1. Introduction
Donepezil hydrochloride (DH) (melting point: 220–222 °C) is a reversible inhibitor of acetylcholinesterase. It produces its therapeutic effects by increasing the concentration of acetylcholine and enhancing the cholinergic function in the brain. It is used for the treatment of mild-to-moderate dementia associated with Alzheimer’s disease [
1]. The aqueous solubility of DH is ≥20 mg/mL. Therefore, it is considered to be a highly water-soluble active pharmaceutical ingredient (API). Ethyl cellulose N7 (EC-N7) is a non-swellable cellulose ethyl ether and an insoluble polymeric carrier. Cationic Eudragit
® RS PO (E-RS) is another example of a hydrophobic/insoluble carrier [
2]. The lipid matrix of Compritol
® 888 ATO (glyceryl behenate United States Pharmacopoeia-National Formulary [USP-NF]) (C-888) is a pH-independent carrier. It has a melting point of around 74 °C and a hydrophile–lipophile balance (HLB) of 2. All these hydrophobic carriers including EC-N7, E-RS, and C-888 are used to extend the release of water-soluble APIs [
2,
3,
4].
Hot-melt extrusion (HME) is a common technology that can be applied to several pharmaceutical preparations, especially to solid dispersions. There are some parameters of HME that should be considered, such as the feeding rate, shearing force, temperature, die geometry, barrel design, and screw rotating speed. With the optimization of these parameters, the formulators can contribute to the production of a desired final product with a preferable drug release profile and a uniformity of size, shape, and drug content. HME offers some advantages over other conventional methods. For example, it is a one-step, solvent-free process that has a continuous operation and a scalable process that has fewer processing steps [
5]. In addition, it requires no compression, and it can improve bioavailability due to the dispersion of the drug at the molecular level in such a dosage form [
5,
6,
7]. The growing demand for versatile continuous processes in the pharmaceutical industry has made HME a preferable technique in their production lines. The reductions of waste, energy, and costs are the advantages of continuous process compared to a single batch process such as heat fusion (melt and mix methods) [
8,
9,
10]. Some conventional methods like sintering techniques have been investigated [
11]. Therefore, these techniques could mimic the residence time when the screw speed of the extruder is decreased. The main limitation of HME in the pharmaceutical industry is that a higher energy input is required to produce products compared to other techniques and may exclude some thermolabile compounds due to high processing temperatures. However, in the case of lipid matrices such as C-888, there is no need for high-energy input since most of them have relatively lower melting points compared to other hydrophobic carriers such as EC-N7 and E-RS. It is widely accepted that HME technology is an innovative and feasible approach in the preparation of various pharmaceutical systems such as mini tablets, granules, immediate and modified release tablets, oral fast-dissolving systems, and transdermal drug delivery systems [
6,
7]. HME technology can also be used for the taste masking of bitter APIs by incorporating them into hydrophobic matrices [
5,
6,
7].
Formulations factors that are based on different ratios of API/hydrophobic carriers and mixtures of two hydrophobic carriers with the API and with or without a pore-former, e.g., mannitol (MNT), can play a critical role in the final product characteristics. MNT can be used to produce different extended release formulations, as well as to produce different physicochemical characteristics for highly soluble drugs like DH. Controlling either formulation parameters (e.g., the ratio of DH/carrier and formulation composition) or processing parameters (e.g., processing temperature and screw speed) could play a significant role on the behavior of the drug release profile, as well as physicochemical properties of the drug within the matrix. The impact of fillers on drug content and the effects of the processing temperatures, formulation composition, tablet disintegration, DH/carrier ratio, and compression force have not been investigated yet. In a previous study, the influence of some of these parameters on the release behavior of a different API (diclofenac sodium) was studied [
4,
12].
Lipid aging could lead to instability during storage. It is the main disadvantage when using lipids in pharmaceutical formulations, as their physical properties could change during storage. Some of these effects are an increase of melting ranges, melting enthalpy, pore formation on the surface, rheological changes, and a decrease in tensile strength [
13].
The main objectives of this study were to develop and evaluate the sustained-release DH formulations using hydrophobic carriers such as C-888, EC-N7, and E-RS while utilizing HME technology in order to avoid the side effects of DH (nightmare, insomnia, anxiety, nausea, emesis and/or diarrhea) associated with an initial sharp spike in blood DH levels for immediate release formulations. Formulations that provide the immediate release of DH are administered once a day. However, the peak plasma concentrations are reached in 2–5 h, resulting in an initial sharp spike in blood plasma levels [
14]. This sharp spike could cause undesirable cholinergic side effects, as mentioned above [
1,
14]. In addition, the specific aims of this study were to study the effect of the extrusion temperature on a drug’s physical state and drug release profile from the lipid carrier and compare it to a conventionally processed formulation. The extrusion of pre-mixed tablet excipients (i.e., microcrystalline cellulose (MCC) and MNT) was considered to be a critical aspect in this study. Co-processing tablet excipients is expected to be a cost-effective process since there are fewer manufacturing steps, e.g., the blending of the tablet’s excipients. However, it is mandatory to investigate the effect of the added excipients on tablet’s physico-mechanical properties (e.g., drug content and tablet content uniformity, hardness, friability, and dissolution profile). To the best of our knowledge, there has been no published work on DH via HME technology.
2. Materials and Methods
2.1. Materials
DH USP was purchased from Wuhan Hengheda Pharm Co. (Wuhan, China). C-888 was kindly given by Gattefossé (Saint-Priest, Lyon, France). EC-N7 was obtained from Ashland (Ashland Aqualon Functional Ingredients, Wilmington, DE, USA). E-RS was kindly gifted by Evonik (Parsippany, NJ, USA), and Avicel® PH 102 (MCC) was received from FMC Biopolymers (1735 Market Street, Philadelphia, PA, USA). Magnesium stearate was purchased from Spectrum Chemicals (Gardena, CA, USA). Pearlitol® (MNT) was received as a gift sample from Roquette America Inc. (Keokuk, IA, USA). Stearic acid was purchased from Mallinckrodt Chemical Ltd. (Bedminster Township, NJ, USA). Marketed Aricept® tablets were purchased from a local university pharmacy shop (Oxford, MS, USA). All chemicals and solvents utilized for analysis in the study were of analytical grade and were obtained either from Spectrum Chemicals (Gardena, CA, USA) or Thermo Fisher Scientific (Waltham, MA, USA).
2.2. HPLC Method for Analysis of DH
An in-house developed, reversed-phase HPLC-based analytical method was used for the determination and quantification of DH in all in vitro samples. This method was validated according to a previous research group [
15]. The HPLC equipped with a UV detector, a Waters Symmetry shield, a C18 column (250 × 4.6 mm, 5 µm particle size), and an isocratic mode of elution. The mobile phase was a mixture of methanol, a 0.02 M phosphate buffer, and trimethylamine (50:50:0.5,
v/
v/
v). The phosphate buffer was prepared by dissolving 2.4 g of monobasic sodium phosphate in 900 mL of water, mixing with 10 mL of trimethylamine, and adjusting to pH 2.7 ± 0.5 with phosphoric acid. The mobile phase was filtered through a nylon membrane (pore size: 0.45 µm) and degassed before use. Chromatography was performed at room temperature. The flow rate and run time for analysis were 1.0 mL/min and 15 min, respectively. In these conditions, the DH retention time (t
R) was around 9 min. The injection volume was 20 µL, and ultraviolet detection was at a wavelength (λ
max) of 268 nm. The column temperature was 40 °C. Finally, the acquired data were processed using the Empower 2 build 2154 software (Waters Inc., Mount Holly, NJ, USA).
2.3. HME Processing Method
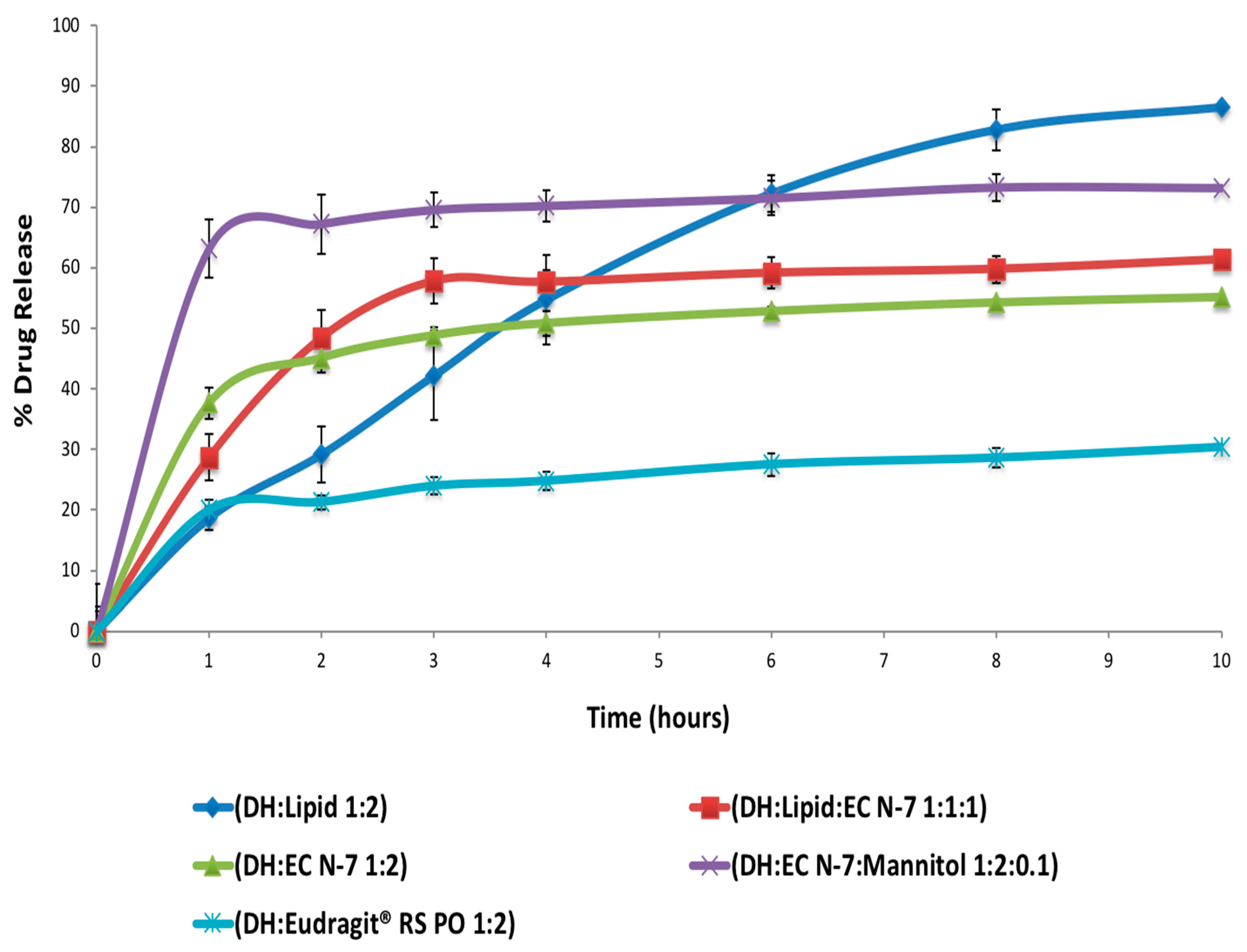
For the screening purposes, three different carriers (C-888, EC-N7, and E-RS) were used (alone or in combination) at different ratios and processing parameters (
Table 1,
Table 2 and
Table 3) to evaluate their effect on sustaining of the DH release. The materials were blended using a V-shell blender (GlobePharma, New Brunswick, NJ, USA, Maxiblend
®) with and without MNT as a pore-former, and samples were analyzed for drug content and content uniformity.
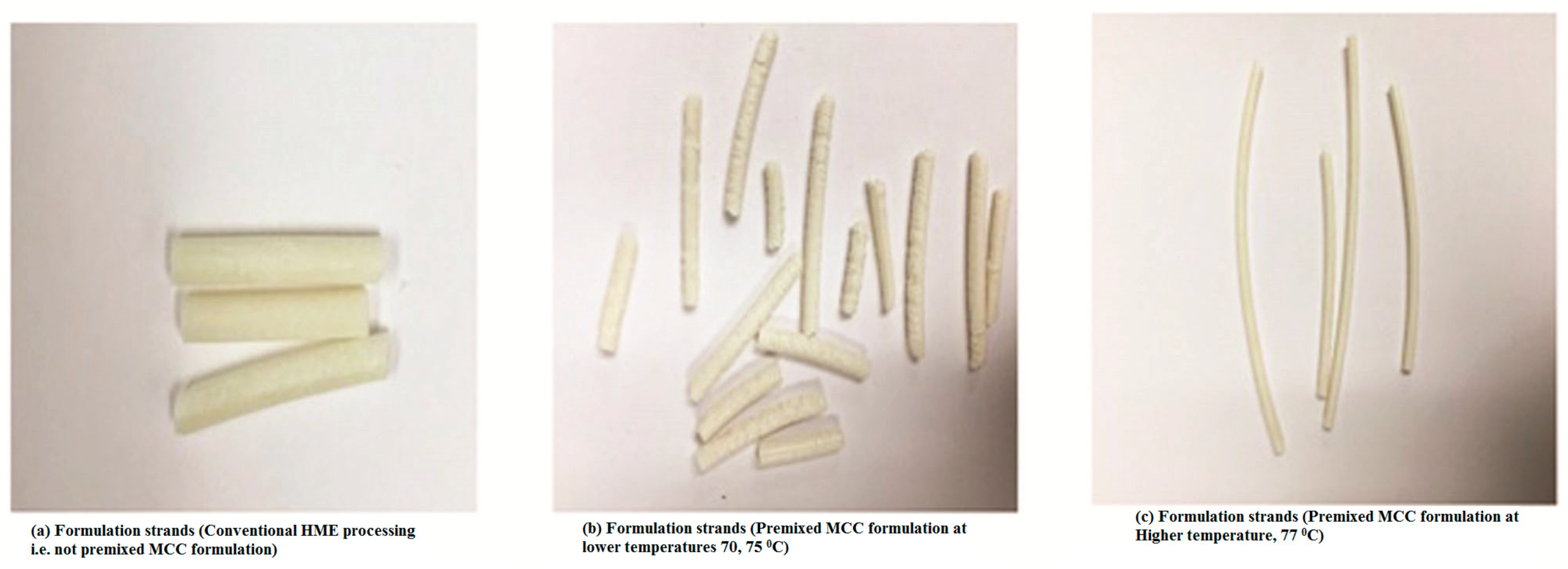
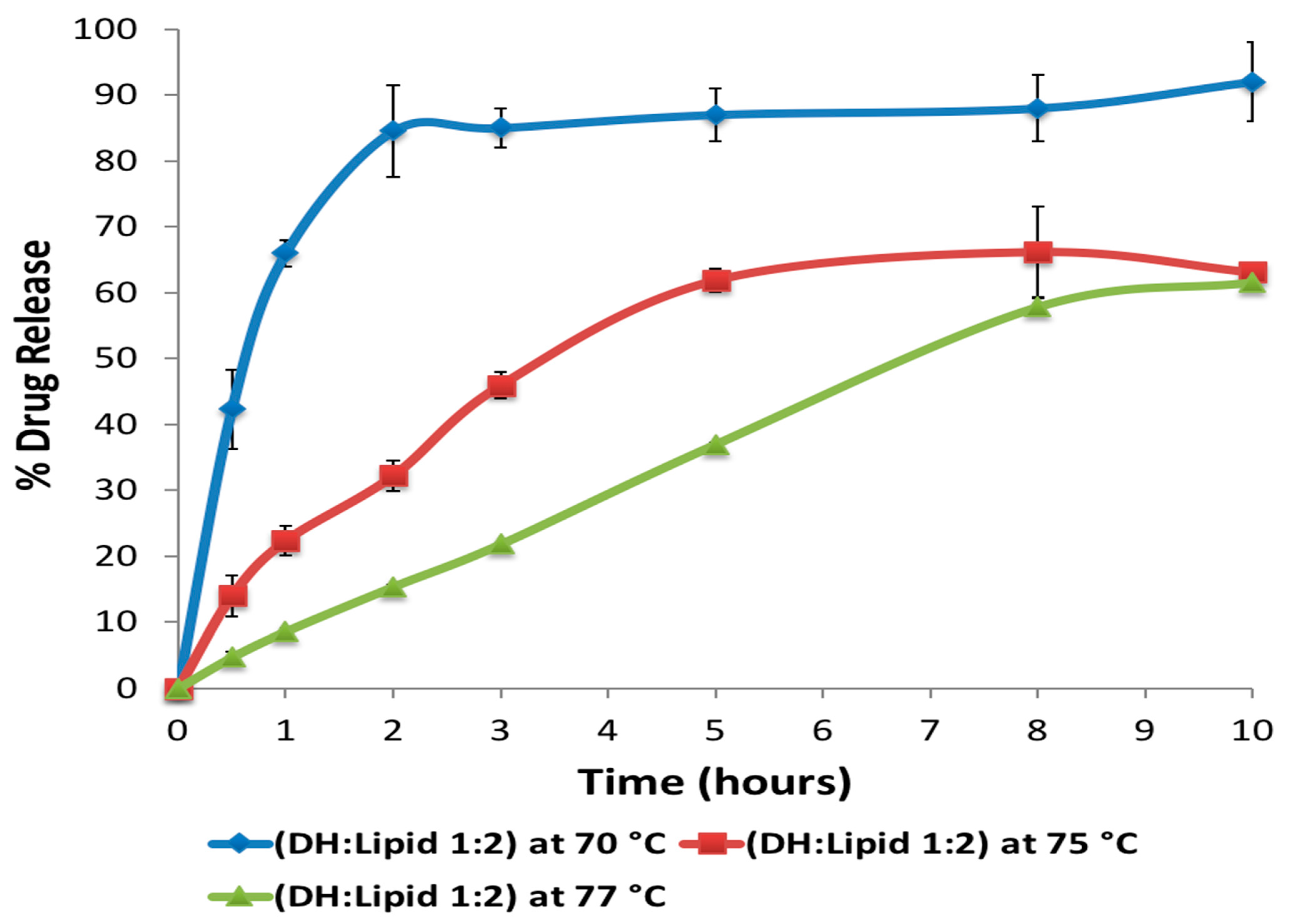
Prior to HME, the materials were sieved using a size #35 USP mesh to remove any aggregated and agglomerated particles. The blended materials were extruded on a 16 mm rotating twin-screw extruder (ThermoFisher Scientific Co., Waltham, MA, USA) utilizing a round-shaped die to produce rod extrudates (EXTs). In addition, to study the effect of processing temperature on the lipid, the materials were run through the heated barrel with a screw rotating speed of 50 rpm, and the temperature for all zones of the extruder was set at 70, 75, or 77 °C except for the last zones and the die, which were set at 70 °C to cool down and solidify the EXTs (
Table 4). The reason different temperatures were set was to study the effect of temperature on the drug, lipid crystallinity, and drug release profile. The processing temperature profile and screw speed were based on the physical properties of hydrophobic carriers in the preliminary studies. The resulting EXTs were further processed using a comminuting mill (Fitzpatrick, Model “L1A”) and then sieved using a size #35 USP mesh.
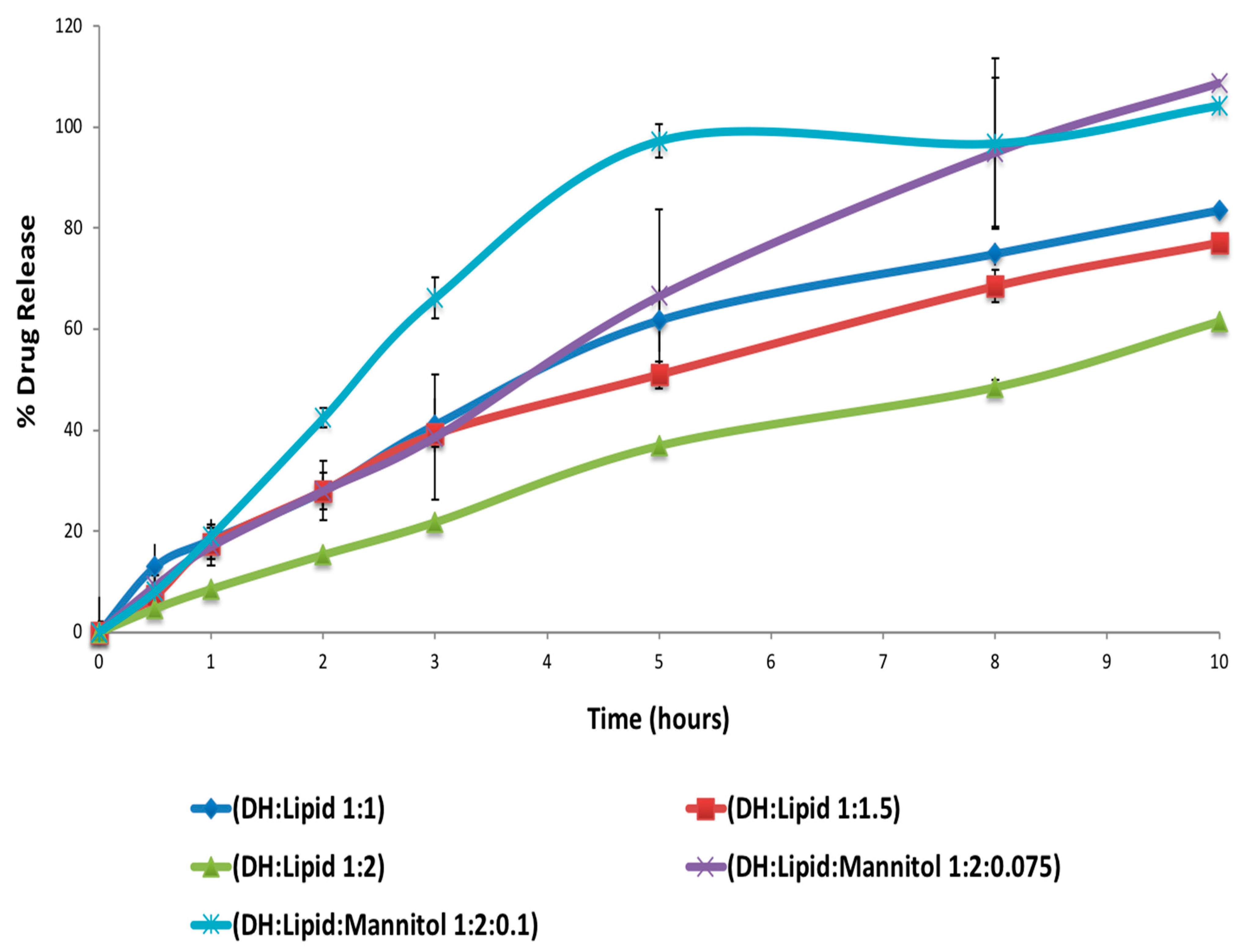
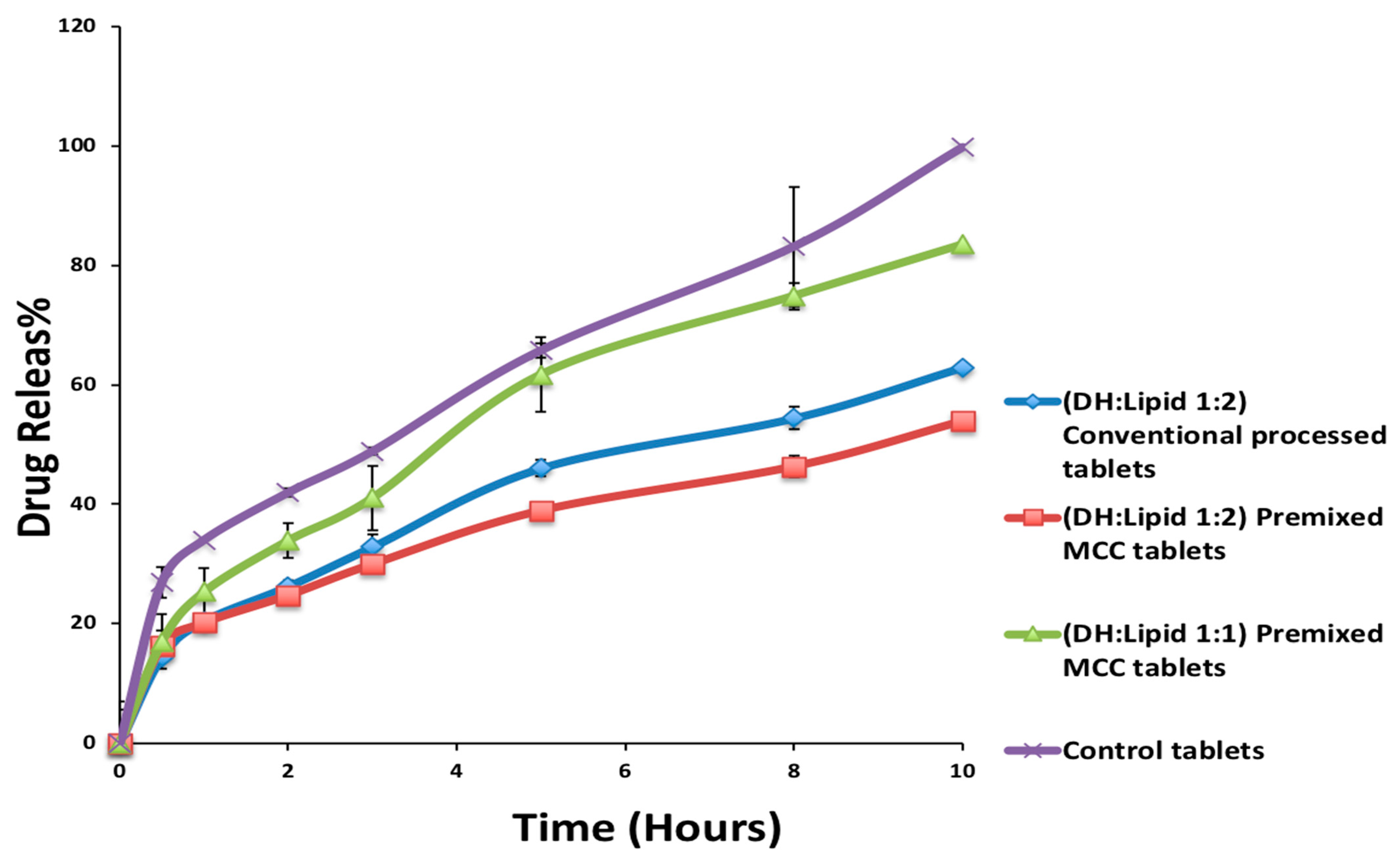
Another part of this study was to improve the drug content and content uniformity of the optimized formulation from the screening stage based on the drug release profile, which is referred to as DH:lipid. As shown in
Table 5 and
Table 6, DH (33.3–50%
w/
w) (1:2 and 1:1 ratios) and C-888 were blended and extruded using the same procedure and processing conditions, as previously mentioned during the screening stage but with and without premixed MCC as a tablet diluent while extruding the blend to observe the effect of the pre-mixed MCC on drug content, content uniformity, and release profile.
2.4. Conventional Heat Fusion Processing Method
The conventional heat fusion processing method is also known as the melt and mix method. This method was used for C-888 to compare its performance with the formulation from HME. In this processing method, C-888 was melted in porcelain casseroles at 100 °C until a homogeneous melt was obtained, and then DH was added and manually mixed to the homogenous melt. The mixture was kept to gradually cool down with continuous stirring until a congealed mass was obtained. Then, the mass melt was milled using the same milling method as mentioned before in HME-processed formulations. Subsequently, the mass was grounded, pulverized, and sieved using a size #35 USP mesh. All processed powders were stored in a desiccator at room temperature till further use.
2.5. TGA and Differential Scanning Calorimetry (DSC) Analysis
The thermal stability of DH and the carriers at the employed processing temperatures was determined by TGA analysis (TGA, Pyris 1 TGA Perkin Elmer, Shelton, CT, USA). A Perkin Elmer Pyris 1 TGA-running Pyris manager software (PerkinElmer Life and Analytical Sciences, Shelton, CT, USA) was utilized for TGA analysis. Drugs and excipients were evaluated for thermal stability at high temperatures. Around 3–4 mg of the sample were weighed and heated from 25 to 250 °C at a 10 °C/min heating rate under an atmosphere of nitrogen, and the TGA spectra for each sample were recorded.
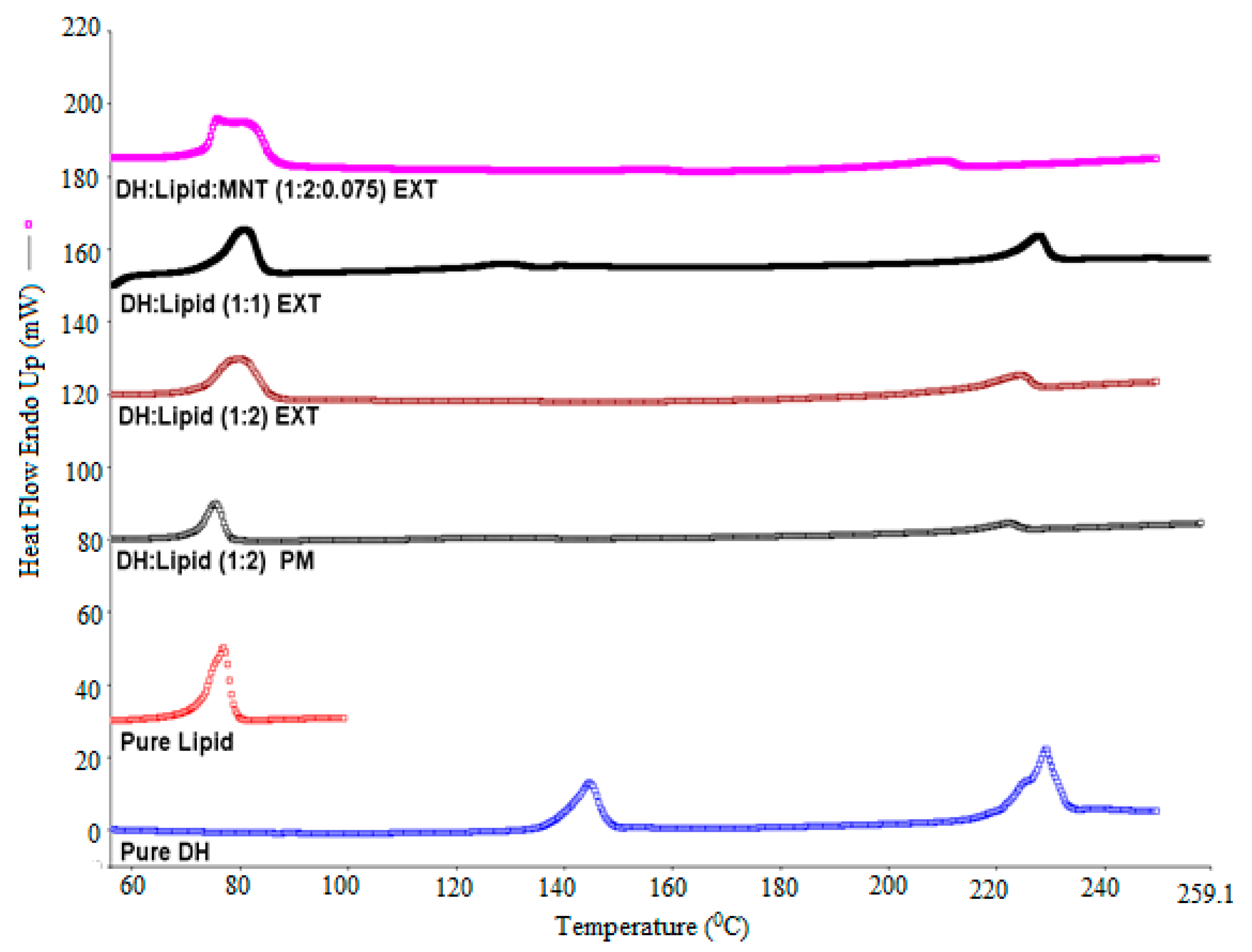
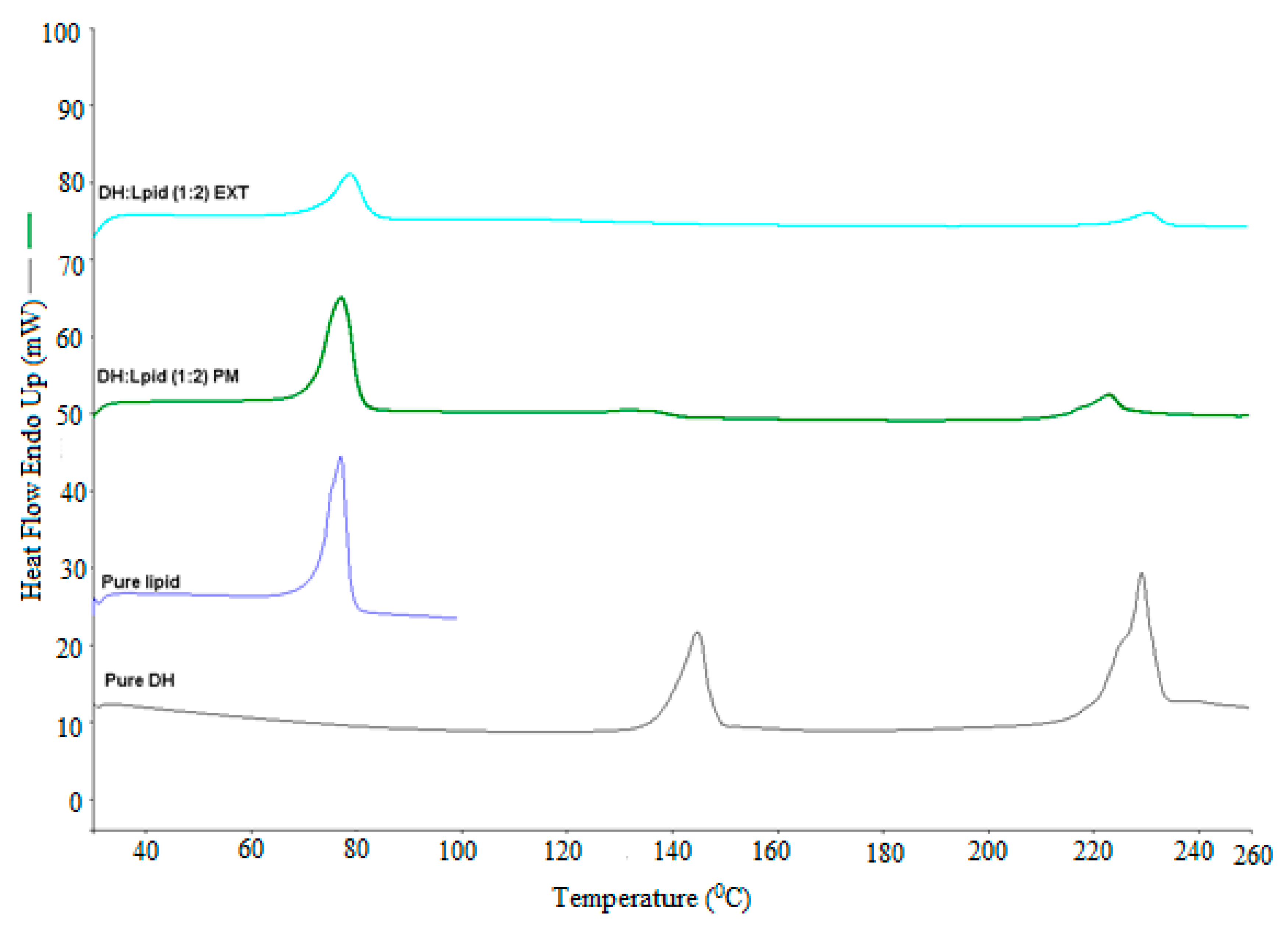
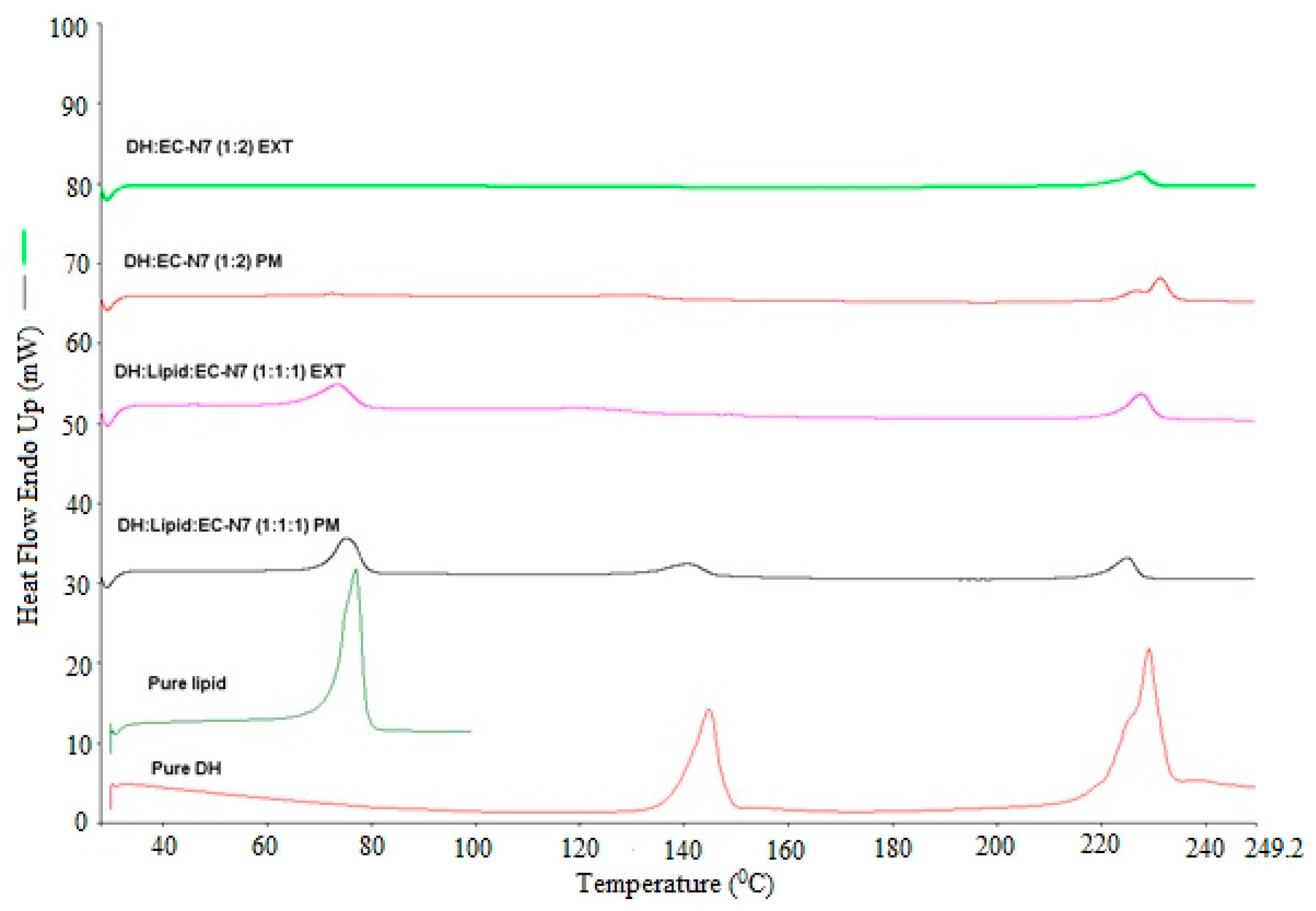
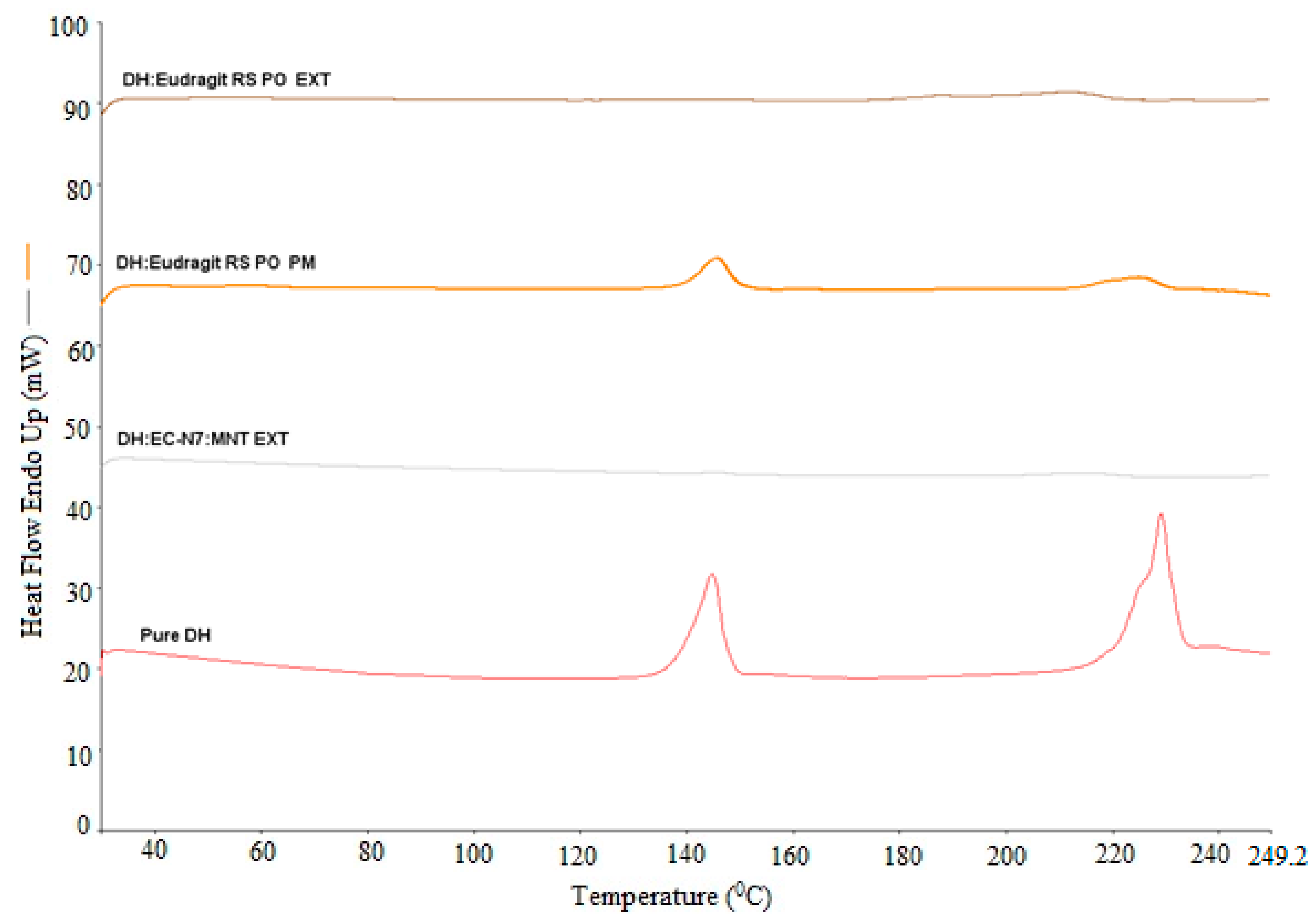
Differential scanning calorimetry (DSC) (DSC, Diamond DSC, Perkin Elmer, Shelton, CT, USA) was utilized to determine the physical states of different API/carrier ratios in each physical mixture and EXT. The physical states of the pure drug, the pure carrier, physical mixtures, and EXTs were studied. The heating range used was from 30 to 250 °C at a heating rate of 20 °C/min under a nitrogen atmosphere. Due to the low melting point of the lipid, it was heated from 30 to 100 °C at the same scanning rate. The accurately weighed amount (4–5 mg) of each sample was taken in an aluminum pan and hermetically sealed. The Pyris manager software (Shelton, CT, USA) was utilized for the data processing and analysis. It was used to analyze the generated data to evaluate the crystallinity. The DSC was calibrated initially with an indium reference. In addition, the drug/lipid miscibility was estimated using the Hoftyzer and Krevelen approaches and the Hansen solubility parameters [
16].
2.6. SEM
The surface morphology of the pure drug and milled EXTs was studied using SEM. The samples were mounted on adhesive carbon tapes placed on aluminum stubs. Gold was used to coat the samples by a Hummer® 6.2 sputtering system (Anatech LTD, Springfield, VA, USA) in a high vacuum evaporator. A scanning electron microscope operating at an accelerating voltage of 0.8 kV was used for imaging (JEOL, JSM-5600, Tokyo, Japan). The morphology of formulations might have reflected the crystallinity status of the DH.
2.7. Solubility Parameter Calculation for Optimized Formulation
Solubility/miscibility is an important parameter in solid dispersion formulations. The Hansen solubility parameters can be obtained from the literature. However, they can also be calculated through Molecular Modeling Pro, v6.2.8 (ChemSW, Fairfield, CA, USA). This software can be conveniently used to calculate the solubility parameters for certain pharmaceutical materials by inputting their corresponding melting points and molecular structures.
2.8. Physico-Mechanical Evaluation for Milled EXTs for Optimized Lipid Formulation
The bulk and tapped density was calculated by measuring the volume of a 5 g milled EXT in a 10 mL graduated cylinder [
17]. Firstly, the initial occupied bulk volume in the cylinder was noted. Secondly, after tapping the cylinder 100 times, the tap volume was noted. The bulk density
and tap density
were calculated by dividing the weight of milled EXTs over the respective volume. Using following equation (Equation (1)), the Carr’s compressibility index (
CI) was calculated:
The Hausner ratio (HR) was obtained by dividing by .
2.9. Tablet Processing
The milled EXTs were mixed with the tableting excipients of MCC and magnesium stearate (
Table 5 and
Table 6) and compressed into tablets equivalent to 23 mg of DH with compaction forces of 600 and 1500 psi, respectively. Tablets were compressed on a manual tablet press using a 10 mm round concave punch to a final tablet weight of 300 mg. The tablets, having luminous surfaces, were stored in a plastic bottle until further use. Tablets were evaluated for thickness, hardness, friability, and in vitro release profiles. In addition, drug content and tablet content uniformity tests were also conducted. The DH content was analyzed using the HPLC-UV method described in above section. For improving the drug content, the pre-mixed MCC formulations were compressed on single punch tablet press (MCTMI, Globe pharma, New Brunswick, NJ, USA) using an 8 mm round flat punch with a compaction force of 1500 psi. Magnesium stearate (1%) was added as a lubricant just before the direct compression process, and each compressed tablet weighed 200 mg to mimic the weight of Aricept
®, which is the commercial product of DH. Aricept
® was used as a control formulation in this study.
2.10. Tablet Characterization
Some standard characterization tests were performed on tablets manufactured from extruded materials. The thickness and diameter of the tablets were measured using a micrometer. Disintegration testing was performed via a disintegration apparatus. Friability testing was performed in a friabilator (Roche Friabilator, Basel, Switzerland). To test the hardness of the tablets, a VarianVK200 (Agilent technologies, 13000 Weston Pkwy, Cary, NC, USA) hardness tester was used. A tablet content uniformity test was achieved by a drug assay via the HPLC method. The tablet weight uniformity test was performed by measuring each tablet after direct compression via the Mettler Toledo scale (Columbus, OH, USA). Six replicates were tested (n = 6).
2.11. In Vitro Release Studies
In vitro release studies for either milled EXTs or tablet equivalent to 23 mg of DH were performed using a USP type-II dissolution apparatus with 900 mL of a pH 6.8 phosphate buffer (simulated intestinal fluid) at 37 ± 0.5 °C and 50 rpm for 10 h [
18,
19]. Due to the extensive use of simulated intestinal fluid for the drug release study of oral and sustained release formulations, it was used as the biorelevant dissolution media for the drug release study in this work [
18,
19,
20]. All dissolution samples were analyzed using an HPLC-UV system (
n = 3). The paddle speed was set at 50 rpm. The samples were collected at every 0.5, 1, 2, 3, 5, 8, and 10 h, filtered, and then analyzed using an HPLC-UV method. The withdrawn amount for the sample was compensated for by adding an equal amount of drug-free fresh dissolution media back to the dissolution vessel at each time point. The release profile for percent release was plotted against time for each formulation. The model-independent similarity factor
2 was used to compare the dissolution profiles among different formulations. In addition, the statistical analysis was conducted using ANOVA to compare the release profiles. It is well-known that if the calculated
2 value is between 50 and 100, then the profiles are considered to be similar [
20]. In addition, the drug release kinetics of different EXTs were evaluated using different kinetic models such as a zero order model, first order model, the Higuchi model, the Hixson–Crowell model, and the Korsmeyer–Peppas model, as described in the literature [
21].
2.12. Statistical Analysis
The data were expressed as the mean ± standard deviation (SD). The confidence interval was chosen to be 95%, and a statistically significant difference was determined at a minimal level of significance of 0.05 using Student’s t-test.
4. Conclusions
DH can be formulated in a solvent-free, one-step, cost-effective, and continuous process using C-888 as a sustained release matrix with MNT as a hydrophilic pore-former. It was confirmed that DH in a lipid matrix was immiscible, and it preserved its crystallinity during the extrusion process. However, some fractions of DH were solubilized in the lipid matrix with no endothermic peaks, whereas the formulations with MNT were not able to maintain DH crystallinity. The optimized formulation (DH:lipid:mannitol; 1:2:0.075) demonstrated a sustained, stable, and complete release in 10 h. In vivo investigations are required to correlate with in vitro dissolution studies for the development of optimum oral sustained release matrix tablets of DH, which is the scope of a future study. The prepared tablets showed good post-compression characteristics regarding hardness, friability, weight variation, content uniformity, and desired and reproducible drug release profiles compared to the reference commercial product. It was concluded from the dissolution profiles that the HME technique can be utilized for the manufacturing of DH sustained release tablets via the co-processing of tablet excipients with API/C-888. Tailoring the ratio of C-888 and MNT in the formulations, along with an appropriate extrusion temperature profile, resulted in a modification of the release of DH and the obtainment of a preferable release pattern.
The co-extrusion of tablet excipient (MCC) with an API/matrix-former could be a problem-solving approach for the development of lipid-based hydrophilic APIs. This could contribute to a reduction of processing steps, such as blending, as well as improving the integrity of lipid-based tablets. The HME processing temperature profile plays key factor in the dissolution behavior of DH. It was demonstrated that the co-extrusion of tablet excipients within an API/lipid matrix resulted in different dissolution behaviors and tablet properties. Though DH was in the amorphous form in the case of the formulation that contained hydrophilic diluents/fillers such as MNT and MCC, the release was sustained and stable. The only explanation is that these hydrophilic excipients were embedded within the C-888 matrix. This new insight shows a new trend in terms of preparing a developed and continuous tableting process (e.g., mini matrices) in the solid lipid extrusion field. This insight could give useful information in terms of product development.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}