Differential Effects of 1α,25-Dihydroxyvitamin D3 on the Expressions and Functions of Hepatic CYP and UGT Enzymes and Its Pharmacokinetic Consequences In Vivo

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. 1,25(OH)2D3 Treatment in Rats
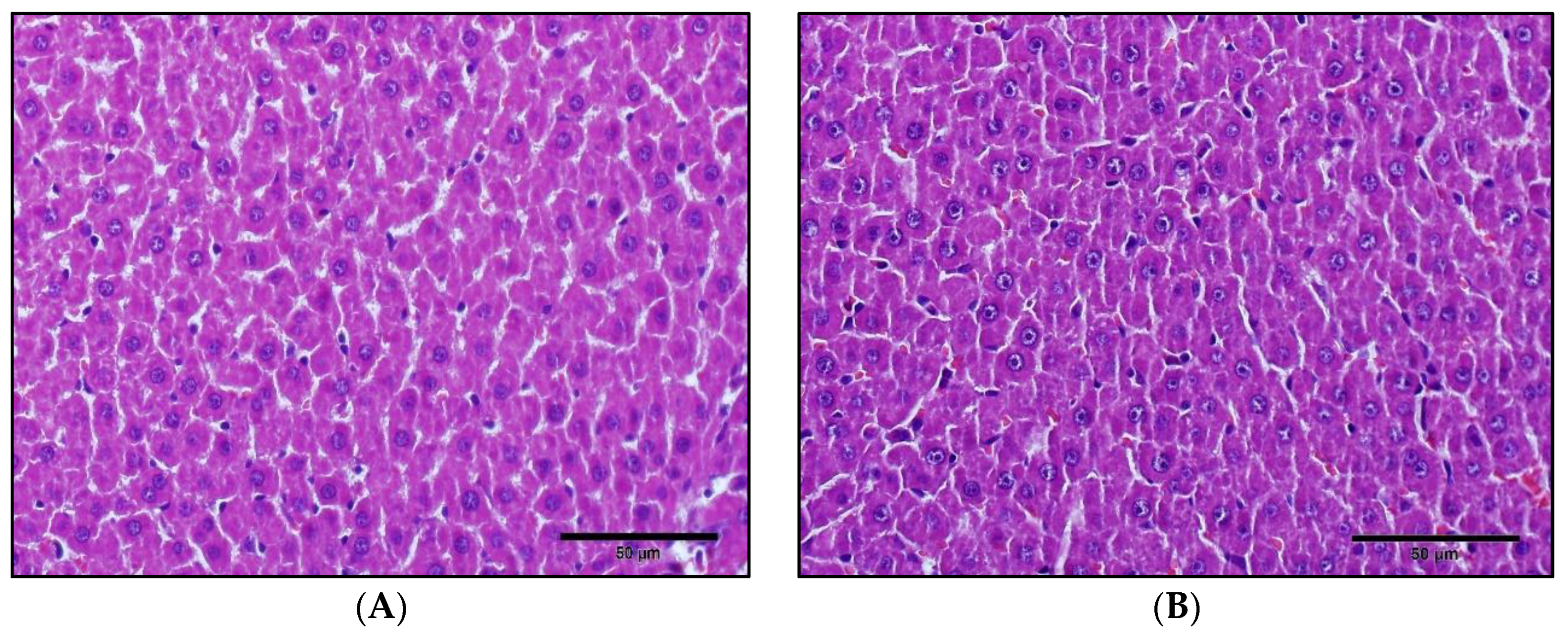
2.3. Liver Histology
2.4. Serum Chemistry
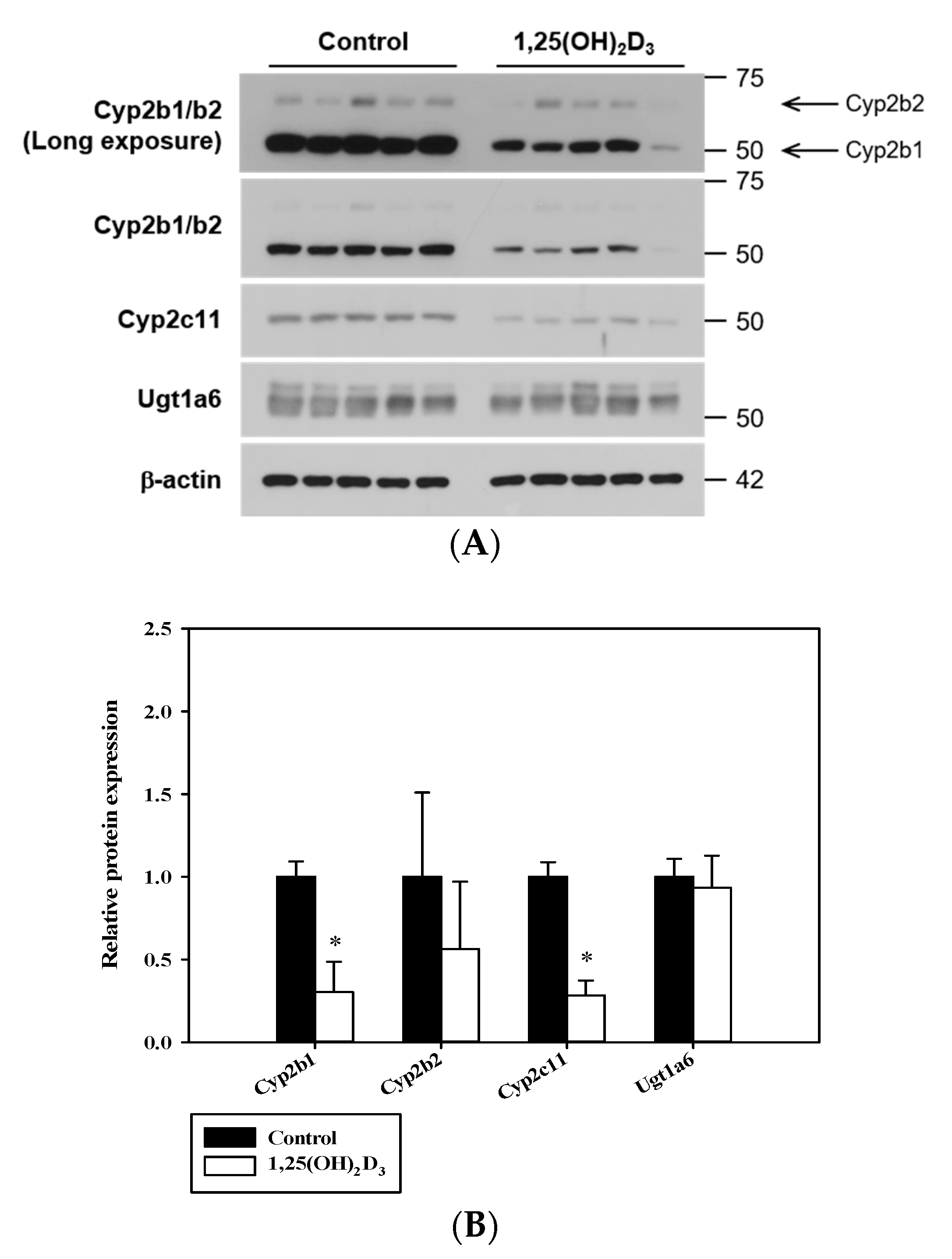
2.5. Preparation of Proteins and Western Blotting
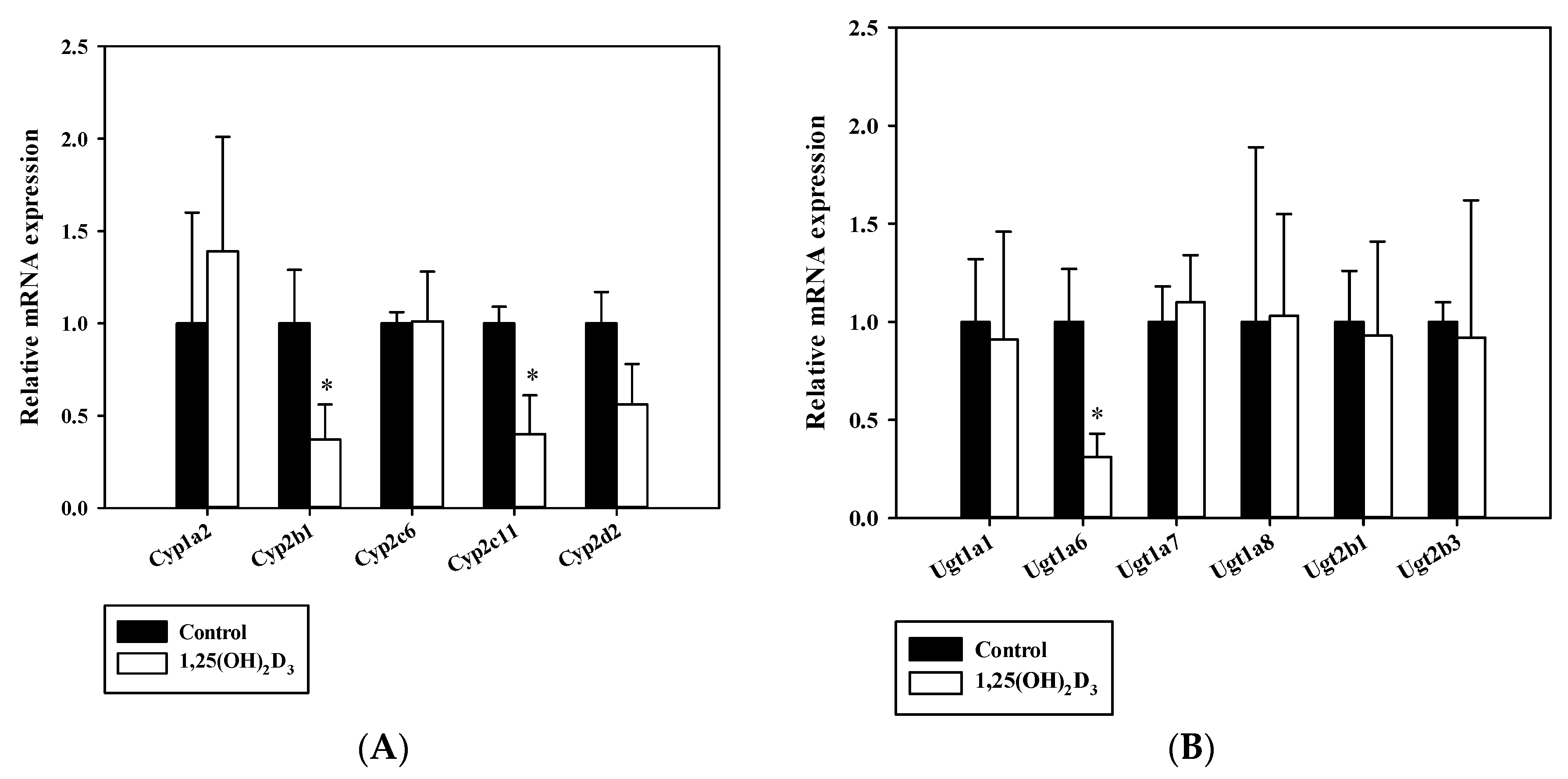
2.6. Real-Time Quantitative Polymerase Chain Reaction
2.7. In Vitro Metabolic Study Using Rat Liver Microsomes (RLMs)
2.8. In Vitro Plasma Protein Binding Study
2.9. In Vivo Pharmacokinetic Study in Rats
2.10. Sample Preparation
2.11. UPLC-DAD Analysis
2.12. LC-MS/MS Analysis
2.13. Pharmacokinetic Analysis
2.14. Statistical Analysis
3. Results
3.1. Effects of 1,25(OH)2D3 on Liver Histology, Serum Chemistry and on The mRNA/Protein Expression Levels of Hepatic Cyps and Ugts in Rats
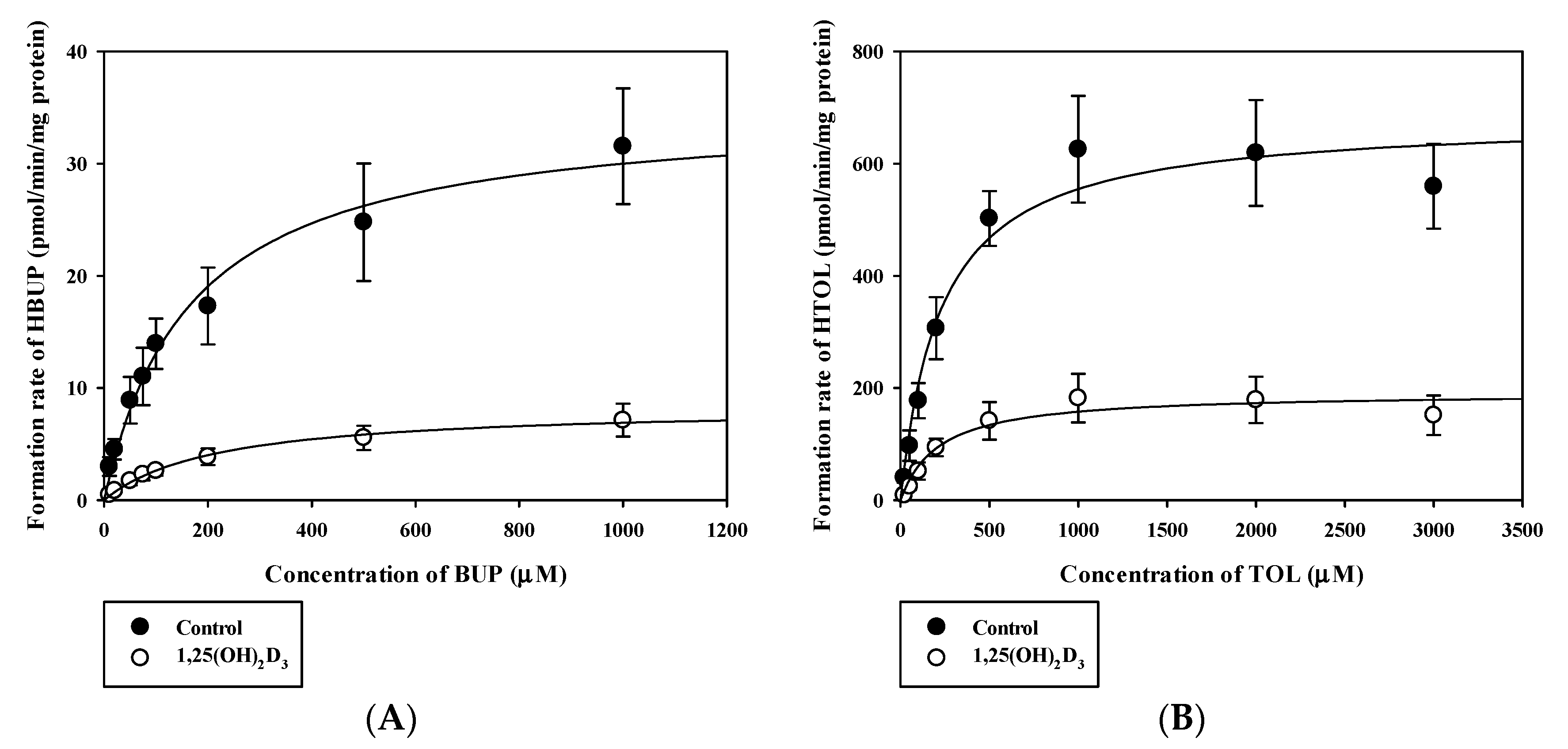
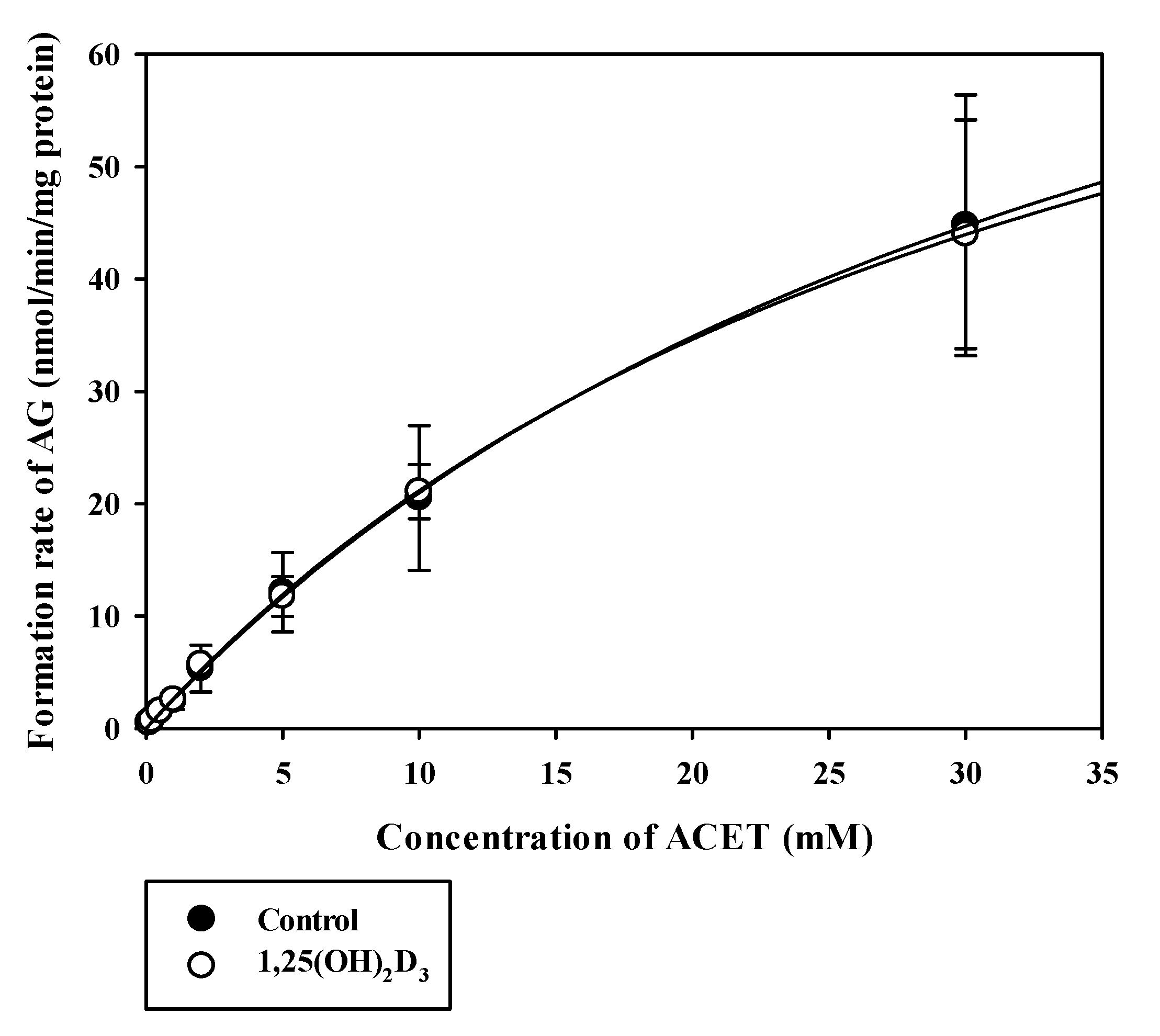
3.2. Effects of 1,25(OH)2D3 on the Functional Activities of CYPs and UGTs in RLMs
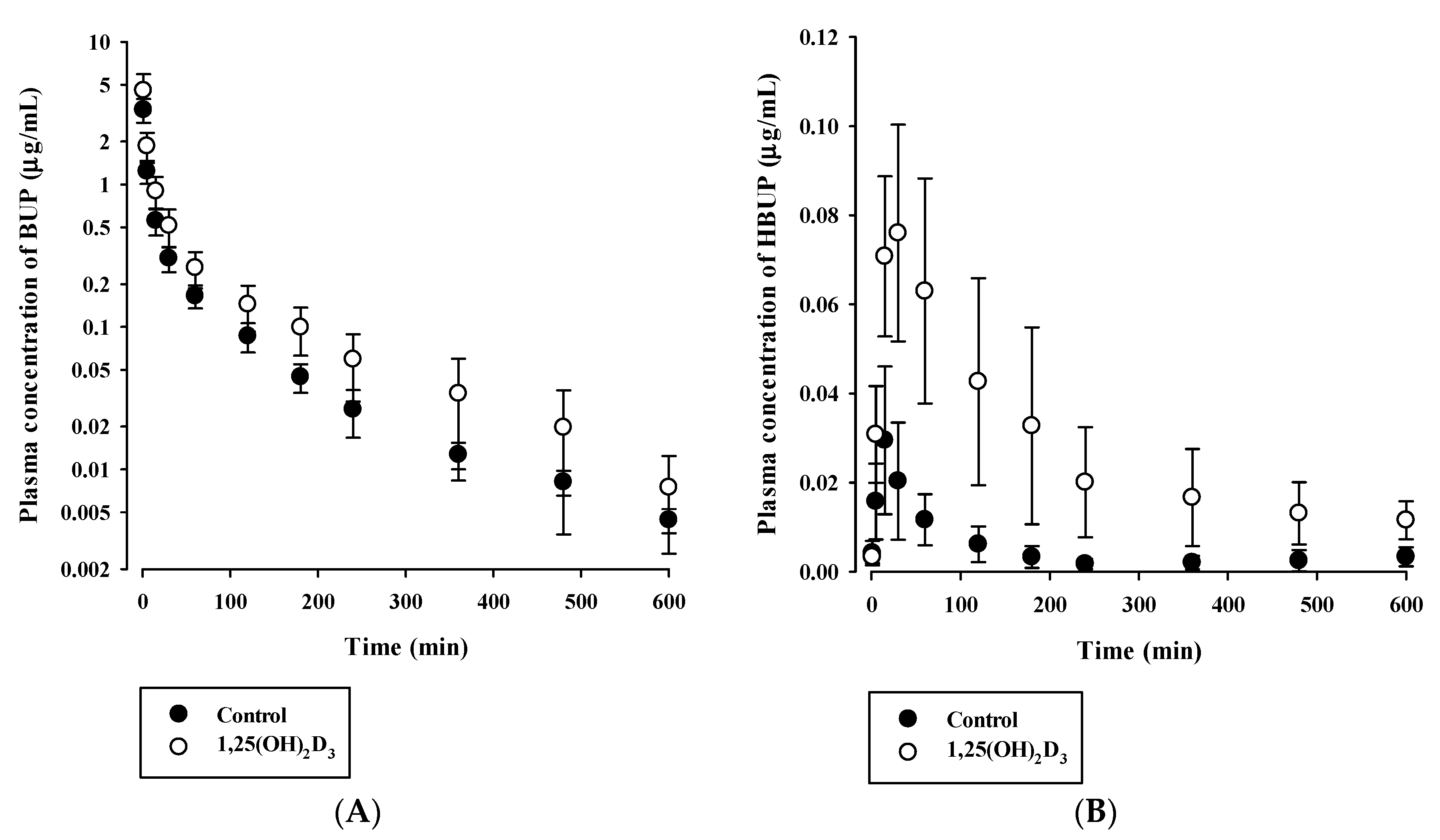
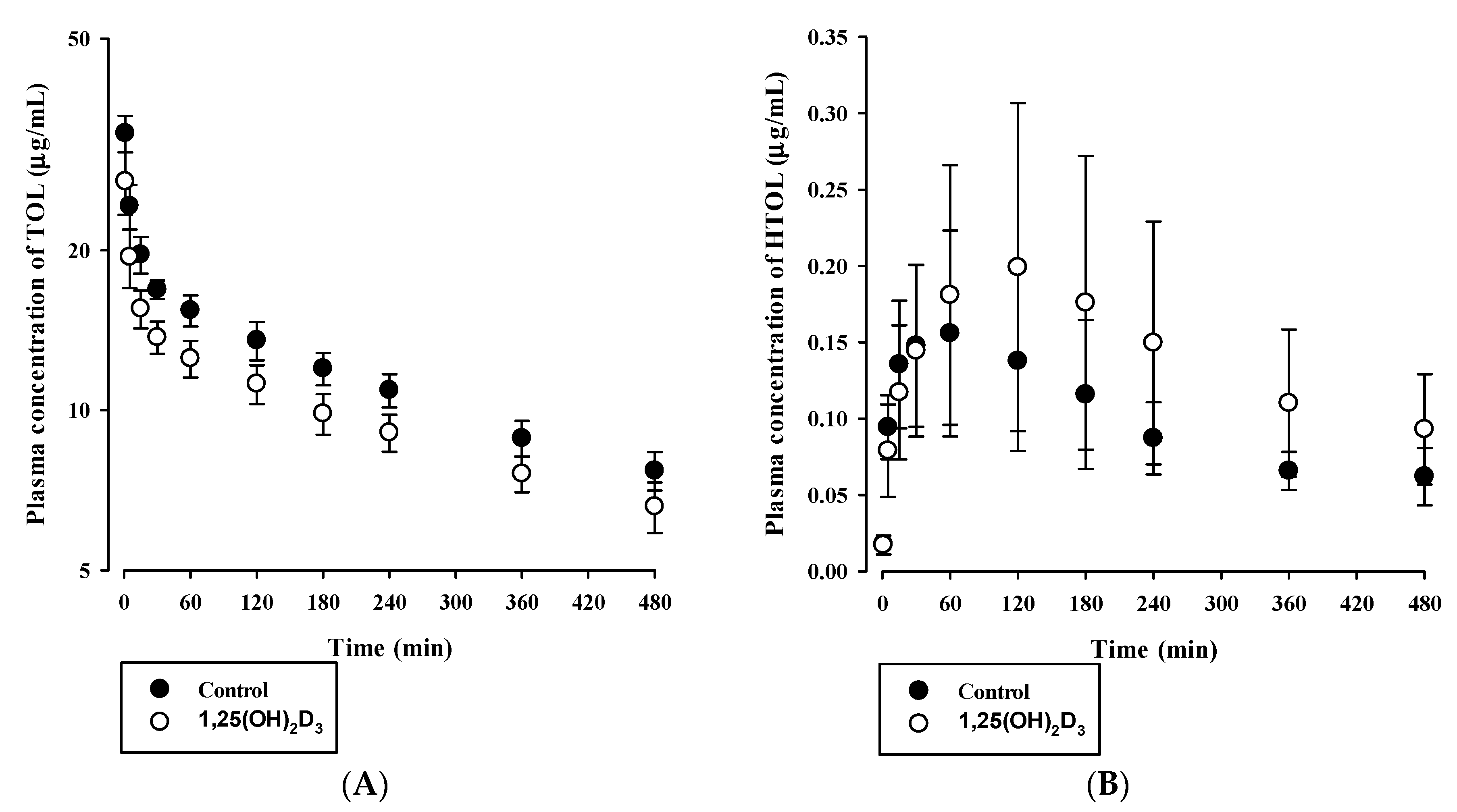
3.3. Effects of 1,25(OH)2D3 on the Pharmacokinetics of BUP and TOL
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Borel, P.; Caillaud, D.; Cano, N.J. Vitamin D bioavailability: State of the art. Crit. Rev. Food Sci. Nutr. 2015, 55, 1193–1205. [Google Scholar] [PubMed]
- Maeng, H.J.; Doan, T.N.K.; Yoon, I.S. Differential regulation of intestinal and hepatic CYP3A by 1alpha,25-dihydroxyvitamin D3: Effects on in vivo oral absorption and disposition of buspirone in rats. Drug Dev. Res. 2019, 80, 333–342. [Google Scholar] [PubMed]
- Dusso, A.S.; Brown, A.J. Mechanism of vitamin D action and its regulation. Am. J. Kidney Dis. 1998, 32, S13–S24. [Google Scholar] [PubMed]
- Ben-Eltriki, M.; Deb, S.; Guns, E.S. Calcitriol in combination therapy for prostate cancer: Pharmacokinetic and pharmacodynamic interactions. J. Cancer 2016, 7, 391–407. [Google Scholar] [PubMed] [Green Version]
- Beer, T.M.; Myrthue, A. Calcitriol in cancer treatment: From the lab to the clinic. Mol. Cancer Ther. 2004, 3, 373–381. [Google Scholar] [PubMed]
- Van Etten, E.; Decallonne, B.; Verlinden, L.; Verstuyf, A.; Bouillon, R.; Mathieu, C. Analogs of 1alpha,25-dihydroxyvitamin D3 as pluripotent immunomodulators. J. Cell Biochem. 2003, 88, 223–226. [Google Scholar] [PubMed]
- Gil, A.; Plaza-Diaz, J.; Mesa, M.D. Vitamin D: Classic and Novel Actions. Ann. Nutr. Metab. 2018, 72, 87–95. [Google Scholar] [PubMed]
- Choi, M.S.; Kim, Y.C.; Maeng, H.J. Therapeutic targets of vitamin D receptor ligands and their pharmacokinetic effects by modulation of transporters and metabolic enzymes. J. Pharm. Invest. 2020, 50, 1–6. [Google Scholar]
- Fan, J.; Liu, S.; Du, Y.; Morrison, J.; Shipman, R.; Pang, K.S. Up-regulation of transporters and enzymes by the vitamin D receptor ligands, 1alpha,25-dihydroxyvitamin D3 and vitamin D analogs, in the Caco-2 cell monolayer. J. Pharmacol. Exp. Ther. 2009, 330, 389–402. [Google Scholar]
- Maeng, H.J.; Chapy, H.; Zaman, S.; Pang, K.S. Effects of 1α,25-dihydroxyvitamin D3 on transport and metabolism of adefovir dipivoxil and its metabolites in Caco-2 cells. Eur. J. Pharm. Sci. 2012, 46, 149–166. [Google Scholar]
- Kim, Y.C.; Kim, I.B.; Noh, C.K.; Quach, H.P.; Yoon, I.S.; Chow, E.C.Y.; Kim, M.; Jin, H.E.; Cho, K.H.; Chung, S.J.; et al. Effects of 1alpha,25-dihydroxyvitamin D3, the natural vitamin D receptor ligand, on the pharmacokinetics of cefdinir and cefadroxil, organic anion transporter substrates, in rat. J. Pharm. Sci. 2014, 103, 3793–3805. [Google Scholar] [PubMed]
- Yoon, I.S.; Son, J.H.; Kim, S.B.; Choi, M.K.; Maeng, H.J. Effects of 1alpha,25-Dihydroxyvitamin D3 on intestinal absorption and disposition of adefovir dipivoxil and its metabolite, adefovir, in rats. Biol. Pharm. Bull. 2015, 38, 1732–1737. [Google Scholar] [PubMed] [Green Version]
- Chow, E.C.; Durk, M.R.; Cummins, C.L.; Pang, K.S. 1 alpha,25-dihydroxyvitamin D3 up-regulates P-glycoprotein via the vitamin D receptor and not farnesoid X receptor in both fxr(−/−) and fxr(+/+) mice and increased renal and brain efflux of digoxin in mice in vivo. J. Pharmacol. Exp. Ther. 2011, 337, 846–859. [Google Scholar] [PubMed] [Green Version]
- Xu, Y.; Iwanaga, K.; Zhou, C.; Cheesman, M.J.; Farin, F.; Thummel, K.E. Selective induction of intestinal CYP3A23 by 1alpha,25-dihydroxyvitamin D3 in rats. Biochem. Pharmacol. 2006, 72, 385–392. [Google Scholar] [PubMed]
- Khan, A.A.; Draqt, B.D.; Porte, R.J.; Groothuis, G.M. Regulation of VDR expression in rat and human intestine and liver--consequences for CYP3A expression. Toxicol. In Vitro 2010, 24, 822–829. [Google Scholar] [PubMed]
- Chow, E.C.; Maeng, H.J.; Liu, S.; Khan, A.A.; Groothuis, G.M.; Pang, K.S. 1alpha,25-Dihydroxyvitamin D(3) triggered vitamin D receptor and farnesoid X receptor-like effects in rat intestine and liver in vivo. Biopharm. Drug Dispos. 2009, 30, 457–475. [Google Scholar] [PubMed]
- Schmiedlin-Ren, P.; Thummel, K.E.; Fisher, J.M.; Paine, M.F.; Lown, K.S.; Watkins, P.B. Expression of enzymatically active CYP3A4 by Caco-2 cells grown on extracellular matrix-coated permeable supports in the presence of 1alpha,25-dihydroxyvitamin D3. Mol. Pharmacol. 1997, 51, 741–754. [Google Scholar] [PubMed] [Green Version]
- Schwartz, J.B. Effects of vitamin D supplementation in atorvastatin-treated patients: A new drug interaction with an unexpected consequence. Clin. Pharmacol. Ther. 2009, 85, 198–203. [Google Scholar]
- Chow, E.C.; Magomedova, L.; Quach, H.P.; Patel, R.; Durk, M.R.; Fan, J.; Maeng, H.J.; Irondi, K.; Anakk, S.; Moore, D.D.; et al. Vitamin D receptor activation down-regulates the small heterodimer partner and increases CYP7A1 to lower cholesterol. Gastroenterology 2014, 146, 1048–1059. [Google Scholar]
- Drocourt, L.; Ourlin, J.C.; Pascussi, J.M.; Maurel, P.; Vilarem, M.J. Expression of CYP3A4, CYP2B6, and CYP2C9 is regulated by the vitamin D receptor pathway in primary human hepatocytes. J. Biol. Chem. 2002, 277, 25125–25132. [Google Scholar]
- Chow, E.C.; Durk, M.R.; Maeng, H.J.; Pang, K.S. Comparative effects of 1alpha-hydroxyvitamin D3 and 1,25-dihydroxyvitamin D3 on transporters and enzymes in fxr(+/+) and fxr(-/-) mice. Biopharm. Drug Dispos. 2013, 34, 402–416. [Google Scholar] [PubMed]
- Wang, X.; Wang, H.; Shen, B.; Overholser, B.R.; Cooper, B.R.; Lu, Y.; Tang, H.; Zhou, C.; Sun, X.; Zhong, L.; et al. 1-Alpha, 25-dihydroxyvitamin D3 alters the pharmacokinetics of mycophenolic acid in renal transplant recipients by regulating two extrahepatic UDP-glucuronosyltransferases 1A8 and 1A10. Transl. Res. 2016, 178, 54–62. [Google Scholar] [PubMed] [Green Version]
- US Food and Drug Administration (FDA). In Vitro Drug Interaction Studies—Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry. 2020. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/vitro-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions (accessed on 21 January 2020).
- Pekthong, D.; Desbans, C.; Martin, H.; Richert, L. Bupropion hydroxylation as a selective marker of rat CYP2B1 catalytic activity. Drug Metab. Dispos. 2012, 40, 32–38. [Google Scholar] [PubMed] [Green Version]
- Fukuno, S.; Nagai, K.; Kasahara, K.; Mizobata, Y.; Omotani, S.; Hatsuda, Y.; Myotoku, M.; Konishi, H. Altered tolbutamide pharmacokinetics by a decrease in hepatic expression of CYP2C6/11 in rats pretreated with 5-fluorouracil. Xenobiotica 2018, 48, 53–59. [Google Scholar] [PubMed]
- Kessler, F.K.; Kessler, M.R.; Auyeung, D.J.; Ritter, J.K. Glucuronidation of acetaminophen catalyzed by multiple rat phenol UDP-glucuronosyltransferases. Drug Metab. Dispos. 2002, 30, 324–330. [Google Scholar]
- Maeng, H.J.; Durk, M.R.; Chow, E.C.; Ghoneim, R.; Pang, K.S. 1α, 25-Dihydroxyvitamin D3 on intestinal transporter function: Studies with the rat everted intestinal sac. Biopharm. Drug Dispos. 2011, 32, 112–125. [Google Scholar]
- Kawase, A.; Fujii, A.; Negoro, A.; Akai, R. Differences in cytochrome P450 and nuclear receptor mRNA. levels in liver and small intestines between SD and DA rats. Drug Metab. Pharmacokinet. 2008, 23, 196–206. [Google Scholar]
- Alkharfy, K.M.; Poloyac, S.M.; Congiu, M.; Desmond, P.V.; Frye, R.F. Effect of the acute phase response induced by endotoxin administration on the expression and activity of UGT isoforms in rats. Drug Metab. Lett. 2008, 2, 248–255. [Google Scholar]
- Yoon, I.S.; Choi, M.K.; Kim, J.S.; Shim, C.K.; Chung, S.J.; Kim, D.D. Pharmacokinetics and first-pass elimination of metoprolol in rats: Contribution of intestinal first-pass extraction to low bioavailability of metoprolol. Xenobiotica 2011, 41, 243–251. [Google Scholar]
- Cho, S.J.; Kim, S.B.; Cho, H.J.; Chong, S.; Chung, S.J.; Kang, I.M.; Lee, J.I.; Yoon, I.S.; Kim, D.D. Effects of nonalcoholic fatty liver disease on hepatic CYP2B1 and in vivo bupropion disposition in rats fed a high-fat or methionine/choline-deficient diet. J. Agric. Food Chem. 2016, 64, 5598–5606. [Google Scholar]
- Choi, M.R.; Kwon, M.H.; Cho, Y.Y.; Choi, H.D.; Kim, Y.C.; Kang, H.E. Pharmacokinetics of tolbutamide and its metabolite 4-hydroxy tolbutamide in poloxamer 407-induced hyperlipidemic rats. Biopharm. Drug Dispos. 2014, 35, 264–274. [Google Scholar]
- Eagling, V.A.; Tjia, J.F.; Back, D.J. Differential selectivity of cytochrome P450 inhibitors against probe substrates in human and rat liver microsomes. Br. J. Clin. Pharmacol. 1998, 45, 107–114. [Google Scholar] [PubMed] [Green Version]
- Meunier, C.J.; Verbeeck, R.K. Glucuronidation of R- and S-ketoprofen, acetaminophen, and diflunisal by liver microsomes of adjuvant-induced arthritic rats. Drug Metab. Dispos. 1999, 27, 26–31. [Google Scholar] [PubMed]
- Berger, U.; Wilson, P.; McClelland, R.A.; Colston, K.; Haussler, M.R.; Pike, J.W.; Coombes, R.C. Immunocytochemical detection of 1,25-dihydroxyvitamin D receptors in normal human tissues. J. Clin. Endocrinol. Metab. 1988, 67, 607–613. [Google Scholar] [PubMed]
- Sandgren, M.E.; Bronnegard, M.; DeLuca, H.F. Tissue distribution of the 1,25-dihydroxyvitamin D3 receptor in the male rat. Biochem. Biophys. Res. Commun. 1991, 181, 611–616. [Google Scholar] [PubMed]
- Cheng, S.; So, W.Y.; Zhang, D.; Cheng, Q.; Boucher, B.J.; Leung, P.S. Calcitriol reduces hepatic triglyceride accumulation and glucose output through Ca2+/CaMKKβ/AMPK activation under insulin-resistant conditions in type 2 diabetes mellitus. Curr. Mol. Med. 2016, 16, 747–758. [Google Scholar] [PubMed]
- Han, H.; Cui, M.; You, X.; Chen, M.; Piao, X.; Jin, G. A role of 1,25(OH)2D3 supplementation in rats with nonalcoholic steatohepatitis induced by choline-deficient diet. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 556–561. [Google Scholar]
- Connarn, J.N.; Flowers, S.; Kelly, M.; Luo, R.; Ward, K.M.; Harrington, G.; Moncion, I.; Kamali, M.; McInnis, M.; Feng, M.R.; et al. Pharmacokinetics and pharmacogenomics of bupropion in three different formulations with different release kinetics in healthy human volunteers. AAPS J. 2017, 19, 1513–1522. [Google Scholar]
- Sachinvala, N.D.; Teramoto, N.; Stergiou, A. Proposed neuroimmune roles of dimethyl fumarate, bupropion, S-adenosylmethionine, and vitamin D3 in affording a chronically Ill patient sustained relief from inflammation and major depression. Brain Sci. 2020, 10, 600. [Google Scholar]
- Nishimura, N.; Naora, K.; Uemura, T.; Hirano, H.; Iwamoto, K. Transepithelial permeation of tolbutamide across the human intestinal cell line, Caco-2. Drug Metab. Pharmacokinet. 2004, 19, 48–54. [Google Scholar]
- Takanaga, H.; Murakami, H.; Koyabu, N.; Matsuo, H.; Naito, M.; Tsuruo, T.; Sawada, Y. Efflux transport of tolbutamide across the blood-brain barrier. J. Pharm. Pharmacol. 1998, 50, 1027–1033. [Google Scholar] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Control | 1,25(OH)2D3 |
|---|---|---|
| Total protein (g/dL) | 5.60 ± 0.30 | 5.73 ± 0.25 |
| Albumin (g/dL) | 3.73 ± 0.12 | 3.97 ± 0.15 |
| sGOT (IU/L) | 156 ± 16 | 158 ± 35 |
| sGPT (IU/L) | 22.3 ± 3.5 | 22.7 ± 3.2 |
| Kinetic Parameters | BUP (Cyp2b1) | TOL (Cyp2c11) | ||
|---|---|---|---|---|
| Control | 1,25(OH)2D3 | Control | 1,25(OH)2D3 | |
| Vmax a | 35.20 ± 6.62 | 8.41 ± 1.76 * | 683.37 ± 94.05 | 191.57 ± 44.01 * |
| Km b | 171.70 ± 39.46 | 219.80 ± 29.37 | 232.83 ± 51.33 | 214.63 ± 23.83 |
| CLint c | 0.21 ± 0.06 | 0.038 ± 0.01 * | 3.00 ± 0.63 | 0.89 ± 0.18 * |
| Kinetic Parameters | ACET (Ugt1a6/1a7/1a8) | |
|---|---|---|
| Control | 1,25(OH)2D3 | |
| Vmax a | 104.04 ± 17.57 | 115.12 ± 75.10 |
| Km b | 41.06 ± 0.89 | 44.18 ± 34.79 |
| CLint c | 2.64 ± 0.89 | 2.77 ± 0.40 |
| Pharmacokinetic Parameters | Control | 1,25(OH)2D3 |
|---|---|---|
| BUP | ||
| AUC (μg·min/mL) | 54.38 ± 7.65 | 96.31 ± 23.65 * |
| t1/2 (min) | 155.4 ± 25.9 | 134.7 ± 48.70 |
| CL (mL/min/kg) | 93.64 ± 14.24 | 54.60 ± 13.37 * |
| CLR (mL/min/kg) | 0.662 ± 0.635 | 1.053 ± 0.407 |
| CLNR (mL/min/kg) | 93.08 ± 13.73 | 53.55 ± 13.02 * |
| MRT (min) | 71.44 ± 10.29 | 88.43 ± 19.63 |
| Vss (×103 mL/kg) | 7.815 ± 1.953 | 5.403 ± 1.867 |
| Ae0-24h (% of dose) | 0.719 ± 0.613 | 1.791 ± 0.031 * |
| HBUP | ||
| AUC (μg·min/mL) | 4.610 ± 1.898 | 19.77 ± 9.04 * |
| t1/2 (min) | 160.4 ± 37.6 | 197.0 ± 54.2 |
| Tmax (min) | 15 | 15 (15–60) |
| Cmax (µg/mL) | 0.046 ± 0.032 | 0.068 ± 0.021 |
| CLR (mL/min/kg) | 0.222 ± 0.124 | 0.077 ± 0.031 * |
| Ae0-24h (% of dose) | 0.015 ± 0.007 | 0.026 ± 0.008 * |
| AUCHBUP/AUCBUP | 0.090 ± 0.065 | 0.209 ± 0.084 * |
| Pharmacokinetic Parameters | Control | 1,25(OH)2D3 |
|---|---|---|
| TOL | ||
| AUC (×103 μg·min/mL) | 10.807 ± 1.877 | 10.184 ± 2.623 |
| t1/2 (min) | 462.7 ± 153.9 | 563.8 ± 207.5 |
| CL (mL/min/kg) | 0.189 ± 0.027 | 0.206 ± 0.045 |
| CLR (μL/min/kg) | 0.208 ± 0.103 | 0.039 ± 0.015 * |
| CLNR (mL/min/kg) | 0.189 ± 0.030 | 0.206 ± 0.045 |
| MRT (min) | 657.4 ± 204.5 | 795.4 ± 290.5 |
| Vss (mL/kg) | 119.9 ± 15.6 | 153.1 ± 19.5 * |
| Ae0-24h (% of dose) | 0.124 ± 0.059 | 0.024 ± 0.016 * |
| HTOL | ||
| AUC (μg·min/mL) | 100.8 ± 65.2 | 151.9 ± 52.0 |
| t1/2 (min) | 521.3 ± 499.0 | 629.7 ± 491.6 |
| Tmax (min) | 60 (15–120) | 120 (60–240) |
| Cmax (µg/mL) | 0.167 ± 0.067 | 0.207 ± 0.119 |
| CLR (mL/min/kg) | 2.713 ± 0.570 | 0.873 ± 0.134 * |
| Ae0-24h (% of dose) | 9.356 ± 0.471 | 5.539 ± 2.305 * |
| AUCHTOL/AUCTOL | 0.0097 ± 0.070 | 0.0155 ± 0.0059 |
| Organs | Control | 1,25(OH)2D3 |
|---|---|---|
| TOL | ||
| Liver | 0.133 ± 0.002 | 0.166 ± 0.019 * |
| Kidney | 0.139 ± 0.008 | 0.154 ± 0.014 |
| Brain | 0.021 ± 0.001 | 0.030 ± 0.004 * |
| Spleen | 0.169 ± 0.030 | 0.175 ± 0.045 * |
| Heart | 0.120 ± 0.003 | 0.184 ± 0.036 * |
| HTOL | ||
| Liver | 0.831 ± 0.152 | 0.657 ± 0.129 |
| Kidney | 5.335 ± 1.127 | 7.768 ± 0.014 * |
| Brain | 0.020 ± 0.008 | 0.019 ± 0.004 |
| Spleen | 0.137 ± 0.010 | 0.163 ± 0.008 * |
| Heart | 0.175 ± 0.013 | 0.252 ± 0.036 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doan, T.N.K.; Vo, D.-K.; Kim, H.; Balla, A.; Lee, Y.; Yoon, I.-S.; Maeng, H.-J. Differential Effects of 1α,25-Dihydroxyvitamin D3 on the Expressions and Functions of Hepatic CYP and UGT Enzymes and Its Pharmacokinetic Consequences In Vivo. Pharmaceutics 2020, 12, 1129. https://doi.org/10.3390/pharmaceutics12111129
Doan TNK, Vo D-K, Kim H, Balla A, Lee Y, Yoon I-S, Maeng H-J. Differential Effects of 1α,25-Dihydroxyvitamin D3 on the Expressions and Functions of Hepatic CYP and UGT Enzymes and Its Pharmacokinetic Consequences In Vivo. Pharmaceutics. 2020; 12(11):1129. https://doi.org/10.3390/pharmaceutics12111129
Chicago/Turabian StyleDoan, Trang Nguyen Kieu, Dang-Khoa Vo, Hyojung Kim, Anusha Balla, Yunjong Lee, In-Soo Yoon, and Han-Joo Maeng. 2020. "Differential Effects of 1α,25-Dihydroxyvitamin D3 on the Expressions and Functions of Hepatic CYP and UGT Enzymes and Its Pharmacokinetic Consequences In Vivo" Pharmaceutics 12, no. 11: 1129. https://doi.org/10.3390/pharmaceutics12111129