Jauvin, D.; Chrétien, J.; Pandey, S.K.; Martineau, L.; Revillod, L.; Bassez, G.; Lachon, A.; McLeod, A.R.; Gourdon, G.; Wheeler, T.M.; et al. Targeting DMPK with antisense oligonucleotide improves muscle strength in myotonic dystrophy type 1 mice. Mol. Ther. Nucleic Acids 2017, 7, 465–474. [Google Scholar] [CrossRef] [Green Version]
- Ousterout, D.G.; Kabadi, A.M.; Thakore, P.I.; Majoros, W.H.; Reddy, T.E.; Gersbach, C.A. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat. Commun. 2015, 6, 6244. [Google Scholar] [CrossRef] [Green Version]
- Ousterout, D.G.; Kabadi, A.M.; Thakore, P.I.; Perez-Pinera, P.; Brown, M.T.; Majoros, W.H.; Reddy, T.E.; Gersbach, C.A. Correction of dystrophin expression in cells from Duchenne Muscular Dystrophy patients through genomic excision of exon 51 by Zinc Finger Nucleases. Mol. Ther. 2015, 23, 523–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ousterout, D.G.; Perez-Pinera, P.; Thakore, P.I.; Kabadi, A.M.; Brown, M.T.; Qin, X.; Fedrigo, O.; Mouly, V.; Tremblay, J.P.; Gersbach, C.A. Reading frame correction by targeted genome editing restores dystrophin expression in cells from Duchenne Muscular Dystrophy patients. Mol. Ther. 2013, 21, 1718–1726. [Google Scholar] [CrossRef] [Green Version]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Nance, M.E.; Hakim, C.H.; Yang, N.N.; Duan, D. Nanotherapy for Duchenne muscular dystrophy. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2018, 10, e1472. [Google Scholar] [CrossRef] [PubMed]
- Amitai, G.; Sorek, R. CRISPR-Cas adaptation: Insights into the mechanism of action. Nat. Rev. Microbiol. 2016, 14, 67–76. [Google Scholar] [CrossRef]
- Kleinstiver, B.P.; Prew, M.S.; Tsai, S.Q.; Topkar, V.V.; Nguyen, N.T.; Zheng, Z.; Gonzales, A.P.W.; Li, Z.; Peterson, R.T.; Yeh, J.-R.J.; et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 2015, 523, 481–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Hao, L.; Wang, H.; Santostefano, K.; Thapa, A.; Cleary, J.; Li, H.; Guo, X.; Terada, N.; Ashizawa, T.; et al. Therapeutic genome editing for myotonic dystrophy type 1 using CRISPR/Cas9. Mol. Ther. 2018, 26, 2617–2630. [Google Scholar] [CrossRef] [Green Version]
- Naso, M.F.; Tomkowicz, B.; Perry, W.L.; Strohl, W.R. Adeno-associated virus (AAV) as a vector for gene therapy. BioDrugs 2017, 31, 317–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Abudayyeh, O.O.; Gootenberg, J.S.; Franklin, B.; Koob, J.; Kellner, M.J.; Ladha, A.; Joung, J.; Kirchgatterer, P.; Cox, D.B.T.; Zhang, F. A Cytosine deaminase for programmable single-base RNA editing. Science 2019, 365, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.-M.; Koo, T.; Kim, K.; Lim, K.; Baek, G.; Kim, S.-T.; Kim, H.S.; Kim, D.-E.; Lee, H.; Chung, E.; et al. Adenine base editing in mouse embryos and an adult mouse model of Duchenne Muscular Dystrophy. Nat Biotechnol 2018, 36, 536–539. [Google Scholar] [CrossRef]
- Jiang, T.; Henderson, J.M.; Coote, K.; Cheng, Y.; Valley, H.C.; Zhang, X.-O.; Wang, Q.; Rhym, L.H.; Cao, Y.; Newby, G.A.; et al. Chemical modifications of adenine base editor mRNA and guide RNA expand its application scope. Nat Commun 2020, 11, 1979. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Knapp, S. New opportunities for kinase drug repurposing and target discovery. Br. J. Cancer 2018, 118, 936–937. [Google Scholar] [CrossRef] [Green Version]
- Wittenstein, A.; Caspi, M.; David, Y.; Shorer, Y.; Nadar-Ponniah, P.T.; Rosin-Arbesfeld, R. Serum starvation enhances nonsense mutation readthrough. J. Mol. Med. 2019, 97, 1695–1710. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, H.M.; Siddiqui, M.A.; Kanneganti, S.; Sharmin, N.; Chowdhury, M.W.; Nasim, M.T. Aminoglycoside-mediated promotion of translation readthrough occurs through a non-stochastic mechanism that competes with translation termination. Hum. Mol. Genet. 2018, 27, 373–384. [Google Scholar] [CrossRef] [Green Version]
- Shimizu-Motohashi, Y.; Komaki, H.; Motohashi, N.; Takeda, S.; Yokota, T.; Aoki, Y. Restoring dystrophin expression in Duchenne muscular dystrophy: Current status of therapeutic approaches. J. Pers. Med. 2019, 9, 1. [Google Scholar] [CrossRef] [Green Version]
- Hayward, R.S.; Harding, J.; Molloy, R.; Land, L.; Longcroft-Neal, K.; Moore, D.; Ross, J.D.C. Adverse effects of a single dose of gentamicin in adults: A systematic review. Br. J. Clin. Pharmacol. 2018, 84, 223–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friesen, W.J.; Johnson, B.; Sierra, J.; Zhuo, J.; Vazirani, P.; Xue, X.; Tomizawa, Y.; Baiazitov, R.; Morrill, C.; Ren, H.; et al. The minor gentamicin complex component, X2, is a potent premature stop codon readthrough molecule with therapeutic potential. PLoS ONE 2018, 13, e0206158. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, À.L.; Konieczny, P.; Llamusi, B.; Delgado-Pinar, E.; Borrell, J.I.; Teixidó, J.; García-España, E.; Pérez-Alonso, M.; Estrada-Tejedor, R.; Artero, R. In silico discovery of substituted pyrido[2,3-d] pyrimidines and pentamidine-like compounds with biological activity in myotonic dystrophy models. PLoS ONE 2017, 12, e0178931. [Google Scholar] [CrossRef] [Green Version]
- Díaz, M.V.; Miranda, M.R.; Campos-Estrada, C.; Reigada, C.; Maya, J.D.; Pereira, C.A.; López-Muñoz, R. Pentamidine exerts in vitro and in vivo anti Trypanosoma cruzi activity and inhibits the polyamine transport in Trypanosoma cruzi. Acta Trop. 2014, 134, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Coonrod, L.A.; Nakamori, M.; Wang, W.; Carrell, S.; Cameron, L.; Bodner, M.J.; Siboni, R.B.; Docter, A.G.; Haley, M.M.; Charles, A.; et al. Reducing levels of toxic RNA with small molecules. ACS Chem. Biol. 2013, 8, 2528–2537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27. [Google Scholar] [CrossRef]
- Zhang, G.; Budker, V.; Williams, P.; Subbotin, V.; Wolff, J.A. Efficient expression of naked DNA delivered intraarterially to limb muscles of nonhuman primates. Hum. Gene Ther. 2001, 12, 427–438. [Google Scholar] [CrossRef]
- Hagstrom, J.E.; Hegge, J.; Zhang, G.; Noble, M.; Budker, V.; Lewis, D.L.; Herweijer, H.; Wolff, J.A. A facile nonviral method for delivering genes and siRNAs to skeletal muscle of mammalian limbs. Mol. Ther. 2004, 10, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Gref, R.; Minamitake, Y.; Peracchia, M.T.; Trubetskoy, V.; Torchilin, V.; Langer, R. Biodegradable long-circulating polymeric nanospheres. Science 1994, 263, 1600–1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longmire, M.; Choyke, P.L.; Kobayashi, H. Clearance properties of nano-sized particles and molecules as imaging agents: Considerations and caveats. Nanomedicine 2008, 3, 703–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafson, H.H.; Holt-Casper, D.; Grainger, D.W.; Ghandehari, H. Nanoparticle uptake: The phagocyte problem. Nano Today 2015, 10, 487–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.H.; Lee, B.-J. Protein corona: A new approach for nanomedicine design. Int. J. Nanomed. 2017, 12, 3137–3151. [Google Scholar] [CrossRef] [Green Version]
- Tenzer, S.; Docter, D.; Kuharev, J.; Musyanovych, A.; Fetz, V.; Hecht, R.; Schlenk, F.; Fischer, D.; Kiouptsi, K.; Reinhardt, C.; et al. Rapid formation of plasma protein corona critically affects nanoparticle pathophysiology. Nat. Nanotechnol. 2013, 8, 772–781. [Google Scholar] [CrossRef]
- Stano, A.; Nembrini, C.; Swartz, M.A.; Hubbell, J.A.; Simeoni, E. Nanoparticle size influences the magnitude and quality of mucosal immune responses after intranasal immunization. Vaccine 2012, 30, 7541–7546. [Google Scholar] [CrossRef] [PubMed]
- Hoshyar, N.; Gray, S.; Han, H.; Bao, G. The effect of nanoparticle size on in vivo pharmacokinetics and cellular interaction. Nanomedicine 2016, 11, 673–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlasova, I.I.; Kapralov, A.A.; Michael, Z.P.; Burkert, S.C.; Shurin, M.R.; Star, A.; Shvedova, A.A.; Kagan, V.E. Enzymatic oxidative biodegradation of nanoparticles: Mechanisms, significance and applications. Toxicol. Appl. Pharmacol. 2016, 299, 58–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tidball, J.G.; Welc, S.S.; Wehling-Henricks, M. Immunobiology of inherited muscular dystrophies. Compr. Physiol. 2018, 8, 1313–1356. [Google Scholar] [CrossRef]
- Smoak, M.M.; Mikos, A.G. Advances in biomaterials for skeletal muscle engineering and obstacles still to overcome. Mater. Today Bio 2020, 7, 100069. [Google Scholar] [CrossRef] [PubMed]
- Evers, M.M.; Toonen, L.J.A.; van Roon-Mom, W.M.C. Antisense oligonucleotides in therapy for neurodegenerative disorders. Adv. Drug Deliv. Rev. 2015, 87, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Kauffman, K.J.; Webber, M.J.; Anderson, D.G. Materials for non-viral intracellular delivery of messenger RNA therapeutics. J. Control. Release 2016, 240, 227–234. [Google Scholar] [CrossRef]
- Parlea, L.; Puri, A.; Kasprzak, W.; Bindewald, E.; Zakrevsky, P.; Satterwhite, E.; Joseph, K.; Afonin, K.A.; Shapiro, B.A. Cellular delivery of RNA nanoparticles. ACS Comb. Sci. 2016, 18, 527–547. [Google Scholar] [CrossRef] [PubMed]
- Wahane, A.; Waghmode, A.; Kapphahn, A.; Dhuri, K.; Gupta, A.; Bahal, R. Role of lipid-based and polymer-based non-viral vectors in nucleic acid delivery for next-generation gene therapy. Molecules 2020, 25, 2866. [Google Scholar] [CrossRef] [PubMed]
- Mastria, E.M.; Cai, L.Y.; Kan, M.J.; Li, X.; Schaal, J.L.; Fiering, S.; Gunn, M.D.; Dewhirst, M.W.; Nair, S.K.; Chilkoti, A. Nanoparticle formulation improves doxorubicin efficacy by enhancing host antitumor immunity. J. Control. Release 2018, 269, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.-M.; Choi, Y.H.; Tu, M.-J. RNA drugs and RNA targets for small molecules: Principles, progress, and challenges. Pharmacol. Rev. 2020, 72, 862–898. [Google Scholar] [CrossRef]
- Givens, B.E.; Naguib, Y.W.; Geary, S.M.; Devor, E.J.; Salem, A.K. Nanoparticle based delivery of CRISPR/Cas9 genome editing therapeutics. AAPS J. 2018, 20, 108. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, L.; Liu, H.; Cheng, K. Delivery strategies of the CRISPR-Cas9 gene-editing system for therapeutic applications. J. Control. Release 2017, 266, 17–26. [Google Scholar] [CrossRef]
- Min, Y.-L.; Bassel-Duby, R.; Olson, E.N. CRISPR correction of Duchenne muscular dystrophy. Annu. Rev. Med. 2019, 27, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.H.; Schray, R.C.; Sirsi, S.R.; Lutz, G.J. Nanopolymers improve delivery of exon skipping oligonucleotides and concomitant dystrophin expression in skeletal muscle of mdx mice. BMC Biotechnol. 2008, 8, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirsi, S.R.; Schray, R.C.; Wheatley, M.A.; Lutz, G.J. Formulation of polylactide-co-glycolic acid nanospheres for encapsulation and sustained release of poly(ethylene imine)-poly(ethylene glycol) copolymers complexed to oligonucleotides. J. Nanobiotechnol. 2009, 7, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Wu, B.; Lu, P.; Cloer, C.; Tucker, J.D.; Lu, Q. Polyethylenimine-modified Pluronics (PCMs) improve morpholino oligomer delivery in cell culture and dystrophic mdx mice. Mol. Ther. 2013, 21, 210–216. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Wu, B.; Lu, P.; Tucker, J.D.; Milazi, S.; Shah, S.N.; Lu, Q.L. Pluronic–PEI copolymers enhance exon-skipping of 2′-O-methyl phosphorothioate oligonucleotide in cell culture and dystrophic mdx mice. Gene Ther. 2014, 21, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Tewari, M.; Pajerowski, J.D.; Cai, S.; Sen, S.; Williams, J.; Sirsi, S.; Lutz, G.; Discher, D.E. Polymersome delivery of siRNA and antisense oligonucleotides. J. Control. Release 2009, 134, 132–140. [Google Scholar] [CrossRef] [Green Version]
- Rimessi, P.; Sabatelli, P.; Fabris, M.; Braghetta, P.; Bassi, E.; Spitali, P.; Vattemi, G.; Tomelleri, G.; Mari, L.; Perrone, D.; et al. Cationic PMMA nanoparticles bind and deliver antisense oligoribonucleotides allowing restoration of dystrophin expression in the mdx mouse. Mol. Ther. 2009, 17, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Ferlini, A.; Sabatelli, P.; Fabris, M.; Bassi, E.; Falzarano, S.; Vattemi, G.; Perrone, D.; Gualandi, F.; Maraldi, N.M.; Merlini, L.; et al. Dystrophin restoration in skeletal, heart and skin arrector pili smooth muscle of mdx mice by ZM2 NP–AON complexes. Gene Ther. 2010, 17, 432–438. [Google Scholar] [CrossRef]
- Bassi, E.; Falzarano, S.; Fabris, M.; Gualandi, F.; Merlini, L.; Vattemi, G.; Perrone, D.; Marchesi, E.; Sabatelli, P.; Sparnacci, K.; et al. Persistent dystrophin protein restoration 90 days after a course of intraperitoneally administered naked 2′OMePS AON and ZM2 NP-AON complexes in mdx Mice. J. Biomed. Biotechnol. 2012, 2012, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Wu, B.; Tucker, J.D.; Bollinger, L.E.; Lu, P.; Lu, Q. Poly(ester amine) composed of polyethylenimine and Pluronic enhance delivery of antisense oligonucleotides in vitro and in dystrophic mdx mice. Mol. Ther. Nucleic Acids 2016, 5, e341. [Google Scholar] [CrossRef]
- Wang, M.; Tucker, J.D.; Lu, P.; Wu, B.; Cloer, C.; Lu, Q. Tris[2-(acryloyloxy)ethyl]isocyanurate cross-linked low-molecular-weight polyethylenimine as gene delivery carriers in cell culture and dystrophic mdx mice. Bioconjug. Chem. 2012, 23, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Itaka, K.; Osada, K.; Morii, K.; Kim, P.; Yun, S.-H.; Kataoka, K. Polyplex nanomicelle promotes hydrodynamic gene introduction to skeletal muscle. J. Control. Release 2010, 143, 112–119. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, P.-C.; Mao, H.-Q.; Leong, K.W. Enhanced gene expression in mouse muscle by sustained release of plasmid DNA using PPE-EA as a carrier. Gene Ther. 2002, 9, 1254–1261. [Google Scholar] [CrossRef] [Green Version]
- Kinouchi, N.; Ohsawa, Y.; Ishimaru, N.; Ohuchi, H.; Sunada, Y.; Hayashi, Y.; Tanimoto, Y.; Moriyama, K.; Noji, S. Atelocollagen-mediated local and systemic applications of myostatin-targeting siRNA increase skeletal muscle mass. Gene Ther. 2008, 15, 1126–1130. [Google Scholar] [CrossRef] [Green Version]
- Márquez-Miranda, V.; Abrigo, J.; Rivera, J.C.; Araya-Duran, I.; Aravena, J.; Simon, F.; Pacheco, N.; Gonzalez-Nilo, F.D.; Cabello-Verrugio, C. The complex of PAMAM-OH dendrimer with angiotensin (1-7) prevented the disuse-induced skeletal muscle atrophy in mice. Int. J. Nanomed. 2017, 12, 1985–1999. [Google Scholar] [CrossRef] [Green Version]
- Negishi, Y.; Ishii, Y.; Shiono, H.; Akiyama, S.; Sekine, S.; Kojima, T.; Mayama, S.; Kikuchi, T.; Hamano, N.; Endo-Takahashi, Y.; et al. Bubble liposomes and ultrasound exposure improve localized morpholino oligomer delivery into the skeletal muscles of dystrophic mdx mice. Mol. Pharm. 2014, 11, 1053–1061. [Google Scholar] [CrossRef] [PubMed]
- Koebis, M.; Kiyatake, T.; Yamaura, H.; Nagano, K.; Higashihara, M.; Sonoo, M.; Hayashi, Y.; Negishi, Y.; Endo-Takahashi, Y.; Yanagihara, D.; et al. Ultrasound-enhanced delivery of morpholino with bubble liposomes ameliorates the myotonia of myotonic dystrophy model mice. Sci. Rep. 2013, 3, 2242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afzal, E.; Zakeri, S.; Keyhanvar, P.; Bagheri, M.; Mahjoubi, P.; Asadian, M.; Omoomi, N.; Dehqanian, M.; Ghalandarlaki, N.; Darvishmohammadi, T.; et al. Nanolipodendrosome-loaded glatiramer acetate and myogenic differentiation 1 as augmentation therapeutic strategy approaches in muscular dystrophy. Int. J. Nanomed. 2013, 8, 2943–2960. [Google Scholar] [CrossRef] [Green Version]
- Turjeman, K.; Yanay, N.; Elbaz, M.; Bavli, Y.; Gross, M.; Rabie, M.; Barenholz, Y.; Nevo, Y. Liposomal steroid nano-drug is superior to steroids as-is in mdx mouse model of Duchenne muscular mystrophy. Nanomedicine 2019, 16, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Yukihara, M.; Ito, K.; Tanoue, O.; Goto, K.; Matsushita, T.; Matsumoto, Y.; Masuda, M.; Kimura, S.; Ueoka, R. Effective drug delivery system for Duchenne muscular dystrophy using hybrid liposomes including gentamicin along with reduced toxicity. Biol. Pharm. Bull. 2011, 34, 712–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bibee, K.P.; Cheng, Y.; Ching, J.K.; Marsh, J.N.; Li, A.J.; Keeling, R.M.; Connolly, A.M.; Golumbek, P.T.; Myerson, J.W.; Hu, G.; et al. Rapamycin nanoparticles target defective autophagy in muscular dystrophy to enhance both strength and cardiac function. FASEB J. 2014, 28, 2047–2061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, T.; Cheng, Q.; Min, Y.-L.; Olson, E.N.; Siegwart, D.J. Systemic nanoparticle delivery of CRISPR-Cas9 ribonucleoproteins for effective tissue specific genome editing. Nat. Commun. 2020, 11, 3232. [Google Scholar] [CrossRef]
- Lee, K.; Conboy, M.; Park, H.M.; Jiang, F.; Kim, H.J.; Dewitt, M.A.; Mackley, V.A.; Chang, K.; Rao, A.; Skinner, C.; et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat. Biomed. Eng. 2017, 1, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Järver, P.; O’Donovan, L.; Gait, M.J. A chemical view of oligonucleotides for exon skipping and related drug applications. Nucleic Acid Ther. 2014, 24, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Wagner, E. History of polymeric gene delivery systems. Top. Curr. Chem. 2017, 375, 26. [Google Scholar] [CrossRef] [PubMed]
- Boussif, O.; Lezoualc’h, F.; Zanta, M.A.; Mergny, M.D.; Scherman, D.; Demeneix, B.; Behr, J.P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: Polyethylenimine. Proc. Natl. Acad. Sci. USA 1995, 92, 7297–7301. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.L.; Mann, C.J.; Lou, F.; Bou-Gharios, G.; Morris, G.E.; Xue, S.; Fletcher, S.; Partridge, T.A.; Wilton, S.D. Functional amounts of dystrophin produced by skipping the mutated exon in the mdx dystrophic mouse. Nat. Med. 2003, 9, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.L.; Bou-Gharios, G.; Partridge, T.A. Non-viral gene delivery in skeletal muscle: A protein factory. Gene Ther. 2003, 10, 131–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, J.H.; Sirsi, S.R.; Latta, D.R.; Lutz, G.J. Induction of dystrophin expression by exon skipping in mdx mice following intramuscular injection of antisense oligonucleotides complexed with PEG–PEI copolymers. Mol. Ther. 2006, 14, 88–96. [Google Scholar] [CrossRef]
- Lutz, G.J.; Sirsi, S.R.; Williams, J.H. PEG-PEI copolymers for oligonucleotide delivery to Cells and tissues. Methods Mol. Biol. 2008, 433, 141–158. [Google Scholar] [CrossRef] [PubMed]
- Castaldello, A.; Brocca-Cofano, E.; Voltan, R.; Triulzi, C.; Altavilla, G.; Laus, M.; Sparnacci, K.; Ballestri, M.; Tondelli, L.; Fortini, C.; et al. DNA prime and protein boost immunization with innovative polymeric cationic core-shell nanoparticles elicits broad immune responses and strongly enhance cellular responses of HIV-1 tat DNA vaccination. Vaccine 2006, 24, 5655–5669. [Google Scholar] [CrossRef]
- Summerton, J.; Weller, D. Morpholino antisense oligomers: Design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997, 7, 187–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, C.M.; Harris, E.N. Antisense oligonucleotides: Treatment strategies and cellular internalization. RNA Dis. 2016, 3, e1393. [Google Scholar] [CrossRef]
- Järver, P.; Zaghloul, E.M.; Arzumanov, A.A.; Saleh, A.F.; McClorey, G.; Hammond, S.M.; Hällbrink, M.; Langel, Ü.; Smith, C.I.E.; Wood, M.J.A.; et al. Peptide nanoparticle delivery of charge-neutral splice-switching morpholino oligonucleotides. Nucleic Acid Ther. 2015, 25, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Takizawa, T.; Negishi, Y.; Hagisawa, K.; Tanaka, K.; Sawamura, K.; Utoguchi, N.; Nishioka, T.; Maruyama, K. Gene delivery by combination of novel liposomal bubbles with perfluoropropane and ultrasound. J. Control. Release 2007, 117, 130–136. [Google Scholar] [CrossRef]
- Negishi, Y.; Endo, Y.; Fukuyama, T.; Suzuki, R.; Takizawa, T.; Omata, D.; Maruyama, K.; Aramaki, Y. Delivery of siRNA into the cytoplasm by liposomal bubbles and ultrasound. J. Control. Release 2008, 132, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Tros de Ilarduya, C.; Sun, Y.; Düzgüneş, N. Gene delivery by lipoplexes and polyplexes. Eur. J. Pharm. Sci. 2010, 40, 159–170. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Cullis, P.R.; van der Meel, R. Lipid nanoparticles enabling gene therapies: From concepts to clinical utility. Nucleic Acid Ther. 2018, 28, 146–157. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Sharili, A.S.; Connelly, J.; Gautrot, J.E. Highly stable RNA capture by dense cationic polymer brushes for the design of cytocompatible, serum-stable siRNA delivery vectors. Biomacromolecules 2018, 19, 606–615. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Myhre, J.L.; Chen, S.; Tam, Y.Y.C.; Danescu, A.; Richman, J.M.; Cullis, P.R. Design of lipid nanoparticles for in vitro and in vivo delivery of plasmid DNA. Nanomedicine 2017, 13, 1377–1387. [Google Scholar] [CrossRef]
- Sano, A.; Maeda, M.; Nagahara, S.; Ochiya, T.; Honma, K.; Itoh, H.; Miyata, T.; Fujioka, K. Atelocollagen for protein and gene delivery. Adv. Drug Deliv. Rev. 2003, 55, 1651–1677. [Google Scholar] [CrossRef] [PubMed]
- Ochiya, T.; Nagahara, S.; Sano, A.; Itoh, H.; Terada, M. Biomaterials for gene delivery: Atelocollagen-mediated controlled release of molecular medicines. Curr. Gene Ther. 2001, 1, 31–52. [Google Scholar] [CrossRef]
- Wang, J.; Mao, H.-Q.; Leong, K.W. A novel biodegradable gene carrier based on polyphosphoester. J. Am. Chem. Soc. 2001, 123, 9480–9481. [Google Scholar] [CrossRef]
- Itaka, K.; Yamauchi, K.; Harada, A.; Nakamura, K.; Kawaguchi, H.; Kataoka, K. Polyion complex micelles from plasmid DNA and poly(ethylene glycol)-poly(L-lysine) block copolymer as serum-tolerable polyplex system: Physicochemical properties of micelles relevant to gene transfection efficiency. Biomaterials 2003, 24, 4495–4506. [Google Scholar] [CrossRef]
- Parvathaneni, V.; Kulkarni, N.S.; Muth, A.; Gupta, V. Drug repurposing: A promising tool to accelerate the drug discovery process. Drug Discov. Today 2019, 24, 2076–2085. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Markoutsa, E.; Li, J.; Xu, P. Repurposing disulfiram for cancer therapy via targeted nanotechnology through enhanced tumor mass penetration and disassembly. Acta Biomater. 2018, 68, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Amini, M.A.; Abbasi, A.Z.; Cai, P.; Lip, H.; Gordijo, C.R.; Li, J.; Chen, B.; Zhang, L.; Rauth, A.M.; Wu, X.Y. Combining tumor microenvironment modulating nanoparticles with doxorubicin to enhance chemotherapeutic efficacy and boost antitumor immunity. J. Natl. Cancer Inst. 2019, 111, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Park, J.; Shim, M.K.; Um, W.; Yoon, H.Y.; Ryu, J.H.; Lim, D.-K.; Kim, K. Recent advances and challenges of repurposing nanoparticle-based drug delivery systems to enhance cancer immunotherapy. Theranostics 2019, 9, 7906–7923. [Google Scholar] [CrossRef] [PubMed]
- Gregoriou, Y.; Gregoriou, G.; Yilmaz, V.; Kapnisis, K.; Prokopi, M.; Anayiotos, A.; Strati, K.; Dietis, N.; Constantinou, A.I.; Andreou, C. Resveratrol loaded polymeric micelles for theranostic targeting of breast cancer cells. Nanotheranostics 2021, 5, 113–124. [Google Scholar] [CrossRef]
- Dunant, P.; Walter, M.C.; Karpati, G.; Lochmüller, H. Gentamicin fails to increase dystrophin expression in dystrophin-deficient muscle. Muscle Nerve 2003, 27, 624–627. [Google Scholar] [CrossRef] [PubMed]
- Zammit, P.S. Function of the myogenic regulatory factors Myf5, MyoD, Myogenin and MRF4 in skeletal muscle, satellite cells and regenerative myogenesis. Semin. Cell Dev. Biol. 2017, 72, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, S.N. Sirolimus: Its discovery, biological properties, and mechanism of action. Transplant. Proc. 2003, 35, 7S–14S. [Google Scholar] [CrossRef]
- Flaim, S.F. Pharmacokinetics and side effects of perfluorocarbon-based blood substitutes. Artif. Cells. Blood Substit. Immobil. Biotechnol. 1994, 22, 1043–1054. [Google Scholar] [CrossRef]
- Spahn, D.R. Blood substitutes. Artificial oxygen carriers: Perfluorocarbon emulsions. Crit. Care 1999, 3, R93–R97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, A.; Tanaka, H.; Sakurai, Y.; Tange, K.; Nakai, Y.; Ohkawara, T.; Takeda, H.; Harashima, H.; Akita, H. Effect of particle size on their accumulation in an inflammatory lesion in a dextran sulfate sodium (DSS)-induced colitis model. Int. J. Pharm. 2016, 509, 118–122. [Google Scholar] [CrossRef]
- Chen, K.-H.; Lundy, D.J.; Toh, E.K.-W.; Chen, C.-H.; Shih, C.; Chen, P.; Chang, H.-C.; Lai, J.J.; Stayton, P.S.; Hoffman, A.S.; et al. Nanoparticle distribution during systemic inflammation is size-dependent and organ-specific. Nanoscale 2015, 7, 15863–15872. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Luther, D.C.; Lee, Y.W.; Nagaraj, H.; Scaletti, F.; Rotello, V.M. Delivery approaches for CRISPR/Cas9 therapeutics in vivo: Advances and challenges. Expert Opin. Drug Deliv. 2018, 15, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A review of the challenges and approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Jiang, Z.; Saha, K.; Kim, C.S.; Kim, S.T.; Landis, R.F.; Rotello, V.M. Gold nanoparticles for nucleic acid delivery. Mol. Ther. 2014, 22, 1075–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chithrani, B.D.; Ghazani, A.A.; Chan, W.C.W. Determining the size and shape dependence of gold nanoparticle uptake into mammalian cells. Nano Lett. 2006, 6, 662–668. [Google Scholar] [CrossRef]
- Arandel, L.; Polay Espinoza, M.; Matloka, M.; Bazinet, A.; De Dea Diniz, D.; Naouar, N.; Rau, F.; Jollet, A.; Edom-Vovard, F.; Mamchaoui, K.; et al. Immortalized human myotonic dystrophy muscle cell lines to assess therapeutic compounds. Dis. Model. Mech. 2017, 10, 487–497. [Google Scholar] [CrossRef] [Green Version]
- Matloka, M.; Klein, A.F.; Rau, F.; Furling, D. Cells of matter—in vitro models for myotonic dystrophy. Front. Neurol. 2018, 9, 361. [Google Scholar] [CrossRef] [PubMed]
- Bigot, A.; Klein, A.F.; Gasnier, E.; Jacquemin, V.; Ravassard, P.; Butler-Browne, G.; Mouly, V.; Furling, D. Large CTG repeats trigger P16-dependent premature senescence in myotonic dystrophy type 1 muscle precursor cells. Am. J. Pathol. 2009, 174, 1435–1442. [Google Scholar] [CrossRef] [Green Version]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Renault, V.; Piron-Hamelin, G.; Forestier, C.; DiDonna, S.; Decary, S.; Hentati, F.; Saillant, G.; Butler-Browne, G.S.; Mouly, V. Skeletal muscle regeneration and the mitotic clock. Exp. Gerontol. 2000, 35, 711–719. [Google Scholar] [CrossRef]
- Renna, L.V.; Cardani, R.; Botta, A.; Rossi, G.; Fossati, B.; Costa, E.; Meola, G. Premature senescence in primary muscle cultures of myotonic dystrophy type 2 is not associated with P16 induction. Eur. J. Histochem. 2014, 58, 2444. [Google Scholar] [CrossRef] [PubMed]
- Thornell, L.-E.; Lindstöm, M.; Renault, V.; Klein, A.; Mouly, V.; Ansved, T.; Butler-Browne, G.; Furling, D. Satellite cell dysfunction contributes to the progressive muscle atrophy in myotonic dystrophy type 1. Neuropathol. Appl. Neurobiol. 2009, 35, 603–613. [Google Scholar] [CrossRef]
- Liang, R.; Dong, W.; Shen, X.; Peng, X.; Aceves, A.G.; Liu, Y. Modeling myotonic dystrophy 1 in C2C12 myoblast cells. J. Vis. Exp. 2016, 113, 54078. [Google Scholar] [CrossRef]
- Mamchaoui, K.; Trollet, C.; Bigot, A.; Negroni, E.; Chaouch, S.; Wolff, A.; Kandalla, P.K.; Marie, S.; Di Santo, J.; St Guily, J.L.; et al. Immortalized pathological human myoblasts: Towards a universal tool for the study of neuromuscular disorders. Skelet. Muscle 2011, 1, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, M.S.; Carlson, M.E.; Conboy, I.M. Differentiation rather than aging of muscle stem cells abolishes their telomerase activity. Biotechnol. Prog. 2009, 25, 1130–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massenet, J.; Gitiaux, C.; Magnan, M.; Cuvellier, S.; Hubas, A.; Nusbaum, P.; Dilworth, F.J.; Desguerre, I.; Chazaud, B. Derivation and characterization of immortalized human muscle satellite cell clones from muscular dystrophy patients and healthy individuals. Cells 2020, 9, 1780. [Google Scholar] [CrossRef]
- Chaouch, S.; Mouly, V.; Goyenvalle, A.; Vulin, A.; Mamchaoui, K.; Negroni, E.; Di Santo, J.; Butler-Browne, G.; Torrente, Y.; Garcia, L.; et al. Immortalized skin fibroblasts expressing conditional MyoD as a renewable and reliable source of converted human muscle cells to assess therapeutic strategies for muscular mystrophies: Validation of an exon-skipping approach to restore dystrophin in Duchenne muscular dystrophy Cells. Hum. Gene Ther. 2009, 20, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.T.; Kizana, E.; Yates, J.D.; Lo, H.P.; Yang, N.; Wu, Z.H.; Alexander, I.E.; North, K.N. Dystrophinopathy carrier determination and detection of protein deficiencies in muscular dystrophy using lentiviral MyoD-forced myogenesis. Neuromuscul. Disord. 2007, 17, 276–284. [Google Scholar] [CrossRef]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duval, K.; Grover, H.; Han, L.-H.; Mou, Y.; Pegoraro, A.F.; Fredberg, J.; Chen, Z. Modeling physiological events in 2D vs. 3D cell culture. Physiology 2017, 32, 266–277. [Google Scholar] [CrossRef]
- Knight, E.; Przyborski, S. Advances in 3D cell culture technologies enabling tissue-like structures to be created in vitro. J. Anat. 2015, 227, 746–756. [Google Scholar] [CrossRef] [Green Version]
- Jeffries, G.D.M.; Xu, S.; Lobovkina, T.; Kirejev, V.; Tusseau, F.; Gyllensten, C.; Singh, A.K.; Karila, P.; Moll, L.; Orwar, O. 3D micro-organisation printing of mammalian cells to generate biological tissues. Sci. Rep. 2020, 10, 19529. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, B.; Gong, L.; An, C.; Lin, J.; Li, Q.; Jiang, D.; Jin, K.; Mechakra, A.; Bunpetch, V.; et al. Dissecting cell diversity and connectivity in skeletal muscle for myogenesis. Cell Death Dis. 2019, 10, 427. [Google Scholar] [CrossRef]
- Juhas, M.; Engelmayr, G.C.; Fontanella, A.N.; Palmer, G.M.; Bursac, N. Biomimetic Engineered muscle with capacity for vascular integration and functional maturation in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, 5508–5513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borselli, C.; Cezar, C.A.; Shvartsman, D.; Vandenburgh, H.H.; Mooney, D.J. The role of multifunctional delivery scaffold in the ability of cultured myoblasts to promote muscle regeneration. Biomaterials 2011, 32, 8905–8914. [Google Scholar] [CrossRef] [Green Version]
- McLean, I.C.; Schwerdtfeger, L.A.; Tobet, S.A.; Henry, C.S. Powering ex vivo tissue models in microfluidic systems. Lab. Chip 2018, 18, 1399–1410. [Google Scholar] [CrossRef]
- Gowing, G.; Svendsen, S.; Svendsen, C.N. Ex vivo gene therapy for the treatment of neurological disorders. Prog. Brain Res. 2017, 230, 99–132. [Google Scholar] [CrossRef] [PubMed]
- Mobini, S.; Song, Y.H.; McCrary, M.W.; Schmidt, C.E. Advances in ex vivo models and lab-on-a-chip devices for neural tissue engineering. Biomaterials 2019, 198, 146–166. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.R.; Meyer, G.A. Skeletal muscle explants: Ex-vivo models to study cellular behavior in a complex tissue environment. Connect. Tissue Res. 2020, 61, 248–261. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Wu, X.; Young, A.T.; Haynes, C.L. Microfluidics-based in vivo mimetic systems for the study of cellular biology. Acc. Chem. Res. 2014, 47, 1165–1173. [Google Scholar] [CrossRef] [PubMed]
- Carton, F.; Calderan, L.; Malatesta, M. Incubation under fluid dynamic conditions markedly improves the structural preservation in vitro of explanted skeletal muscles. Eur. J. Histochem. 2017, 61, 2862. [Google Scholar] [CrossRef] [Green Version]
- Carosio, S.; Barberi, L.; Rizzuto, E.; Nicoletti, C.; Del Prete, Z.; Musarò, A. Generation of ex vivo-vascularized muscle engineered tissue (X-MET). Sci. Rep. 2013, 3, 1420. [Google Scholar] [CrossRef] [PubMed]
- Fischer, H.C.; Chan, W.C. Nanotoxicity: The growing need for in vivo study. Curr. Opin. Biotechnol. 2007, 18, 565–571. [Google Scholar] [CrossRef]
- Bostrom, M.; O’Keefe, R. What experimental approaches (eg, in vivo, in vitro, tissue retrieval) are effective in investigating the biologic effects of particles? J. Am. Acad. Orthop. Surg. 2008, 16, S63–S67. [Google Scholar] [CrossRef] [Green Version]
- McGreevy, J.W.; Hakim, C.H.; McIntosh, M.A.; Duan, D. Animal models of Duchenne muscular dystrophy: From basic mechanisms to gene therapy. Dis. Model. Mech. 2015, 8, 195–213. [Google Scholar] [CrossRef] [Green Version]
- Wells, D.J. Tracking progress: An update on animal models for Duchenne muscular dystrophy. Dis. Model. Mech. 2018, 11, dmm035774. [Google Scholar] [CrossRef] [Green Version]
- Collins, C.A.; Morgan, J.E. Duchenne’s muscular dystrophy: Animal models used to investigate pathogenesis and develop therapeutic strategies. Int. J. Exp. Pathol. 2003, 84, 165–172. [Google Scholar] [CrossRef]
- Bulfield, G.; Siller, W.G.; Wight, P.A.; Moore, K.J. X Chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. USA 1984, 81, 1189–1192. [Google Scholar] [CrossRef] [Green Version]
- van Putten, M.; Putker, K.; Overzier, M.; Adamzek, W.A.; Pasteuning-Vuhman, S.; Plomp, J.J.; Aartsma-Rus, A. Natural disease history of the D2-mdx mouse model for Duchenne muscular dystrophy. FASEB J. 2019, 33, 8110–8124. [Google Scholar] [CrossRef] [Green Version]
- Desguerre, I.; Arnold, L.; Vignaud, A.; Cuvellier, S.; Yacoub-Youssef, H.; Gherardi, R.K.; Chelly, J.; Chretien, F.; Mounier, R.; Ferry, A.; et al. A new model of experimental fibrosis in hindlimb skeletal muscle of adult mdx mouse mimicking muscular dystrophy. Muscle Nerve 2012, 45, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Ng, R.; Banks, G.B.; Hall, J.K.; Muir, L.A.; Ramos, J.N.; Wicki, J.; Odom, G.L.; Konieczny, P.; Seto, J.; Chamberlain, J.R.; et al. Animal models of muscular dystrophy. Prog. Mol. Biol. Transl. Sci. 2012, 105, 83–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornegay, J.N. The golden retriever model of Duchenne muscular dystrophy. Skelet. Muscle 2017, 7, 9. [Google Scholar] [CrossRef]
- Kawahara, G.; Karpf, J.A.; Myers, J.A.; Alexander, M.S.; Guyon, J.R.; Kunkel, L.M. Drug screening in a zebrafish model of Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2011, 108, 5331–5336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widrick, J.J.; Kawahara, G.; Alexander, M.S.; Beggs, A.H.; Kunkel, L.M. Discovery of novel therapeutics for muscular dystrophies using zebrafish phenotypic screens. J. Neuromuscul. Dis. 2019, 6, 271–287. [Google Scholar] [CrossRef] [Green Version]
- Sicot, G.; Gomes-Pereira, M. RNA toxicity in human disease and animal models: From the uncovering of a new mechanism to the development of promising therapies. Biochim. Biophys. Acta 2013, 1832, 1390–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes-Pereira, M.; Cooper, T.A.; Gourdon, G. Myotonic dystrophy mouse models: Towards rational therapy development. Trends Mol. Med. 2011, 17, 506–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guiraud-Dogan, C.; Huguet, A.; Gomes-Pereira, M.; Brisson, E.; Bassez, G.; Junien, C.; Gourdon, G. DM1 CTG expansions affect insulin receptor isoforms expression in various tissues of transgenic mice. Biochim. Biophys. Acta 2007, 1772, 1183–1191. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Miller, J.W.; Mankodi, A.; Kanadia, R.N.; Yuan, Y.; Moxley, R.T.; Swanson, M.S.; Thornton, C.A. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum. Mol. Genet. 2006, 15, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Mankodi, A.; Logigian, E.; Callahan, L.; McClain, C.; White, R.; Henderson, D.; Krym, M.; Thornton, C.A. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 2000, 289, 1769–1773. [Google Scholar] [CrossRef] [Green Version]
- Mankodi, A.; Takahashi, M.P.; Jiang, H.; Beck, C.L.; Bowers, W.J.; Moxley, R.T.; Cannon, S.C.; Thornton, C.A. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol. Cell 2002, 10, 35–44. [Google Scholar] [CrossRef]
- Seznec, H.; Agbulut, O.; Sergeant, N.; Savouret, C.; Ghestem, A.; Tabti, N.; Willer, J.C.; Ourth, L.; Duros, C.; Brisson, E.; et al. Mice transgenic for the human myotonic dystrophy region with expanded CTG repeats display muscular and brain abnormalities. Hum. Mol. Genet. 2001, 10, 2717–2726. [Google Scholar] [CrossRef] [Green Version]
- Hinman, M.N.; Richardson, J.I.; Sockol, R.A.; Aronson, E.D.; Stednitz, S.J.; Murray, K.N.; Berglund, J.A.; Guillemin, K. Zebrafish mbnl mutants model physical and molecular phenotypes of myotonic dystrophy. bioRxiv 2020, 665380. [Google Scholar] [CrossRef]
- Machuca-Tzili, L.E.; Buxton, S.; Thorpe, A.; Timson, C.M.; Wigmore, P.; Luther, P.K.; Brook, J.D. Zebrafish deficient for Muscleblind-like 2 exhibit features of myotonic dystrophy. Dis. Model. Mech. 2011, 4, 381–392. [Google Scholar] [CrossRef] [Green Version]
- Carton, F.; Chevalier, Y.; Nicoletti, L.; Tarnowska, M.; Stella, B.; Arpicco, S.; Malatesta, M.; Jordheim, L.P.; Briançon, S.; Lollo, G. Rationally designed hyaluronic acid-based nano-complexes for pentamidine delivery. Int. J. Pharm. 2019, 568, 118526. [Google Scholar] [CrossRef] [PubMed]
- Stella, B.; Andreana, I.; Zonari, D.; Arpicco, S. Pentamidine-loaded lipid and polymer nanocarriers as tunable anticancer drug delivery systems. J. Pharm. Sci. 2020, 109, 1297–1302. [Google Scholar] [CrossRef]
- Falzarano, M.S.; Bassi, E.; Passarelli, C.; Braghetta, P.; Ferlini, A. Biodistribution studies of polymeric nanoparticles for drug delivery in mice. Hum Gene Ther. 2014, 25, 927–928. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.; Yue, F.; Qiu, J.; Deng, M.; Kuang, S. Polymeric nanoparticles functionalized with muscle-homing peptides for targeted delivery of phosphatase and tensin homolog inhibitor to skeletal muscle. Acta Biomater. 2020, 118, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Herweijer, H.; Wolff, J.A. Gene therapy progress and prospects: Hydrodynamic gene delivery. Gene Ther. 2007, 14, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Kumbhari, V.; Li, L.; Piontek, K.; Ishida, M.; Fu, R.; Khalil, B.; Garrett, C.M.; Liapi, E.; Kalloo, A.N.; Selaru, F.M. Successful liver-directed gene delivery by ERCP-guided hydrodynamic injection (with Videos). Gastrointest. Endosc. 2018, 88, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Le Guen, Y.T.; Le Gall, T.; Midoux, P.; Guégan, P.; Braun, S.; Montier, T. Gene transfer to skeletal muscle using hydrodynamic limb vein injection: Current applications, hurdles and possible optimizations. J. Gene Med. 2020, 22, e3150. [Google Scholar] [CrossRef] [PubMed]
- Ashfaq, U.A.; Riaz, M.; Yasmeen, E.; Yousaf, M. Recent advances in nanoparticle-based targeted drug-delivery systems against cancer and role of tumor microenvironment. Crit. Rev. Ther. Drug Carrier Syst. 2017, 34, 317–353. [Google Scholar] [CrossRef]
- Pietersz, G.A.; Wang, X.; Yap, M.L.; Lim, B.; Peter, K. Therapeutic targeting in nanomedicine: The future lies in recombinant antibodies. Nanomedicine 2017, 12, 1873–1889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arahata, K.; Ishiura, S.; Ishiguro, T.; Tsukahara, T.; Suhara, Y.; Eguchi, C.; Ishiharat, T.; Nonaka, I.; Ozawa, E.; Sugita, H. Immunostaining of skeletal and cardiac muscle surface membrane with antibody against Duchenne muscular dystrophy peptide. Nature 1988, 333, 861–863. [Google Scholar] [CrossRef]
- Zhang, R.; Kim, A.S.; Fox, J.M.; Nair, S.; Basore, K.; Klimstra, W.B.; Rimkunas, R.; Fong, R.H.; Lin, H.; Poddar, S.; et al. Mxra8 is a receptor for multiple arthritogenic alphaviruses. Nature 2018, 557, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Poon, W.; Zhang, X.; Bekah, D.; Teodoro, J.G.; Nadeau, J.L. Targeting B16 tumors in vivo with peptide-conjugated gold nanoparticles. Nanotechnology 2015, 26, 285101. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, C.; Tagalakis, A.D.; Manunta, M.D.; Hart, S.L.; Khaw, P.T. Receptor-targeted liposome-peptide-siRNA nanoparticles represent an efficient delivery system for MRTF silencing in conjunctival fibrosis. Sci. Rep. 2016, 6, 21881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajau, R.; Rohani, R.; Abdul Hamid, S.S.; Adam, Z.; Mohd Janib, S.N.; Salleh, M.Z. Surface functionalisation of poly-APO- b -polyol ester cross-linked copolymers as core–shell nanoparticles for targeted breast cancer therapy. Sci. Rep. 2020, 10, 21704. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-Y.; Yuan, Z.; Cao, Z.; Wang, B.; Qiao, C.; Li, J.; Xiao, X. A muscle-targeting peptide displayed on AAV2 improves muscle tropism on systemic delivery. Gene Ther. 2009, 16, 953–962. [Google Scholar] [CrossRef] [Green Version]
- Seow, Y.; Yin, H.; Wood, M.J.A. Identification of a novel muscle targeting peptide in mdx mice. Peptides 2010, 31, 1873–1877. [Google Scholar] [CrossRef]
- Tsoumpra, M.K.; Fukumoto, S.; Matsumoto, T.; Takeda, S.; Wood, M.J.A.; Aoki, Y. Peptide-conjugate antisense based splice-correction for Duchenne muscular dystrophy and other neuromuscular diseases. EBioMedicine 2019, 45, 630–645. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Zhao, J.; Han, G.; Zhang, Y.; Dong, X.; Cao, L.; Wang, Q.; Moulton, H.M.; Yin, H. Effective dystrophin restoration by a novel muscle-homing peptide-morpholino conjugate in dystrophin-deficient mdx mice. Mol. Ther. 2014, 22, 1333–1341. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Approach | Active Agent | Target | Pathology | Limitations | Development Phase | Reference |
|---|---|---|---|---|---|---|
| Antisense oligonucleotides | Etelprisen | mRNA | DMD | Rapid degradation by exonuclease Low cellular uptake Activation of immune systemInflammatory effects | FDA approved (Clin. Trial NCT02255552) | [77] |
| Drisapersen | Phase III (Clin. Trial NCT01254019) | [78,79] | ||||
| cEt ASO | CUG/CCUGexp | DM | Preclinical studies | [80,81] | ||
| CRISPR/Cas9 | Not approved | DNA | DMD | Higher accumulation in proliferating cells than in fully differentiated cells Rapid degradation | Preclinical studies | [82,83,84,85] |
| Not approved | CTGexp DNA | DM | Uncompleted repair of protein expression Low transfection efficiency | Preclinical studies | [86] | |
| Small molecules | Aminoglycoside antibiotics | Non-sense mutations on mRNA | DMD | Ototoxicity Nephrotoxicity | Preclinical studies | [87,88,89] |
| Ataluren | Non-sense mutations on mRNA | DMD | High dosage required | EMA approved (Clin. Trial NCT01826487) | [90,91] | |
| Pentamidine | CUGexp RNA | DM | Nephrotoxicity Off-label use | Preclinical studies | [18,92] | |
| Furamidine and erytromicin | CTGexp DNA | DM | Off-label use | Preclinical studies | [93,94,95] | |
| ISOX and vorinostat | MBNL1-splicing factors | DM1 | Off-label use | Preclinical studies | [96] |
| Class of Nanocarriers | Nanocarrier Composition | Muscle Pathology | Loaded Molecules | Therapeutic Target | Mouse Model | Advantages and Limitations | Admin. Route | Ref. |
|---|---|---|---|---|---|---|---|---|
| Polymeric | PEI-PEG | DMD | 2′-OMe ASO | Dystrophin pre-mRNA | mdx | (+) high dystrophin-positive fibers increased (+) long term residual efficacy over 6 weeks (-) low general transfection efficiency | I.M. | [163] |
| PEI-PEG/PLGA | DMD | 2′-OMe ASO | Dystrophin pre-mRNA | mdx | (-) no improvement compared to PEI-PEG-ASO | I.M. | [164] | |
| PEI-Pluronic® | DMD | PMO ASO | Dystrophin pre-mRNA | mdx | (+) dystrophin-positive fibers increased up to 4-fold after I.M. (+) dystrophin-positive fibers increased up to 3-fold in all skeletal muscles after I.V. (+) dystrophin-positive fibers increased up to 5-fold in heart after I.V. (+) low muscle tissue, liver and kidney toxicity (-) mild general transfection efficiency | I.M./I.V. | [165] | |
| DMD | 2′-OMe ASO | Dystrophin pre-mRNA | mdx | (+) dystrophin-positive fibers increased up to 10-fold | I.M. | [166] | ||
| PEG-polycaprolactone PEG-(polylactic acid) | DMD | PMO ASO | Dystrophin pre-mRNA | mdx | (+) dystrophin-positive fibers increased up to 3-fold (+) low muscle tissue toxicity (-) mild general transfection efficiency | I.M. | [167] | |
| PMMA | DMD | 2′-OMe ASO | Dystrophin pre-mRNA | mdx | (+) dystrophin-positive fibers increased up to 7-fold (-) slow biodegradability | I.P. | [168] | |
| PMMA/NIPAM | DMD | 2′-OMe ASO | Dystrophin pre-mRNA | mdx | (+) dystrophin-positive fibers increased up to 4-fold (+) body-wide dystrophin restoration after I.V. (+) exon-skipping level enhanced up to 20-fold (+) long term residual efficacy over 90 days | I.P./I.V. | [169,170] | |
| PEA | DMD | 2′-OMe ASO | Dystrophin pre-mRNA | mdx | (+) dystrophin-positive fibers increased up to 3–10-fold | I.M. | [171] | |
| DMD | PMO ASO | Dystrophin pre-mRNA | mdx | (+) dystrophin-positive fibers increased up to 3-fold after I.M. (+) body-wide dystrophin-positive fibers increased up to 3-fold after I.V. | I.M./I.V. | [171] | ||
| Muscle atrophy/ DMD | pDNA | Cell nucleus | mdx | (+) transfection efficiency enhanced up to 6-fold | I.M. | [172] | ||
| PLys-PEG | Muscle atrophy | pDNA | Cell nucleus | Balb/c | (+) transfection efficiency enhanced up to 10-fold | I.V. | [173] | |
| PPE-EA | Muscle atrophy | pDNA | Cell nucleus | Balb/c | (+) transfection efficiency enhanced up to 13-fold (+) long term residual efficacy over 14 days | I.M. | [174] | |
| Atelocollagen | Muscle atrophy/ DMD | siRNA | Cytoplasm | mdx | (+) higher mass muscle increase | I.M./I.V. | [175] | |
| PAMAM-OH | Muscle atrophy | Angiotensin (1–7) | Cytoplasm | Balb/c | (+) higher anti-atrophic effects | I.P. | [176] | |
| Lipidic | PEG-bubble liposomes | DMD | PMO ASO | Dystrophin pre-mRNA | mdx | (+) dystrophin-positive fibers increased up to 1.5-fold (+) exon-skipping level enhanced up to 5-fold | I.M. | [177] |
| DM1 | PMO ASO | Clcn1 pre-mRNA | HSALR | (+) increased expression of Clcn1 protein up to 1.4-fold | I.M. | [178] | ||
| Nanolipodendrosomes | DMD | MyoD and GA | Cytoplasm | SW-1 | (+) slight mass muscle increase | I.M. | [179] | |
| Nanoliposomes | DMD | Glucocorticoide | Cell nucleus | mdx | (+) lower inflammatory induced response (+) lower bone catabolic effects | I.V. | [180] | |
| Hybrid liposomes DMPC and (C12(EO)23) | DMD | Gentamicin | Ribosomes | mdx | (+) dystrophin-positive fibers increased up to 4-fold (+) lower ototoxicity and nephrotoxicity | I.P. | [181] | |
| Perfluorocarbon | DMD | Rapamycin | mTORC1 complex | mdx | (+) high muscle strength increase (+) high cardiac contractile performance increase | I.V. | [182] | |
| Lipid NPs | DMD | CRISPR/Cas9 | Dystrophin DNA sequence | ΔEx44 | (+) dystrophin expression restored up to 5% | I.M. | [183] | |
| Inorganic | Gold | DMD | CRISPR/Cas9 | Dystrophin DNA sequence | mdx | (+) HDR in the dystrophin gene enhanced up to 18-fold | I.M. | [184] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andreana, I.; Repellin, M.; Carton, F.; Kryza, D.; Briançon, S.; Chazaud, B.; Mounier, R.; Arpicco, S.; Malatesta, M.; Stella, B.; et al. Nanomedicine for Gene Delivery and Drug Repurposing in the Treatment of Muscular Dystrophies. Pharmaceutics 2021, 13, 278. https://doi.org/10.3390/pharmaceutics13020278
Andreana I, Repellin M, Carton F, Kryza D, Briançon S, Chazaud B, Mounier R, Arpicco S, Malatesta M, Stella B, et al. Nanomedicine for Gene Delivery and Drug Repurposing in the Treatment of Muscular Dystrophies. Pharmaceutics. 2021; 13(2):278. https://doi.org/10.3390/pharmaceutics13020278
Chicago/Turabian StyleAndreana, Ilaria, Mathieu Repellin, Flavia Carton, David Kryza, Stéphanie Briançon, Bénédicte Chazaud, Rémi Mounier, Silvia Arpicco, Manuela Malatesta, Barbara Stella, and et al. 2021. "Nanomedicine for Gene Delivery and Drug Repurposing in the Treatment of Muscular Dystrophies" Pharmaceutics 13, no. 2: 278. https://doi.org/10.3390/pharmaceutics13020278