
Challenges of Dissolution Methods Development for Soft Gelatin Capsules

Abstract
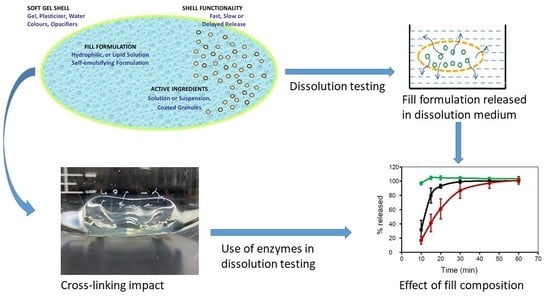
:
1. Introduction
2. Advantages and Disadvantages of SGCs
- (1)
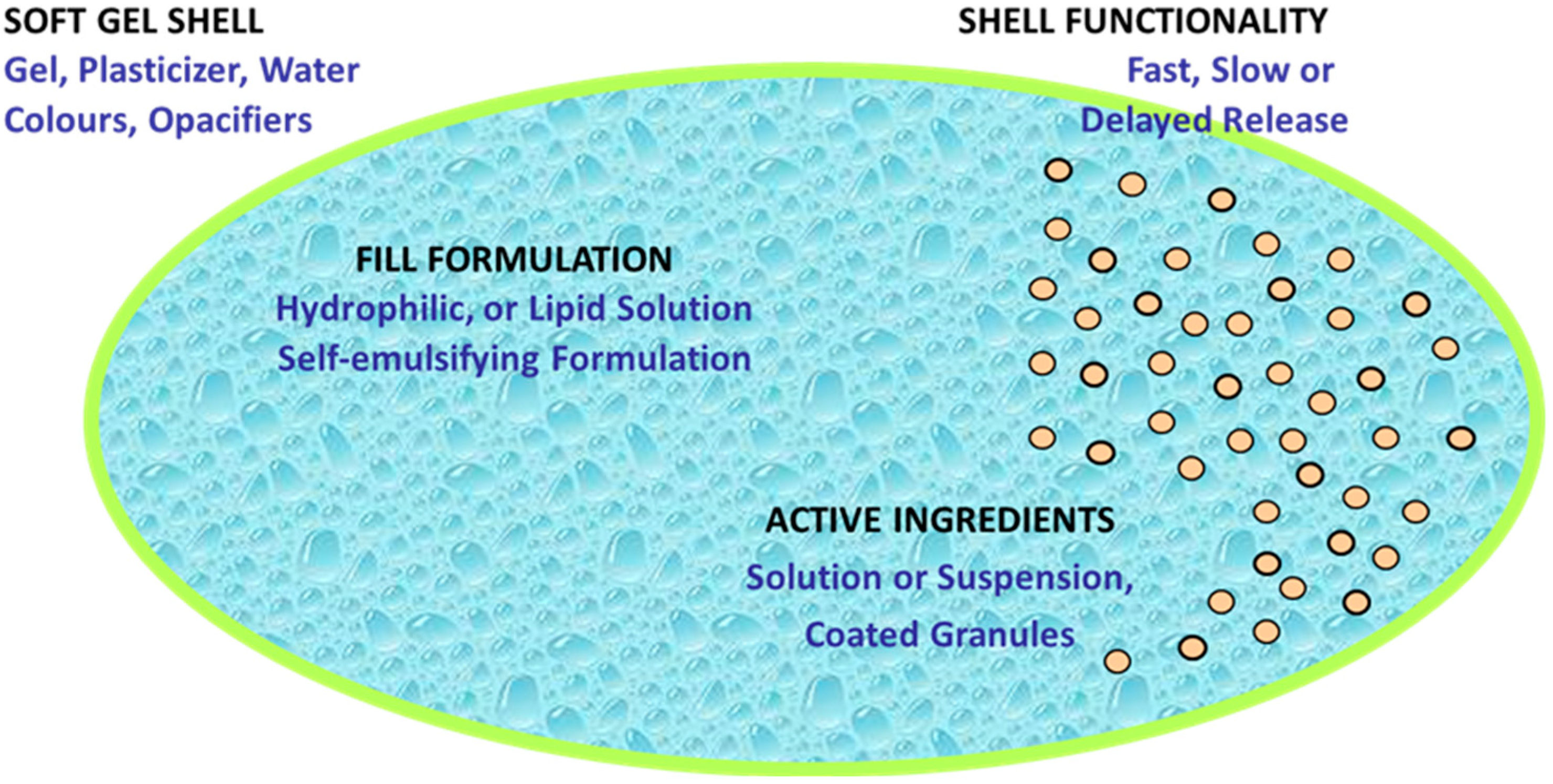
- Consumer Preference: SGCs dosage forms were developed to conceal the unpleasant taste and odor of drugs. Compared to tablets, SGCs are more comfortable to swallow when used with water because the soft gelatin capsule is self-lubricating [16,17]. SGCs look more appealing and enjoyable to consumers as they can easily be produced in various shapes, sizes, and colors (Figure 1), and different drug delivery system like chewable softgels [18] and meltable softgels [19].
- (2)
- Technical Advantages: SGCs have high dosage accuracy and uniformity [16,17] as well as higher consistent manufacturing requirements and product stability. It is possible to deliver an API with a higher degree of accuracy and greater consistency between different manufacturing lots due to more accurate compounding, blending, and dispensing of liquid fill materials. SGC products tend to have higher stability as the entire encapsulation process can be done under inert conditions to protect drugs against oxidation and degradation. This is especially important for drugs that are subject to hydrolytic and oxidative degradation.
- (3)
- Safety Aspects: The tight sealing of the gelatin shell protects the fill material from air and environmental contaminations. The shell can be formulated to block ultraviolet (UV) as well as visible light. Also, SGC formulation helps to avoid dust handling contaminations and enhances operator safety [17].
- (4)
- Bioavailability Advantages: SGCs can increase the bioavailability of poorly soluble drugs by improving solubility and enhancing absorption within the GIT [20,21]. Water-insoluble drugs are formulated in form of SGCs using lipophilic vehicles as a portion of fill material, greatly enhancing uptake of such drugs within the GIT [22].
- (1)
- SGC technology is believed to be relatively expensive to produce, and this can increase prices to consumers. Many pharmaceutical companies do not have the specialized equipment necessary to fill SGCs, and most of them rely on contract laboratories/manufacturers for their supplies.
- (2)
- Unlike solid dosage forms, SGCs can also be affected by humidity and microbial contamination. This can cause product stability issues if the medications are not kept in sealed containers or a cool and dry place.
- (3)
- Depending on the nature of the drug that is dissolved within the lipophilic vehicle, there is a chance that the drug could migrate into the shell of the capsule. This migration can cause issues during absorption within the body, as the release rate of the drug would be altered. Likewise, SGCs are not generally capable of holding water-based liquids, as there is a possibility that the medication could diffuse out of the soft-gelatin capsule [16,17].
- (4)
- Another issue concerning the use of soft gelatin drug products is the fact that some groups have dietary restrictions that prevent them from consuming animal products found in SGCs. Gelatin is primarily made from bones, skins, and other parts of animals such as pigs and cows. Because capsule shells are made from animal parts, many vegetarians also opt not to use them. Due to this, there is emerging technologies on animal-free substitute gelatin capsules made from seaweed extract or other sources, but they are generally more expensive and harder to find [23].
- (5)
- (6)
- Alkaline or acidic solutions are not good candidates for soft gelatin fill because they can cause hydrolysis and leakage of the gelatin shell unless their pH is adjusted to neutral [20].
3. Influence of Physicochemical Properties of Gelatin on the Dissolution of SGC Drug Products
3.1. Gel or Bloom Strength
3.2. Gel Viscosity
3.3. Molecular Weight Distribution
3.4. Setting Point
4. Manufacture of SGCs
- (1)
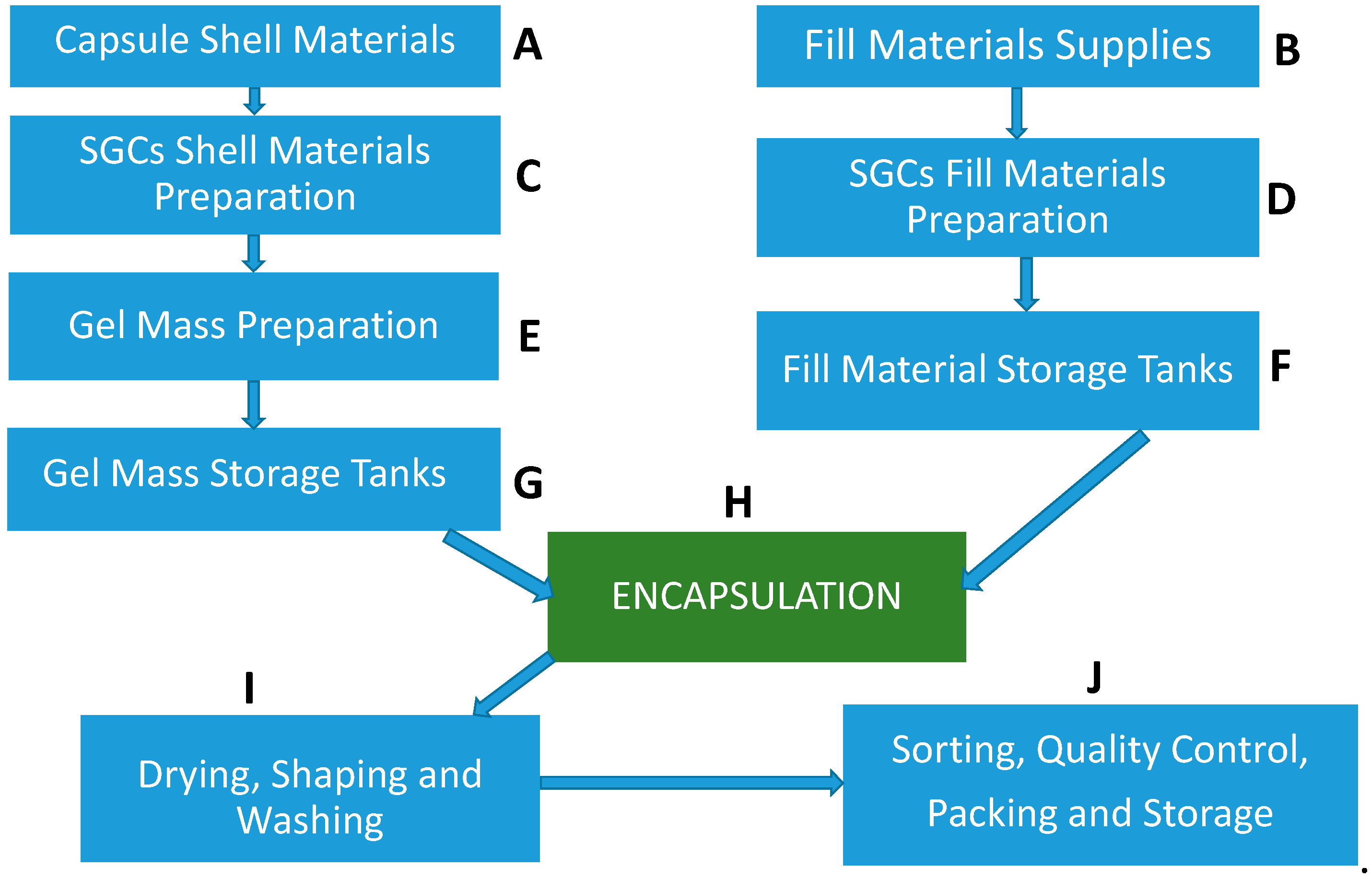
- Raw materials such as raw gelatin, plasticizers, and purified water are mixed under vacuum conditions at a temperature of about 70 °C depending on gel formulation (Figure 3C,D). Any undissolved gel is removed by filtration.
- (2)
- All compounds required for a specific formulation are added and mixed under appropriate vacuum and nitrogen blanket conditions. During the mixing process, samples are taken from a different area of the vessel to ensure homogeneity of the mixture and for viscosity control. The vessel size depends on the development of the commercial stage of a drug product; for example, gelatin melting tanks can range from small scale (e.g., 100 L) to large scale (e.g., 1200 L), while gelatin service tanks can range from 50 L (lab scale) to 300 L (large scale) [49]. These vessels are connected to a mill to reduce particle size to below 180 µm for the encapsulation process.
- (3)
- The mixture is stored in the receiver containers (Figure 3F,G).
- (4)
- When fill material and gel are ready, the encapsulation with soft gelatin will start (Figure 3H). Gel and fill material are connected to the encapsulation machine through heated tubing and under the nitrogen atmosphere. After encapsulation, SGCs enter a tumbler for the initial drying process. Then, they are stored in a drying tunnel for several days, depending on formulation and capsule size, to dry further. Most of the moisture is removed during this stage, and capsules’ moisture content and hardness is measured at the end of this step.
- (5)
- After capsules are dry to the desired level, they are sorted and graded (Figure 3I,J)—grading is especially critical and important for blister production.
- (6)
- Capsules are polished with a cloth soaked with isopropanol for a defined period and then with a dry cloth for the same amount of time. In the end, capsules are printed and packaged (Figure 3J).
5. Shell and Fill Formulation of SGCs
- (1)
- It should be able to dissolve API completely and prevent precipitation of excipients and API during manufacturing and throughout the shelf-life period.
- (2)
- It must be able to stabilize fill material and be compatible with gelatin shell formulation.
- (3)
- It is desired that the developed fill formulation optimizes the physical and chemical stability of the API.
6. Dissolution Methods Development and Considerations for SGCs
6.1. Dissolution Definition
6.2. Dissolution Rate
6.3. Solubility
6.4. Disintegration and Rupture Tests
6.5. Practical Concepts of Developing a Dissolution Method
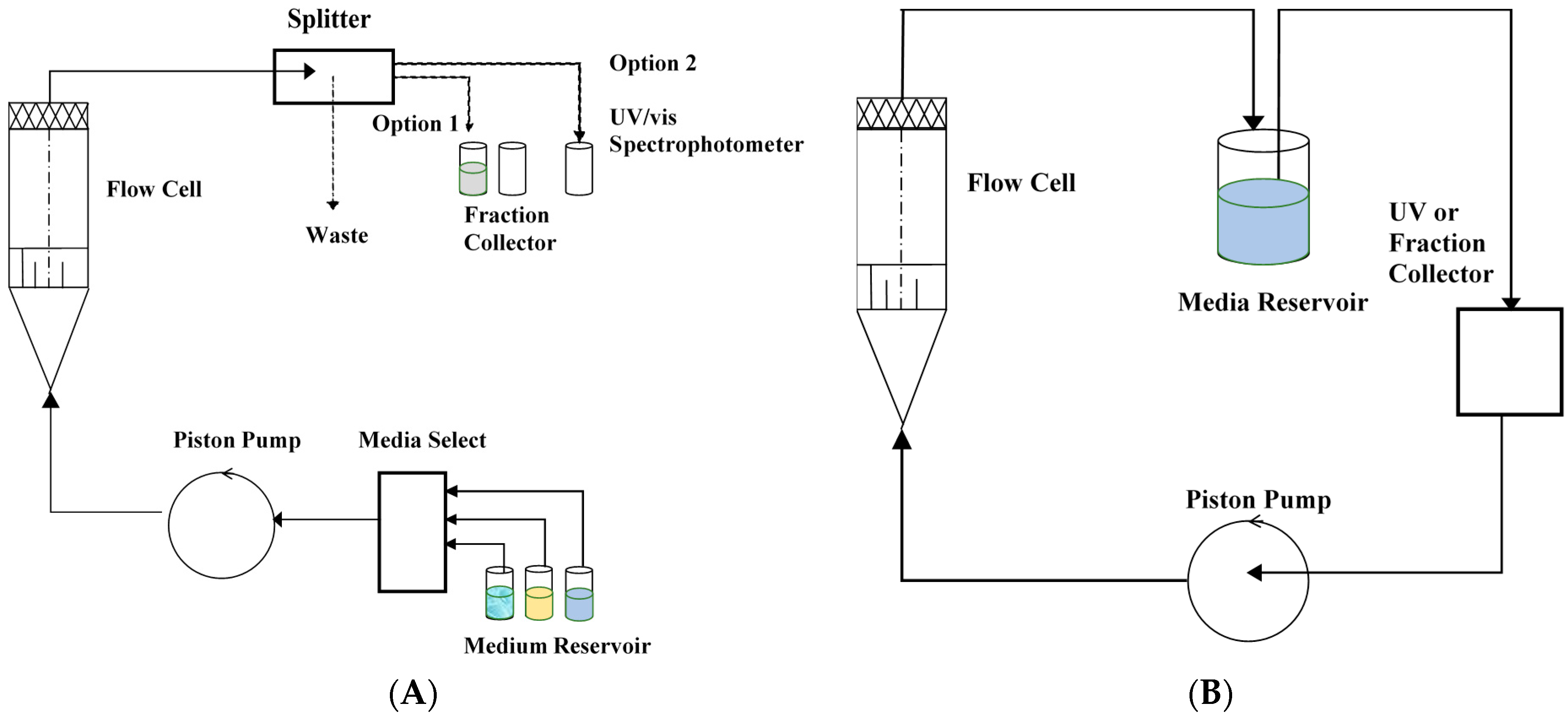
7. Flow-Through Dissolution Methods
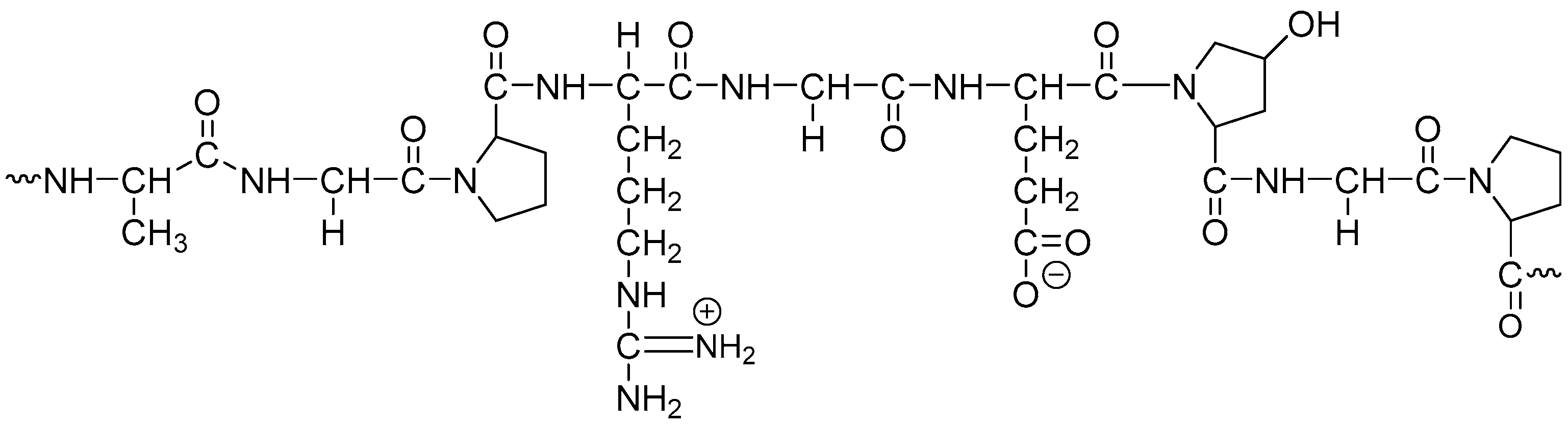
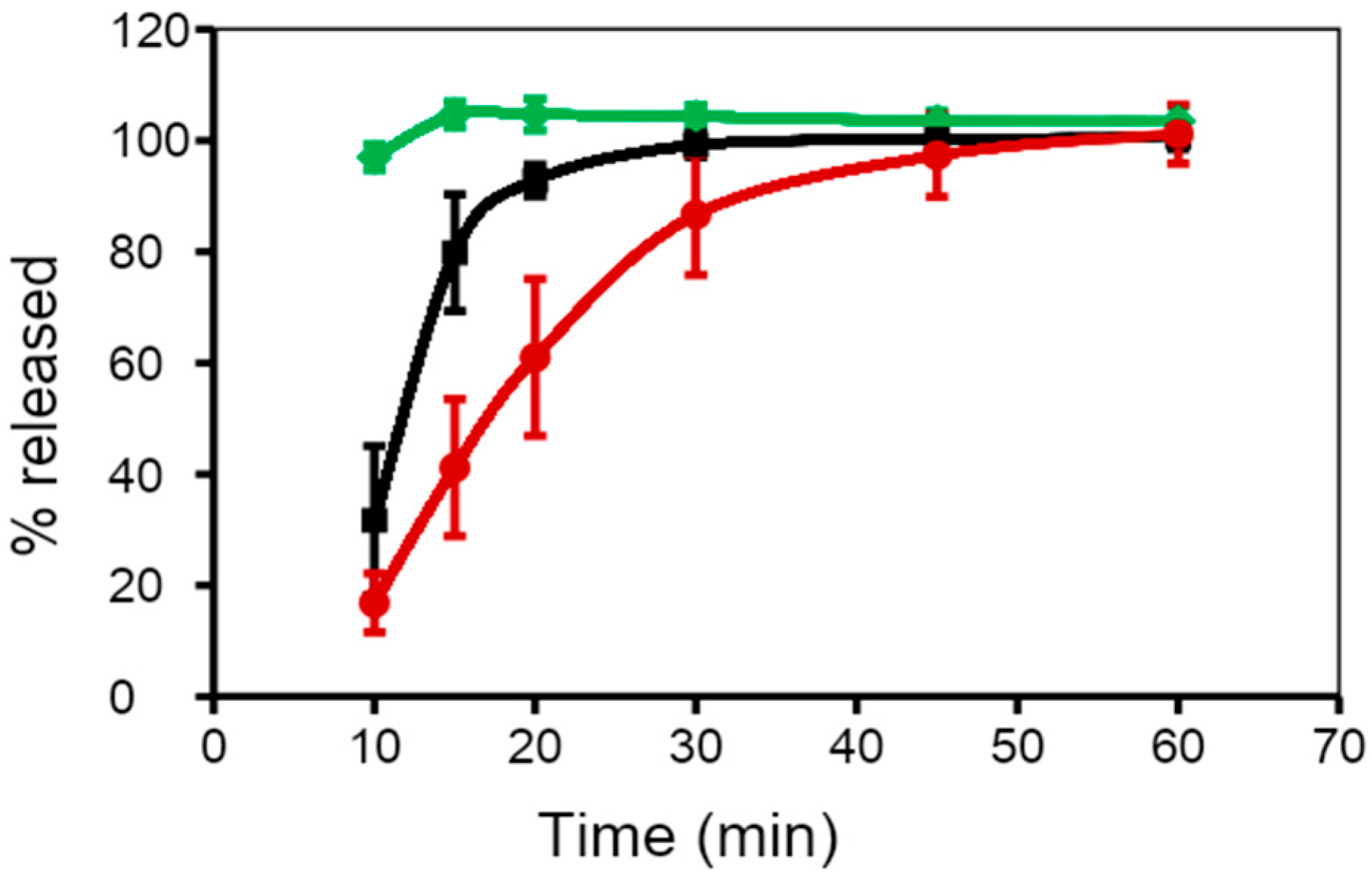
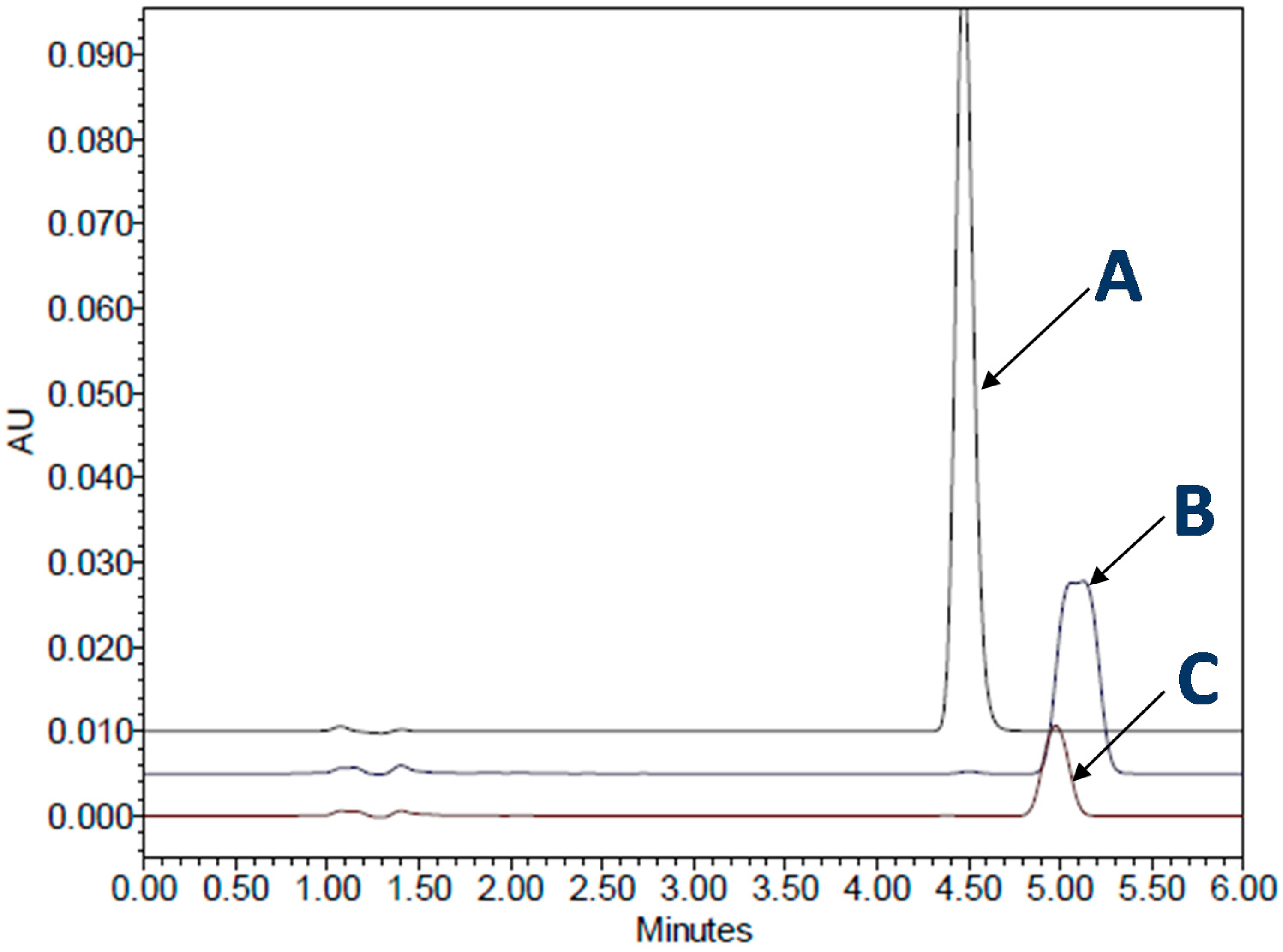
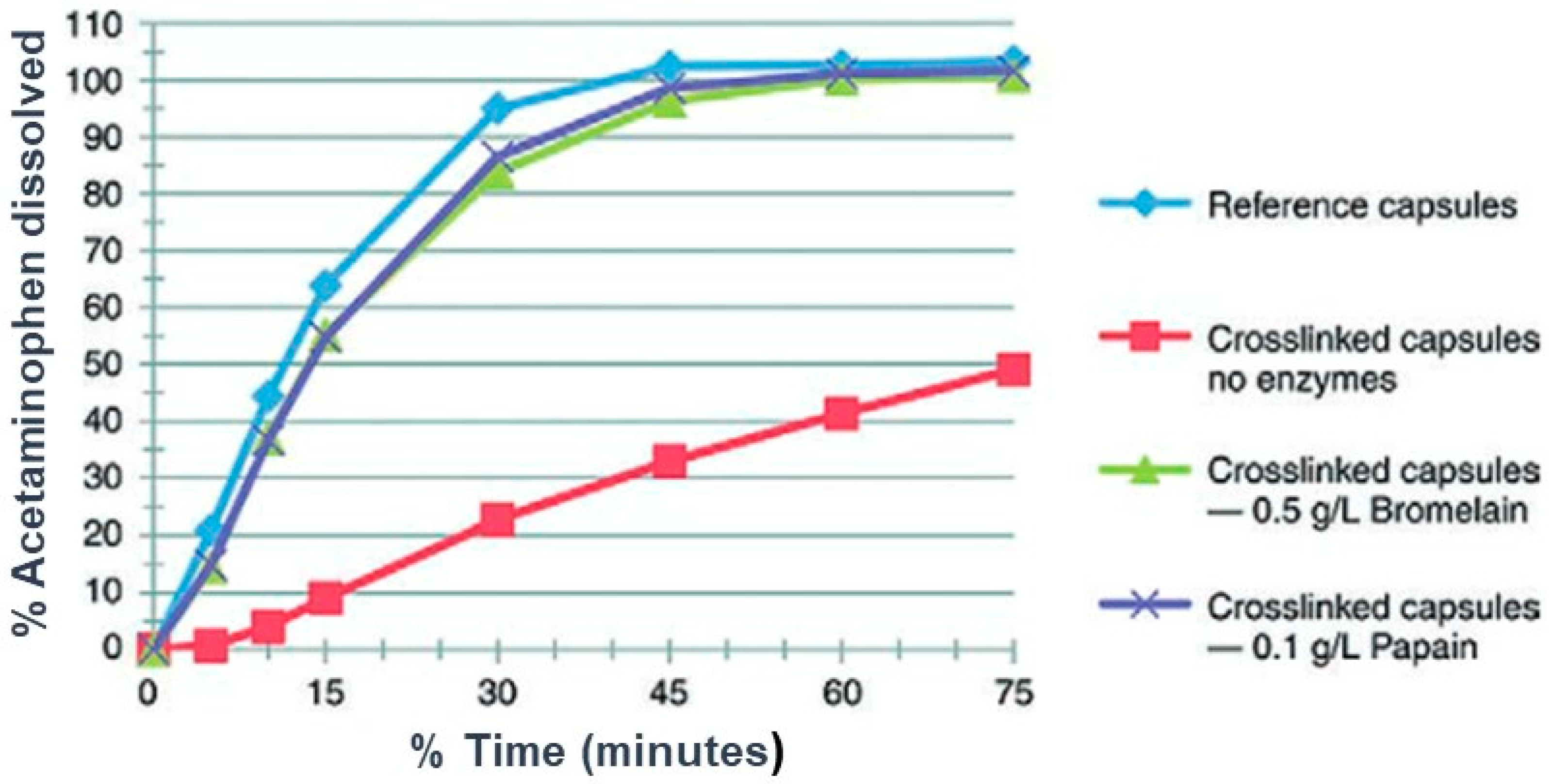
8. Effect of Cross-Linking on the Dissolution Properties of SGCs
9. Effect of Digestive Enzymes on Cross-linking of SGCs
10. Conclusions and Future Prospects
- The solubility of the API
- Compatibility of API with soft gel fill materials
- Nature of capsule shell
- Type of surfactants in the dissolution medium
- The need for sinkers depends on whether the SGCs are floating or moving within the dissolution medium
- The design of SGCs, i.e., coated or non-coated SGCs
- Stability of the API in the dissolution medium
- Nature of fill components, i.e., hydrophilic, suspension, lipophilic, or co-solvents
- Dealing with drug products whose gelatin is already cross-linked
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kawabata, Y.; Wada, K.; Nakatani, M.; Yamada, S.; Onoue, S. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: Basic approaches and practical applications. Int. J. Pharm. 2011, 420, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gershanik, T.; Benita, S. Self-dispersing lipid formulations for improving oral absorption of lipophilic drugs. Eur. J. Pharm. Biopharm. 2000, 50, 179–188. [Google Scholar] [CrossRef]
- Pouton, C.W.; Porter, C.J.H. Formulation of lipid-based delivery systems for oral administration: Materials, methods and strategies. Adv. Drug Deliv. Rev. 2008, 60, 625–637. [Google Scholar] [CrossRef]
- Rane, S.S.; Anderson, B.D. What determines drug solubility in lipid vehicles: Is it predictable? Adv. Drug Deliv. Rev. 2008, 60, 638–656. [Google Scholar] [CrossRef] [PubMed]
- Sha, X.; Yan, G.; Wu, Y.; Li, J.; Fang, X. Effect of self-microemulsifying drug delivery systems containing Labrasol on tight junctions in Caco-2 cells. Eur. J. Pharm. Sci. 2005, 24, 477–486. [Google Scholar] [CrossRef]
- Woo, J.S.; Song, Y.-K.; Hong, J.-Y.; Lim, S.-J.; Kim, C.-K. Reduced food-effect and enhanced bioavailability of a self-microemulsifying formulation of itraconazole in healthy volunteers. Eur. J. Pharm. Sci. 2008, 33, 159–165. [Google Scholar] [CrossRef]
- Gursoy, R.N.; Benita, S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed. Pharmacother. 2004, 58, 173–182. [Google Scholar] [CrossRef]
- Trevaskis, N.L.; Charman, W.N.; Porter, C.J.H. Lipid-based delivery systems and intestinal lymphatic drug transport: A mechanistic update. Adv. Drug Deliv. Rev. 2008, 60, 702–716. [Google Scholar] [CrossRef] [PubMed]
- Cole, E.T.; Cadé, D.; Benameur, H. Challenges and opportunities in the encapsulation of liquid and semi-solid formulations into capsules for oral administration. Adv. Drug Deliv. Rev. 2008, 60, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kesarla, R.; Omri, A. Formulation strategies to improve the bioavailability of poorly absorbed drugs with special emphasis on self-emulsifying systems. Int. Sch. Res. Not. 2013, 2013, 848043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Zhang, l.; Zhang, M.; Pang, Y.; Li, Z.; Zhao, A.; Feng, J. Self-emulsifying drug delivery system and the applications in herbal drugs. Drug Deliv. 2015, 22, 475–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattaram, V.A.; Graefe, U.; Kohlert, C.; Veit, M.; Derendorf, H. Pharmacokinetics and bioavailability of herbal medicinal products. Phytomedicine 2002, 9 (Suppl. 3), 1–33. [Google Scholar] [CrossRef] [PubMed]
- Sachetto, J.-P.; Bufton, R.; Buser, T. Soft Gelatin Capsule Comprising Omega-3 Polyunsaturated Fatty Acid; Tillotts Pharma Ag: Ziefen, Switzerland, 2005; p. 15. [Google Scholar]
- Pissinati, R.; Oliveira, W.P. Enteric coating of soft gelatin capsules by spouted bed: Effect of operating conditions on coating efficiency and on product quality. Eur. J. Pharm. Biopharm. 2003, 55, 313–321. [Google Scholar] [CrossRef]
- Siewert, M.; Dressman, J.; Brown, C.; Shah, V. FIP/AAPS Guidelines for dissolution/In vitro release testing of novel/special dosage forms. AAPS PharmSciTech. 2003, 4, 43–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augsburger, L.L. Hard and Soft Shell Capsules. In Modern Pharmaceutics; Banker, G.S., Rhodes, C.T., Eds.; Marcel Dekker, Inc.: New York, NY, USA, 2002; pp. 512–575. [Google Scholar]
- Reich, G. Formulation and physical properties of soft capsules. In Pharmaceutical Capsules; Podczeck, F., Jones, B.E., Eds.; Pharmaceutical Press: London, UK, 2004; pp. 201–212. [Google Scholar]
- Hassan, E.M.; Kindt, W.W.; Gordon, R. Chewable Soft Capsule; Patheon Softgels Inc.: High Point, NC, USA, 2012. [Google Scholar]
- Schwab, E. Hot Melt-Filled Soft Capsules; Swiss Caps Rechte und Lizenzen AG: Kirchberg, Switzerland, 2010; p. 15. [Google Scholar]
- Bekerman, T.; Golenser, J.; Domb, A. Cyclosporin nanoparticulate lipospheres for oral administration. J. Pharm. Sci. 2004, 93, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Lissy, M.; Scallion, R.; Stiff, D.D.; Moore, K. Pharmacokinetic comparison of an oral diclofenac potassium liquid-filled soft gelatin capsule with a diclofenac potassium tablet. Expert Opin. Pharmacother. 2010, 11, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Bende, G.; Biswal, S.; Bhad, P.; Chen, Y.; Salunke, A.; Winter, S.; Wagner, R.; Sunkara, G. Relative bioavailability of diclofenac potassium from softgel capsule versus powder for oral solution and immediate-release tablet formulation. Clin. Pharm. Drug Dev. 2016, 5, 76–82. [Google Scholar] [CrossRef]
- Fonkwe, L.G.; Archibald, D.A.; Gennadios, A. Non-Gelatin Capsule Shell Formulation in Greensboro, NC; Patheon Softgels Inc.: High Point, NC, USA, 2005. [Google Scholar]
- Jones, B.E.; Podczeck, F.; Lukas, P. Capsule shell manufacture. In Pharmaceutical Dosage Forms: Capsules; Augsburger, L.L., Hoag, S.W., Eds.; Taylor & Francis Group: London, UK, 2018; pp. 75–110. [Google Scholar]
- Duconseille, A.; Astruc, T.; Quintana, N.; Meersman, F.; Sante-Lhoutellier, V. Gelatin structure and composition linked to hard capsule dissolution: A review. Food Hydrocoll. 2015, 43, 360–376. [Google Scholar] [CrossRef]
- Lin, L.; Regenstein, J.M.; Lv, S.; Lu, J.; Jiang, S. An overview of gelatin derived from aquatic animals: Properties and modification. Trends Food Sci. Technol. 2017, 68, 102–112. [Google Scholar] [CrossRef]
- Stegemann, S. Non-gelatin based capsules. In Pharmaceutical Dosage Forms: Capsules; Augsburger, L., Hoag, S., Eds.; CRC Press: Boca Raton, FL, USA, 2017; pp. 110–130. [Google Scholar]
- Kommareddy, S.; Shenoy, D.; Amiji, M. Gelatin Nanoparticles and Their Biofunctionalization. In Nanotechnologies for the Life Sciences, Vol. 1 Biofunctionalization of Nanomaterials; Kumar, F., Ed.; Wiley-VCH: Weinheim, Germany, 2007; pp. 330–352. [Google Scholar]
- Djagny, K.B.; Wang, Z.; Xu, S. Gelatin: A Valuable Protein for Food and Pharmaceutical Industries: Review. Crit. Rev. Food Sci. Nutr. 2001, 41, 481–492. [Google Scholar] [CrossRef]
- Jongjareonrak, A.; Benjakul, S.; Visessanguan, W.; Tanaka, M. Skin gelatin from bigeye snapper and brownstripe red snapper: Chemical compositions and effect of microbial transglutaminase on gel properties. Food Hydrocoll. 2006, 20, 1216–1222. [Google Scholar] [CrossRef]
- Lai, J.-Y.; Lin, P.-K.; Hsiue, G.-H.; Cheng, H.-Y.; Huang, S.-J.; Li, Y.-T. Low bloom strength gelatin as a carrier for potential use in retinal sheet encapsulation and transplantation. Biomacromolecules 2008, 10, 310–319. [Google Scholar] [CrossRef]
- Remuñán-López, C.; Bodmeier, R. Effect of formulation and process variables on the formation of chitosan-gelatin coacervates. Int. J. Pharm. 1996, 135, 63–72. [Google Scholar] [CrossRef]
- Osorio, F.A.; Bilbao, E.; Bustos, R.; Alvarez, F. Effects of concentration, bloom degree, and pH on gelatin melting and gelling temperatures using small amplitude oscillatory rheology. Int. J. Food Prop. 2007, 10, 841–851. [Google Scholar] [CrossRef]
- Hoare, T.R.; Kohane, D.S. Hydrogels in drug delivery: Progress and challenges. Polymer 2008, 49, 1993–2007. [Google Scholar] [CrossRef] [Green Version]
- Hom, F.S.; Veresh, S.A.; Miskel, J.J. Soft gelatin capsules I: Factors affecting capsule shell dissolution rate. J. Pharm. Sci. 1973, 62, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Qiao, C.; Chen, G.; Li, Y.; Li, T. Viscosity properties of gelatin in solutions of monovalent and divalent Salts. Korea-Aust. Rheol. J. 2013, 25, 227–231. [Google Scholar] [CrossRef]
- Davis, C.E.; Oakes, E.T.; Browne, H.H. Viscosity of gelatin solutions. J. Am. Chem. Soc. 1921, 43, 1526–1538. [Google Scholar] [CrossRef]
- Horský, J.; Švantner, J. Gelatin renaturation and viscosity of dilute gelatin solutions. Polym. Int. 1993, 32, 159–164. [Google Scholar] [CrossRef]
- Cho, S.M.; Gu, Y.S.; Kim, S.B. Extracting optimization and physical properties of yellowfin tuna (Thunnus albacares) skin gelatin compared to mammalian gelatins. Food Hydrocoll. 2005, 19, 221–229. [Google Scholar] [CrossRef]
- Lim, Y.P.; Mohammad, A.W. Physicochemical properties of mammalian melatin in relation to membrane process requirement. Food Bioprocess Tech. 2009, 4, 304–311. [Google Scholar] [CrossRef]
- Ninan, G.; Joseph, J.; Abubacker, Z. Physical, mechanical, and barrier properties of carp and mammalian skin gelatin films. J. Food Sci. 2010, 75, E620–E626. [Google Scholar] [CrossRef] [PubMed]
- Ninan, G.; Joseph, J.; Aliyamveettil, Z. A comparative study on the physical, chemical and functional properties of carp skin and mammalian gelatins. J. Food Sci. Technol. 2014, 51, 2085–2091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Digenis, G.A.; Gold, T.B.; Shah, V.P. Cross-linking of gelatin capsules and its relevance to their in vitro-in vivo performance. J. Pharm. Sci. 1994, 83, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.; Grohens, Y.; Vandanjon, L.; Bourseau, P.; Balnois, E.; Levesque, G. A comparative study of the rheological and structural properties of gelatin gels of mammalian and fish origins. In Macromolecular Symposia; WILEY-VCH Verlag: Weinheim, Germany, 2003; Volume 203, pp. 331–338. [Google Scholar]
- Inal, O.; Yapar, E.A. Effect of mechanical properties on the release of meloxicam from poloxamer gel bases. Indian J. Pharm. Sci. 2013, 75, 700–706. [Google Scholar] [PubMed]
- Meyer, J.D.; Manning, M.C. Hydrophobic ion pairing: Altering the solubility properties of biomolecules. Pharm. Res. 1998, 15, 188–193. [Google Scholar] [CrossRef]
- Overholt, S.M. Chewable Soft Capsule; Banner Pharmacaps, Inc.: High Point, NC, USA, 2000. [Google Scholar]
- Gullapalli, R. Soft gelatin capsules (Softgels). J. Pharm. Sci. 2010, 99, 4107–4148. [Google Scholar] [CrossRef] [PubMed]
- Joysun Pharm. Softgel Making Machine. Available online: https://www.jspharma.net/category/softgel-capsule-auxiliary-equipment/ (accessed on 28 October 2020).
- Armstrong, N.A.; James, K.C.; Pugh, W.K. Drug migration into soft gelatin capsule shells and its effect on in-vitro availability. J. Pharm. Pharmacol. 1984, 36, 361–365. [Google Scholar] [CrossRef]
- Serajuddin, A.T.; Sheen, P.C.; Augustine, M.A. Water migration from soft gelatin capsule shell to fill material and its effect on drug solubility. J. Pharm. Sci. 1986, 75, 62–64. [Google Scholar] [CrossRef]
- Eldridge, J.E.; Ferry, J.D. Studies of the cross-linking process in gelatin gels. III. Dependence of melting point on concentration and molecular weight. J. Phys. Chem. 1954, 58, 992–995. [Google Scholar] [CrossRef]
- Meyer, M.C.; Straughn, A.B.; Mhatre, R.M.; Hussain, A.; Shah, V.P.; Bottom, C.B.; Cole, E.T.; Lesko, L.L.; Mallinowski, H.; Williams, R.L. The effect of gelatin cross-linking on the bioequivalence of hard and soft gelatin acetaminophen capsules. Pharm. Res. 2000, 17, 962–966. [Google Scholar] [CrossRef] [PubMed]
- Constantinides, P.P. Lipid microemulsions for improving drug dissolution and oral absorption: Physical and biopharmaceutical aspects. Pharm. Res. 1995, 12, 1561–1572. [Google Scholar] [CrossRef] [PubMed]
- Pillay, V.; Fassihi, R. A new method for dissolution studies of lipid-filled capsules employing nifedipine as a model drug. Pharm. Res. 1999, 16, 333–337. [Google Scholar] [CrossRef]
- Felton, L.A.; Shah, N.H.; Zhang, G.; Infeld, M.H.; Malick, A.W.; McGinity, J.W. Physical-mechanical properties of film-coated soft gelatin capsules. Int. J. Pharm. 1996, 127, 203–211. [Google Scholar] [CrossRef]
- Nazzal, S.; Wang, Y. Characterization of soft gelatin capsules by thermal analysis. Int. J. Pharm. 2001, 230, 35–45. [Google Scholar] [CrossRef]
- Agrawal, A.; Kumar, A.; Gide, P. Formulation of solid self-nanoemulsifying drug delivery systems using N -methyl pyrrolidone as cosolvent. Drug Dev. Ind. Pharm. 2014, 41, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Dokania, S.; Joshi, A.K. Self-microemulsifying drug delivery system (SMEDDS)—Challenges and road ahead. Drug Deliv. 2015, 22, 675–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beg, S.; Sandhu, P.S.; Batra, R.S.; Khurana, R.K.; Singh, B. QbD-based systematic development of novel optimized solid self-nanoemulsifying drug delivery systems (SNEDDS) of lovastatin with enhanced biopharmaceutical performance. Drug Deliv. 2015, 22, 765–784. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.D.; Park, Y.J. In vitro and in vivo evaluation of a self-microemulsifying drug delivery system for the poorly soluble drug fenofibrate. Arch. Pharm. Res. 2014, 37, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Felice, B.; Prabhakaran, M.P.; Rodriguez, A.P.; Ramakrishna, S. Drug delivery vehicles on a nano-engineering perspective. Mater. Sci. Eng. C Mater. Biol. Appl. 2014, 41, 178–195. [Google Scholar] [CrossRef] [PubMed]
- Damian, F.; Harati, M.; Pathak, V.; Schwartzenhauer, J.; Durham, D.; Quiquero, V.; Van Cauwenberghe, O.; Wettig, S.D. Development of a discriminating dissolution method for immediate-release soft gelatin capsules containing a BCS Class II compound. Dissolut. Technol. 2006, 23, 6–11. [Google Scholar] [CrossRef]
- Poppe, J. Gelatin. In Thickening and Gelling Agents for Food; Imeson, A., Ed.; Springer: Boston, MA, USA, 1997; pp. 144–168. [Google Scholar]
- Felton, L.A.; Haase, M.M.; Shah, N.H.; Zhang, G.; Infeld, M.H.; Malick, A.W.; McGinity, J.W. Physical and enteric properties of soft gelatin capsules coated with eudragit® L 30 D-55. Int. J. Pharm. 1995, 113, 17–24. [Google Scholar] [CrossRef]
- Hom, F.S.; Veresh, S.A.; Ebert, W.R. Soft gelatin capsules II: Oxygen permeability study of capsule shells. J. Pharm. Sci. 1975, 64, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Muñoz, P.; Villalobos, R.; Chiralt, A. Effect of cross-linking using aldehydes on properties of glutenin-rich films. Food Hydrocoll. 2004, 18, 403–411. [Google Scholar] [CrossRef]
- Corma, A.; Hamid, S.B.A.; Iborra, S.; Velty, A. Surfactants from biomass: A two-step cascade reaction for the synthesis of sorbitol fatty acid esters using solid acid catalysts. ChemSusChem 2008, 1, 85–90. [Google Scholar] [CrossRef]
- Buice, R.G., Jr.; Gold, T.B.; Lodder, R.A.; Digenis, G.A. Determination of moisture in intact gelatin capsules by near-infrared spectrophotometry. Pharm. Res. 1995, 12, 161–163. [Google Scholar] [CrossRef]
- Pandey, P.; Hamey, R.; Bindra, D.S.; Huang, Z.; Mathias, N.; Eley, T.; Crison, J.; Yan, B.; Perrone, R.; Vemavarapu, C. From bench to humans: Formulation development of a poorly water soluble drug to mitigate food effect. AAPS PharmSciTech 2014, 15, 407–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreton, R.C.; Armstrong, N.A. The effect of film composition on the diffusion of ethanol through soft gelatin films. Int. J. Pharm. 1998, 161, 123–131. [Google Scholar] [CrossRef]
- Rowe, R.C.; Sheskey, P.; Owen, S.C. Handbook of Pharmaceutical Excipients, 5th ed.; Pharmaceutical Press: London, UK, 2006. [Google Scholar]
- Vanin, F.; Sobral, P.; Menegalli, F.; Carvalho, R.; Habitante, A. Effects of plasticizers and their concentrations on thermal and functional properties of gelatin-based films. Food Hydrocoll. 2005, 19, 899–907. [Google Scholar] [CrossRef]
- Noyes, A.A.; Whitney, W.R. The rate of solution of solid substances in their own solutions. J. Am. Chem. Soc. 1897, 19, 930–934. [Google Scholar] [CrossRef] [Green Version]
- Azarmi, S.; Roa, W.; Lobenberg, R. Current perspectives in dissolution testing of conventional and novel dosage forms. Int. J. Pharm. 2007, 328, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Costa, P.; Lobo, J.M.S. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef]
- Dokoumetzidis, A.; Macheras, P. A century of dissolution research: From Noyes and Whitney to the biopharmaceutics classification system. Int. J. Pharm. 2006, 321, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siepmann, J.; Siepmann, F. Mathematical modeling of drug dissolution. Int. J. Pharm. 2013, 453, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Van den Mooter, G. The use of amorphous solid dispersions: A formulation strategy to overcome poor solubility and dissolution rate. Drug Discov. Today Technol. 2012, 9, e79–e85. [Google Scholar] [CrossRef]
- Damian, F.; Blaton, N.; Desseyn, H.; Clou, K.; Augustijns, P.; Naesens, L.; Balzarini, J.; Kinget, R.; Van den Mooter, G. Solid state properties of pure UC-781 and solid dispersions with polyvinylpyrrolidone (PVP K30). J. Pharm. Pharmacol. 2001, 53, 1109–1116. [Google Scholar] [CrossRef] [PubMed]
- Siepmann, J.; Peppas, N.A. Modeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC). Adv. Drug Deliv. Rev. 2001, 48, 139–157. [Google Scholar] [CrossRef]
- Singh, A.; Worku, Z.A.; Van den Mooter, G. Oral formulation strategies to improve solubility of poorly water-soluble drugs. Expert Opin. Drug Deliv. 2011, 8, 1361–1378. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.; Goldman, D.; Krumme, M.; Rohan, L.C.; Smoot, S.; Friend, D.R. Advances in development, scale-up and manufacturing of microbicide gels, films, and tablets. Antivir. Res. 2010, 88 (Suppl. 1), S19–S29. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, T. Rate of release of medicaments from ointment bases containing drugs in suspension. J. Pharm. Sci. 1961, 50, 874–875. [Google Scholar] [CrossRef]
- Higuchi, T. Mechanism of sustained-actionmedication, theoretical analysis of rate of release of of solid drugs dispersed in solid matrices. J. Pharm. Sci. 1963, 52, 1145–1149. [Google Scholar] [CrossRef]
- Wang, T.T.; Kwei, T.K.; Frisch, H.L. Diffusion in glassy polymers. III. J. Polym. Sci. 1969, 7, 2019–2028. [Google Scholar] [CrossRef]
- Ritger, P.L.; Peppas, N.A. A simple equation for description of solute release I. Fickian and non-fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. J. Control. Release 1987, 5, 23–36. [Google Scholar] [CrossRef]
- Peppas, N.A.; Sahlin, J.J. A simple equation for the description of solute release. III. Coupling of diffusion and relaxation. Int. J. Pharm. 1989, 57, 169–172. [Google Scholar] [CrossRef]
- Connors, K.A.; Mecozzi, S. Thermodynamics of Pharmaceutical Systems: An Introduction to Theory and Applications, 2nd ed.; Wiley: Hoboken, NJ, USA, 2010; pp. 182–200. [Google Scholar]
- Gupta, A.; Hunt, R.L.; Shah, R.B.; Sayeed, V.A.; Khan, M.A. Disintegration of highly soluble immediate release tablets: A surrogate for dissolution. AAPS PharmSciTech 2009, 10, 495–499. [Google Scholar] [CrossRef] [Green Version]
- The United States Pharmacopeia and National Formulary (USP 42, NF-37) <701> Disintegration; The United States Pharmacopeial Convenction, Inc.: Rockville, MD, USA, 2019.
- The United States Pharmacopeia and National Formulary (USP 38, NF-33) <2040> Disintegration and Dissolution of Dietary Supplements; The United States Pharmacopeial Convention Inc.: Rockville, MD, USA, 2015; pp. 1774–1781.
- Almukainzi, M.; Salehi, M.; Chacra, N.A.; Löbenberg, R. Comparison of the rupture and disintegration tests for soft-shell capsules. Dissolut. Technol. 2011, 18, 21–25. [Google Scholar] [CrossRef]
- Bachour, G.; Bou-Chacra, N.A.; Löbenberg, R. Evaluation of the rupture test for stability studies of soft-shell capsules. Dissolut. Technol. 2017, 24, 16–19. [Google Scholar] [CrossRef]
- Brown, C.K.; Friedel, H.D.; Barker, A.R.; Buhse, L.F.; Keitel, S.; Cecil, T.L.; Kraemer, J.; Morris, J.M.; Reppas, C.; Stickelmeyer, M.P.; et al. FIP/AAPS joint workshop report: Dissolution/in vitro release testing of novel/special dosage forms. AAPS PharmSciTech 2011, 12, 782–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The United States Pharmacopeia and National Formulary (USP 42, NF-37) <711> Dissolution; The United States Pharmacopeial Convenction, Inc.: Rockville, MD, USA, 2019.
- Food and Drug Administration: Guidance for Industry: Dissolution Testing of Immediate Release Solid Oral Dosage Forms; US Department of Health and Human Services: Rockville, MD, USA, 1997. Available online: https://www.fda.gov/media/70936/download (accessed on 3 February 2021).
- Wang, Q.; Gray, V. Handbook of Pharmaceutical Analysis by HPLC. In HPLC in Dissolution Testing; Ahuja, S., Dong, M.W., Eds.; Academic Press: San Diego, CA, USA, 2005; pp. 379–400. [Google Scholar]
- USP 4 Flow-Through Dissolution Systems; SOTAX Corporation: Hopkinton, MA, USA; Available online: http://disotax.com.br/uploads/9/2/1/2/92120604/ce_7smart_mb10023en-01_2.pdf (accessed on 3 February 2021).
- Shohin, I.E.; Grebenkin, D.; Malashenko, E.A.; Stanishevskii, Y.; Ramenskaya, G.V. A brief review of the FDA dissolution methods database. Dissolut. Technol. 2016, 23, 6–10. [Google Scholar] [CrossRef]
- Gautam, J.; Schott, H. Interaction of anionic compounds with gelatin I: Binding studies. J. Pharm. Sci. 1994, 83, 922–930. [Google Scholar] [CrossRef] [PubMed]
- Amidon, G.L.; Lennernas, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinno, J.; Oh, D.; Crison, J.R.; Amidon, G.L. Dissolution of ionizable water-insoluble drugs: The combined effect of pH and surfactant. J. Pharm. Sci. 2000, 89, 268–274. [Google Scholar] [CrossRef] [Green Version]
- Gibaldi, M.; Feldman, S. Mechanisms of surfactant effects on drug absorption. J. Pharm. Sci. 1970, 59, 579–589. [Google Scholar] [CrossRef]
- Damian, F.; Blaton, N.; Naesens, L.; Balzarini, J.; Kinget, R.; Augustijns, P.; Van den Mooter, G. Physicochemical characterization of solid dispersions of the antiviral agent UC-781 with polyethylene glycol 6000 and Gelucire 44/14. Eur. J. Pharm. Sci. 2000, 10, 311–322. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, E.; Yang, J.; Cao, Z. Strategies to improve micelle stability for drug delivery. Nano Res. 2018, 11, 4985–4998. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Malayev, V.; Rao, V.; Hussain, M. Effect of sodium lauryl sulfate in dissolution media on dissolution of hard gelatin capsule shells. Pharm. Res. 2004, 21, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Pennings, F.H.; Kwee, B.L.; Vromans, H. Influence of enzymes and surfactants on the disintegration behavior of cross-linked hard gelatin capsules during dissolution. Drug Dev. Ind. Pharm. 2006, 32, 33–37. [Google Scholar] [CrossRef]
- Vertzoni, M.; Augustijns, P.; Grimm, M.; Koziolek, M.; Lemmens, G.; Parrott, N.; Pentafragka, C.; Reppas, C.; Rubbens, J.; Van Den Abeele, J.; et al. Impact of regional differences along the gastrointestinal tract of healthy adults on oral drug absorption: An UNGAP review. Eur. J. Pharm. Sci. 2019, 134, 153–175. [Google Scholar] [CrossRef]
- Dressman, J.B.; Amidon, G.L.; Reppas, C.; Shah, V.P. Dissolution testing as a prognostic tool for oral drug absorption: Immediate release dosage forms. Pharm. Res. 1998, 15, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Vertzoni, M.; Dressman, J.; Butler, J.; Hempenstall, J.; Reppas, C. Simulation of fasting gastric conditions and its importance for the in vivo dissolution of lipophilic compounds. Eur. J. Pharm. Biopharm. 2005, 60, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Klein, S. The use of biorelevant dissolution media to forecast the in vivo performance of a drug. AAPS J. 2010, 12, 397–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galia, E.; Nicolaides, E.; Hörter, D.; Löbenberg, R.; Reppas, C.; Dressman, J.B. Evaluation of various dissolution media for predicting in vivo performance of class I and II drugs. Pharm. Res. 1998, 15, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Maharaj, A.R.; Edginton, A.N.; Fotaki, N. Assessment of age-related changes in pediatric gastrointestinal solubility. Pharm. Res. 2016, 33, 52–71. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.M. Gastric hypochlorhydria and achlorhydria in older adults. JAMA 1997, 278, 1659–1660. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.; Marques, M. Question and answer section. Dissolut. Technol. 2013, 20. [Google Scholar] [CrossRef]
- Soltero, R.A.; Hoover, J.M.; Jones, T.F.; Standish, M. Effects of sinker shapes on dissolution profiles. J. Pharm. Sci. 1989, 78, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Monterroza, D.; De León, L.P. Development of a USP Apparatus 3 Dissolution Method for Progesterone Soft Gelatin Capsules. Softigel by Procaps. Available online: http://softigel.com/ArchivosSoftigel/Softigel/a4/a4eea097-1bdf-4456-beed-f59f60ffa658.pdf (accessed on 28 December 2020).
- FDA Dissolution Methods Database. Available online: https://www.accessdata.fda.gov/scripts/cder/dissolution/index.cfm (accessed on 28 October 2020).
- Miller, D.A.; Gamba, M.; Sauer, D.; Purvis, T.P.; Clemens, N.T.; Williams, R.O. Evaluation of the USP dissolution test method A for enteric-coated articles by planar laser-induced fluorescence. Int. J. Pharm. 2007, 330, 61–72. [Google Scholar] [CrossRef]
- Porter, S.C.; Ridgway, K. The permeability of enteric coatings and the dissolution rates of coated tablets. J. Pharm. Pharmacol. 1982, 34, 5–8. [Google Scholar] [CrossRef]
- Guo, H.X.; Heinämäki, J.; Yliruusi, J. Diffusion of a freely water-soluble drug in aqueous enteric-coated pellets. AAPS PharmSciTech 2002, 3, 97–104. [Google Scholar] [CrossRef] [Green Version]
- 123. The United States Pharmacopeia <711> Dissolution: Stage 6 Harmonization; The United States Pharmacopeial Convenction, Inc.: Rockville, MD, USA, 2011; Available online: https://www.usp.org/sites/default/files/usp/document/harmonization/gen-method/stage_6_monograph_25_feb_2011.pdf (accessed on 3 February 2021).
- Zhao, H.; Cafiero, S.; Williams, Z.; Bynum, K. Practical considerations for the development of a robust two-step dissolution test for enteric-coated immediate-and extended-release solid oral dosage formulations. Dissolut. Technol. 2011, 18, 6–10. [Google Scholar] [CrossRef]
- Lo, L.; Lu, X.; Lloyd, D. Dissolution testing of a controlled-release capsule formulation: Challenges and solutions using a semi-automated dissolution system. Dissolut. Technol. 2013, 20, 6–12. [Google Scholar] [CrossRef]
- The European Medicines Agency. 2017: Dissolution Specification for Generic Oral Immediate Release Products; The European Medicines Agency: London, UK, 2017. [Google Scholar]
- Gray, V.A.; Cole, E.; Toma, J.M.D.R.; Ghidorsi, L.; Guo, J.-H.; Han, J.-H.; Han, F.; Hosty, C.T.; Kochling, J.D.; Kraemer, J.; et al. Use of enzymes in the dissolution testing of gelatin capsules and gelatin-coated tablets-Revisions to dissolution <711> and disintegration and dissolution of dietary supplements <2040>. Dissolut. Technol. 2014, 21, 6–19. [Google Scholar] [CrossRef]
- Marques, M.R.C. Enzymes in the dissolution testing of gelatin capsules. AAPS PharmSciTech 2014, 15, 1410–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchais, H.; Cayzeele, G.; Legendre, J.Y.; Skiba, M.; Arnaud, P. Cross-linking of hard gelatin carbamazepine capsules: Effect of dissolution conditions on in vitro drug release. Eur. J. Pharm. Sci. 2003, 19, 129–132. [Google Scholar] [CrossRef]
- Song, X.; Cui, Y.; Xie, M. Gelatin capsule shell cross-linking: Tier II dissolution method development in the presence of sodium lauryl sulfate. Pharm. Technol. 2011, 35, 62–68. [Google Scholar]
- Nishimura, H.; Hayashi, C.; Aiba, T.; Okamoto, I.; Miyamoto, Y.; Nakade, S.; Takeda, K.; Kurosaki, Y. Application of the correlation of in vitro dissolution behavior and in vivo plasma concentration profile (IVIVC) for soft-gel capsules-a pointless pursuit? Biol. Pharm. Bull. 2007, 30, 2221–2225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, R.C.; Dias, C.L.; Donato, E.M.; Martins, L.A.; Bergold, A.M.; Fröehlich, P.E. Development and validation of dissolution test for ritonavir soft gelatin capsules based on in vivo data. Int. J. Pharm. 2007, 338, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Donato, E.M.; Martins, L.A.; Fröehlich, P.E.; Bergold, A.M. Development and validation of dissolution test for lopinavir, a poorly water-soluble drug, in soft gel capsules, based on in vivo data. J. Pharm. Biomed. Anal. 2008, 47, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Möller, H.; Wirbitzki, E. Regulatory aspects of modified release dosage forms: Special cases of dissolution testing using the flow-through system. Boll. Chim. Farm. 1993, 132, 105–115. [Google Scholar] [PubMed]
- Rawat, A.; Burgess, D.J. USP apparatus 4 method for in vitro release testing of protein loaded microspheres. Int. J. Pharm. 2011, 409, 178–184. [Google Scholar] [CrossRef]
- Zolnik, B.S.; Raton, J.-L.; Burgess, D.J. Application of USP apparatus 4 and in situ fiber optic analysis to microsphere release testing. Dissolut. Technol. 2005, 12, 11–14. [Google Scholar] [CrossRef]
- Heng, D.; Cutler, D.J.; Chan, H.K.; Yun, J.; Raper, J.A. What is a suitable dissolution method for drug nanoparticles? Pharm. Res. 2008, 25, 1696–1701. [Google Scholar] [CrossRef]
- Nir, I.; Lu, X. In situ UV fiber optics for dissolution testing—What, why, and where we are after 30 years. Dissolut. Technol. 2018, 25, 70–77. [Google Scholar] [CrossRef]
- The United States Pharmacopeia and National Formulary (USP 31, NF-26) <711> Dissolution; The United States Pharmacopeial Convenction, Inc.: Rockville, MD, USA, 2008.
- Hu, J.; Kyad, A.; Ku, V.; Zhou, P.; Cauchon, N. A comparison of dissolution testing on lipid soft gelatin capsules using USP apparatus 2 and apparatus 4. Dissolut. Technol. 2005, 12, 6–9. [Google Scholar] [CrossRef]
- Neisingh, S.E.; Sam, A.P.; de Nijs, H. A Dissolution method for hard and soft gelatin capsules containing testosterone undecanoate in oleic acid. Drug Dev. Ind. Pharm. 1986, 12, 651–663. [Google Scholar] [CrossRef]
- Lu, X.; Shah, P. Dissolution of gelatin capsules: Evidence and confirmation of cross-linking. Dissolut. Technol. 2017, 24, 6–21. [Google Scholar] [CrossRef]
- Carstensen, J.T.; Rhodes, C.T. Pellicule formation in gelatin capsules. Drug Dev. Ind. Pharm. 1993, 19, 2709–2712. [Google Scholar] [CrossRef]
- Gold, T.B.; Buice, R.G., Jr.; Lodder, R.A.; Digenis, G.A. Detection of formaldehyde-induced crosslinking in soft elastic gelatin capsules using near-infrared spectrophotometry. Pharm. Dev. Technol. 1998, 3, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Rao, K.V.; Venugopal, K.; Manikandan, R. Alteration in dissolution characteristic of gelatin containing formulations: A review of the problem, test methods, and solutions. Pharm. Technol. 2002, 26, 36–58. [Google Scholar]
- Gold, T.; Smith, S.; Digenis, G. Studies on the influence of pH and pancreatin on 13C-formaldehyde-induced gelatin cross-links using Nuclear Magnetic Resonance. Pharm. Dev. Technol. 1996, 1, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Albert, K.; Peters, B.; Bayer, E. Crosslinking of gelatin with formaldehyde: A 13C NMR study. Z. Nat. B 1986, 41, 351–358. [Google Scholar] [CrossRef]
- Albert, K.; Bayer, E.; Wörsching, A.; Vögele, H. Investigation of the hardening reaction of gelatin with 13C labeled formaldehyde by solution and solid state 13C NMR spectroscopy. Z. Nat. B 1991, 46, 385–389. [Google Scholar] [CrossRef]
- Cooper, J.W., Jr.; Ansel, H.C.; Cadwallader, D.E. Liquid and solid solution interactions of primary certified colorants with pharmaceutical gelatins. J. Pharm. Sci. 1973, 62, 1156–1164. [Google Scholar] [CrossRef] [PubMed]
- Murthy, K.S.; Enders, N.A.; Fawzi, M.B. Dissolution stability of hard-shell capsule products, part I: The effect of exaggerated storage conditions. Pharm. Technol. 1989, 13, 72–84. [Google Scholar]
- Tengroth, C.; Gasslander, U.; Andersson, F.O.; Jacobsson, S.P. Cross-linking of gelatin capsules with formaldehyde and other aldehydes: An FTIR spectroscopy study. Pharm. Dev. Technol. 2005, 10, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Chafetz, L.; Hong, W.H.; Tsilifonis, D.C.; Taylor, A.K.; Philip, J. Decrease in the rate of capsule dissolution due to formaldehyde from polysorbate 80 autoxidation. J. Pharm. Sci. 1984, 73, 1186–1187. [Google Scholar] [CrossRef] [PubMed]
- Bottom, C.B.; Clark, M.; Carstensen, J.T. Dissolution testing of soft shell capsules-acetaminophen and nifedipine. J. Pharm. Sci. 1997, 86, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Schwier, J.R.; Cooke, G.G.; Hartauer, K.J.; Yu, L. Rayon: A source of furfural· a reactive aldehyde capable of insolubilizing gelatin capsules. Pharm. Technol. 1993, 17, 78–80. [Google Scholar]
- Ofner, C.M.; Bubnis, W.A. Chemical and swelling evaluations of amino group crosslinking in gelatin and modified gelatin matrices. Pharm. Res. 1996, 13, 1821–1827. [Google Scholar] [CrossRef]
- Adesunloye, T.A.; Stach, P.E. Effect of glycine/citric acid on the dissolution stability of hard gelatin capsules. Drug Dev. Ind. Pharm. 1998, 24, 493–500. [Google Scholar] [CrossRef]
- El-Fattah, S.A.; Khalil, S.A.H. Variations in dissolution rates of sugar-coated chlorpromazine tablets. Int. J. Pharm. 1984, 18, 225–234. [Google Scholar] [CrossRef]
- Ray-Johnson, M.L.; Jackson, I.M. Temperature-related incompatibility between gelatin and calcium carbonate in sugar-coated tablets. J. Pharm. Pharmacol. 1976, 28, 309–310. [Google Scholar] [CrossRef]
- Guyot, M.; Fawaz, F.; Maury, M. In vitro release of theophylline from cross-linked gelatin capsules. Int. J. Pharm. 1996, 144, 209–216. [Google Scholar] [CrossRef]
- Bindra, D.S.; Williams, T.D.; Stella, V.J. Degradation of O6-Benzylguanine in Aqueous Polyethylene Glycol 400 (PEG 400) Solutions: Concerns with Formaldehyde in PEG 400. Pharm. Res. 1994, 11, 1060–1064. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Kozlowski, B.M.; Chang, E.P. Analysis of aldehydes in excipients used in liquid/semi-solid formulations by gas chromatography-negative chemical ionization mass spectrometry. J. Chromatogr. A 2007, 1160, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Hamburger, R.; Azaz, E.; Donbrow, M. Autoxidation of polyoxyethylenic non-ionic surfactants and of polyethylene glycols. Pharm. Acta Helv. 1975, 50, 10–17. [Google Scholar]
- McGinity, J.W.; Hill, J.A.; La Via, A.L. Influence of peroxide impurities in polyethylene glycols on drug stability. J. Pharm. Sci. 1975, 64, 356–357. [Google Scholar] [CrossRef]
- Donbrow, M.; Azaz, E.; Pillersdorf, A. Autoxidation of polysorbates. J. Pharm. Sci. 1978, 67, 1676–1681. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.M.; Taylor, W.F. Degradation of fenprostalene in polyethylene glycol 400 solution. J. Pharm. Sci. 1984, 73, 1414–1417. [Google Scholar] [CrossRef]
- Wasylaschuk, W.R.; Harmon, P.A.; Wagner, G.; Harman, A.B.; Templeton, A.C.; Xu, H.; Reed, R.A. Evaluation of hydroperoxides in common pharmaceutical excipients. J. Pharm. Sci. 2007, 96, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Erlich, L.; Yu, D.; Pallister, D.A.; Levinson, R.S.; Gole, D.G.; Wilkinson, P.A.; Erlich, R.E.; Reeve, L.E.; Viegas, T.X. Relative bioavailability of danazol in dogs from liquid-filled hard gelatin capsules. Int. J. Pharm. 1999, 179, 49–53. [Google Scholar] [CrossRef]
- Rowe, R.C.; Sheskey, P.J.; Quinn, M.E. Handbook of Pharmaceutical Excipients, 6th ed.; Pharmaceutical Press: London, UK; American Pharmacists Association: Washington, DC, USA, 2009. [Google Scholar]
- Bühler, V. Kollidon®-Polyvinylpyrrolidone Excipients for the Pharmaceutical Industry, 9th ed.; BASF: Ludwigshafen, Germany, 2008. [Google Scholar]
- Hartauer, K.; Bucko, J.; Cooke, G.; Mayer, R. The effect of rayon coiler on the dissolution stability of hard-shell gelatin capsules. Pharm. Technol. 1993, 17, 76–83. [Google Scholar]
- Caliph, S.M.; Charman, W.N.; Porter, C.J. Effect of short-, medium-, and long-chain fatty acid-based vehicles on the absolute oral bioavailability and intestinal lymphatic transport of halofantrine and assessment of mass balance in lymph-cannulated and non-cannulated rats. J. Pharm. Sci. 2000, 89, 1073–1084. [Google Scholar] [CrossRef]
- Fischler, M.; Frisch, E.P.; Ortengren, B. Plasma concentrations after oral administration of different pharmaceutical preparations of clomethiazole. Acta Pharm. Suec. 1973, 10, 483–492. [Google Scholar] [PubMed]
- Yannas, I.V.; Tobolsky, A.V. Cross-linking of gelatine by dehydration. Nature 1967, 215, 509–510. [Google Scholar] [CrossRef] [PubMed]
- Welz, M.M.; Ofner, C.M. Examination of self-crosslinked gelatin as a hydrogel for controlled release. J. Pharm. Sci. 1992, 81, 85–90. [Google Scholar] [CrossRef]
- Singh, S.; Manikandan, R.; Singh, S. Stability testing for gelatin-based formulations: Rapidly evaluating the possibility of a reduction in dissolution rates. Pharm. Technol. 2000, 24, 58–72. [Google Scholar]
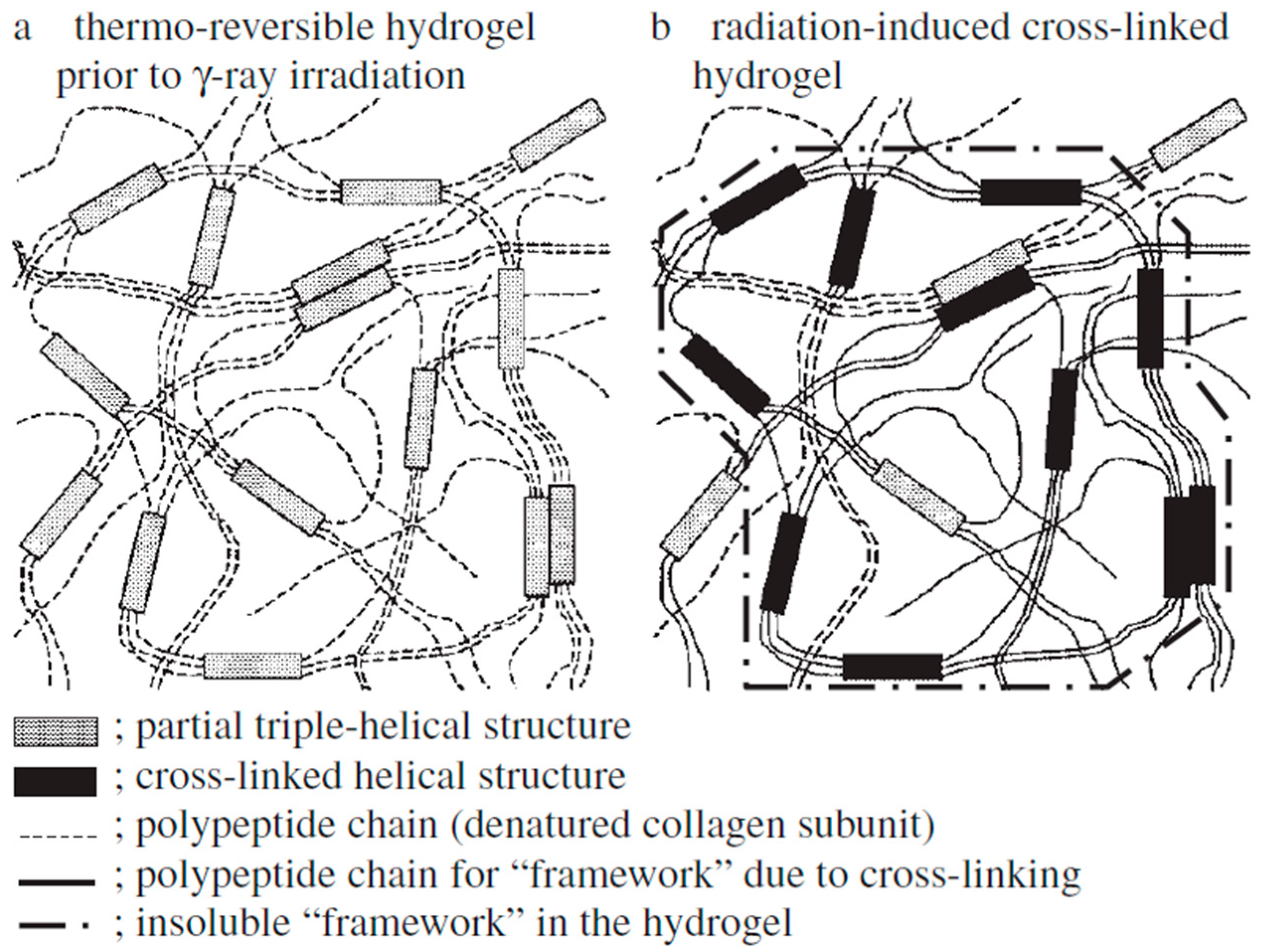
- Bessho, M.; Kojima, T.; Okuda, S.; Hara, M. Radiation-induced cross-linking of gelatin by using γ-rays: Insoluble gelatin hydrogel formation. Bull. Chem. Soc. Jpn. 2007, 80, 979–985. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Brand and Company | Common Uses |
|---|---|---|
| Cyclosporine | Neoral®, Novartis Pharm. Corp | Prevention of organ and bone marrow transplants rejection |
| Dutasteride | Avodart®, GSK Canada | To relieve symptoms of benign prostatic hyperplasia for enlarged prostates |
| Calcitriol | Rocaltrol®, Roche Canada | Management of hypocalcemia and secondary hyperparathyroidism |
| Isotretinoin | Clarus®, Cipher Pharmaceuticals Inc. | Indicated for the treatment of severe forms of acne |
| Progesterone | Prometrium®, Merck | Hormone replacement therapy |
| Valproic acid | Depakene®, Abbott Laboratories | Antiepileptic |
| Testosterone | Andriol®, Merck Canada | Testosterone replacement therapy |
| Ritonavir | Norvir®, Abbott Laboratories | HIV ** treatment |
| Amprenavir | Agenerase®, GlaxoSmithKline | HIV treatment |
| Loratadine | Claritin® liquid Gels, Schering-Plough Canada Inc. | Management of allergies |
| Type of Apparatus | Principle | Common Dosage Forms |
|---|---|---|
| Type 1 | Basket | IR, chewable tablets, DR, ER, suppositories, capsules, floating dosage forms |
| Type 2 | Paddle | IR, ODT, chewable tablets, DR, ER, enteric-coated tablets or capsules |
| Type 3 | Reciprocating Cylinder | CR, chewable tablets and beads |
| Type 4 | Flow-Through Cell | ER, soft and hard gelatin capsules, powder, granules, pellets, suppositories, and implants |
| Type 5 | Paddle Over Disk | Transdermal patches, ointments, and emulsions |
| Type 6 | Rotating Cylinder | Transdermal patches |
| Type 7 | Reciprocating Holder | Transdermal formulations, and ND oral-modified release formulations |
| Drug Product Information | Dissolution Method |
|---|---|
| Cyclosporine (100 mg) | Apparatus 2 at 75 rpm in 1000 mL 0.1 N HCl containing 4 mg of N,N-dimethydodecylamine-N-oxide per mL |
| Dutasteride | Tier 1: Apparatus 2 at 50 rpm in 900 mL 0.1 N HCI with 2% (w/v) SLS. Tier 2: Apparatus 2 at 50 rpm in 0.1 N HCI with pepsin (as per USP) (450 mL) for the first 25 min, followed by addition of 0.1 N HCI with SLS (4% w/v) (450 mL) for the remainder of the dissolution test |
| Isotretinoin | Apparatus 1 with 20 mesh at 100 rpm in 900 mL 0.05 M Potassium Phosphate Buffer, dibasic, pH 7.8, containing 0.5% lauryldimethylamine N-oxide (LDAO) |
| Paricalcitol | Apparatus 1 at 100 rpm in 500 mL in 4 mg/mL of 0.4% lauryldimethylamine N-oxide (LDAO) |
| Ergocalciferol | Apparatus 2 at 100 rpm in 500 mL 0.5 N NaOH with 10% Triton-X-100 |
| Lopinavir/Ritonavir | Apparatus 2 at 50 rpm in 900 mL, Tier 1: 0.05 M Polyoxyethylene 10 Lauryl Ether with 10 mM Sodium Phosphate monobasic (pH 6.8); Tier 2: same as above with not more than (NMT) 1750 USP units/L of Pancreatin |
| Amprenavir | Apparatus 2 at 75 rpm in 900 mL 0.1 N HCl |
| Loratadine | Apparatus 2 with sinker at 75 rpm in 900 mL. Tier 1: 0.1 N HCl with 0.1% Tween 20. Tier 2: 0.1 N HCl with 0.1% Tween 20 with addition of pepsin (as per USP) |
| Ibuprofen | Apparatus 1 at 150 rpm in 900 mL 50 mM Phosphate Buffer, pH 7.2 |
| Material | Reference |
|---|---|
| Aldehydes (furfural, acrolein, formaldehyde, glutaraldehyde, glyceryl aldehyde) | [43,151,152,153,154,155,156] |
| Imines | [143] |
| Ketones | [143] |
| Saccharides (glucose and aldose sugars) | [43] |
| Calcium carbonate | [157,158] |
| Hydrogen peroxide | [150,156] |
| Sulfonic acids and p-toluene sulfonic acid | [150,156] |
| Carbodiimides (1-ethylene 3-(3-dimethylamino propyl) carbodiimide hydrochloride, guanidine hydrochloride) | [155,156] |
| Benzene | [156] |
| Terephthaloyl chloride | [159] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Damian, F.; Harati, M.; Schwartzenhauer, J.; Van Cauwenberghe, O.; Wettig, S.D. Challenges of Dissolution Methods Development for Soft Gelatin Capsules. Pharmaceutics 2021, 13, 214. https://doi.org/10.3390/pharmaceutics13020214
Damian F, Harati M, Schwartzenhauer J, Van Cauwenberghe O, Wettig SD. Challenges of Dissolution Methods Development for Soft Gelatin Capsules. Pharmaceutics. 2021; 13(2):214. https://doi.org/10.3390/pharmaceutics13020214
Chicago/Turabian StyleDamian, Festo, Mohammad Harati, Jeff Schwartzenhauer, Owen Van Cauwenberghe, and Shawn D. Wettig. 2021. "Challenges of Dissolution Methods Development for Soft Gelatin Capsules" Pharmaceutics 13, no. 2: 214. https://doi.org/10.3390/pharmaceutics13020214