
Disclosure of a Promising Lead to Tackle Complicated Skin and Skin Structure Infections: Antimicrobial and Antibiofilm Actions of Peptide PP4-3.1


 ,
,  ,
,  , , , , and
, , , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Peptide Synthesis
2.2. Solutions for In Vitro Assays
2.3. Bacterial Strains and Culture Conditions
2.4. Antibacterial Activity
2.5. Antibiofilm Activity
2.5.1. Crystal Violet Assay
2.5.2. Microscopic Visualization of Biofilms
2.6. Cell Culture Conditions
2.7. Toxicity to Human Keratinocytes
2.8. Antifungal Activity
2.9. Activity in Simulated Wound Fluid
2.10. Statistics and Data Analysis
3. Results and Discussion
3.1. Peptide Synthesis
3.2. Antibacterial Activity
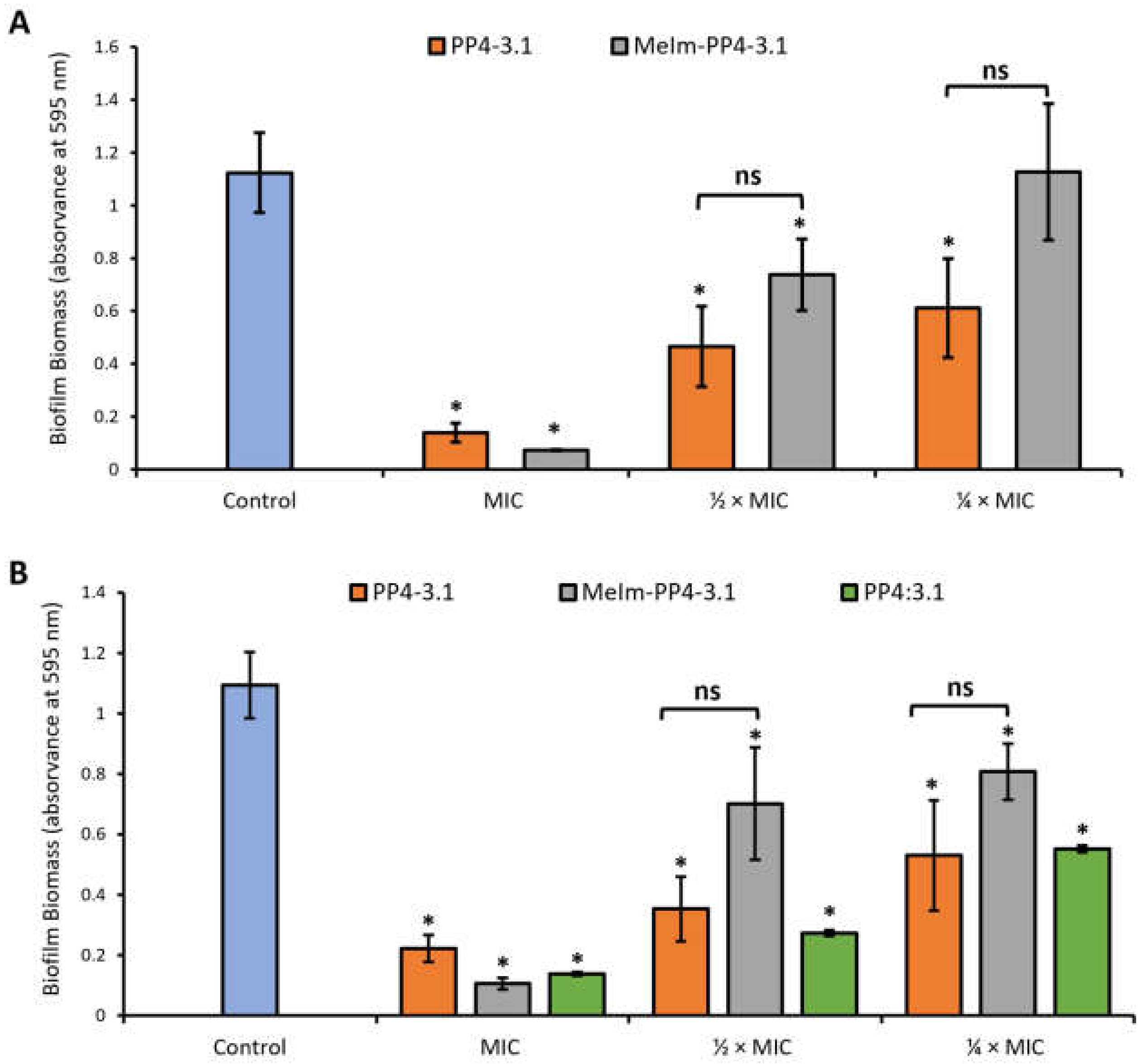
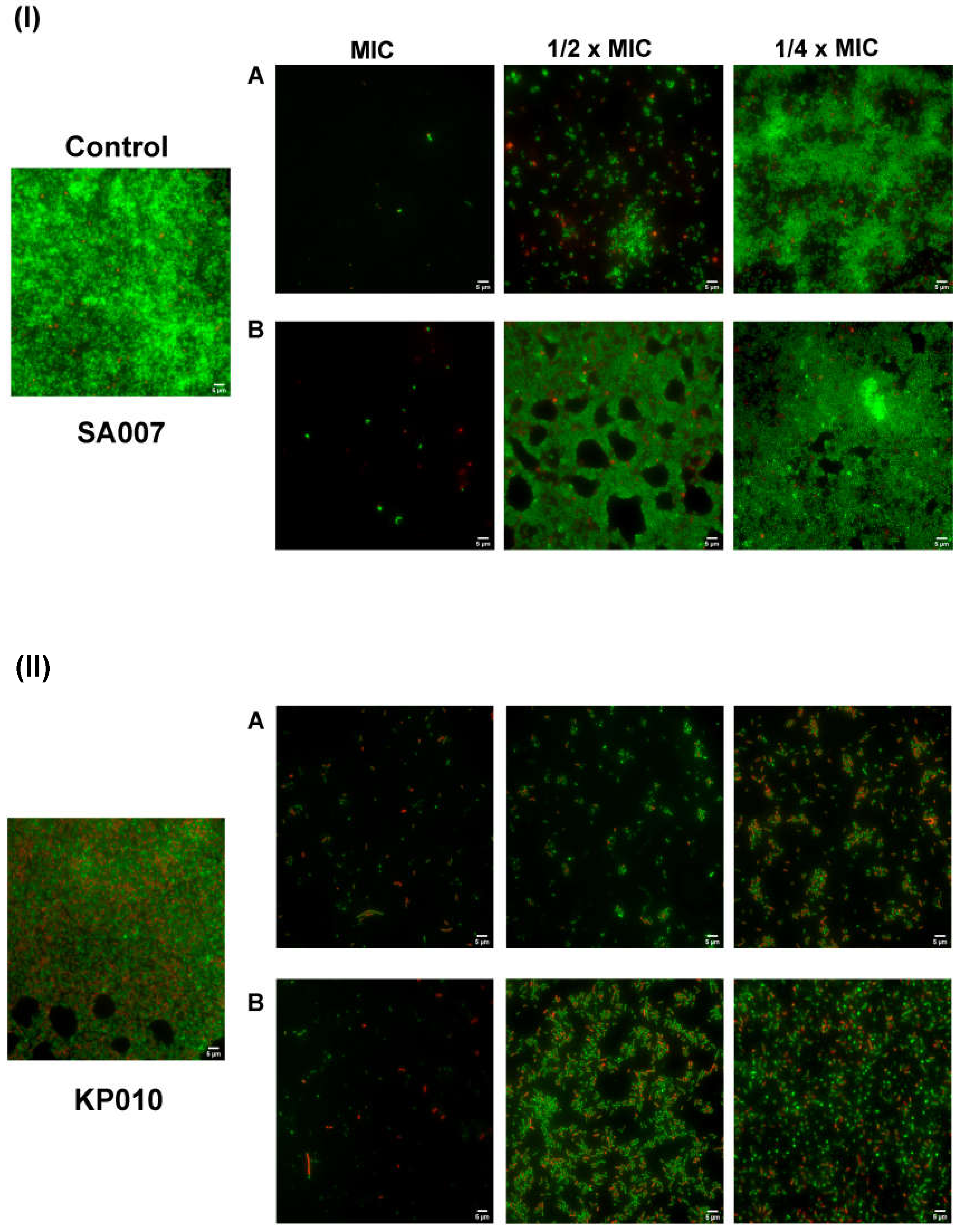
3.3. Antibiofilm Activity
3.3.1. Inhibition of Biofilm Formation by the Crystal Violet Assay
3.3.2. Inhibition of Biofilm Formation: Microscopic Visualization
3.4. Toxicity to Human Keratinocytes
3.5. Antifungal Activity
3.6. Antibacterial Activity in Simulated Wound Fluid
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad bugs, no drugs: No ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, J.; Resistance, R.o.A.; Trust, W. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations; Antimicrobial Resistance: London, UK, 2016; p. 84. [Google Scholar]
- WHO Publishes List of Bacteria for Which New Antibiotics Are Urgently Needed. Available online: https://www.who.int/news-room/detail/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed on 20 August 2021).
- Wang, J.; Dou, X.; Song, J.; Lyu, Y.; Zhu, X.; Xu, L.; Li, W.; Shan, A. Antimicrobial peptides: Promising alternatives in the post feeding antibiotic era. Med. Res. Rev. 2019, 39, 831–859. [Google Scholar] [CrossRef] [PubMed]
- Browne, K.; Chakraborty, S.; Chen, R.; Willcox, M.D.; Black, D.S.; Walsh, W.R.; Kumar, N. A New Era of Antibiotics: The Clinical Potential of Antimicrobial Peptides. Int. J. Mol. Sci. 2020, 21, 7047. [Google Scholar] [CrossRef]
- Pfalzgraff, A.; Brandenburg, K.; Weindl, G. Antimicrobial Peptides and Their Therapeutic Potential for Bacterial Skin Infections and Wounds. Front. Pharmacol. 2018, 9, 281. [Google Scholar] [CrossRef] [PubMed]
- Omar, A.; Wright, J.B.; Schultz, G.; Burrell, R.; Nadworny, P. Microbial Biofilms and Chronic Wounds. Microorganisms 2017, 5, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves, P.J.; Barreto, R.T.; Barrois, B.M.; Gryson, L.G.; Meaume, S.; Monstrey, S.J. Update on the role of antiseptics in the management of chronic wounds with critical colonisation and/or biofilm. Int. Wound J. 2021, 18, 342–358. [Google Scholar] [CrossRef] [PubMed]
- Wolcott, R.D.; Rumbaugh, K.P.; James, G.; Schultz, G.; Phillips, P.; Yang, Q.; Watters, C.; Stewart, P.S.; Dowd, S.E. Biofilm maturity studies indicate sharp debridement opens a time-dependent therapeutic window. J. Wound Care 2010, 19, 320–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, G.; Bjarnsholt, T.; James, G.A.; Leaper, D.J.; McBain, A.J.; Malone, M.; Stoodley, P.; Swanson, T.; Tachi, M.; Wolcott, R.D.; et al. Consensus guidelines for the identification and treatment of biofilms in chronic nonhealing wounds. Wound Repair Regen. 2017, 25, 744–757. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.; Wrobel, J.S.; Maceachern, M.P.; Boles, B.R. Collagen-based wound dressings for the treatment of diabetes-related foot ulcers: A systematic review. Diabetes Metab. Syndr. Obes. Targets Ther. 2013, 6, 17–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew-Steiner, S.S.; Roy, S.; Sen, C.K. Collagen in Wound Healing. Bioengineering 2021, 8, 63. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.; Bessa, L.J.; Fernandes, I.; Ferraz, R.; Mateus, N.; Gameiro, P.; Teixeira, C.; Gomes, P. Turning a Collagenesis-Inducing Peptide Into a Potent Antibacterial and Antibiofilm Agent Against Multidrug-Resistant Gram-Negative Bacteria. Front. Microbiol. 2019, 10, 1915. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.; Bessa, L.J.; Correia, P.; Fernandes, I.; Ferraz, R.; Gameiro, P.; Teixeira, C.; Gomes, P. “Clicking” an Ionic Liquid to a Potent Antimicrobial Peptide: On the Route towards Improved Stability. Int. J. Mol. Sci. 2020, 21, 6174. [Google Scholar] [CrossRef] [PubMed]
- Exner, M.; Bhattacharya, S.; Christiansen, B.; Gebel, J.; Goroncy-Bermes, P.; Hartemann, P.; Heeg, P.; Ilschner, C.; Kramer, A.; Larson, E.; et al. Antibiotic resistance: What is so special about multidrug-resistant Gram-negative bacteria? GMS Hyg. Infect. Control 2017, 12, 1–24. [Google Scholar] [CrossRef]
- Leong, H.N.; Kurup, A.; Tan, M.Y.; Kwa, A.L.H.; Liau, K.H.; Wilcox, M.H. Management of complicated skin and soft tissue infections with a special focus on the role of newer antibiotics. Infect. Drug Resist. 2018, 11, 1959–1974. [Google Scholar] [CrossRef] [Green Version]
- Gunaydin, S.D.; Arikan-Akdagli, S.; Akova, M. Fungal infections of the skin and soft tissue. Curr. Opin. Infect. Dis. 2020, 33, 130–136. [Google Scholar] [CrossRef]
- Kalan, L.; Loesche, M.; Hodkinson, B.P.; Heilmann, K.; Ruthel, G.; Gardner, S.E.; Grice, E.A. Redefining the Chronic-Wound Microbiome: Fungal Communities Are Prevalent, Dynamic, and Associated with Delayed Healing. MBio 2016, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kalan, L.; Grice, E.A. Fungi in the Wound Microbiome. Adv. Wound Care 2018, 7, 247–255. [Google Scholar] [CrossRef]
- Behrendt, R.; White, P.; Offer, J. Advances in Fmoc solid-phase peptide synthesis. J. Pept. Sci. 2016, 22, 4–27. [Google Scholar] [CrossRef] [Green Version]
- Loughrey, S.; Mannion, J.; Matlock, B. Using the NanoDrop One to Quantify Protein and Peptide Preparations at 205 nm. Available online: http://tools.thermofisher.com/content/sfs/brochures/ND-One-Protein-and-Peptide-r16-01-18.pdf (accessed on 20 August 2021).
- Patel, J.B. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically; Approved Standard M7-A9; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2012. [Google Scholar]
- Bessa, L.J.; Eaton, P.; Dematei, A.; Placido, A.; Vale, N.; Gomes, P.; Delerue-Matos, C.; Sa Leite, J.R.; Gameiro, P. Synergistic and antibiofilm properties of ocellatin peptides against multidrug-resistant Pseudomonas aeruginosa. Future Microbiol. 2018, 13, 151–163. [Google Scholar] [CrossRef] [Green Version]
- Coelho, P.; Oliveira, J.; Fernandes, I.; Araújo, P.; Pereira, A.R.; Gameiro, P.; Bessa, L.J. Pyranoanthocyanins Interfering with the Quorum Sensing of Pseudomonas aeruginosa and Staphylococcus aureus. Int. J. Mol. Sci. 2021, 22, 1–20. [Google Scholar] [CrossRef]
- EUCAST. The European Committee on Antimicrobial Susceptibility Testing. In Clinical Breakpoints for Fungi (Candida and Aspergillus Species), Version 10.0; The European Committee on Antimicrobial Susceptibility Testing: Växjö, Sweden, 2020. [Google Scholar]
- EUCAST. The European Committee on Antimicrobial Susceptibility Testing. In Breakpoint Tables for Interpretation of MICs for Antifungal Agents, Version 10.0; The European Committee on Antimicrobial Susceptibility Testing: Växjö, Sweden, 2020. [Google Scholar]
- EUCAST. The European Committee on Antimicrobial Susceptibility Testing. In Method for Susceptibility Testing of Yeasts, Version 7.3.2; The European Committee on Antimicrobial Susceptibility Testing: Växjö, Sweden, 2020. [Google Scholar]
- EUCAST. The European Committee on Antimicrobial Susceptibility Testing. In Antifungal MIC Method for Yeasts, Version 7.3.2; The European Committee on Antimicrobial Susceptibility Testing: Växjö, Sweden, 2020. [Google Scholar]
- Wiegand, I.; Hilpert, K.; Hancock, R.E.W. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Büttner, H.; Mack, D.; Rohde, H. Structural basis of Staphylococcus epidermidis biofilm formation: Mechanisms and molecular interactions. Front. Cell Infect. Microbiol. 2015, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Bruun, T.; Kittang, B.R.; de Hoog, B.J.; Aardal, S.; Flaatten, H.K.; Langeland, N.; Mylvaganam, H.; Vindenes, H.A.; Skrede, S. Necrotizing soft tissue infections caused by Streptococcus pyogenes and Streptococcus dysgalactiae subsp. equisimilis of groups C and G in western Norway. Clin. Microbiol. Infect. 2013, 19, E545–E550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, N.M.; Bessa, L.J.; Buttachon, S.; Costa, P.M.; Buaruang, J.; Dethoup, T.; Silva, A.M.; Kijjoa, A. Antibacterial and antibiofilm activities of tryptoquivalines and meroditerpenes isolated from the marine-derived fungi Neosartorya paulistensis, N. laciniosa, N. tsunodae, and the soil fungi N. fischeri and N. siamensis. Mar. Drugs 2014, 12, 822–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taei, M.; Chadeganipour, M.; Mohammadi, R. An alarming rise of non-albicans Candida species and uncommon yeasts in the clinical samples; a combination of various molecular techniques for identification of etiologic agents. BMC Res. Notes 2019, 12, 779. [Google Scholar] [CrossRef] [Green Version]
- Price, B.L.; Lovering, A.M.; Bowling, F.L.; Dobson, C.B. Development of a Novel Collagen Wound Model To Simulate the Activity and Distribution of Antimicrobials in Soft Tissue during Diabetic Foot Infection. Antimicrob. Agents Chemother. 2016, 60, 6880–6889. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Chung, H.; Shin, Y.P.; Kim, M.-A.; Natarajan, S.; Veerappan, K.; Kim, S.H.; Park, J.; Hwang, J.S. Deciphering Novel Antimicrobial Peptides from the Transcriptome of Papilio xuthus. Insects 2020, 11, 776. [Google Scholar] [CrossRef]
- Schneider, R.; Primon-Barros, M.; Von Borowski, R.G.; Chat, S.; Nonin-Lecomte, S.; Gillet, R.; Macedo, A.J. Pseudonajide peptide derived from snake venom alters cell envelope integrity interfering on biofilm formation in Staphylococcus epidermidis. BMC Microbiol. 2020, 20, 237. [Google Scholar] [CrossRef]
- de Barros, E.; Gonçalves, R.M.; Cardoso, M.H.; Santos, N.C.; Franco, O.L.; Cândido, E.S. Snake Venom Cathelicidins as Natural Antimicrobial Peptides. Front. Pharmacol. 2019, 10, 1415. [Google Scholar] [CrossRef] [Green Version]
- Dell’Olmo, E.; Gaglione, R.; Cesaro, A.; Cafaro, V.; Teertstra, W.R.; de Cock, H.; Notomista, E.; Haagsman, H.P.; Veldhuizen, E.J.A.; Arciello, A. Host defence peptides identified in human apolipoprotein B as promising antifungal agents. Appl. Microbiol. Biotechnol. 2021, 105, 1953–1964. [Google Scholar] [CrossRef] [PubMed]
- Gaglione, R.; Cesaro, A.; Dell’Olmo, E.; Di Girolamo, R.; Tartaglione, L.; Pizzo, E.; Arciello, A. Cryptides Identified in Human Apolipoprotein B as New Weapons to Fight Antibiotic Resistance in Cystic Fibrosis Disease. Int. J. Mol. Sci. 2020, 21, 2049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, F.; Teixeira, C.; Gomes, P.; Martins, M.C.L. Clinical Application of AMPs. In Antimicrobial Peptides: Basics for Clinical Application; Matsuzaki, K., Ed.; Springer: Singapore, 2019; pp. 281–298. [Google Scholar]
- Moretta, A.; Scieuzo, C.; Petrone, A.M.; Salvia, R.; Manniello, M.D.; Franco, A.; Lucchetti, D.; Vassallo, A.; Vogel, H.; Sgambato, A.; et al. Antimicrobial Peptides: A New Hope in Biomedical and Pharmaceutical Fields. Front Cell Infect. Microbiol. 2021, 11, 453. [Google Scholar] [CrossRef]
- Rima, M.; Rima, M.; Fajloun, Z.; Sabatier, J.-M.; Bechinger, B.; Naas, T. Antimicrobial Peptides: A Potent Alternative to Antibiotics. Antibiotics 2021, 10, 1095. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Qian, Y.; Fu, X.; Huang, S.; Li, Y.; Zhou, C. De Novo Design of Triblock Amphiphilic Short Antimicrobial Peptides. ACS Appl. Polym. Mater. 2020, 2, 3988–3992. [Google Scholar] [CrossRef]
- Gong, H.; Hu, X.; Liao, M.; Fa, K.; Ciumac, D.; Clifton, L.A.; Sani, M.-A.; King, S.M.; Maestro, A.; Separovic, F.; et al. Structural Disruptions of the Outer Membranes of Gram-Negative Bacteria by Rationally Designed Amphiphilic Antimicrobial Peptides. ACS Appl. Mater. Interfaces 2021, 13, 16062–16074. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, M.H.; Orozco, R.Q.; Rezende, S.B.; Rodrigues, G.; Oshiro, K.G.N.; Cândido, E.S.; Franco, O.L. Computer-Aided Design of Antimicrobial Peptides: Are We Generating Effective Drug Candidates? Front. Microbiol. 2020, 10, 3097. [Google Scholar] [CrossRef] [PubMed]
- Fleeman, R.M.; Macias, L.A.; Brodbelt, J.S.; Davies, B.W. Defining principles that influence antimicrobial peptide activity against capsulated Klebsiella pneumoniae. PNAS 2020, 117, 27620. [Google Scholar] [CrossRef]
- Simonson, A.W.; Mongia, A.S.; Aronson, M.R.; Alumasa, J.N.; Chan, D.C.; Lawanprasert, A.; Howe, M.D.; Bolotsky, A.; Mal, T.K.; George, C.; et al. Pathogen-specific antimicrobials engineered de novo through membrane-protein biomimicry. Nat. Biomed. Eng. 2021, 5, 467–480. [Google Scholar] [CrossRef]
- Gaglione, R.; Cesaro, A.; Dell’Olmo, E.; Della Ventura, B.; Casillo, A.; Di Girolamo, R.; Velotta, R.; Notomista, E.; Veldhuizen, E.J.A.; Corsaro, M.M.; et al. Effects of human antimicrobial cryptides identified in apolipoprotein B depend on specific features of bacterial strains. Sci. Rep. 2019, 9, 6728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haney, E.F.; Straus, S.K.; Hancock, R.E.W. Reassessing the Host Defense Peptide Landscape. Front. Chem. 2019, 7, 43. [Google Scholar] [CrossRef] [Green Version]
- Chellan, G.; Neethu, K.; Varma, A.K.; Mangalanandan, T.S.; Shashikala, S.; Dinesh, K.R.; Sundaram, K.R.; Varma, N.; Jayakumar, R.V.; Bal, A.; et al. Targeted treatment of invasive fungal infections accelerates healing of foot wounds in patients with Type 2 diabetes. Diabet. Med. 2012, 29, e255–e262. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Peptide a | Sequence b/Structure | MW/Da |
|---|---|---|
| PP4-3.1 | KTTKSKKLLKWLLKLL | 1940.5 |
| MeIm-PP4-3.1 |  | 2144.8 |
| PP4 | KTTKS | 562.7 |
| 3.1 | KKLLKWLLKLL | 1394.9 |
| Bacterial Species | Reference Strain or MDR Isolate | MIC in µM (in µg/mL) a | ||||||
|---|---|---|---|---|---|---|---|---|
| CIP | PP4-3.1 | MeIm-PP4.3.1 | PP4 | 3.1 | PP4:3.1 (1:1) | |||
| Gram-negative | E. coli | ATCC 25922 | 0.012 (0.004) | 1.4 (2.8) | 0.7 (1.5) | >60 | 3.8 | 1.9 |
| P. aeruginosa | ATCC 27853 | 0.18 (0.06) | 1.4 (2.8) | 1.4 (2.9) | >60 | 3.8 | 7.5 | |
| PA004 | 96 (32) | 1.4 (2.8) | 1.4 (2.9) | ND b | ND b | ND b | ||
| K. pneumoniae | ATCC 13883 | 0.75 (0.25) | 1.4 (2.8) | 1.4 (2.9) | >60 | 3.8 | 7.5 | |
| KP010 | 48 (16) | 2.9 (5.6) | 2.8 (5.9) | ND b | ND b | ND b | ||
| Gram-Positive | S. aureus | ATCC 29213 | 1.5 (0.5) | 2.9 (5.6) | 2.8 (5.9) | >60 | 1.9 | 1.9 |
| SA007 | 193 (64) | 1.4 (2.8) | 1.4 (2.9) | ND b | ND b | ND b | ||
| E. faecalis | ATCC 29212 | 0.38 (0.125) | 5.7 (11) | 5.5 (12) | >60 | 3.8 | 1.9 | |
| S. epidermidis | ATCC 14990 | 0.75 (0.25) | 0.8 (1.5) | 0.7 (1.5) | ND b | ND b | ND b | |
| S. pyogenes | ATCC 19615 | 6.04 (2) | 2.7 (11) | 5.5 (12) | ND b | ND b | ND b | |
| Peptides | MIC in µM (µg/mL) | |
|---|---|---|
| S. aureus (SA007) | K. pneumoniae (KP010) | |
| PP4-3.1 | 1.3 (2.8) | 5.7 (11.1) |
| MeIm-PP4-3.1 | 1.4 (2.9) | 10.9 (23.4) |
| PP4:3.1 (1:1) | ND a | 15.0 |
| Peptide | IC50 (µM) a |
|---|---|
| PP4-3.1 | 13.0 ± 1.0 |
| MeIm-PP4-3.1 | 5.7 ± 1.0 |
| PP4:3.1 (1:1) | 11.7 ± 1.1 |
| Peptide | MIC in µM (µg/mL) | ||
|---|---|---|---|
| Candida albicans ATCC 90028 | Candida glabrata ATCC 90030 | Candida parapsilosis ATCC 22019 | |
| PP4-3.1 | 2.9 (5.6) | 11.5 (22.2) | 1.4 (2.8) |
| MeIm-PP4-3.1 | 10.5 (23.4) | 43.7 (93.7) | 2.8 (5.9) |
| PP4 | >60 | >60 | >60 |
| 3.1 | 7.5 | 15 | 3.75 |
| PP4:3.1 (1:1) | 7.5 | 15 | 3.75 |
| Fluconazole | 1.6 (0.5) | 52 (16) | 6.5 (2) |
| Peptide | MHB | SWF | ||
|---|---|---|---|---|
| MIC | MBC | MIC | MBC | |
| PP4-3.1 | 2.1 (4) | 2.1 (4) | 0.3–0.5 (0.5–1) | 1–2.1 (2–4) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomes, A.; Bessa, L.J.; Fernandes, I.; Ferraz, R.; Monteiro, C.; L. Martins, M.C.; Mateus, N.; Gameiro, P.; Teixeira, C.; Gomes, P. Disclosure of a Promising Lead to Tackle Complicated Skin and Skin Structure Infections: Antimicrobial and Antibiofilm Actions of Peptide PP4-3.1. Pharmaceutics 2021, 13, 1962. https://doi.org/10.3390/pharmaceutics13111962
Gomes A, Bessa LJ, Fernandes I, Ferraz R, Monteiro C, L. Martins MC, Mateus N, Gameiro P, Teixeira C, Gomes P. Disclosure of a Promising Lead to Tackle Complicated Skin and Skin Structure Infections: Antimicrobial and Antibiofilm Actions of Peptide PP4-3.1. Pharmaceutics. 2021; 13(11):1962. https://doi.org/10.3390/pharmaceutics13111962
Chicago/Turabian StyleGomes, Ana, Lucinda J. Bessa, Iva Fernandes, Ricardo Ferraz, Cláudia Monteiro, M. Cristina L. Martins, Nuno Mateus, Paula Gameiro, Cátia Teixeira, and Paula Gomes. 2021. "Disclosure of a Promising Lead to Tackle Complicated Skin and Skin Structure Infections: Antimicrobial and Antibiofilm Actions of Peptide PP4-3.1" Pharmaceutics 13, no. 11: 1962. https://doi.org/10.3390/pharmaceutics13111962