
Mechanistic Studies of Antibiotic Adjuvants Reducing Kidney’s Bacterial Loads upon Systemic Monotherapy


Abstract
:1. Introduction
2. Materials and Methods
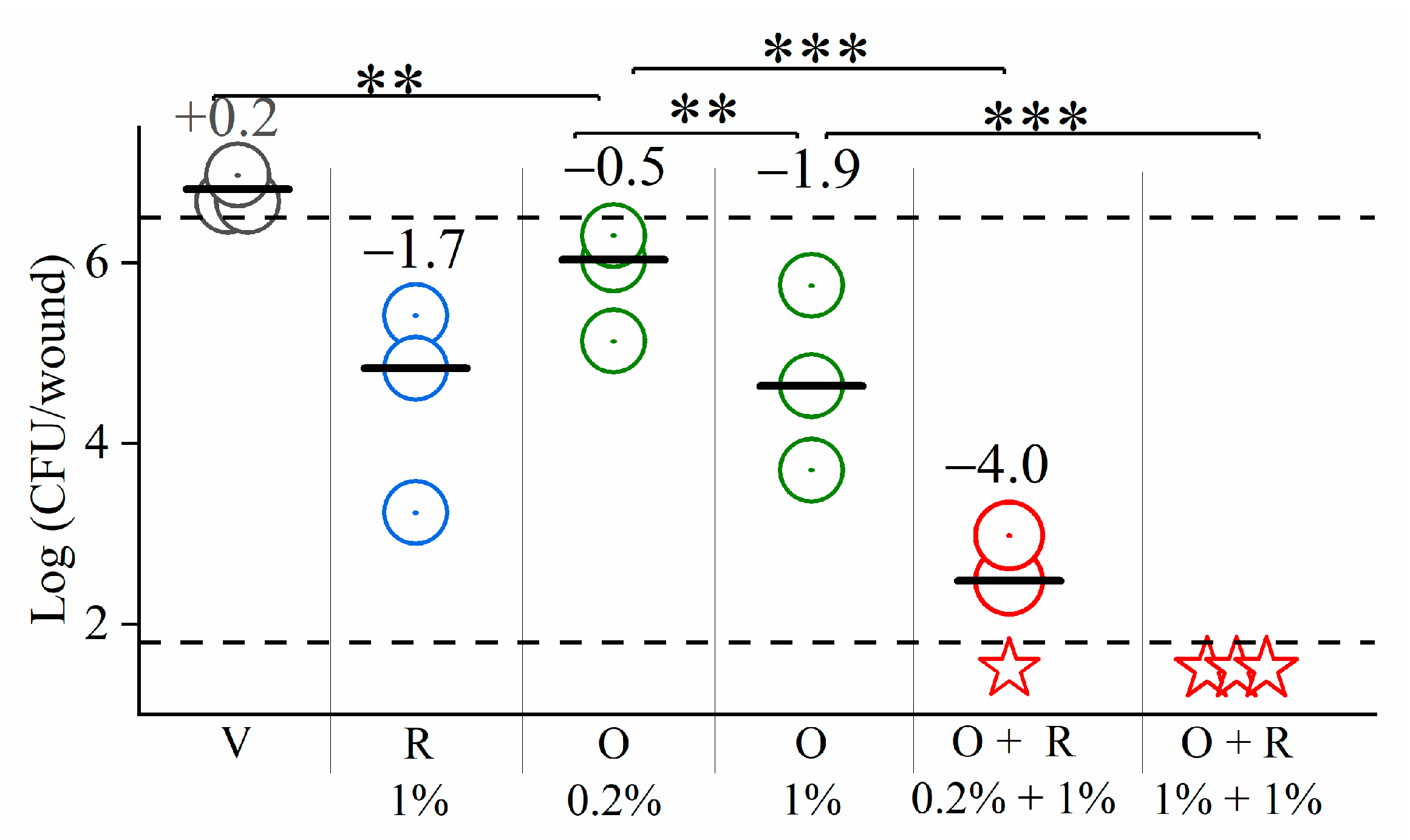
- Excisional skin wound infection model: mice were anesthetized by intraperitoneal administration of a mixture of ketamine 100 mg/kg and xylazine 5 mg/kg in PBS and their backs shaved with electric clippers. The following day mice were similarly anesthetized and were administrated (S.C) 0.05 mg/kg buprenorphine (for pain relief). A 5 mm diameter piece of skin was removed from the middle of the shaved back, with sterile biopsy punch resulting in a full-thickness injury. A total of 20 µL of a mid-logarithmic culture, containing 5 × 106 CFU P. aeruginosa 27853 were applied on the wound. Then, 15 min after inoculation, about 50 µL of hypromellose gel (prepared as described [43]) containing OAC, antibiotic, or their combination were applied on the skin and covered with a piece of medical tape. As a control, the vehicle (drug-free gel) was similarly applied on the skin. After a treatment period of 4 h, about 8 mm diameter of skin surrounding the infected area was removed, suspended in PBS, homogenized, serially diluted 10-fold, and plated for CFU enumeration. The number of viable bacteria was determined after overnight incubation at 37 °C. The lower limit of detection was 50 CFU/wound.
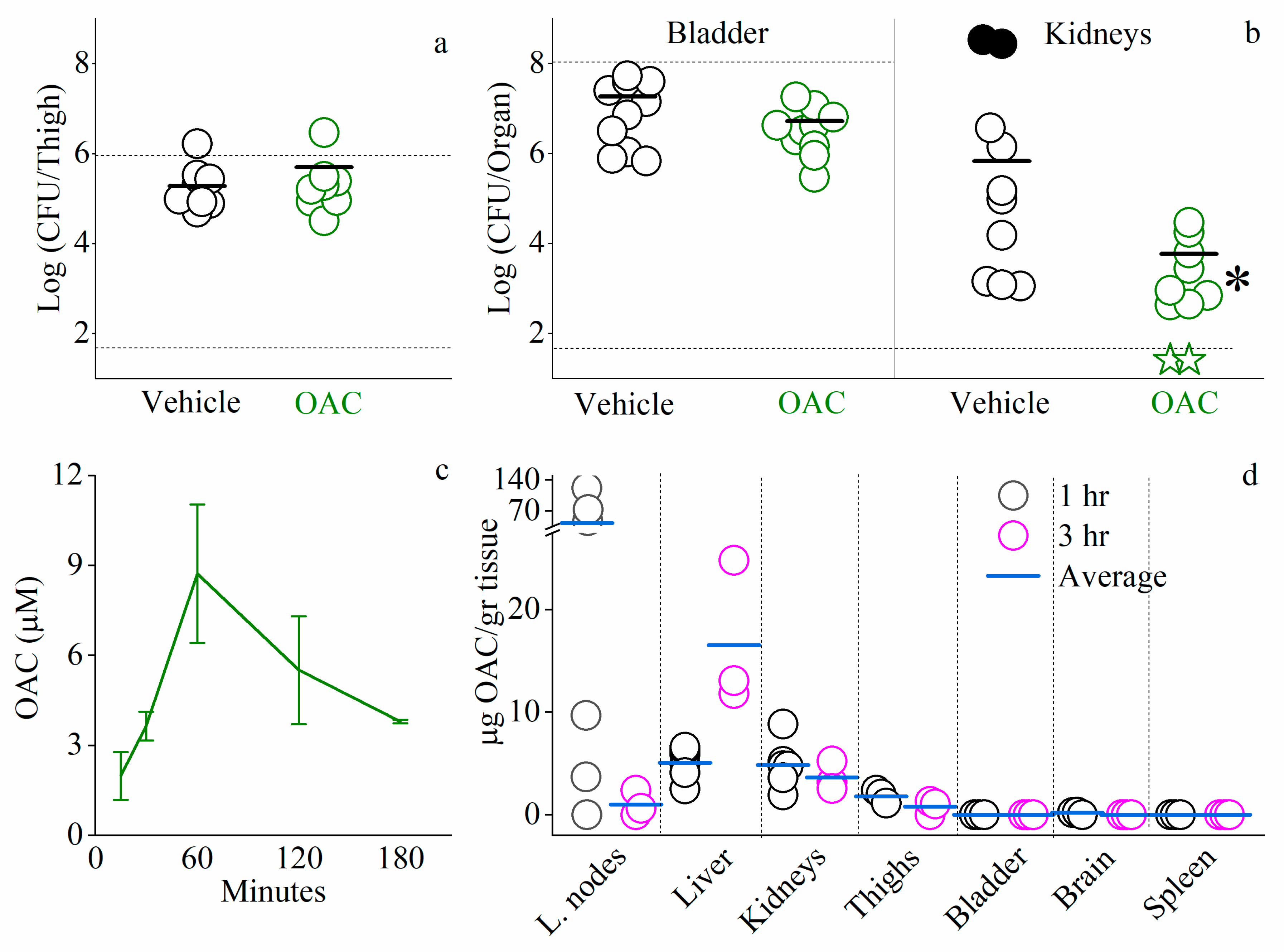
- Thigh infection model (TI): mice were inoculated intramuscularly with 1 × 106 CFU/mouse of mid-logarithmic E. coli 25922 and treated subcutaneously 1 and 3 h after inoculation. Mice were sacrificed 24 h after infection, their thighs excised, homogenized, serially diluted 10-fold, and plated for CFU enumeration. The number of viable bacteria was determined after overnight incubation at 37 °C. The lower limit of detection was 50 CFU/thigh.
- Urinary tract infection model (UTI): mice were anesthetized via intraperitoneal injection of 100 mg/kg ketamine and 5 mg/kg xylazine. Mice penises were lubricated with an analgesic 2% lidocaine gel. Then, mice were infected with 50 µL of 1 × 108 CFU/mouse of E. coli UPEC CFT073, administrated by an intra-urethral injection using a catheter (24 GA, 0.156 IN, 0.7 × 14 mm). Mice were subcutaneously treated with OAC at 1 and 6 h post infection.Mice were sacrificed 24 h post inoculation, the bladder and kidneys were excised, homogenized, serially diluted 10-fold, and plated for CFU enumeration. The number of viable bacteria was determined after overnight incubation at 37 °C. The lower limit of detection was 50 CFU/organ.
Blood Circulating Concentrations and Organ Bio-Distribution of C14(ω5)OOc10O
3. Results and Discussion
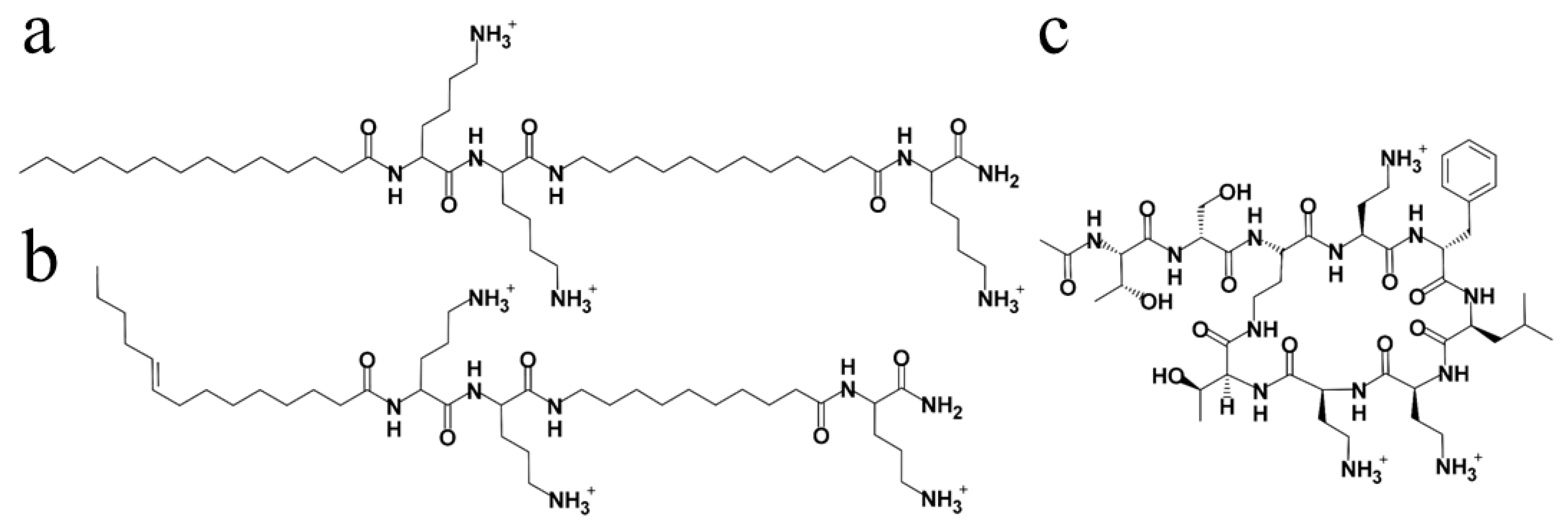
3.1. Derivatives Design and In Vitro Assessment
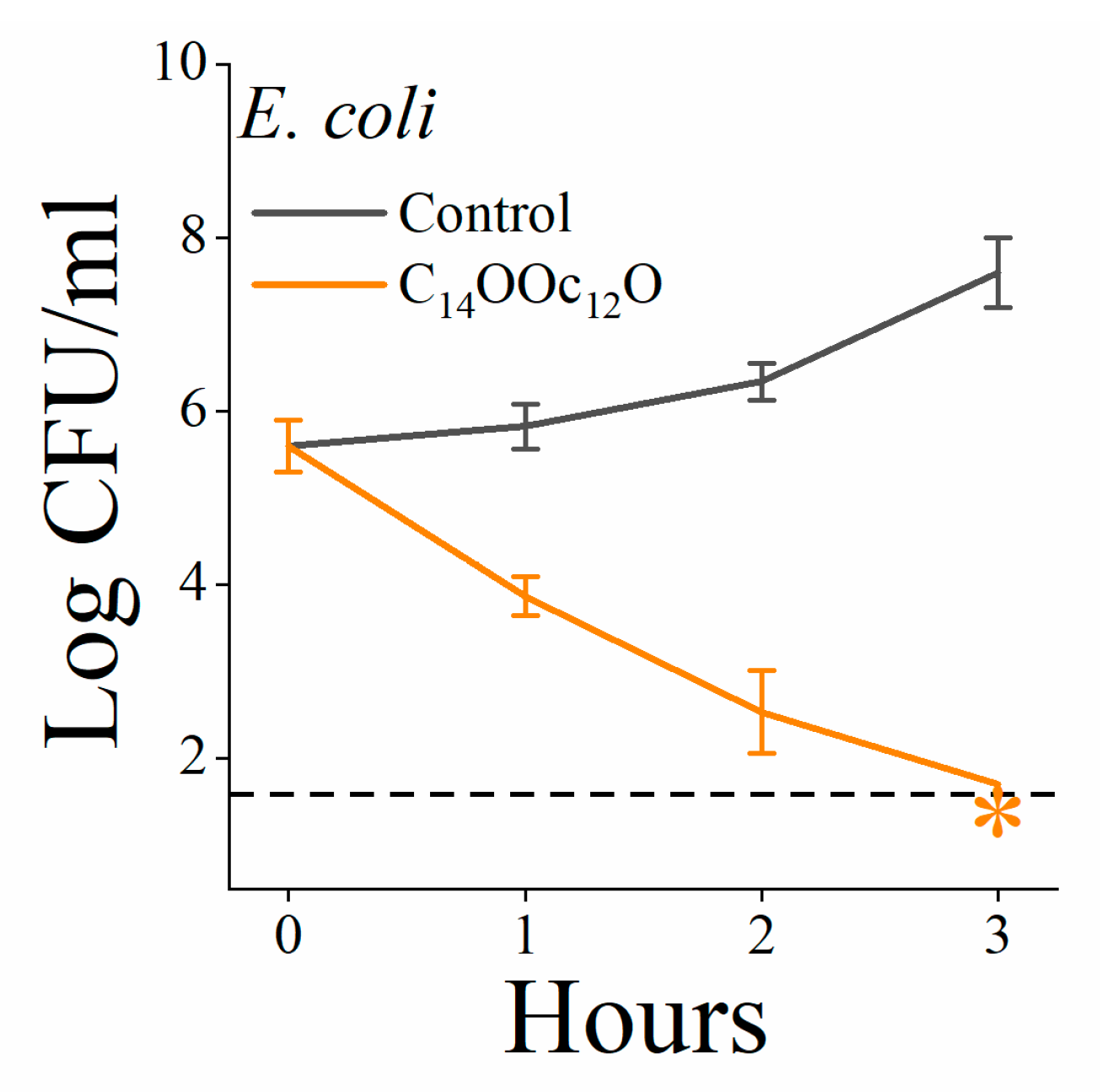
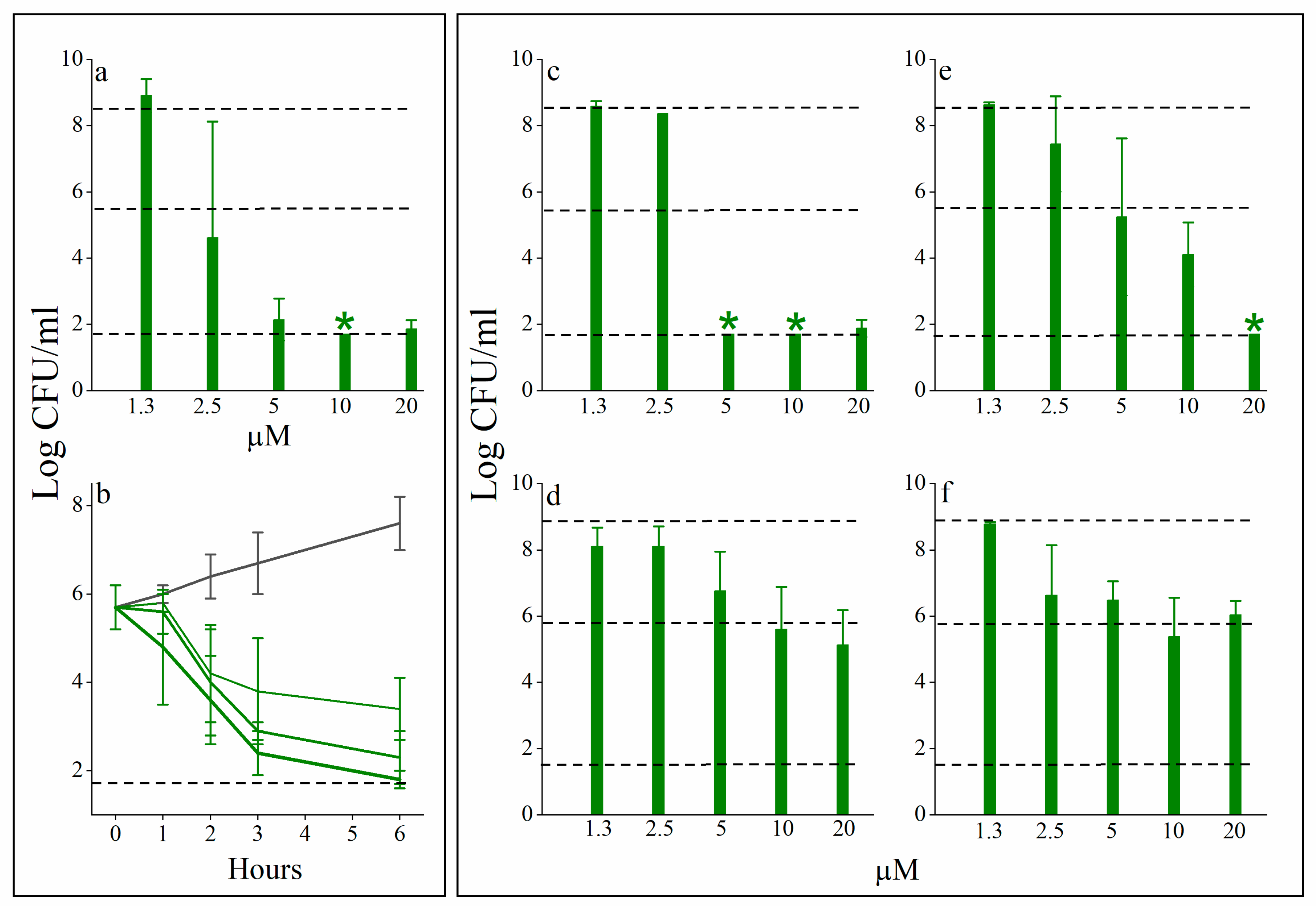
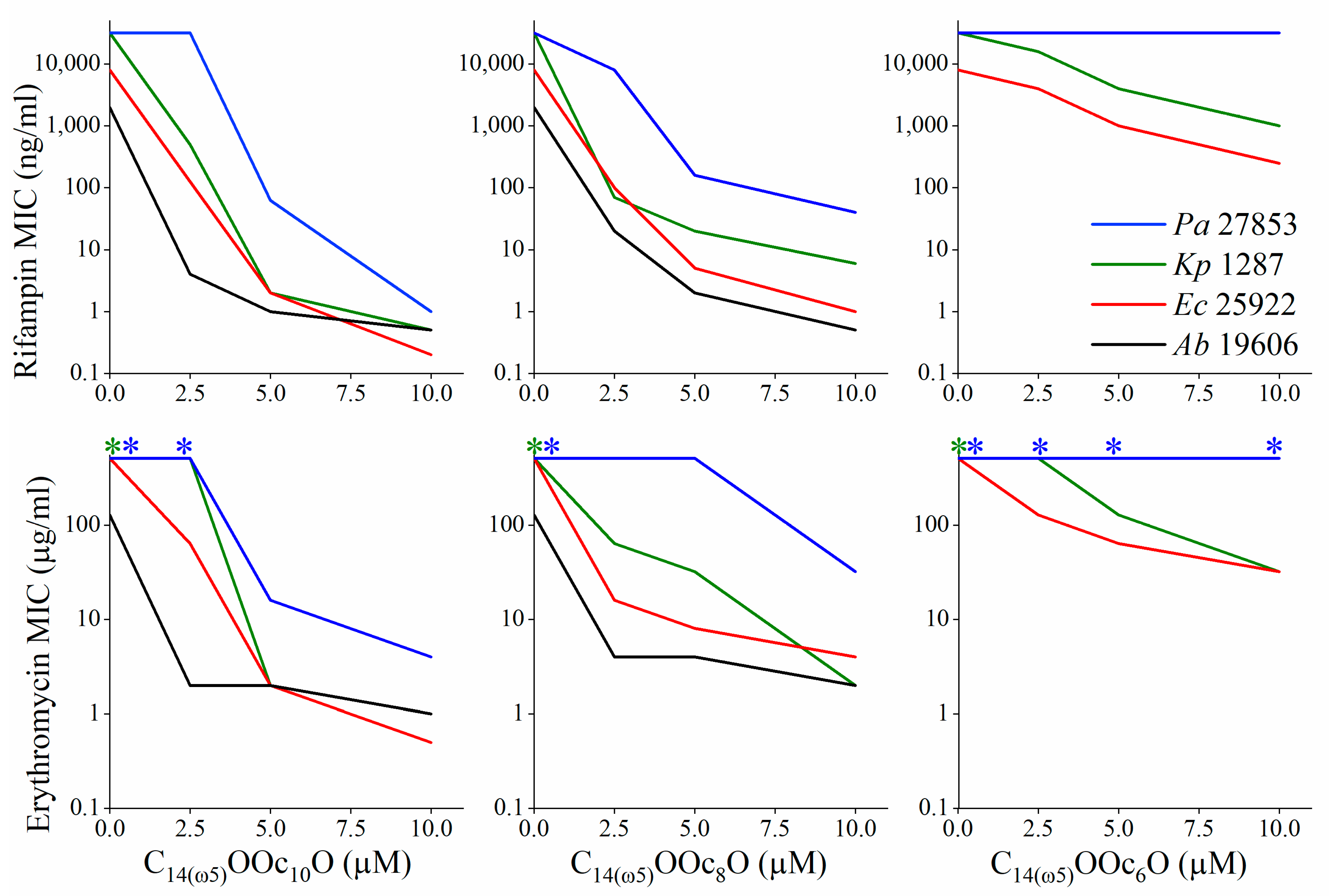
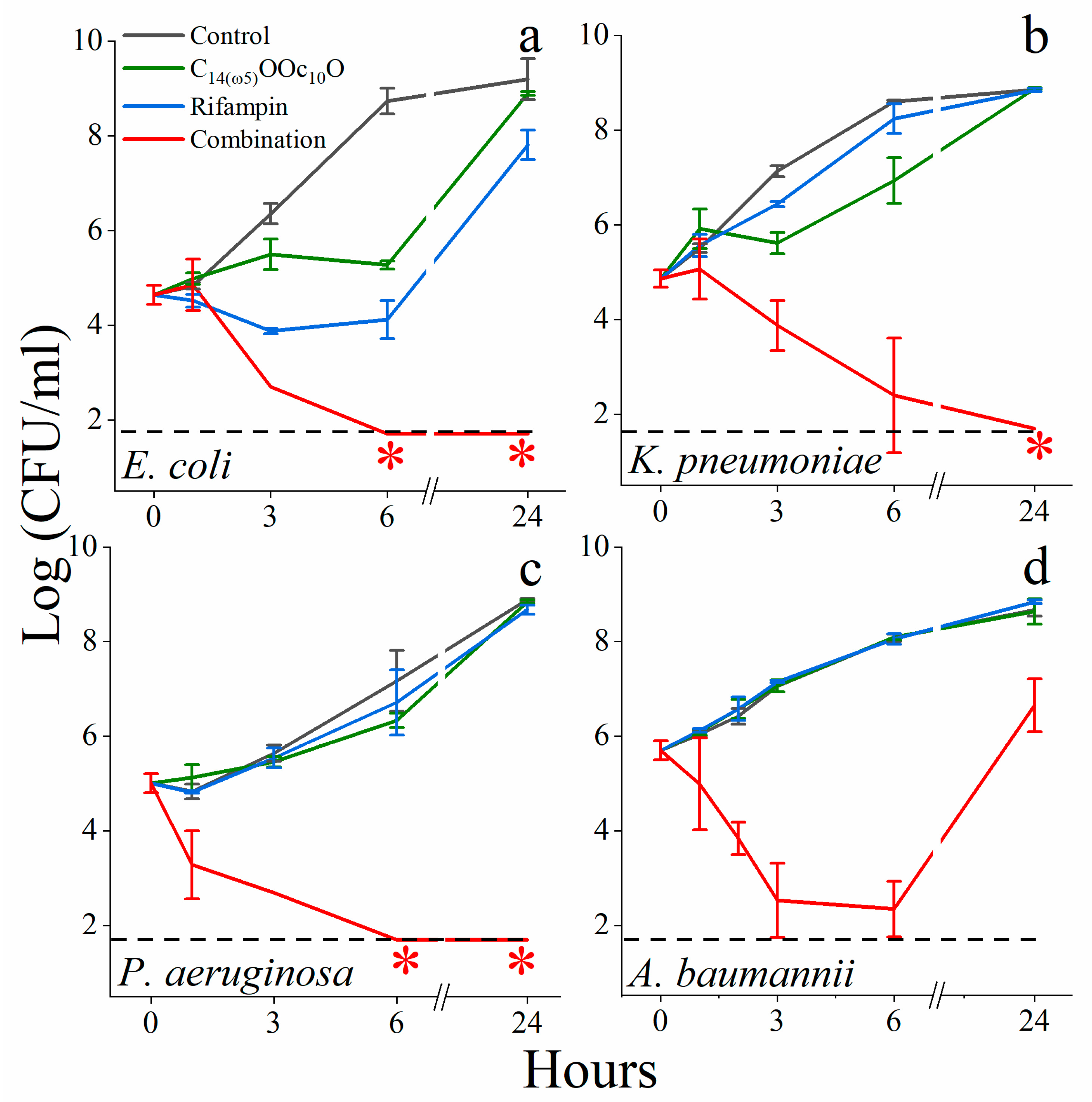
3.2. C14(ω5)OOc10O Is a Remarkable Antibiotics Potentiator against GNB
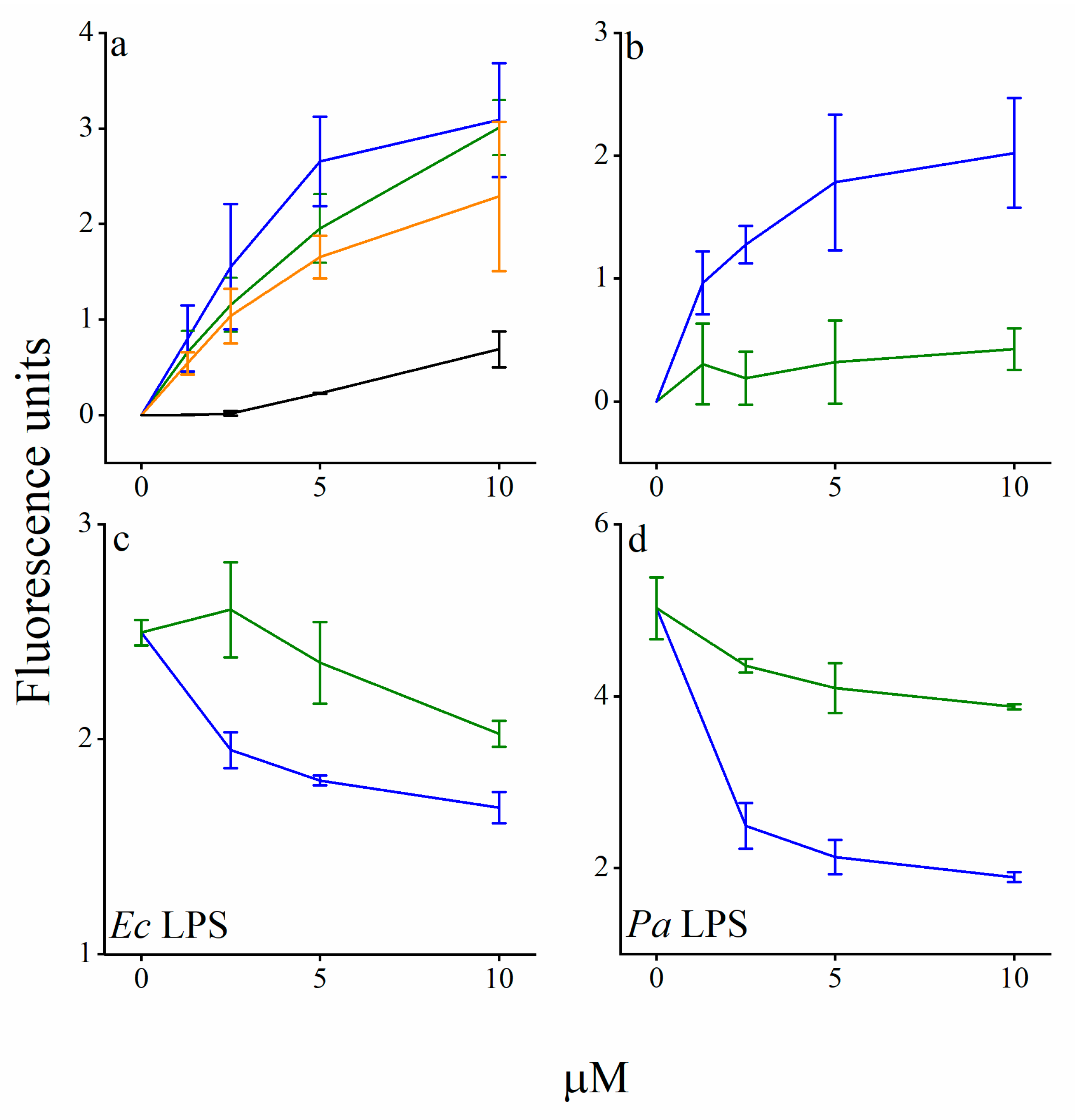
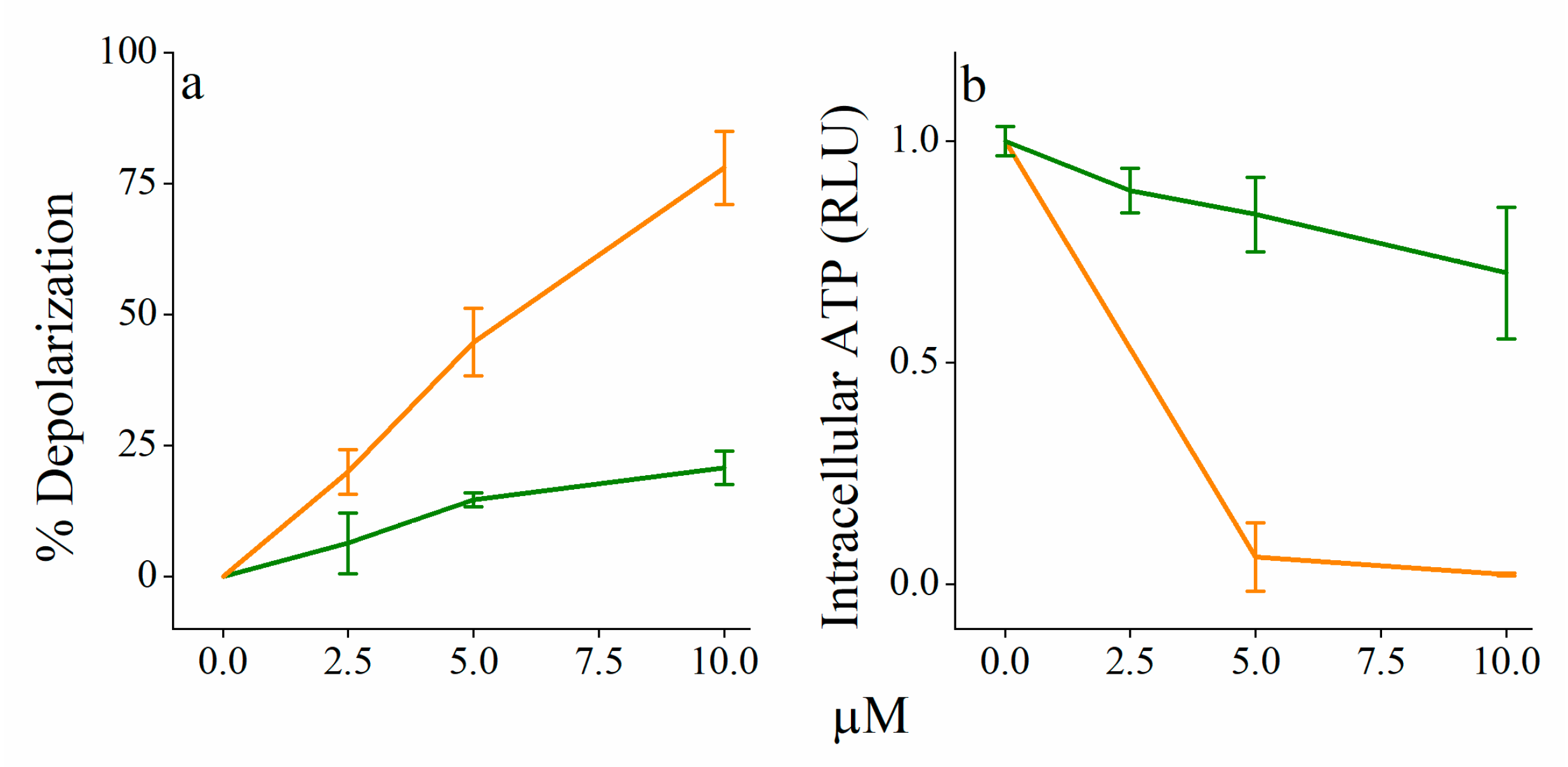
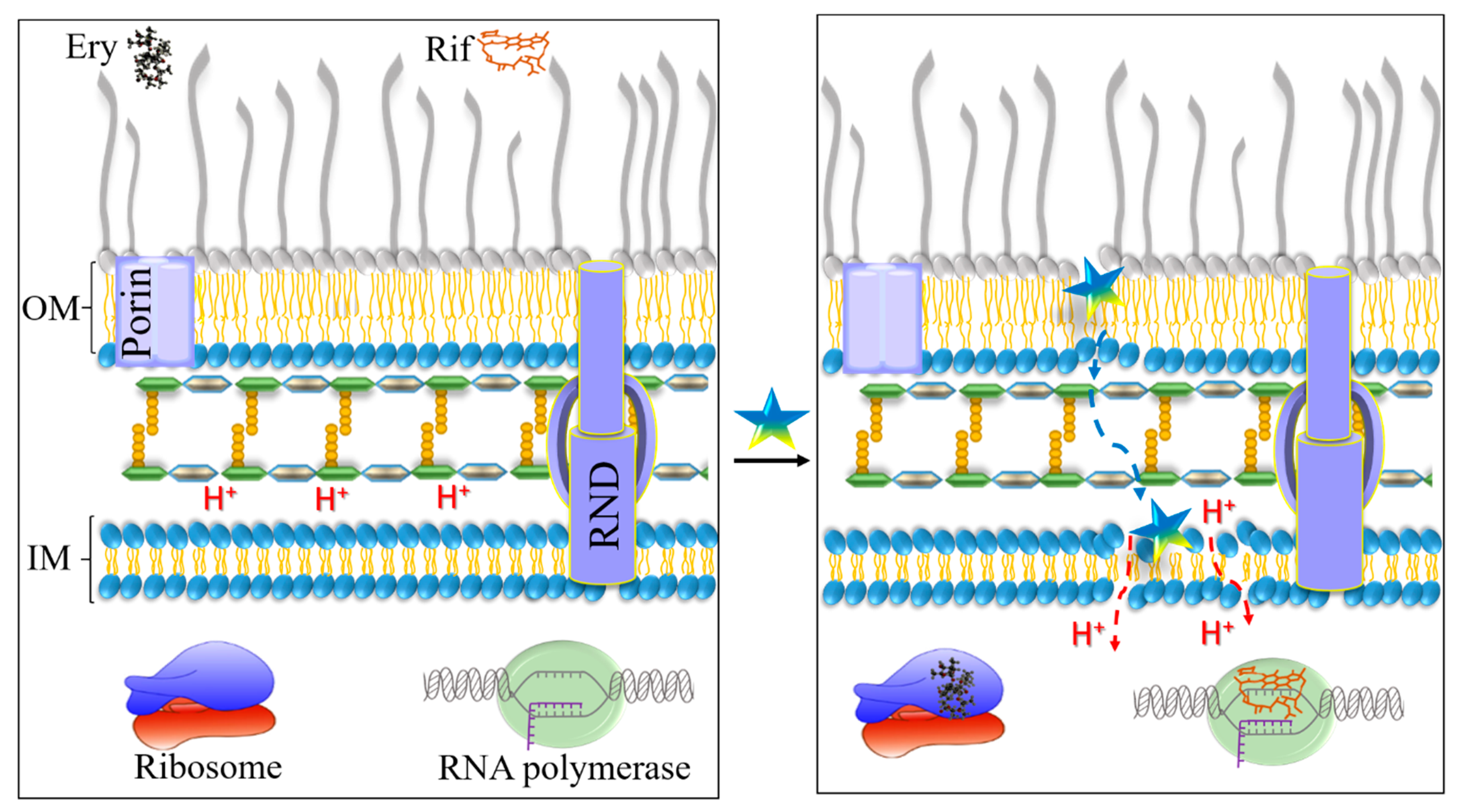
3.3. Mechanistic Studies
3.4. In Vivo Studies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Theuretzbacher, U.; Outterson, K. The global preclinical antibacterial pipeline. Nat. Rev. Microbiol. 2020, 18, 275–285. [Google Scholar] [CrossRef] [Green Version]
- Zabawa, T.P.; Pucci, M.J.; Parr, T.R.; Lister, T. Treatment of Gram-negative bacterial infections by potentiation of antibiotics. Curr. Opin. Microbiol. 2016, 33, 7–12. [Google Scholar] [CrossRef]
- Wright, G.D. Antibiotic Adjuvants: Rescuing Antibiotics from Resistance. Trends Microbiol. 2016, 24, 862–871. [Google Scholar] [CrossRef]
- Masi, M.; Réfregiers, M.; Pos, K.M.; Pagès, J.M. Mechanisms of envelope permeability and antibiotic influx and efflux in Gram-negative bacteria. Nat. Microbiol. 2017, 2, 17001. [Google Scholar] [CrossRef]
- Silver, L.L. A Gestalt approach to Gram-negative entry. Bioorg. Med. Chem. 2016, 24, 6379–6389. [Google Scholar] [CrossRef]
- Lazzaro, B.P.; Zasloff, M.; Rolff, J. Antimicrobial peptides: Application informed by evolution. Science 2020, 368, eaau5480. [Google Scholar] [CrossRef]
- Mookherjee, N.; Anderson, M.A.; Haagsman, H.P.; Davidson, D.J. Antimicrobial host defence peptides: Functions and clinical potential. Nat. Rev. Drug Discov. 2020, 19, 311–332. [Google Scholar] [CrossRef]
- Mishra, B.; Reiling, S.; Zarena, D.; Wang, G. Host defense antimicrobial peptides as antibiotics: Design and application strategies. Curr. Opin. Chem. Biol. 2017, 38, 87–96. [Google Scholar] [CrossRef]
- Epand, R.F.; Maloy, W.L.; Ramamoorthy, A.; Epand, R.M. Probing the “ Charge Cluster Mechanism ” in Amphipathic Helical Cationic. Biochemistry 2010, 49, 4076–4084. [Google Scholar] [CrossRef] [Green Version]
- Epand, R.M.; Rotem, S.; Mor, A.; Berno, B.; Epand, R.F. Bacterial membranes as predictors of antimicrobial potency. J. Am. Chem. Soc. 2008, 130, 14346–14352. [Google Scholar] [CrossRef]
- Westerhoff, H.V.; Juretid, D.; Hendler, R.W.; Zasloff, M. Magainins and the disruption of membrane-linked free-energy transduction. Proc. Natl. Acad. Sci. USA 1989, 86, 6597–6601. [Google Scholar] [CrossRef] [Green Version]
- Kaneti, G.; Meir, O.; Mor, A. Controlling bacterial infections by inhibiting proton-dependent processes. BBA Biomembr. 2015, 1858, 995–1003. [Google Scholar] [CrossRef]
- Hicks, D.B.; Cohen, D.M.; Krulwich, T.A. Reconstitution of Energy-Linked Activities of the Solubilized F1F0 ATP Synthase from Bacillus subtilis. J. Bacteriol. 1994, 176, 4192–4195. [Google Scholar] [CrossRef] [Green Version]
- Wenzel, M.; Chiriac, A.I.; Otto, A.; Zweytick, D.; May, C.; Schumacher, C.; Gust, R.; Albada, H.B.; Penkova, M.; Krämer, U.; et al. Small cationic antimicrobial peptides delocalize peripheral membrane proteins. Proc. Natl. Acad. Sci. USA 2014, 111, 1409–1418. [Google Scholar] [CrossRef] [Green Version]
- Epand, R.F.; Pollard, J.E.; Wright, J.O.; Savage, P.B.; Epand, R.M. Depolarization, bacterial membrane composition, and the antimicrobial action of ceragenins. Antimicrob. Agents Chemother. 2010, 54, 3708–3713. [Google Scholar] [CrossRef] [Green Version]
- Reens, A.L.; Crooks, A.L.; Su, C.C.; Nagy, T.A.; Reens, D.L.; Podoll, J.D.; Edwards, M.E.; Yu, E.W.; Detweiler, C.S. A cell-based infection assay identifies efflux pump modulators that reduce bacterial intracellular load. PLoS Pathog. 2018, 14, e1007115. [Google Scholar] [CrossRef]
- Brandenburg, K.; Heinbockel, L.; Correa, W.; Lohner, K. Peptides with dual mode of action: Killing bacteria and preventing endotoxin-induced sepsis. BBA Biomembr. 2016, 1858, 971–979. [Google Scholar] [CrossRef]
- Kaneti, G.; Sarig, H.; Marjieh, I.; Fadia, Z.; Mor, A. Simultaneous breakdown of multiple antibiotic resistance mechanisms in S. aureus. FASEB J. 2013, 27, 4834–4843. [Google Scholar] [CrossRef]
- Hershkovits, A.S.; Pozdnyakov, I.; Meir, O.; Mor, A. Sub-inhibitory membrane damage undermines Staphylococcus aureus virulence. BBA Biomembr. 2019, 1861, 1172–1179. [Google Scholar] [CrossRef]
- Vaara, M. Agents That Increase the Permeability of the Outer Membrane. Microbiol. Rev. 1992, 56, 395–411. [Google Scholar] [CrossRef]
- Baker, K.R.; Jana, B.; Hansen, A.M.; Nielsen, H.M.; Franzyk, H.; Guardabassi, L. Repurposing azithromycin and rifampicin against gram-negative pathogens by combination with peptidomimetics. Front. Cell. Infect. Microbiol. 2019, 9, 236. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Nonejuie, P.; Munguia, J.; Hollands, A.; Olson, J.; Dam, Q.; Kumaraswamy, M.; Rivera, H.; Corriden, R.; Rohde, M.; et al. Azithromycin Synergizes with Cationic Antimicrobial Peptides to Exert Bactericidal and Therapeutic Activity Against Highly Multidrug-Resistant Gram-Negative Bacterial Pathogens. EBioMedicine 2015, 2, 690–698. [Google Scholar] [CrossRef] [Green Version]
- Vaara, M.; Vaara, T. Sensitization of Gram-negative bacteria to antibiotics and complement by a nontoxic oligopeptide. Nature 1983, 303, 526–528. [Google Scholar] [CrossRef]
- Ruden, S.; Rieder, A.; Chis Ster, I.; Schwartz, T.; Mikut, R.; Hilpert, K. Synergy Pattern of Short Cationic Antimicrobial Peptides Against Multidrug-Resistant Pseudomonas aeruginosa. Front. Microbiol. 2019, 10, 2740. [Google Scholar] [CrossRef] [Green Version]
- Taylor, P.W. Bactericidal and bacteriolytic activity of serum against gram-negative bacteria. Microbiol. Rev. 1983, 47, 46–83. [Google Scholar] [CrossRef]
- Radzishevsky, I.S.; Rotem, S.; Bourdetsky, D.; Navon-Venezia, S.; Carmeli, Y.; Mor, A. Improved antimicrobial peptides based on acyl-lysine oligomers. Nat. Biotechnol. 2007, 25, 657–659. [Google Scholar] [CrossRef]
- Livne, L.; Kovachi, T.; Sarig, H.; Epand, R.F.; Zaknoon, F.; Epand, R.M.; Mor, A. Design and Characterization of a Broad -Spectrum Bactericidal Acyl-lysyl Oligomer. Chem. Biol. 2009, 16, 1250–1258. [Google Scholar] [CrossRef] [Green Version]
- Radzishevsky, I.S.; Kovachi, T.; Porat, Y.; Ziserman, L.; Zaknoon, F.; Danino, D.; Mor, A. Structure-Activity Relationships of Antibacterial Acyl-Lysine Oligomers. Chem. Biol. 2008, 15, 354–362. [Google Scholar] [CrossRef] [Green Version]
- Jammal, J.; Zaknoon, F.; Kaneti, G.; Hershkovits, A.S.; Mor, A. Sensitization of Gram-Negative Bacilli to Host Antibacterial Proteins. J. Infect. Dis. 2017, 215, 1599–1607. [Google Scholar] [CrossRef] [Green Version]
- Jammal, J.; Zaknoon, F.; Mor, A. Eliciting improved antibacterial efficacy of host proteins in the presence of antibiotics. FASEB J. 2018, 32, 369–376. [Google Scholar] [CrossRef] [Green Version]
- Jammal, J.; Zaknoon, F.; Kaneti, G.; Goldberg, K.; Mor, A. Sensitization of Gram-negative bacteria to rifampin and OAK combinations. Sci. Rep. 2015, 5, srep09216. [Google Scholar] [CrossRef] [Green Version]
- Meir, O.; Zaknoon, F.; Cogan, U.; Mor, A. A broad-spectrum bactericidal lipopeptide with anti-biofilm properties. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Sarig, H.; Livne, L.; Held-Kuznetsov, V.; Zaknoon, F.; Ivankin, A.; Gidalevitz, D.; Mor, A. A miniature mimic of host defense peptides with systemic antibacterial efficacy. FASEB J. 2010, 24, 1904–1913. [Google Scholar] [CrossRef] [Green Version]
- Okusu, H.; Ma, D.; Nikaido, H. AcrAB efflux pump plays a major role in the antibiotic resistance phenotype of Escherichia coli multiple-antibiotic-resistance (Mar) mutants. J. Bacteriol. 1996, 178, 306–308. [Google Scholar] [CrossRef] [Green Version]
- Redfield, R. Antibiotic Resistance Threats in the United States; CDC: Atlanta, GA, USA, 2019.
- Zaknoon, F.; Sarig, H.; Rotem, S.; Livne, L.; Ivankin, A.; Gidalevitz, D.; Mor, A. Antibacterial properties and mode of action of a short acyl-lysyl oligomer. Antimicrob. Agents Chemother. 2009, 53, 3422–3429. [Google Scholar] [CrossRef] [Green Version]
- Zaknoon, F.; Goldberg, K.; Sarig, H.; Epand, R.F.; Epand, R.M.; Mor, A. Antibacterial properties of an oligo-acyl-lysyl hexamer targeting gram-negative species. Antimicrob. Agents Chemother. 2012, 56, 4827–4832. [Google Scholar] [CrossRef] [Green Version]
- Hancock, R.E.W.; Farmer, S.W.; Li, Z.; Poole, K. Interaction of aminoglycosides with the outer membranes and purified lipopolysaccharide and OmpF porin of Escherichia coli. Antimicrob. Agents Chemother. 1991, 35, 1309–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daugelavicius, R.; Bakiene, E.; Bamford, D.H. Stages of polymyxin B interaction with the Escherichia coli cell envelope. Antimicrob. Agents Chemother. 2000, 44, 2969–2978. [Google Scholar] [CrossRef] [Green Version]
- Brown, P.; Dawson, M.J. Development of new polymyxin derivatives for multi-drug resistant Gram-negative infections. J. Antibiot. 2017, 70, 386–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehrer, R.I.; Barton, A.; Ganz, T. Concurrent assessment of inner and outer membrane permeabilization and bacteriolysis in E. coli by multiple-wavelength spectrophotometry. J. Immunol. Methods 1988, 108, 153–158. [Google Scholar] [CrossRef]
- Moore, R.; Bates, N.C.; Hancock, R.E.W. Interaction of polycationic antibiotics with Pseudomonas aeruginosa Lipopolysaccharide and lipid A studied by using dansyl-polymyxin. Antimicrob. Agents Chemother. 1986, 29, 496–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haisma, E.M.; Göblyös, A.; Ravensbergen, B.; Adriaans, A.E.; Cordfunke, R.A.; Schrumpf, J.; Limpens, R.W.A.L.; Schimmel, K.J.M.; Den Hartigh, J.; Hiemstra, P.S.; et al. Antimicrobial peptide P60.4Ac-containing creams and gel for eradication of methicillin-resistant Staphylococcus aureus from cultured skin and airway epithelial surfaces. Antimicrob. Agents Chemother. 2016, 60, 4063–4072. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Dou, X.; Song, J.; Lyu, Y.; Zhu, X.; Xu, L.; Li, W.; Shan, A. Antimicrobial peptides: Promising alternatives in the post feeding antibiotic era. Med. Res. Rev. 2019, 39, 831–859. [Google Scholar] [CrossRef] [PubMed]
- Wiradharma, N.; Sng, M.Y.S.; Khan, M.; Ong, Z. Rationally Designed α -Helical Broad-Spectrum Antimicrobial Peptides with Idealized Facial Amphiphilicity. Macromol. Rapid Commun. 2013, 34, 74–80. [Google Scholar] [CrossRef]
- Hollmann, A.; Martínez, M.; Noguera, M.E.; Augusto, M.T.; Disalvo, A.; Santos, N.C.; Semorile, L.; Maffía, P.C. Role of amphipathicity and hydrophobicity in the balance between hemolysis and peptide-membrane interactions of three related antimicrobial peptides. Colloids Surf. B Biointerfaces 2016, 141, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Vaara, M.; Fox, J.; Loidl, G.; Siikanen, O.; Apajalahti, J.; Hansen, F.; Frimodt-Møller, N.; Nagai, J.; Takano, M.; Vaara, T. Novel polymyxin derivatives carrying only three positive charges are effective antibacterial agents. Antimicrob. Agents Chemother. 2008, 52, 3229–3236. [Google Scholar] [CrossRef] [Green Version]
- Vaara, M.; Siikanen, O.; Apajalahti, J.; Fox, J.; Frimodt-Møller, N.; He, H.; Poudyal, A.; Li, J.; Nation, R.L.; Vaara, T. A novel polymyxin derivative that lacks the fatty acid tail and carries only three positive charges has strong synergism with agents excluded by the intact outer membrane. Antimicrob. Agents Chemother. 2010, 54, 3341–3346. [Google Scholar] [CrossRef] [Green Version]
- Mares, J.; Kumaran, S.; Gobbo, M.; Zerbe, O. Interactions of lipopolysaccharide and polymyxin studied by NMR spectroscopy. J. Biol. Chem. 2009, 284, 11498–11506. [Google Scholar] [CrossRef] [Green Version]
- Clinical and Laboratory Standards Institute M100-S26. Performance Standards for Antimicrobial Susceptibility Testing, 26th ed.; Approved Standard; CLSI: Wayne, PA, USA, 2015. [Google Scholar]
- Corbett, D.; Wise, A.; Langley, T.; Skinner, K.; Trimby, E.; Birchall, S.; Dorali, A.; Sandiford, S.; Williams, J.; Warn, P.; et al. Potentiation of antibiotic activity by a novel cationic peptide: Potency and spectrum of activity of SPR741. Antimicrob. Agents Chemother. 2017, 61, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Scocchi, M.; Mardirossian, M.; Runti, G.; Benincasa, M. Non-Membrane Permeabilizing Modes of Action of Antimicrobial Peptides on Bacteria. Curr. Top. Med. Chem. 2015, 16, 76–88. [Google Scholar] [CrossRef]
- Lobritz, M.A.; Belenky, P.; Porter, C.B.M.; Gutierrez, A.; Yang, J.H. Antibiotic efficacy is linked to bacterial cellular respiration. Proc. Natl. Acad. Sci. USA 2015, 112, 8173–8180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Maier, E.; Benz, R.; Hancock, R.E.W. Mechanism of interaction of different classes of cationic antimicrobial peptides with planar bilayers and with the cytoplasmic membrane of Escherichia coli. Biochemistry 1999, 38, 7235–7242. [Google Scholar] [CrossRef]
- Boyer, P.D. The ATP synthase—A splendid molecular machine. Annu. Rev. Biochem. 1997, 66, 717–749. [Google Scholar] [CrossRef] [Green Version]
- Chouchani, E.T.; Kazak, L.; Spiegelman, B.M. New Advances in Adaptive Thermogenesis: UCP1 and Beyond. Cell Metab. 2019, 29, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Du, D.; Wang-Kan, X.; Neuberger, A.; van Veen, H.W.; Pos, K.M.; Piddock, L.J.V.; Luisi, B.F. Multidrug efflux pumps: Structure, function and regulation. Nat. Rev. Microbiol. 2018, 16, 523–539. [Google Scholar] [CrossRef] [PubMed]
- Shafer, W.M.; Qu, X.D.; Waring, A.J.; Lehrer, R.I. Modulation of Neisseria gonorrhoeae susceptibility to vertebrate antibacterial peptides due to a member of the resistance/nodulation/division efflux pump family. Proc. Natl. Acad. Sci. USA 1998, 95, 1829–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zgurskaya, H.I.; Nikaido, H. Bypassing the periplasm: Reconstitution of the AcrAB multidrug efflux pump of Escherichia coli. Proc. Natl. Acad. Sci. USA 1999, 96, 7190–7195. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, K.; Sarig, H.; Zaknoon, F.; Epand, R.F.; Epand, R.M.; Mor, A. Sensitization of gram-negative bacteria by targeting the membrane potential. FASEB J. 2013, 27, 3818–3826. [Google Scholar] [CrossRef]
- Trimble, M.J.; Mlynarcik, P.; Kolar, M.; Hancock, R.E.W. Polymyxin: Alternative Mechanisms of Action. Cold Spring Harb Perspect. Med. 2016, 6, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Hancock, R.E.W. Alterations in structure of the cell envelope. Ann. Rev. Microbiol. 1984, 38, 237–264. [Google Scholar] [CrossRef]
- Gaidukov, L.; Fish, A.; Mor, A. Analysis of Membrane-Binding Properties of Dermaseptin Analogues: Relationships between Binding and Cytotoxicity. Biochemistry 2003, 42, 12866–12874. [Google Scholar] [CrossRef]
- Blair, J.M.A.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J. V Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Laarman, A.J.; Bardoel, B.W.; Ruyken, M.; Fernie, J.; Milder, F.J.; Strijp, J.A.G.; van Suzan, H.; Rooijakkers, M.; Strijp, J.A.G.; van Rooijakkers, S.H.M. Pseudomonas aeruginosa Alkaline Protease Blocks Complement Activation via the Classical and Lectin Pathways. J. Immunol. 2012, 188, 386–393. [Google Scholar] [CrossRef] [Green Version]
- Mangoni, M.L.; Mcdermott, A.M.; Zasloff, M. Antimicrobial peptides and wound healing: Biological and therapeutic considerations. Exp. Dermatol. 2016, 25, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Pfalzgraff, A.; Brandenburg, K.; Weindl, G. Antimicrobial peptides and their therapeutic potential for bacterial skin infections and wounds. Front. Pharmacol. 2018, 9, 1–23. [Google Scholar] [CrossRef]
- Fernandes, P.B.; Hardy, D.J.; McDaniel, D.; Hanson, C.W.; Swanson, R.N. In vitro and in vivo activities of clarithromycin against Mycobacterium avium. Antimicrob. Agents Chemother. 1989, 33, 1531–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Y.; Da, Y.; Zhao, Y.; Qi, S.; Liu, L.; Lu, L.; Luo, Q.; Zhang, Z.H. Melittin-lipid nanoparticles target to lymph nodesand elicit a systemic anti-tumor immune response. Nat. Commun. 2020, 11, 1110. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lipopeptide Sequence | H (%) | CAC (µM) | HC50 (µM) | MIC (µM) | |
|---|---|---|---|---|---|
| LB a Medium | Human b Plasma | ||||
| * C14KKc12K | 55 | 20 ± 5 | 12 ± 1 | 3–6 | >20 |
| C14OOc12O | 55 | 15 ± 1 | 14 ± 4 | 3–6 | >20 |
| C14OOc10O | 53 | 45 ± 14 | 28 ± 2 | 12.5–25 | 10–20 |
| C14(ω5)OOc10O | 50 | >100 | >100 | >50 | 2.5–5 |
| C14(ω5)OOc8O | 48 | >100 | >100 | >50 | 2.5–5 |
| C14(ω5)OOc6O | 47 | >100 | >100 | >50 | 5 |
| OOc12O | 24 | >100 | >100 | >50 | >20 |
| Sensitization Factor at 8 µg/mL | |||
|---|---|---|---|
| Bacteria | C14(ω5)OOc10O | C10OOc12O | SPR741 |
| Kp | 64,000 | 8000 [30] | 32 [51] |
| Ec | 32,000 | 16,000 [30] | 8192 [51] |
| Pa | 32,000 | 1000 | 5 [48] |
| Ab | 4000 | 4000 | 256 [48] |
| Tested Compound | MIC (µM) | |
|---|---|---|
| Ag100 | Ag100A | |
| LL-37 | 22.2 | 1.1 |
| Erythromycin | 174.4 | 10.9 |
| C14(ω5)OOc10O | 25 | 6.2 |
| C14OOc12O | 3.1 | 3.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaknoon, F.; Meir, O.; Mor, A. Mechanistic Studies of Antibiotic Adjuvants Reducing Kidney’s Bacterial Loads upon Systemic Monotherapy. Pharmaceutics 2021, 13, 1947. https://doi.org/10.3390/pharmaceutics13111947
Zaknoon F, Meir O, Mor A. Mechanistic Studies of Antibiotic Adjuvants Reducing Kidney’s Bacterial Loads upon Systemic Monotherapy. Pharmaceutics. 2021; 13(11):1947. https://doi.org/10.3390/pharmaceutics13111947
Chicago/Turabian StyleZaknoon, Fadia, Ohad Meir, and Amram Mor. 2021. "Mechanistic Studies of Antibiotic Adjuvants Reducing Kidney’s Bacterial Loads upon Systemic Monotherapy" Pharmaceutics 13, no. 11: 1947. https://doi.org/10.3390/pharmaceutics13111947