
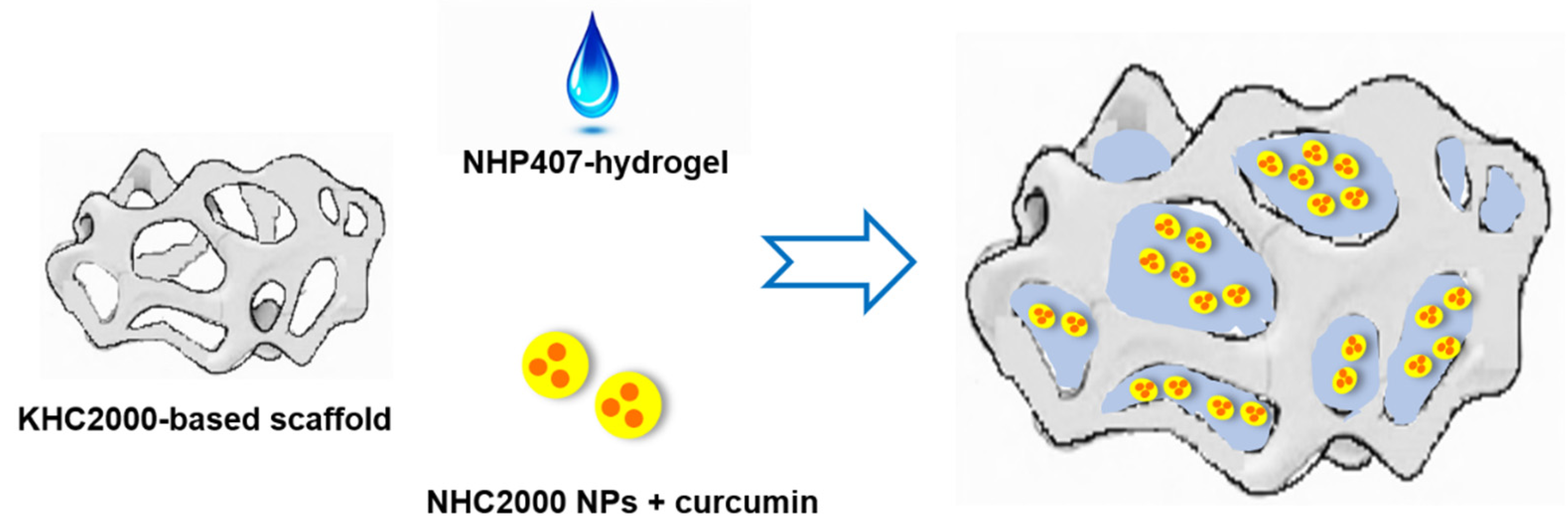
Designing Multifunctional Devices for Regenerative Pharmacology Based on 3D Scaffolds, Drug-Loaded Nanoparticles, and Thermosensitive Hydrogels: A Proof-of-Concept Study


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Poly(urethane) Synthesis and Characterization
2.2.1. Synthesis Protocol
2.2.2. Poly(urethane) Nomenclature
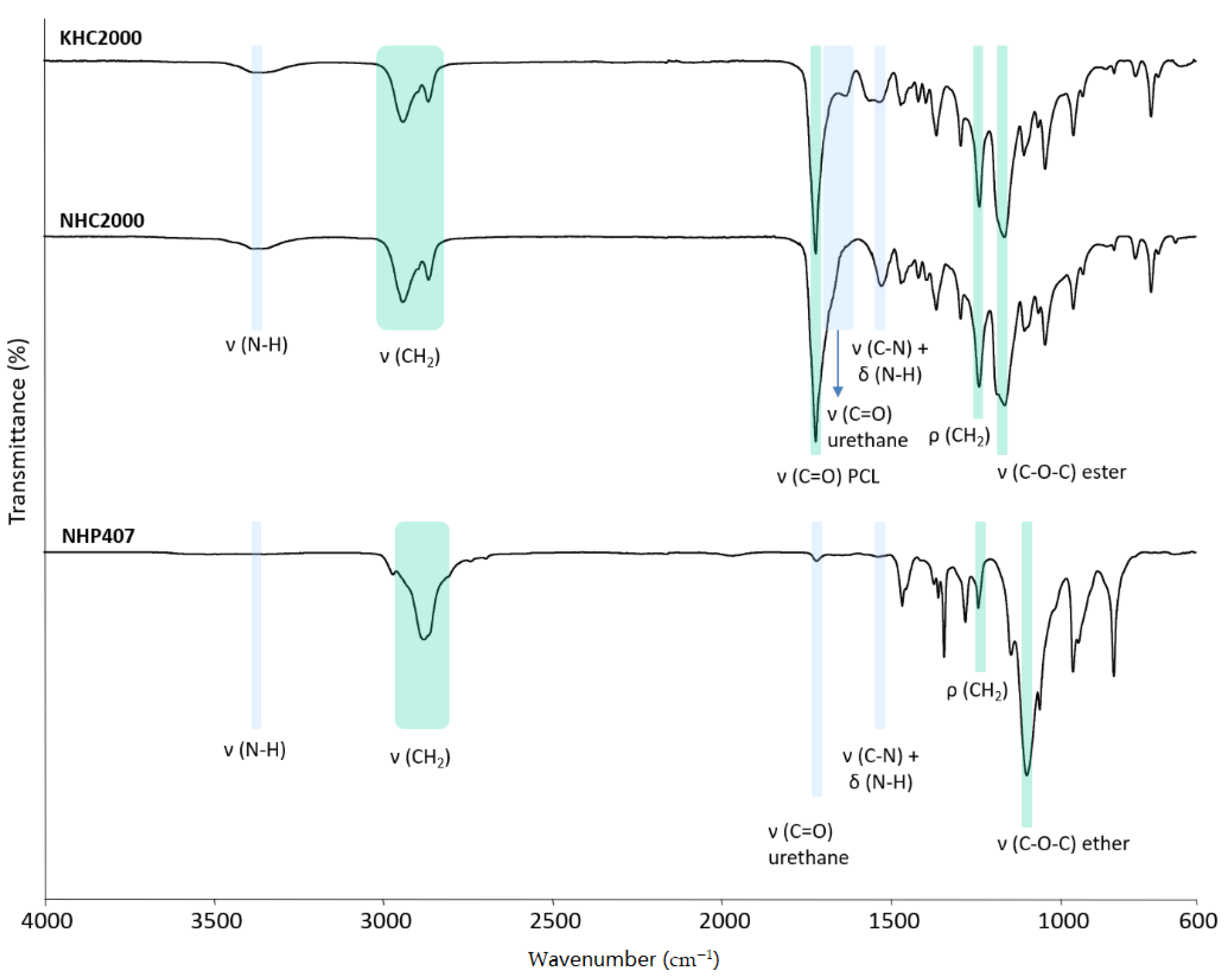
2.2.3. Chemical Characterization
2.3. Scaffold Fabrication and Characterization
2.3.1. Scaffold Fabrication through Thermally Induced Phase Separation (TIPS)
2.3.2. Scaffold Characterization
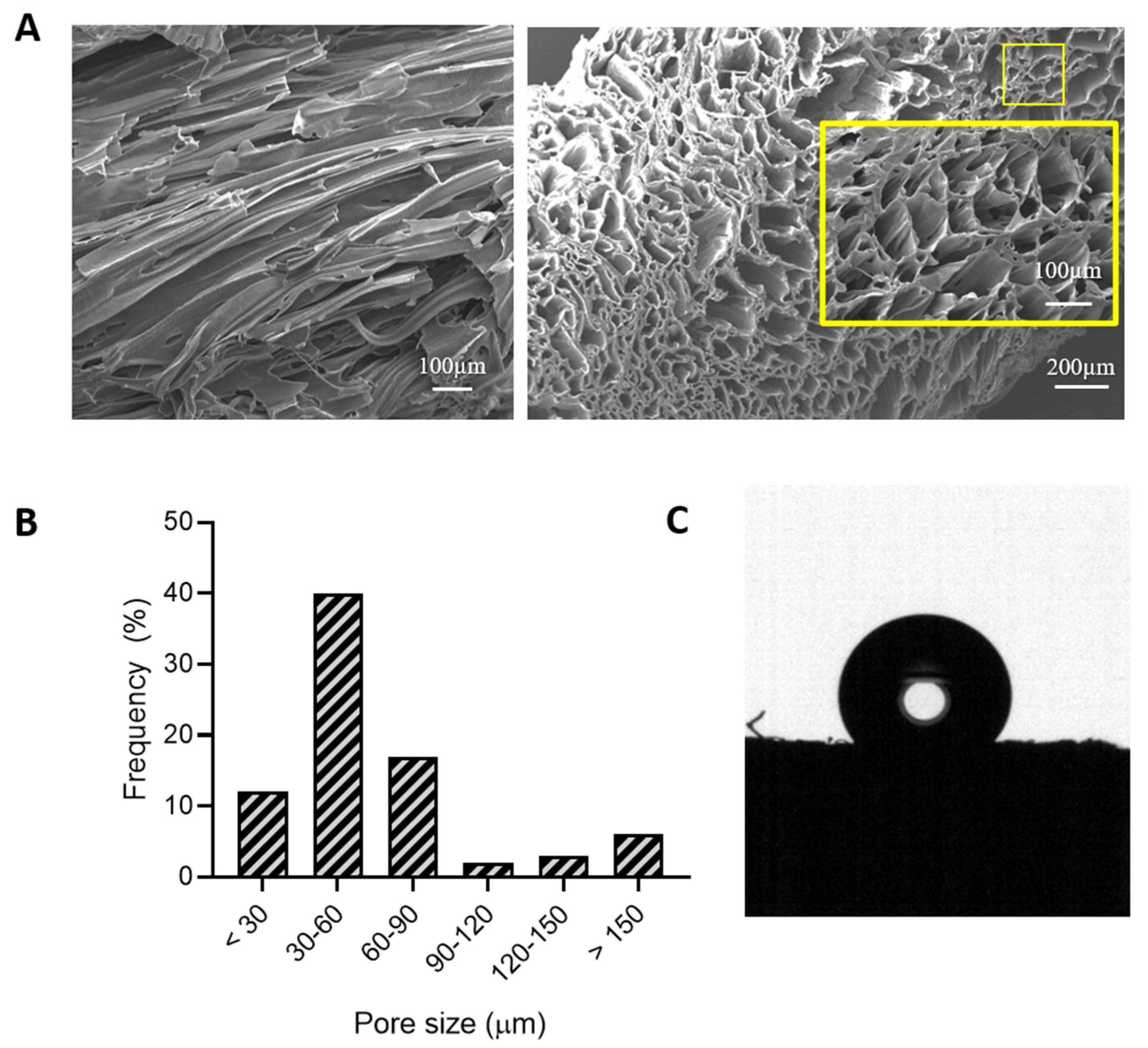
Morphology and Porosity Measurements
Contact Angle Measurements
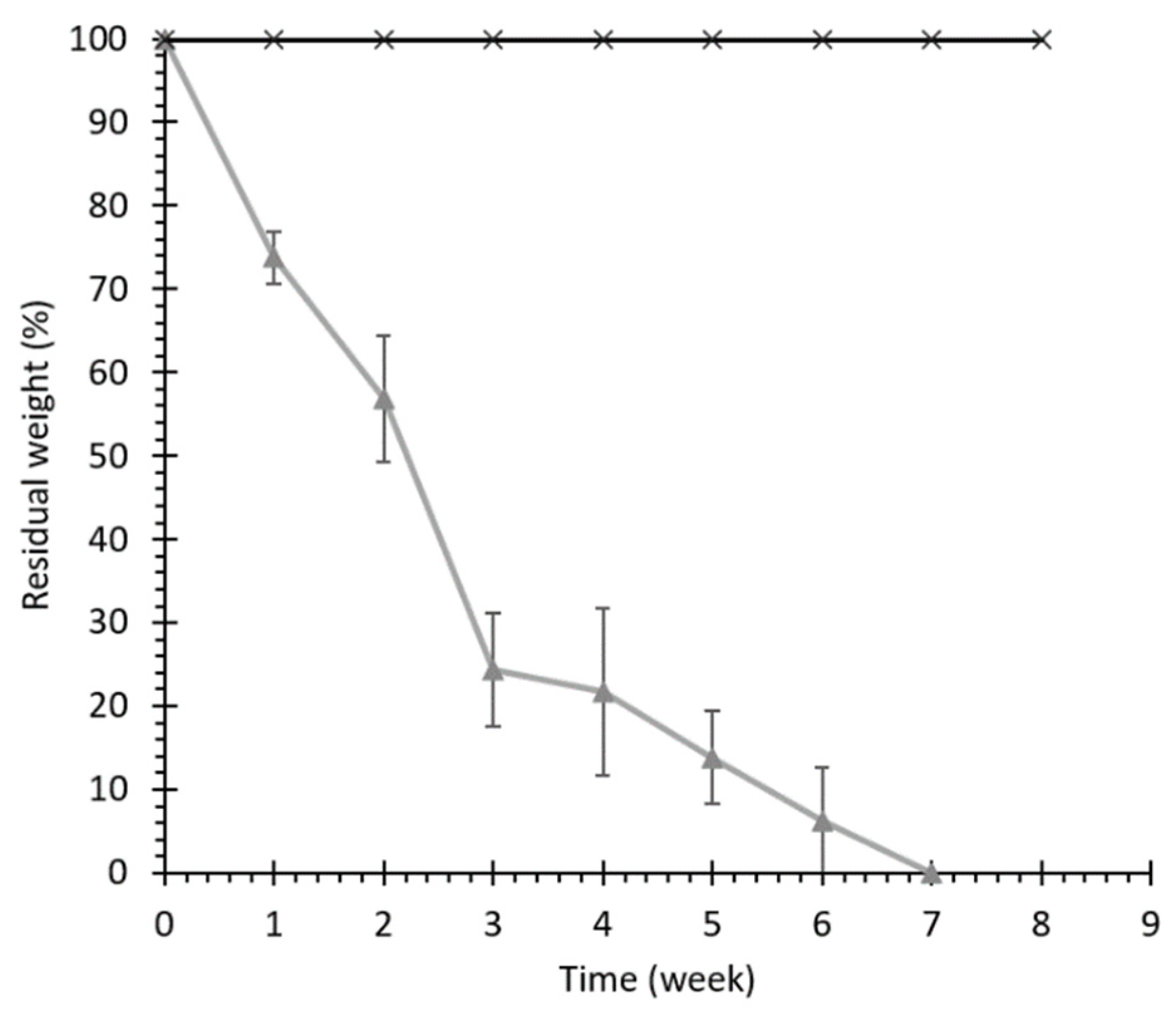
Hydrolytic and Enzymatic Degradation Tests
Mechanical Properties
2.4. Hydrogel Preparation and Characterization
2.5. Hydrogel-Loaded Scaffolds
2.5.1. Assembly Procedure
2.5.2. Nutrient Permeability Test
2.6. Nanoparticle Preparation and Characterization
2.6.1. Preparation of nanoparticles (NPs)
2.6.2. NP Characterization
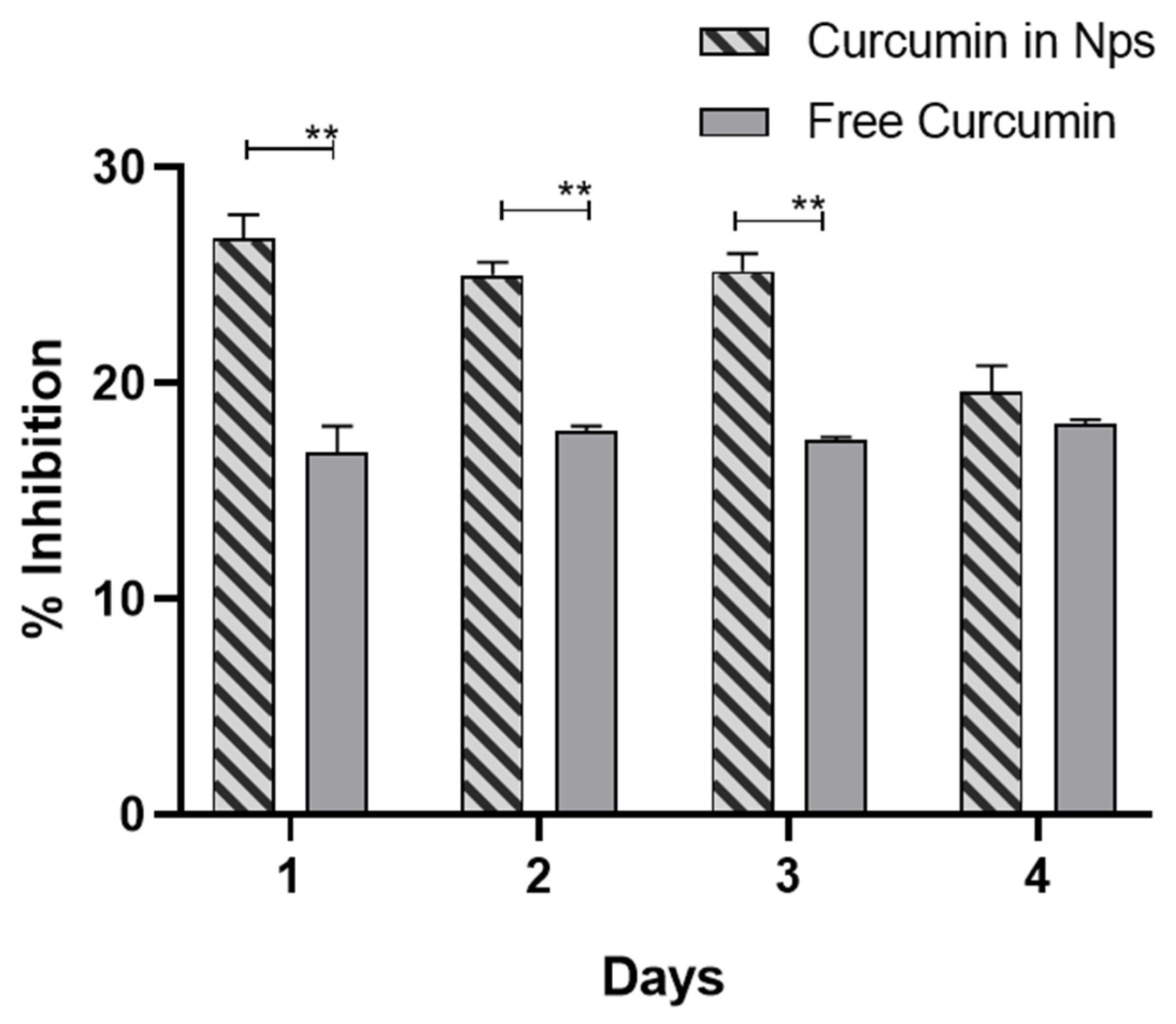
2.6.3. Antioxidant Activity of Released Curcumin
2.7. Preparation and Characterization of NP-Loaded Hydrogels
2.8. Statistical Analysis
3. Results and Discussion
3.1. Poly(urethane) (PU) Synthesis
3.2. Scaffold Characterization
3.2.1. Morphology, Porosity, and Surface Characterization
3.2.2. Hydrolytic and Enzymatic Degradation
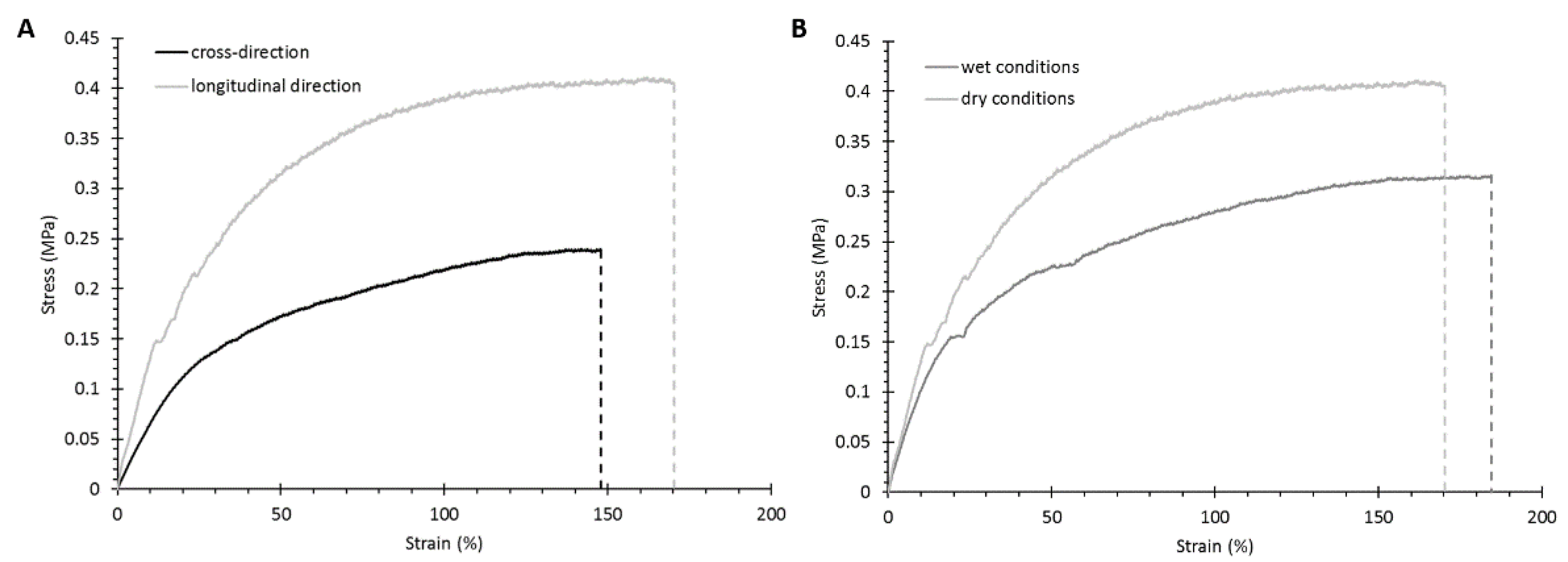
3.2.3. Mechanical Properties
3.3. Hydrogel Characterization
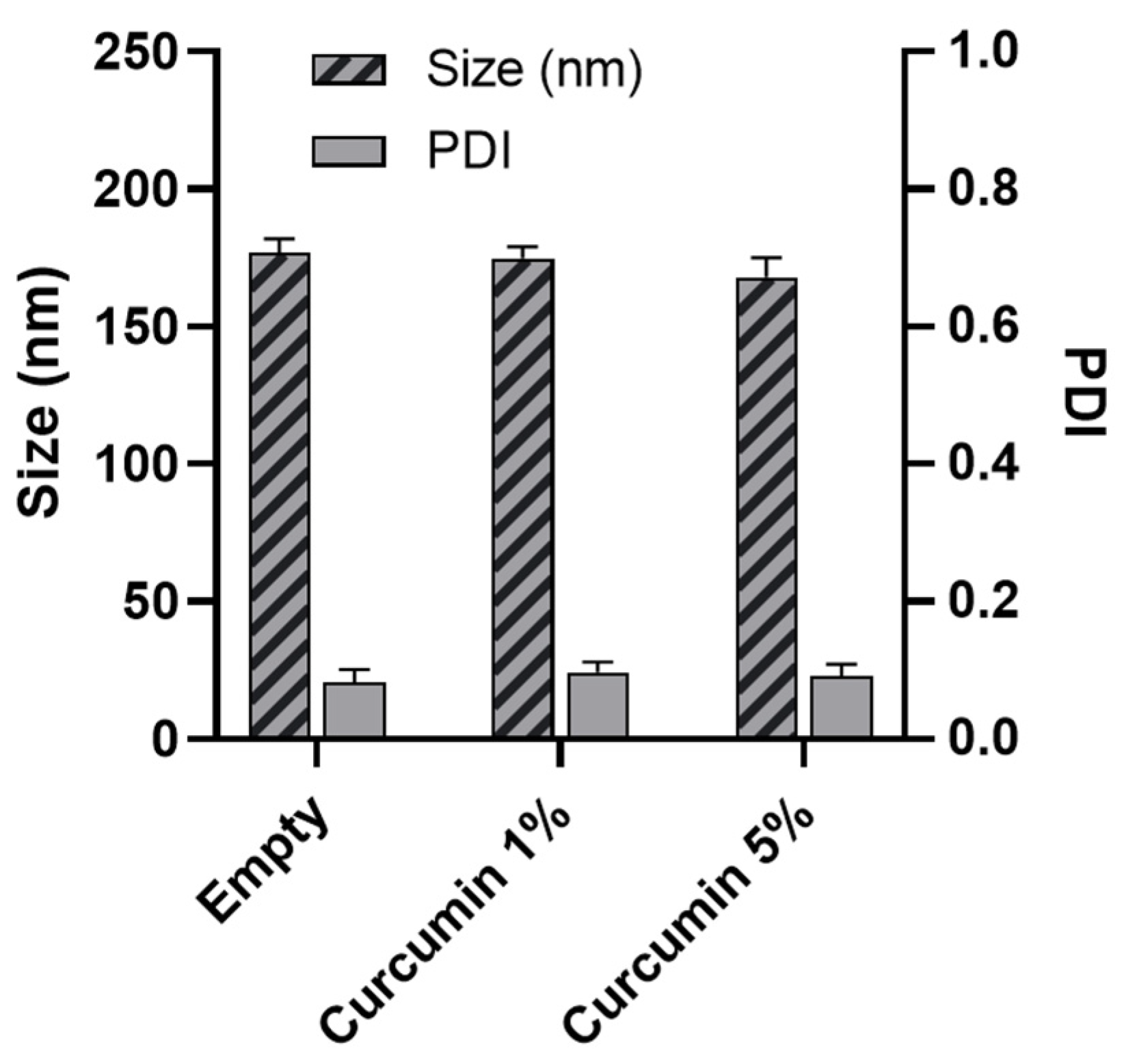
3.4. Nanoparticle Characterization
3.5. Characterization of Hydrogel Embedded with Nanoparticles
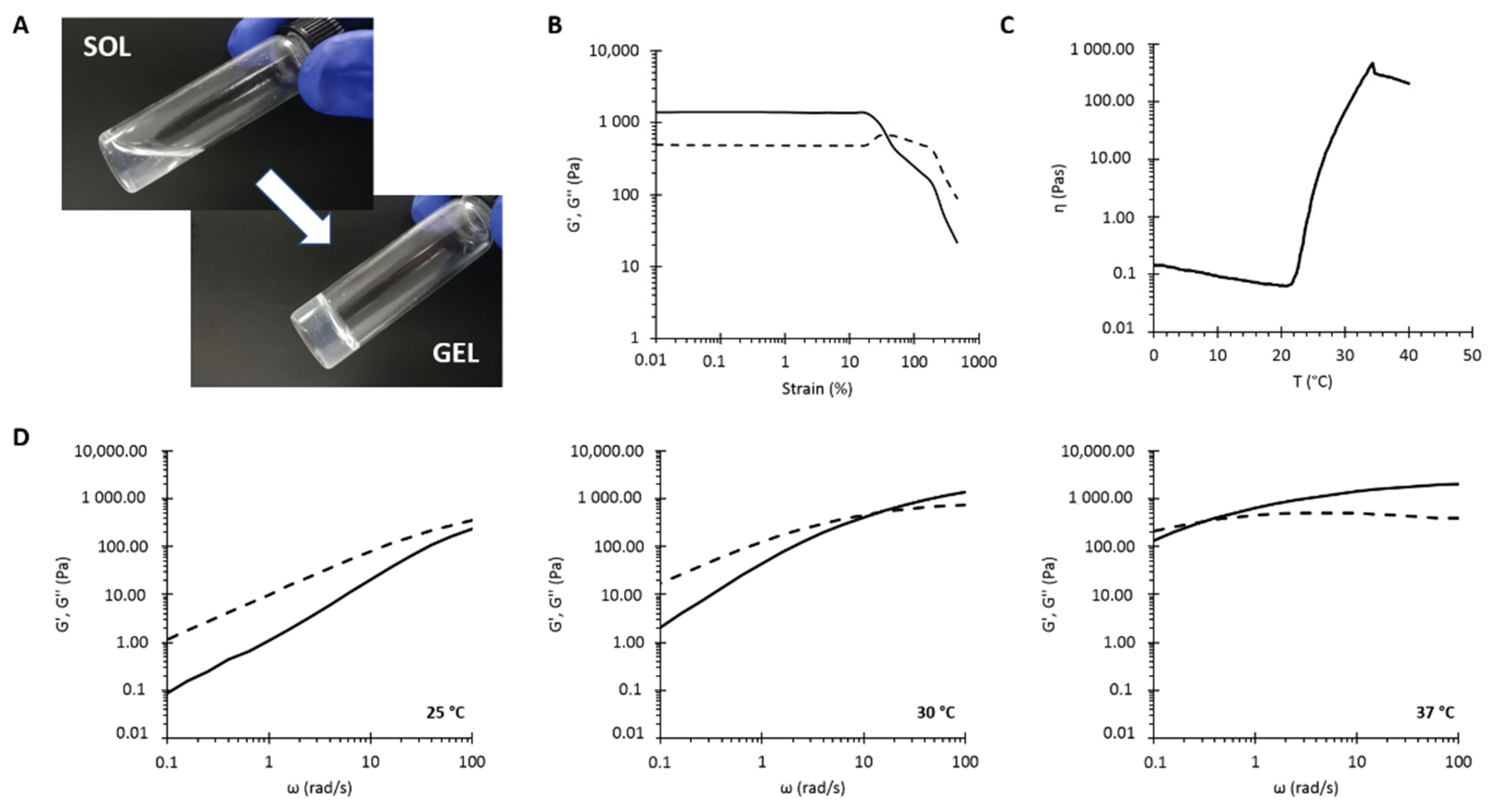
3.5.1. Study of the Sol-to-Gel Transition
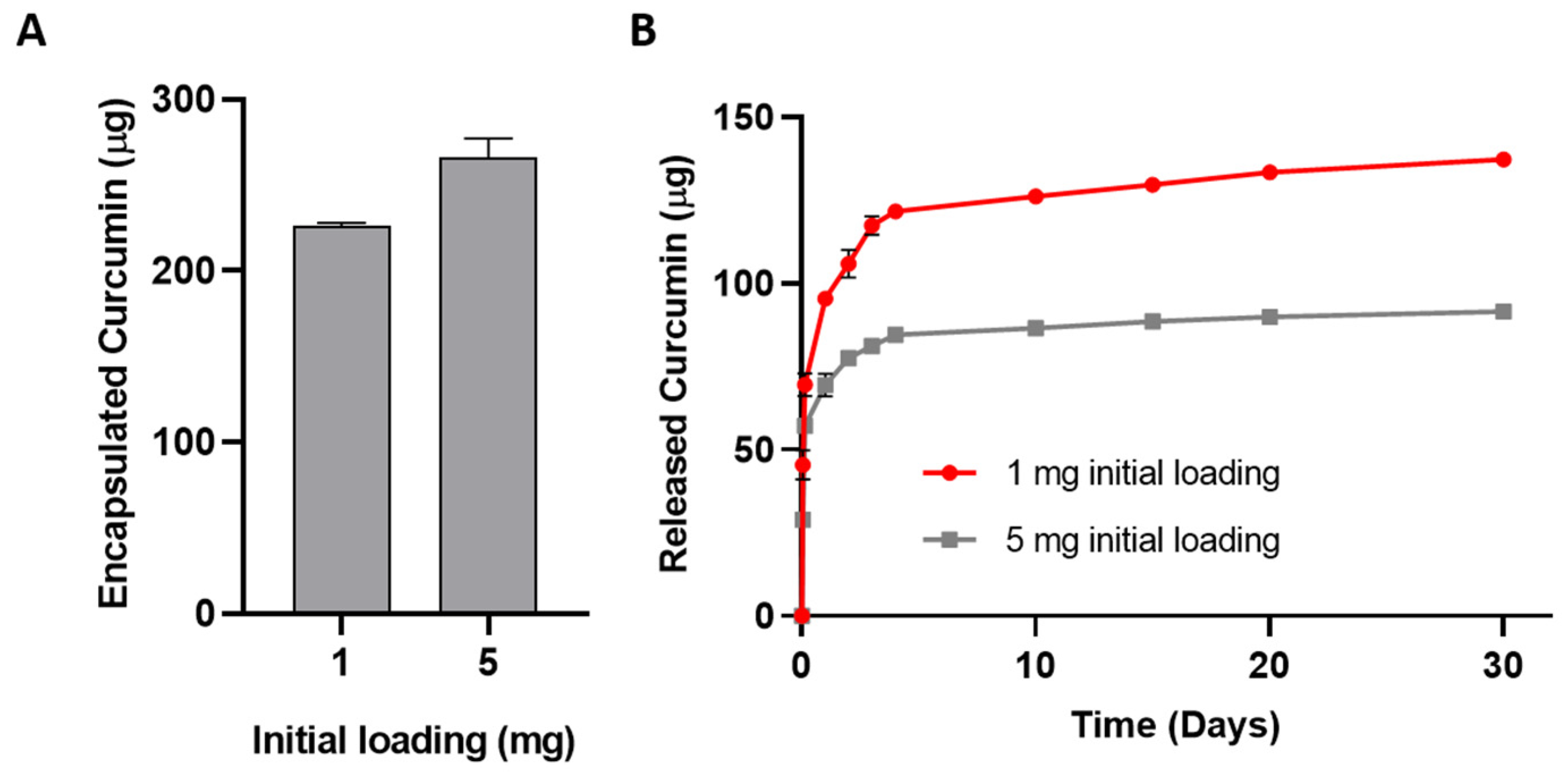
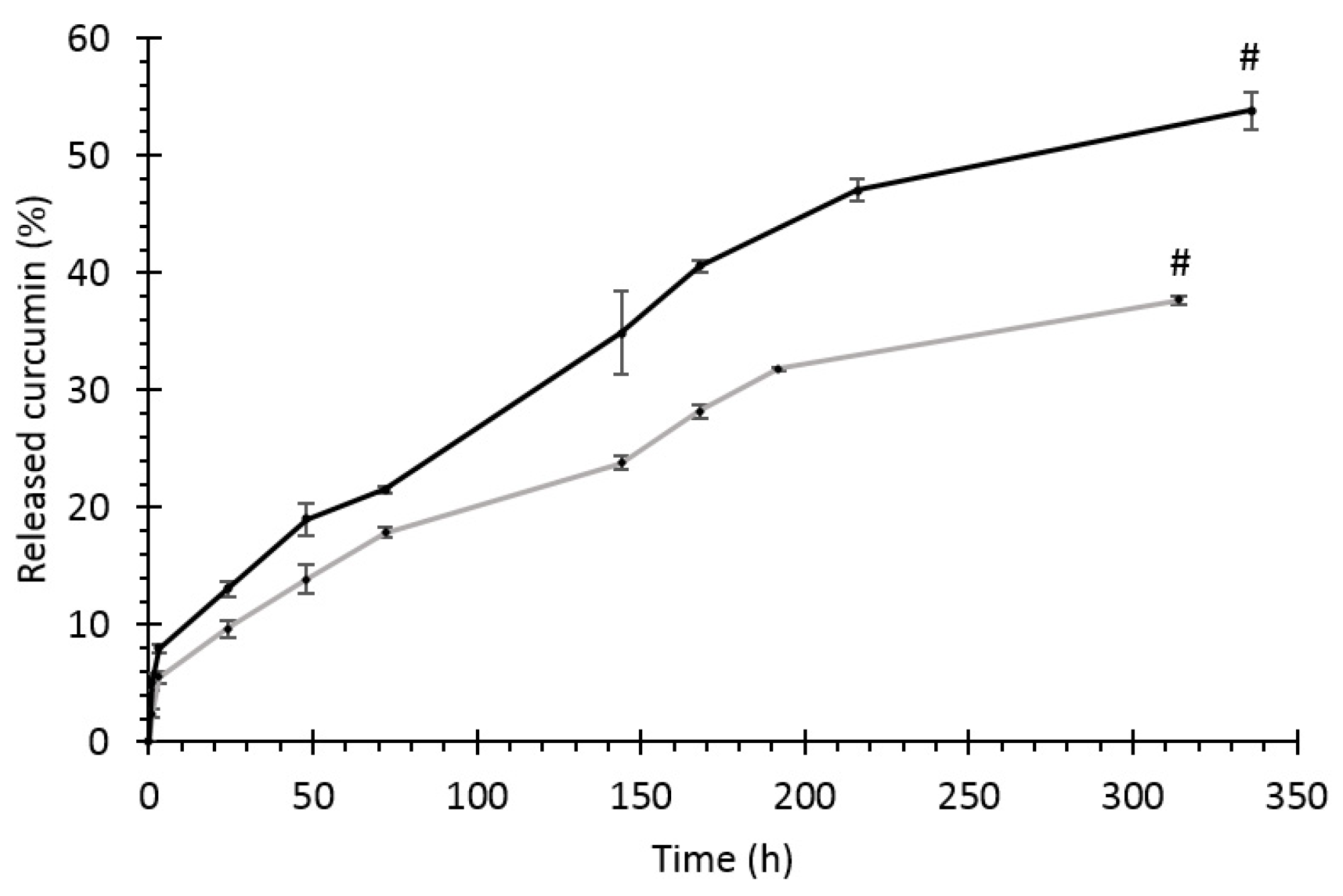
3.5.2. Drug Release Profile
3.6. Assembly of Multifunctional NHP407_NPs-Loaded Scaffolds
3.6.1. Hydrogel Absorption by KHC2000 Scaffolds
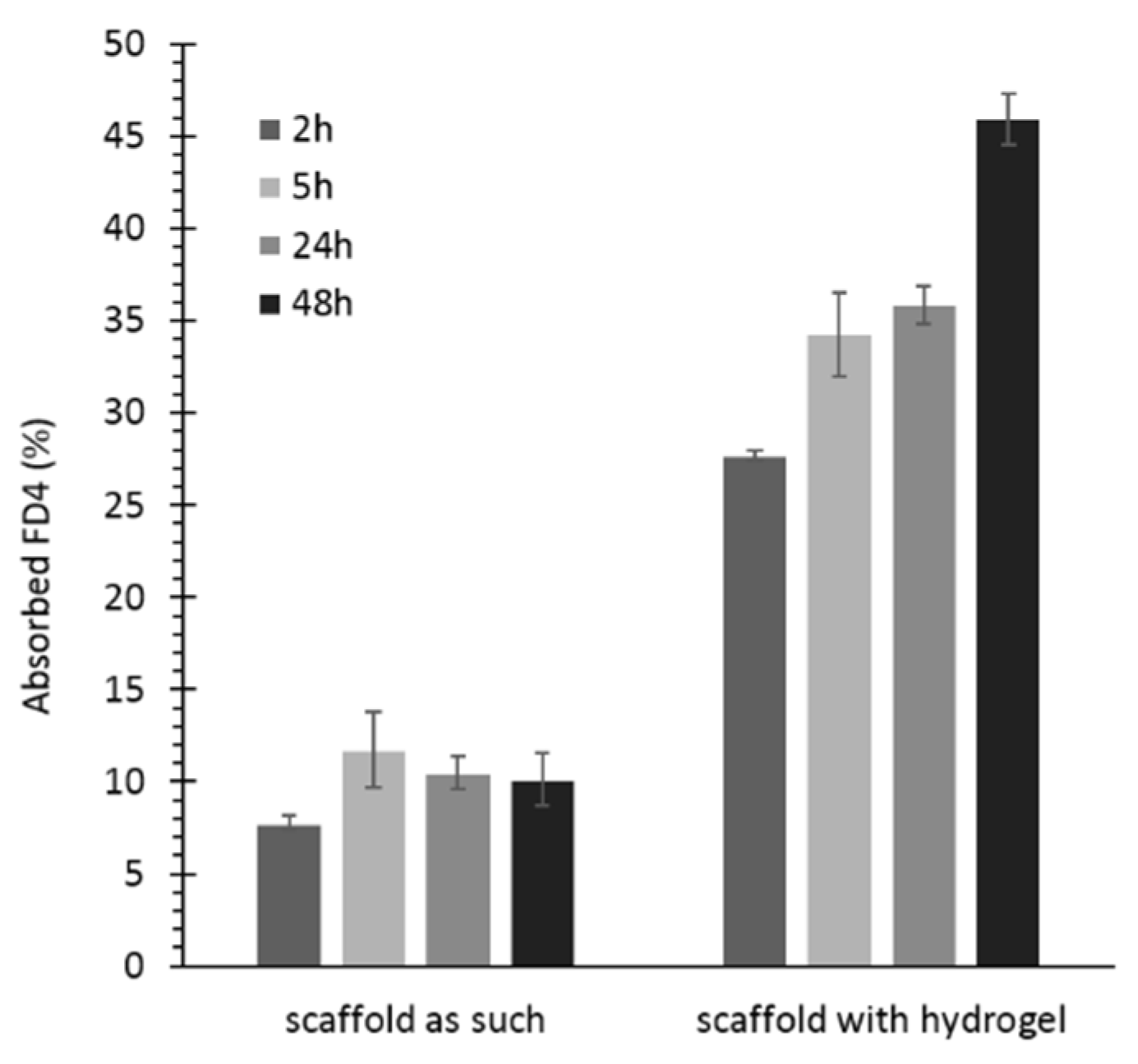
3.6.2. Permeability Test
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Salgado, A.J.; Oliveira, J.M.; Martins, A.; Teixeira, F.G.; Silva, N.A.; Neves, N.M.; Sousa, N.; Reis, R.L. Tissue engineering and regenerative medicine. Int. Rev. Neurobiol. 2013, 108, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Frey, B.M.; Zeisberger, S.M.; Hoerstrup, S.P. Tissue engineering and regenerative medicine-New initiatives for individual treatment offers. Transfus. Med. Hemother. 2016, 43, 318–319. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Evans, D. Tissue engineering: The future of stem cells. Top. Tissue Eng. 2005, 2, 1–21. [Google Scholar]
- Caddeo, S.; Boffito, M.; Sartori, S. Tissue engineering approaches in the design of healthy and pathological in vitro tissue models. Front. Bioeng. Biotechnol. 2017, 5, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silvestri, A.; Boffito, M.; Sartori, S.; Ciardelli, G. Biomimetic materials and scaffolds for myocardial tissue regeneration. Macromol. Biosci. 2013, 13, 984–1019. [Google Scholar] [CrossRef]
- Boffito, M.; Sartori, S.; Ciardelli, G. Polymeric scaffolds for cardiac tissue engineering: Requirements and fabrication technologies. Polym. Int. 2013, 63, 2–11. [Google Scholar] [CrossRef]
- Kai, D.; Prabhakaran, M.P.; Jin, G.; Ramakrishna, S. Guided orientation of cardiomyocytes on electrospun aligned nanofibers for cardiac tissue engineering. J. Biomed. Mater. Res. Part B Appl. Biomater. 2011, 98, 379–386. [Google Scholar] [CrossRef]
- Parrag, I.C.; Zandstra, P.W.; Woodhouse, K.A. Fiber alignment and coculture with fibroblasts improves the differentiated phenotype of murine embryonic stem cell-derived cardiomyocytes for cardiac tissue engineering. Biotechnol. Bioeng. 2011, 109, 813–822. [Google Scholar] [CrossRef]
- Andersson, K.-E.; Christ, G.J. Regenerative pharmacology: The future is now. Mol. Interv. 2007, 7, 79–86. [Google Scholar] [CrossRef]
- Christ, G.J.; Saul, J.M.; Furth, M.E.; Andersson, K.-E. The pharmacology of regenerative medicine. Pharmacol. Rev. 2013, 65, 1091–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, K.; Klibanov, A.M.; Langer, R. Protein stability in controlled-release systems. Nat. Biotechnol. 2000, 18, 24–25. [Google Scholar] [CrossRef]
- Kumari, A.; Yadav, S.K.; Yadav, S.C. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf. B. 2010, 75, 1–18. [Google Scholar] [CrossRef]
- Pelss, J.; Loca, D.; Berzina-Cimdina, L.; Locs, J.; Lakevics, V. Release of anticancer drug doxorubicin from biodegradable polymer coated porous hydroxyapatite scaffolds. Adv. Mater. Res. 2011, 284–286, 1770–1773. [Google Scholar] [CrossRef]
- Wang, H.; Deng, Z.; Chen, J.; Qi, X.; Pang, L.; Lin, B.; Adib, Y.T.Y.; Miao, N.; Wang, D.; Zhang, Y.; et al. A novel vehicle-like drug delivery 3D printing scaffold and its applications for a rat femoral bone repairing in vitro and in vivo. Int. J. Biol. Sci. 2020, 16, 1821–1832. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.; Jin, J.; Yang, S.; Li, G. Doxorubicin-loaded PLA/pearl electrospun nanofibrous scaffold for drug delivery and tumor cell treatment. Mater. Res. Express 2017, 4, 075403. [Google Scholar] [CrossRef]
- Zafar, M.; Najeeb, S.; Khurshid, Z.; Vazirzadeh, M.; Zohaib, S.; Najeeb, B.; Sefat, F. Potential of electrospun nanofibers for biomedical and dental applications. Materials 2016, 9, 73. [Google Scholar] [CrossRef]
- Qasim, S.B.; Zafar, M.S.; Najeeb, S.; Khurshid, Z.; Shah, A.H.; Husain, S.; Rehman, I.U. Electrospinning of chitosan-based solutions for tissue engineering and regenerative medicine. Int. J. Mol. Sci. 2018, 19, 407. [Google Scholar] [CrossRef] [Green Version]
- Husain, S.; Al-Samadani, K.H.; Najeeb, S.; Zafar, M.S.; Khurshid, Z.; Zohaib, K.; Qasim, S.B. Chitosan biomaterials for current and potential dental applications. Materials 2017, 10, 602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarwar, M.S.; Huang, Q.; Ghaffar, A.; Abid, M.A.; Zafar, M.S.; Khurshid, Z.; Latif, M. A smart drug delivery system based on biodegradable chitosan/Poly(Allylamine hydrochloride) blend films. Pharmaceutics 2020, 12, 131. [Google Scholar] [CrossRef] [Green Version]
- Çelebier, S.K.; Bozdağ, K.; Pehlivan, S.B.; Demirbilek, M.; Akıncı, M.; Vural, I.; Akdağ, Y.; Yürüker, S.; Ünlü, N. Development of an anti-inflammatory drug-incorporated biomimetic scaffold for corneal tissue engineering. J. Ocul. Pharmacol. Ther. 2020, 36. [Google Scholar] [CrossRef]
- Hsin-Yi, L.; Tsang-Wen, C. Chitosan-coated alginate tissue scaffold constructed by three-dimensional plotting technique used for drug and cell delivery. Front. Bioeng. Biotechnol. 2016, 4. [Google Scholar] [CrossRef]
- Brachi, G.; Ruiz-Ramírez, J.; Dogra, P.; Wang, Z.; Cristini, V.; Ciardelli, G.; Rostomily, R.C.; Ferrari, M.; Mikheev, A.M.; Blanco, E.; et al. Intratumoral injection of hydrogel-embedded nanoparticles enhances retention in glioblastoma. Nanoscale 2020, 12, 23838–23850. [Google Scholar] [CrossRef]
- Dewhurst, R.M.; Scalzone, A.; Buckley, J.; Mattu, C.; Rankin, K.S.; Gentile, P.; Ferreira, A.M. Development of natural-based bone cement for a controlled doxorubicin-drug release. Front. Bioeng. Biotechnol. 2020, 8, 754. [Google Scholar] [CrossRef]
- Boffito, M.; Laurano, R.; Giasafaki, D.; Steriotis, T.; Papadopoulos, A.; Tonda-Turo, C.; Cassino, C.; Charalambopoulou, G.; Ciardelli, G. Embedding ordered mesoporous carbons into thermosensitive hydrogels: A cutting-edge strategy to vehiculate a cargo and control its release profile. Nanomaterials 2020, 10, 2165. [Google Scholar] [CrossRef]
- Pontremoli, C.; Boffito, M.; Fiorilli, S.; Laurano, R.; Torchio, A.; Bari, A.; Tonda-Turo, C.; Ciardelli, G.; Vitale-Brovarone, C. Hybrid injectable platforms for the in situ delivery of therapeutic ions from mesoporous glasses. Chem. Eng. J. 2018, 340, 103–113. [Google Scholar] [CrossRef]
- Shokry, H.; Mattinen, U.; Wiltschka, O.; Niinimäki, J.; Lerche, M.; Levon, K.; Lindén, M.; Sahlgren, C. Mesoporous silica particle-PLA–PANI hybrid scaffolds for cell-directed intracellular drug delivery and tissue vascularization. Nanoscale 2015, 7, 14434–14443. [Google Scholar] [CrossRef]
- He, D.; Zhao, A.; Su, H.; Zhang, Y.; Wang, Y.; Luo, D.; Gao, Y.; Li, J.; Yang, P. An injectable scaffold based on temperature-responsive hydrogel and factor-loaded nanoparticles for application in vascularization in tissue engineering. J. Biomed. Mater. Res. Part A 2019, 107, 2123–2134. [Google Scholar] [CrossRef]
- Asghar, W.; Islam, M.; Wadajkar, A.S.; Wan, Y.; Ilyas, A.; Nguyen, K.T.; Iqbal, S.M. PLGA micro-and nanoparticles loaded into gelatin scaffold for controlled drug release. IEEE Trans. Nanotechnol. 2012, 11, 546–553. [Google Scholar] [CrossRef]
- Nooeaid, P.; Chuysinuan, P.; Pengsuk, C.; Dechtrirat, D.; Lirdprapamongkol, K.; Techasakul, S.; Svasti, J. Polylactic acid microparticles embedded porous gelatin scaffolds with multifunctional properties for soft tissue engineering. J. Sci. Adv. Mater. Devices 2020, 5, 337–345. [Google Scholar] [CrossRef]
- Gentile, P.; Nandagiri, V.K.; Daly, J.; Chiono, V.; Mattu, C.; Tonda-Turo, C.; Ciardelli, G.; Ramtoola, Z. Localised controlled release of simvastatin from porous chitosan–gelatin scaffolds engrafted with simvastatin loaded PLGA-microparticles for bone tissue engineering application. Mater. Sci. Eng. C 2016, 59, 249–257. [Google Scholar] [CrossRef]
- Gentile, P.; Bellucci, D.; Sola, A.; Mattu, C.; Cannillo, V.; Ciardelli, G. Composite scaffolds for controlled drug release: Role of the polyurethane nanoparticles on the physical properties and cell behaviour. J. Mech. Behav. Biomed. Mater. 2015, 44, 53–60. [Google Scholar] [CrossRef]
- Ferreira, A.M.; Mattu, C.; Ranzato, E.; Ciardelli, G. Bioinspired porous membranes containing polymer nanoparticles for wound healing. J. Biomed. Mater. Res. Part A 2014, 102. [Google Scholar] [CrossRef]
- Smith, I.O.; Liu, X.H.; Smith, L.A.; Ma, P.X. Nanostructured polymer scaffolds for tissue engineering and regenerative medicine. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2009, 1, 226–236. [Google Scholar] [CrossRef] [Green Version]
- Sartori, S.; Boffito, M.; Serafini, P.; Caporale, A.; Silvestri, A.; Bernardi, E.; Sassi, M.P.; Boccafoschi, F.; Ciardelli, G. Synthesis and structure–property relationship of polyester-urethanes and their evaluation for the regeneration of contractile tissues. React. Funct. Polym. 2013, 73, 1366–1376. [Google Scholar] [CrossRef]
- Gioffredi, E.; Boffito, M.; Calzone, S.; Giannitelli, S.M.; Rainer, A.; Trombetta, M.; Mozetic, P.; Chiono, V. Pluronic F127 hydrogel characterization and biofabrication in cellularized constructs for tissue engineering applications. Procedia CIRP 2016, 49, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Mattu, C.; Pabari, R.; Boffito, M.; Sartori, S.; Ciardelli, G.; Ramtoola, Z. Comparative evaluation of novel biodegradable nanoparticles for the drug targeting to breast cancer cells. Eur. J. Pharm. Biopharm. 2013, 85, 463–472. [Google Scholar] [CrossRef]
- Riboldi, S.A.; Sampaolesi, M.; Neuenschwander, P.; Cossu, G.; Mantero, S. Electrospun degradable polyesterurethane membranes: Potential scaffolds for skeletal muscle tissue engineering. Biomaterials 2005, 26, 4606–4615. [Google Scholar] [CrossRef] [Green Version]
- Levy-Mishali, M.; Zoldan, J.; Levenberg, S. Effect of scaffold stiffness on myoblast differentiation. Tissue Eng. Part A 2009, 15, 935–944. [Google Scholar] [CrossRef]
- Boffito, M.; Gioffredi, E.; Chiono, V.; Calzone, S.; Ranzato, E.; Martinotti, S.; Ciardelli, G. Novel polyurethane-based thermosensitive hydrogels as drug release and tissue engineering platforms: Design and in vitro characterization. Polym. Int. 2016, 65, 756–769. [Google Scholar] [CrossRef]
- Boffito, M.; Bernardi, E.; Sartori, S.; Ciardelli, G.; Sassi, M.P. A mechanical characterization of polymer scaffolds and films at the macroscale and nanoscale. J. Biomed. Mater. Res. Part A 2014, 103, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Fujimoto, K.L.; Sacks, M.S.; Wagner, W.R. Preparation and characterization of highly porous, biodegradable polyurethane scaffolds for soft tissue applications. Biomaterials 2005, 26, 3961–3971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, M.; Malinen, M.M.; Lauren, P.; Lou, Y.-R.; Kuisma, S.W.; Kanninen, L.; Lille, M.; Corlu, A.; GuGuen-Guillouzo, C.; Ikkala, O.; et al. Nanofibrillar cellulose hydrogel promotes three-dimensional liver cell culture. J. Control. Release 2012, 164, 291–298. [Google Scholar] [CrossRef]
- Brand-Williams, W.; Cuvelier, M.; Berset, C. Use of a free radical method to evaluate antioxidant activity. LWT Food Sci. Technol. 1995, 28, 25–30. [Google Scholar] [CrossRef]
- Rahman, M.; Islam, B.; Biswas, M.; Alam, A.H.M.K. In vitro antioxidant and free radical scavenging activity of different parts of Tabebuia pallida growing in Bangladesh. BMC Res. Notes 2015, 8, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Laurano, R.; Abrami, M.; Grassi, M.; Ciardelli, G.; Boffito, M.; Chiono, V. Using poloxamer® 407 as building block of amphiphilic poly(Ether urethane)S: Effect of its molecular weight distribution on thermo-sensitive hydrogel performances in the perspective of their biomedical application. Front. Mater. 2020, 7. [Google Scholar] [CrossRef]
- Areias, A.C.; Ribeiro, C.; Sencadas, V.; Garciagiralt, N.; Diezperez, A.; Ribelles, J.L.G.; Lanceros-Méndez, S. Influence of crystallinity and fiber orientation on hydrophobicity and biological response of poly(l-lactide) electrospun mats. Soft Matter 2012, 8, 5818–5825. [Google Scholar] [CrossRef] [Green Version]
- Herzog, K.; Müller, R.-J.; Deckwer, W.-D. Mechanism and kinetics of the enzymatic hydrolysis of polyester nanoparticles by lipases. Polym. Degrad. Stab. 2006, 91, 2486–2498. [Google Scholar] [CrossRef]
- Boffito, M.; Di Meglio, F.; Vitale, N.; Brancaccio, M.; Tarone, G.; Basoli, F.; Rainer, A.; Trombetta, M.; Ciardelli, G.; Chiono, V.; et al. Surface functionalization of polyurethane scaffolds mimicking the myocardial microenvironment to support cardiac primitive cells. PLoS ONE 2018, 13, e0199896. [Google Scholar] [CrossRef]
- Klouda, E.L.; Vaz, C.M.; Driessen-Mol, A.A.; Baaijens, F.P.T.; Bouten, C.V.C. Effect of biomimetic conditions on mechanical and structural integrity of PGA/P4HB and electrospun PCL scaffolds. J. Mater. Sci. Mater. Med. 2007, 19, 1137–1144. [Google Scholar] [CrossRef]
- Pan, Z.; Ding, J. Poly(lactide- co -glycolide) porous scaffolds for tissue engineering and regenerative medicine. Interface Focus 2012, 2, 366–377. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Gong, Y.; Gao, C. Microstructure and mechanical properties of poly(L-lactide) scaffolds fabricated by gelatin particle leaching method. J. Appl. Polym. Sci. 2005, 98, 1373–1379. [Google Scholar] [CrossRef]
- Anitha, A.; Deepagan, V.; Rani, V.D.; Menon, D.; Nair, S.; Jayakumar, R. Preparation, characterization, in vitro drug release and biological studies of curcumin loaded dextran sulphate–chitosan nanoparticles. Carbohydr. Polym. 2011, 84, 1158–1164. [Google Scholar] [CrossRef]
- Esfandiarpour-Boroujeni, S.; Bagheri-Khoulenjani, S.; Mirzadeh, H.; Amanpour, S. Fabrication and study of curcumin loaded nanoparticles based on folate-chitosan for breast cancer therapy application. Carbohydr. Polym. 2017, 168, 14–21. [Google Scholar] [CrossRef]
- Iodice, C.; Cervadoro, A.; Palange, A.; Key, J.; Aryal, S.; Ramirez, M.R.; Mattu, C.; Ciardelli, G.; O’Neill, B.E.; Decuzzi, P. Enhancing photothermal cancer therapy by clustering gold nanoparticles into spherical polymeric nanoconstructs. Opt. Lasers Eng. 2016, 76, 74–81. [Google Scholar] [CrossRef]
- Mattu, C.; Boffito, M.; Sartori, S.; Ranzato, E.; Bernardi, E.; Sassi, M.P.; Di Rienzo, A.M.; Ciardelli, G. Therapeutic nanoparticles from novel multiblock engineered polyesterurethanes. J. Nanopart. Res. 2012, 14, 1306. [Google Scholar] [CrossRef]
- Rejinold, N.S.; Muthunarayanan, M.; Divyarani, V.; Sreerekha, P.; Chennazhi, K.; Nair, S.; Tamura, H.; Jayakumar, R. Curcumin-loaded biocompatible thermoresponsive polymeric nanoparticles for cancer drug delivery. J. Colloid Interface Sci. 2011, 360, 39–51. [Google Scholar] [CrossRef]
- Kulisic, T.; Radonic, A.; Katalinic, V.; Milos, M. Use of different methods for testing antioxidative activity of oregano essential oil. Food Chem. 2004, 85, 633–640. [Google Scholar] [CrossRef]
- Aoki, T.; Kawashima, M.; Katono, H.; Sanui, K.; Ogata, N.; Okano, T.; Sakurai, Y. Temperature-responsive interpenetrating polymer networks constructed with poly(Acrylic acid) And poly(N,n-dimethylacrylamide). Macromolecules 1994, 27, 947–952. [Google Scholar] [CrossRef]
- Boffito, M.; Torchio, A.; Schmidt-Bleek, K.; Ciardelli, G.; Tonda-Turo, C.; Laurano, R.; Gisbert-Garzarán, M.; Berkmann, J.C.; Cassino, C.; Manzano, M.; et al. Hybrid injectable Sol-gel systems based on thermo-sensitive polyurethane hydrogels carrying PH-sensitive mesoporous silica nanoparticles for the controlled and triggered release of therapeutic agents. Front. Bioeng. Biotechnol. 2020, 8. [Google Scholar] [CrossRef]
- Esmaili, Z.; Bayrami, S.; Dorkoosh, F.A.; Javar, H.A.; Seyedjafari, E.; Zargarian, S.S.; Haddadi-Asl, V. Development and characterization of electrosprayed nanoparticles for encapsulation of Curcumin. J. Biomed. Mater. Res. Part A 2017, 106, 285–292. [Google Scholar] [CrossRef]
- Sivasami, P.; Hemalatha, T. Augmentation of therapeutic potential of curcumin using nanotechnology: Current perspective. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1004–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naksuriya, O.; Van Steenbergen, M.J.; Torano, J.S.; Okonogi, S.; Hennink, W.E. A kinetic degradation study of curcumin in its free form and loaded in polymeric micelles. AAPS J. 2016, 18, 777–787. [Google Scholar] [CrossRef] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composition | Nomenclature | ||
|---|---|---|---|
| Chain Extender | Diisocyanate | Macrodiol | |
| l-lysine ethyl ester (K) | 1,6-hexamethylene diisocyanate (H) | Poly(ε-caprolactone) diol (C2000) | KHC2000 |
| N-Boc serinol (N) | 1,6-hexamethylene diisocyanate (H) | Poly(ε-caprolactone) diol (C2000) | NHC2000 |
| N-Boc serinol (N) | 1,6-hexamethylene diisocyanate (H) | Poloxamer® 407 (P407) | NHP407 |
| Sample | Water Contact Angle | ||
|---|---|---|---|
| KHC2000 | 6 × 104 | 1.6 | 88 ± 0.4 |
| NHC2000 | 4 × 104 | 1.4 | 75 ± 1 |
| NHP407 | 5 × 104 | 1.3 | - |
| Testing Conditions | Young’s Modulus (MPa) | Stress at Break (MPa) | Strain at Break (%) | |||
|---|---|---|---|---|---|---|
| Longitudinal | Cross Section | Longitudinal | Cross Section | Longitudinal | Cross Section | |
| DRY | 2.7 ± 0.7 | 0.8 ± 0.2 | 0.6 ± 0.2 | 0.3 ± 0.04 | 170 ± 19 | 148 ± 14 |
| WET | 1.0 ± 0.3 | - | 0.4 ± 0.05 | - | 186 ± 38 | - |
| Parameters | NHP407 | NHP407_NPs | |
|---|---|---|---|
| Tube-inverting test | LCGT (°C) | 32 °C | 29 °C |
| Gelation time at 37 °C (min) | 7 min | 7 min | |
| Strain sweep test | γL (%) | 18.6% | 29.7% |
| γgel-sol (%) | 35% | 70% | |
| Tonset (°C) | 20.7 °C | 19.4 °C | |
| Temperature ramp test | Initial viscosity (Pa·s) | 0.15 Pa·s | 0.26 Pa·s |
| Minimum viscosity (Pa·s) | 0.06 Pa·s | 0.11 Pa·s | |
| Viscosity @ 25 °C (Pa·s) | 22.58 Pa·s | 5.64 Pa·s | |
| Frequency sweep test | ωCROSS at 25 °C (rad/s) | >100 rad/s | >100 rad/s |
| ωCROSS at 30 °C (rad/s) | 10 rad/s | 12.9 rad/s | |
| ωCROSS at 37 °C (rad/s) | 0.33 rad/s | 0.29 rad/s | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colucci, F.; Mancini, V.; Mattu, C.; Boffito, M. Designing Multifunctional Devices for Regenerative Pharmacology Based on 3D Scaffolds, Drug-Loaded Nanoparticles, and Thermosensitive Hydrogels: A Proof-of-Concept Study. Pharmaceutics 2021, 13, 464. https://doi.org/10.3390/pharmaceutics13040464
Colucci F, Mancini V, Mattu C, Boffito M. Designing Multifunctional Devices for Regenerative Pharmacology Based on 3D Scaffolds, Drug-Loaded Nanoparticles, and Thermosensitive Hydrogels: A Proof-of-Concept Study. Pharmaceutics. 2021; 13(4):464. https://doi.org/10.3390/pharmaceutics13040464
Chicago/Turabian StyleColucci, Francesco, Vanessa Mancini, Clara Mattu, and Monica Boffito. 2021. "Designing Multifunctional Devices for Regenerative Pharmacology Based on 3D Scaffolds, Drug-Loaded Nanoparticles, and Thermosensitive Hydrogels: A Proof-of-Concept Study" Pharmaceutics 13, no. 4: 464. https://doi.org/10.3390/pharmaceutics13040464